 Max Christoph Liebau
Max Christoph Liebau
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Med., 05 October 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.739534
This article is part of the Research TopicThe Role of Mitochondria and Metabolic Rewiring in Kidney DiseasesView all 7 articles
Polycystic kidney diseases (PKD) form a group of severe genetic kidney diseases that are characterized by fibrocystic changes of the kidneys and the liver. The main forms of PKD are autosomal dominant and autosomal recessive polycystic kidney disease (ADPKD, ARPKD). In general, cystic kidney diseases are the most common genetic cause of chronic kidney failure in both children and adults (1).
ARPKD is a disorder that typically is diagnosed very early in life or even prenatally. Typically, the kidneys show bilateral massive enlargement although they keep their reniform shape. Kidney disease results in a need for kidney replacement therapy (KRT) in around 50% of patients in the first two decades of life (2, 3). Liver involvement is obligatory in ARPKD, typically presenting with congenital hepatic fibrosis and portal hypertension or with dilated bile ducts. Both are considered to be a consequence of a defect during development of the bile ducts (“ductal plate malformation”) (4). Adult patients with ARPKD have been described with variable disease manifestations. Hepatic disease may be the leading clinical issue in adults with ARPKD (5).
In ADPKD renal cystogenesis starts early in life or even before birth but does not present with obvious clinical symptoms until adulthood in most patients (1, 6, 7). Liver involvement is typically characterized by liver cysts. Additional extrarenal symptoms in ADPKD include for example intracranial aneurysms, cardiac valve anomalies like mitral prolapse or abdominal wall diverticula in a subset of patients (1, 7). Extrarenal manifestations may be very helpful for clinical differential diagnosis and to distinguish ADPKD from ARPKD.
ADPKD is mainly caused by variants in the genes PKD1 and PKD2 with the large majority of patients carrying PKD1 variants (1). Variants in other genes e.g., with atypical forms of ADPKD have been identified (7). Variants in PKHD1 are the main cause of ARPKD with phenotypical overlay e.g., of kidney disease in patients with other underlying genetic changes (1, 8), including biallelic hypomorphic PKD1 variants.
Genotype-phenotype correlations in ADPKD show more rapid progression of kidney disease in patients with PKD1 variants and especially in those patients carrying variants resulting in protein truncation (7, 9). For ARPKD it was shown by various groups that biallelic truncating variants are associated with severe phenotypes. A recent study extended the findings and revealed that for missense variants the affected region in PKHD1 also seems to be important (10). In a study on 304 children with the clinical diagnosis of ARPKD and detected PKHD1 variants it was found that patients with either two missense variants affecting the amino acids 709–1837 or a null variant and a missense variant in this region less frequently showed progression to chronic kidney failure when being compared during their follow-up when compared to patients with variants affecting other regions of PKHD1. On the other hand patients with variants affecting the amino acids 2625–4074 showed less favorable hepatic outcome (10). The underlying molecular mechanisms of these associations remain to be explored.
Even though many questions remain, much has been learned about the functions of polycystin-1 (PC1) and polycystin-2 (PC2), the proteins encoded by PKD1 and PKD2, respectively. Very briefly summarized, PC1 is a very large transmembrane receptor-like protein with 11 transmembrane domains and a large extracellular part. PC2 is non-selective cation channel and belongs to the family of transient receptor potential ion channels (TRPs). Both PC proteins have been found in a joint protein complex but may also have independent cellular functions (11). Recent work on high-resolution cryo-electron microscopy has found that the transmembrane domains of PC1 and PC2 can assemble in a structure consisting of three PC2 molecules and one PC1 molecule. Still, many questions on how the proteins interact remain unsolved (12).
PC1 and PC2 have been shown to be involved in the regulation of multiple signaling networks that obviously are closely interconnected and cannot be seen as isolated events. There is e.g., dysregulation of intracellular cAMP signaling in ADPKD which itself is controlled by the vasopressin V2 receptor. Insights into a dysregulation of this cascade have led to the establishment of the first targeted treatment in ADPKD for adult patients with the use of the V2 receptor antagonist Tolvaptan (1, 7, 13). Other dysregulated signaling cascades include tyrosine receptor kinase-PKA-SRC signaling, Myc- and STAT-signaling or macrophage-activation (11).
Recently, evidence for a role of PC1 in the regulation of cellular metabolic processes has been accumulating. Details of the work have recently been reviewed and are beyond the scope of this manuscript, but it has been shown that PC1 is involved in the regulation of multiple signaling cascades controlling cellular metabolic processes (14, 15). This includes regulation of mTOR signaling, amino acid and glutamine metabolism as well as changes in fatty acid oxidation (14, 15). The ADPKD proteins PC1 and PC2 seem to be crucial regulators of cellular metabolism including effect on the cellular sensor AMPK, the transcription factor PPARalpha and the regulator of mitochondrial biosynthesis PPARγ Coaktivator 1α (PGC1α) or the tumor suppressor kinase LKB1. A landmark study that served as a starting point for intensive follow-up studies identified a shift of glycolysis and oxidative phosphorylation and a Warburg-effect like effect in PKD cells (16). An increase of glycolysis with altered fatty acid oxidation and mitochondrial function as well as an increase of reactive oxygen species have independently been identified in various cellular and preclinical models as well as in samples from patients (15–22). In addition to these metabolic alterations, changes in mitochondrial structure from PKD cells were identified and fragments of the cytoplasmic PC1 tail or the PC1–PC2 complex have been shown to localize to mitochondria and mitochondria-associated membranes (18, 23).
Importantly, these findings bear therapeutic potential. In a preclinical setting inhibition of cellular glycolysis by the non-metabolizable 2-Deoxyglucose (2-DG) or caloric restriction results in reduced cyst growth in both orthologous and non-orthologous mouse models of ADPKD (15–17, 24). Furthermore, for an activation of PPARα (Fenofibrate), PPARγ (Glitazone) and AMPK (Metformin) positive effects have been described (15). A phase 2 trial recently found that metformin was safe and tolerable in adult patients in early ADPKD stages resulting in a slight reduction of eGFR loss that however did not yet reach significance in this trial of 97 patients (25). A larger trial will be required. Follow-up work on caloric restriction studies revealed that also time-restricted feeding induced positive effects (26). A ketogenic diet but also oral administration of the ketone ß-hydroxybutyrate showed very promising effects on the kidney phenotype in various preclinical PKD models (26). Translation of these preclinical findings into clinical trials is ongoing.
Various lines of evidence point to an overlap of pathogenic factors between ARPKD and ADPKD (Table 1). As previously pointed out patients with (biallelic) PKD1 variants may mimic the ARPKD phenotype, even though ARPKD is typically caused by variants in PKHD1. Furthermore, patients with variants in multiple PKD genes and preclinical rodent models with digenic genetic modifications show more severe phenotypes (27–29). Importantly, the PCK rat, a preclinical model that phenotypically mimics ADPKD, genetically is orthologous to ARPKD. Overlapping dysregulation of signaling cascades has been described, e.g., with STAT3 activation in cyst-lining epithelial cells in both ARPKD and ADPKD (47–49). While the main gene affected in ARPKD has been known for two decades, the function of the PKHD1-encoded protein fibrocystin (FC) remains incompletely understood. FC has been found in joint protein complexes with the ADPKD proteins PC1 and PC2 in extracellular vesicles (50, 51). A physical interaction has also been proposed for FC and PC2 (52) but the data has not been independently confirmed. Overall, ARPKD and ADPKD may not be seen as completely separate diseases but rather as two different ends of a spectrum of disease.
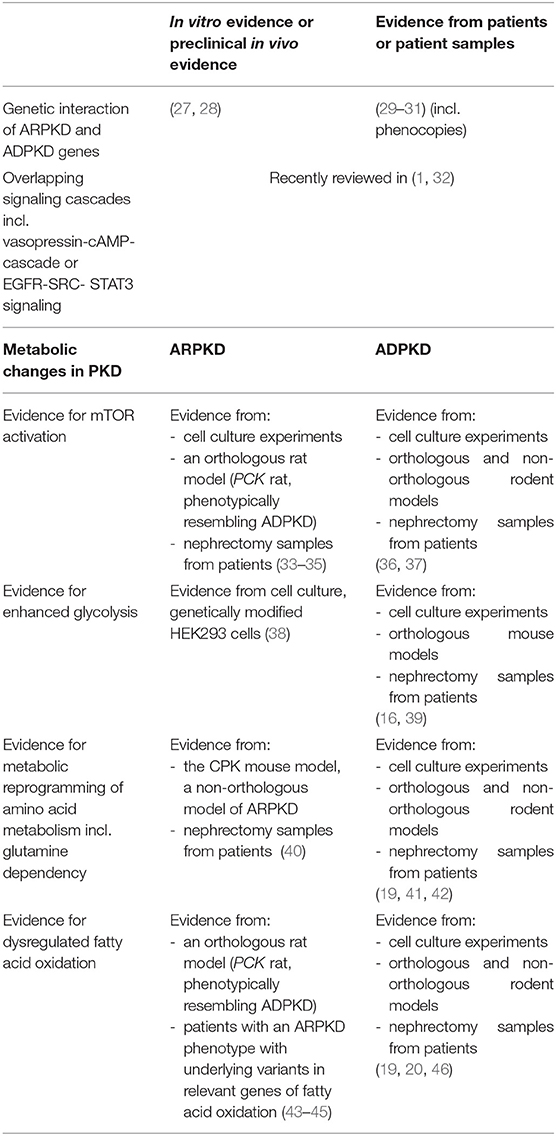
Table 1. Selected evidence for overlapping dysregulated signaling cascades and metabolic changes in ARPKD and ADPKD.
So, what is the evidence suggesting that there could also be a role for mitochondrial dysfunction in the pathogenesis of ARPKD? Data is much more limited for ARPKD than for ADPKD, but first hints of cellular metabolic dysregulation in ARPKD are emerging.
Importantly, an activation of mTOR in cyst-lining renal epithelial cells was described for ARPKD (33). Full length FC was shown to control mTOR activation in vitro with the cytoplasmic tail regulating the function of full-length protein (34). Treatment with NVP-BEZ235, an inhibitor of PI3-kinase and mTOR, had a positive effect on cystic dilatation of the intrahepatic bile ducts in the PCK rat (35).
A positive effect of the PPARγ-agonist pioglitazone on the renal and hepatic phenotype was shown in the PCK rat (43). Gene expression analyses after treatment with pioglitazone pointed to an important modification of fatty acid oxidation by this treatment in the PCK rat (44). Defects in lipid metabolism and fatty acid metabolism were described as modifiers in the same rat model that were associated with a more severe renal phenotype (53). Interestingly, defects of fatty acid oxidation in humans can result in a renal phenotype that resembles ARPKD (4, 45) and need to be considered as clinical differential diagnoses (4). In this context another interesting differential diagnosis of ARPKD are variants in the promoter region of PMM2 encoding Phosphomannomutase 2 that can result in an ARPKD-like kidney phenotype combined with hyperinsulinemic hypoglycemia (54).
A recent publication looked at the effects of truncating PKHD1 variants in HEK293 cells on metabolic aspects. Variants were induced by Crispr/Cas9 and were associated with an acidification of the cell medium, an activation of the Krebs cycle and mild structural changes in mitochondria (38). Even though HEK293 cells may not be fully recapitulating the specifics of human renal tubular cells this finding is of interest.
A challenge in ARPKD research in this context is the fact that orthologous mouse models do not fully recapitulate the human renal phenotypes although various models have shown hepatic effects that are closer to human disease. Non-orthologous models have given important insights (55). The recent description of digenic Pkd1-Pkhd1 mouse that mimic the kidney phenotype of ARPKD was therefore of major interest (28). The experience from previous studies highlight the importance of studying mouse models for ADPKD that show the slowly progressive kidney phenotype as some of the Pkd1 knockout models show rapid progression (56). Crossing the slowly progressive ADPKD Pkd1RC/RC model (representing the hypomorphic human missense variant p.Arg3277Cys) variant with a Pkhd1−/− model (only developing mild kidney disease) resulted in ARPKD-like kidneys in a mouse model. Thus, these models may be helpful to study ARPKD kidney disease in preclinical models and may turn out to be a major step ahead despite being genetically partially different from human disease (28). As an ideal orthologous model is not available such data needs to be complemented e.g., by patient samples. It was interesting to note that a proteomics approach comparing human ARPKD kidneys and control kidneys identified upregulation of many proteins localizing to mitochondria (57) and that an increase of the oxygen consumption rate was noted in HEK293 cells with truncating PKHD1 variants whereas a reduction of mitochondrial mass has been reported in cyst-lining cells from a murine Pkd1 model. Thus, it seems possible that there are specific PKHD1 effects on mitochondria that may partly be different from PKD1 or PKD2 effects. More overlapping data from preclinical models and patient samples as well as functional work on the PKHD1-encoded protein fibrocystin will be required. It is important to note that the examined kidneys in the proteomics study were obtained from young patients obviously suffering from severe disease requiring nephrectomy. Such patients may show rapid and sometimes enormous growth of the kidneys. Obviously, patient samples such as nephrectomy specimens will in many cases give insights into a terminal situation of the kidneys with pronounced chronic kidney disease of the patients and differentiating cause and consequence may be a challenge. A link to clinical courses will thus also be needed. Current national and international data and sample collections (e.g., Rare Renal in the UK, the hepatorenal fibrocystic core center in the US, ARegPKD in Europe) may become important resources for such approaches.
Different from ADPKD, rapid kidney growth in ARPKD typically occurs before birth with some patients showing rapid renal growth very early in life. Indeed, many ARPKD patients are nowadays identified antenatally. A recent clinical publication identified prenatal risk markers for early dialysis dependency in ARPKD (58). These markers included the antenatal detection of enlarged kidneys and have served as the basis for a first phase 3 trials in ARPKD that are currently being initiated (NCT04782258, NCT04786574). In utero, kidneys are exposed to a hypoxic environment as oxygen pressures within the fetal blood are generally lower with the kidneys being located in a lethal corner at the very end of oxygen delivery. It will be interesting to study the changes of FC function under hypoxic conditions even more as e.g., the oxygen-regulated transcription factor HIF1a is known to enhance cystogenesis in an ADPKD model (59). Strikingly, HIF1 activates glycolysis and mitochondrial oxygen consumption (60). Again, there is a link to ADPKD: local hypoxia is a presumed factor contributing to kidney disease progression in ADPKD.
Over the past years, metabolic changes in PKD have become the focus of a highly active field of research. Most of the work has been done on ADPKD but there is first evidence that metabolic changes may also be important in ARPKD disease progression. Deciphering primary changes from secondary changes due to chronic kidney disease may become particularly challenging in ARPKD due to the early development of severe kidney disease in ARPKD. Linking functional cellular work and novel preclinical models to patient genetics, patient disease courses and biosamples seems particularly relevant to obtain a balanced view of causes and consequences in ARPKD disease progression and cellular metabolic changes. Lessons learned from ADPKD may guide the steps to take to achieve progress in our understanding of this severe disease with the potential to open paths for first targeted therapeutic approaches.
The author confirms being the sole contributor of this work and has approved it for publication.
ML is a member of the European Reference Network on Rare Kidney Diseases (ERKNet)—Project ID No. 739532 who was supported by grants of the German Society for Pediatric Nephrology, the European Society for Paediatric Nephrology (ESPN2014.2), and the Marga and Walter Boll-Foundation, and furthermore supported by the German Federal Ministry of Research and Education (BMBF grant 01GM1903B, NEOCYST consortium) and the German Research Council (DFG Li 23975/1).
ML serves on an advisory board of Otsuka Pharmaceuticals as a representative of the University Hospital of Cologne.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primer. (2018) 4:50. doi: 10.1038/s41572-018-0047-y
2. Liebau MC. Early clinical management of autosomal recessive polycystic kidney disease. Pediatr Nephrol Berl Ger. (2021). doi: 10.1007/s00467-021-04970-8. [Epub ahead of print].
3. Guay-Woodford LM. Autosomal recessive polycystic kidney disease: the prototype of the hepato-renal fibrocystic diseases. J Pediatr Genet. (2014) 3:89–101. doi: 10.3233/PGE-14092
4. Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, Cadnapaphornchai MA, Dell KM, et al. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J Pediatr. (2014) 165:611–7. doi: 10.1016/j.jpeds.2014.06.015
5. Burgmaier K, Kilian S, Bammens B, Benzing T, Billing H, Büscher A, et al. Clinical courses and complications of young adults with Autosomal Recessive Polycystic Kidney Disease (ARPKD). Sci Rep. (2019) 9:7919. doi: 10.1038/s41598-019-43488-w
6. De Rechter S, Bammens B, Schaefer F, Liebau MC, Mekahli D. Unmet needs and challenges for follow-up and treatment of autosomal dominant polycystic kidney disease: the paediatric perspective. Clin Kidney J. (2018) 11:i14–26. doi: 10.1093/ckj/sfy088
7. Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet Lond Engl. (2019) 393:919–35. doi: 10.1016/S0140-6736(18)32782-X
8. Bergmann C. ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol Berl Ger. (2015) 30:15–30. doi: 10.1007/s00467-013-2706-2
9. Cornec-Le Gall E, Audrézet M-P, Rousseau A, Hourmant M, Renaudineau E, Charasse C, et al. The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. (2016) 27:942–51. doi: 10.1681/ASN.2015010016
10. Burgmaier K, Brinker L, Erger F, Beck BB, Benz MR, Bergmann C, et al. Refining genotype-phenotype correlations in 304 patients with autosomal recessive polycystic kidney disease and PKHD1 gene variants. Kidney Int. (2021) 100:650–9. doi: 10.1016/j.kint.2021.04.019
11. Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. (2014) 124:2315–24. doi: 10.1172/JCI72272
12. Su Q, Hu F, Ge X, Lei J, Yu S, Wang T, et al. Structure of the human PKD1-PKD2 complex. Science. (2018) 361:eaat9819. doi: 10.1126/science.aat9819
13. Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. (2012) 367:2407–18. doi: 10.1056/NEJMoa1205511
14. Haumann S, Müller R-U, Liebau MC. Metabolic changes in polycystic kidney disease as a potential target for systemic treatment. Int J Mol Sci. (2020) 21:6093. doi: 10.3390/ijms21176093
15. Padovano V, Podrini C, Boletta A, Caplan MJ. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat Rev Nephrol. (2018) 14:678–87. doi: 10.1038/s41581-018-0051-1
16. Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. (2013) 19:488–93. doi: 10.1038/nm.3092
17. Chiaravalli M, Rowe I, Mannella V, Quilici G, Canu T, Bianchi V, et al. 2-Deoxy-d-glucose ameliorates PKD progression. J Am Soc Nephrol. (2016) 27:1958–69. doi: 10.1681/ASN.2015030231
18. Padovano V, Kuo IY, Stavola LK, Aerni HR, Flaherty BJ, Chapin HC, et al. The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol Biol Cell. (2017) 28:261–9. doi: 10.1091/mbc.e16-08-0597
19. Podrini C, Rowe I, Pagliarini R, Costa ASH, Chiaravalli M, Di Meo I, et al. Dissection of metabolic reprogramming in polycystic kidney disease reveals coordinated rewiring of bioenergetic pathways. Commun Biol. (2018) 1:194. doi: 10.1038/s42003-018-0200-x
20. Menezes LF, Lin C-C, Zhou F, Germino GG. Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine. (2016) 5:183–92. doi: 10.1016/j.ebiom.2016.01.027
21. Hajarnis S, Lakhia R, Yheskel M, Williams D, Sorourian M, Liu X, et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun. (2017) 8:14395. doi: 10.1038/ncomms14395
22. Ishimoto Y, Inagi R, Yoshihara D, Kugita M, Nagao S, Shimizu A, et al. Mitochondrial abnormality facilitates cyst formation in autosomal dominant polycystic kidney disease. Mol Cell Biol. (2017) 37:e00337–17. doi: 10.1128/MCB.00337-17
23. Lin C-C, Kurashige M, Liu Y, Terabayashi T, Ishimoto Y, Wang T, et al. A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci Rep. (2018) 8:2743. doi: 10.1038/s41598-018-20856-6
24. Kipp KR, Rezaei M, Lin L, Dewey EC, Weimbs T A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am J Physiol Renal Physiol. (2016) 310:F726–31. doi: 10.1152/ajprenal.00551.2015
25. Perrone RD, Abebe KZ, Watnick T, Althouse A, Hallows KR, Lalama CM, et al. Primary results of the randomized trial of metformin administration in polycystic kidney disease (TAME PKD). Kidney Int. (2021) 100:684–96. doi: 10.1016/j.kint.2021.06.013
26. Torres JA, Kruger SL, Broderick C, Amarlkhagva T, Agrawal S, Dodam JR, et al. Ketosis ameliorates renal cyst growth in polycystic kidney disease. Cell Metab. (2019) 30:1007-23.e5. doi: 10.1016/j.cmet.2019.09.012
27. Garcia-Gonzalez MA, Menezes LF, Piontek KB, Kaimori J, Huso DL, Watnick T, et al. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum Mol Genet. (2007) 16:1940–50. doi: 10.1093/hmg/ddm141
28. Olson RJ, Hopp K, Wells H, Smith JM, Furtado J, Constans MM, et al. Synergistic genetic interactions between Pkhd1 and Pkd1 result in an ARPKD-like phenotype in murine models. J Am Soc Nephrol. (2019) 30:2113–27. doi: 10.1681/ASN.2019020150
29. Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol. (2011) 22:2047–56. doi: 10.1681/ASN.2010101080
30. Durkie M, Chong J, Valluru MK, Harris PC, Ong ACM. Biallelic inheritance of hypomorphic PKD1 variants is highly prevalent in very early onset polycystic kidney disease. Genet Med. (2021) 23:689–97. doi: 10.1038/s41436-020-01026-4
31. Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol. (2010) 21:1097–102. doi: 10.1681/ASN.2009101070
32. Cordido A, Vizoso-Gonzalez M, Garcia-Gonzalez MA. Molecular pathophysiology of autosomal recessive polycystic kidney disease. Int J Mol Sci. (2021) 22:6523. doi: 10.3390/ijms22126523
33. Fischer D-C, Jacoby U, Pape L, Ward CJ, Kuwertz-Broeking E, Renken C, et al. Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol Dial Transplant. (2009) 24:1819–27. doi: 10.1093/ndt/gfn744
34. Wang S, Wu M, Yao G, Zhang J, Zhou J. The cytoplasmic tail of FPC antagonizes the full-length protein in the regulation of mTOR pathway. PLoS ONE. (2014) 9:e95630. doi: 10.1371/journal.pone.0095630
35. Ren XS, Sato Y, Harada K, Sasaki M, Furubo S, Song JY, et al. Activation of the PI3K/mTOR pathway is involved in cystic proliferation of cholangiocytes of the PCK rat. PLoS ONE. (2014) 9:e87660. doi: 10.1371/journal.pone.0087660
36. Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wüthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant. (2006) 21:598–604. doi: 10.1093/ndt/gfi181
37. Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. (2006) 103:5466–71. doi: 10.1073/pnas.0509694103
38. Chumley P, Zhou J, Mrug S, Chacko B, Parant JM, Challa AK, et al. Truncating PKHD1 and PKD2 mutations alter energy metabolism. Am J Physiol Renal Physiol. (2019) 316:F414–25. doi: 10.1152/ajprenal.00167.2018
39. Riwanto M, Kapoor S, Rodriguez D, Edenhofer I, Segerer S, Wüthrich RP. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS ONE. (2016) 11:e0146654. doi: 10.1371/journal.pone.0146654
40. Hwang VJ, Kim J, Rand A, Yang C, Sturdivant S, Hammock B, et al. The cpk model of recessive PKD shows glutamine dependence associated with the production of the oncometabolite 2-hydroxyglutarate. Am J Physiol Renal Physiol. (2015) 309:F492–498. doi: 10.1152/ajprenal.00238.2015
41. Flowers EM, Sudderth J, Zacharias L, Mernaugh G, Zent R, DeBerardinis RJ, et al. Lkb1 deficiency confers glutamine dependency in polycystic kidney disease. Nat Commun. (2018) 9:814. doi: 10.1038/s41467-018-03036-y
42. Trott JF, Hwang VJ, Ishimaru T, Chmiel KJ, Zhou JX, Shim K, et al. Arginine reprogramming in ADPKD results in arginine-dependent cystogenesis. Am J Physiol Renal Physiol. (2018) 315:F1855–68. doi: 10.1152/ajprenal.00025.2018
43. Yoshihara D, Kurahashi H, Morita M, Kugita M, Hiki Y, Aukema HM, et al. PPAR-gamma agonist ameliorates kidney and liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. Am J Physiol Renal Physiol. (2011) 300:F465–474. doi: 10.1152/ajprenal.00460.2010
44. Yoshihara D, Kugita M, Yamaguchi T, Aukema HM, Kurahashi H, Morita M, et al. Global gene expression profiling in PPAR-γ agonist-treated kidneys in an orthologous rat model of human autosomal recessive polycystic kidney disease. PPAR Res. (2012) 2012:695898. doi: 10.1155/2012/695898
45. Hackl A, Mehler K, Gottschalk I, Vierzig A, Eydam M, Hauke J, et al. Disorders of fatty acid oxidation and autosomal recessive polycystic kidney disease-different clinical entities and comparable perinatal renal abnormalities. Pediatr Nephrol Berl Ger. (2017) 32:791–800. doi: 10.1007/s00467-016-3556-5
46. Lakhia R, Yheskel M, Flaten A, Quittner-Strom EB, Holland WL, Patel V. PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am J Physiol Renal Physiol. (2018) 314:F122–31. doi: 10.1152/ajprenal.00352.2017
47. Strubl S, Torres JA, Spindt AK, Pellegrini H, Liebau MC, Weimbs T, et al. Signaling in polycystic kidney disease. Cell Signal. (2020) 72:109639. doi: 10.1016/j.cellsig.2020.109639
48. Dafinger C, Mandel AM, Braun A, Göbel H, Burgmaier K, Massella L, et al. The carboxy-terminus of the human ARPKD protein fibrocystin can control STAT3 signalling by regulating SRC-activation. J Cell Mol Med. (2020) 24:14633–8. doi: 10.1111/jcmm.16014
49. Talbot JJ, Song X, Wang X, Rinschen MM, Doerr N, Lariviere WB, et al. The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J Am Soc Nephrol. (2014) 25:1737–48. doi: 10.1681/ASN.2013091026
50. Hogan MC, Manganelli L, Woollard JR, Masyuk AI, Masyuk TV, Tammachote R, et al. Characterization of PKD protein-positive exosome-like vesicles. J Am Soc Nephrol. (2009) 20:278–88. doi: 10.1681/ASN.2008060564
51. Lea WA, McGreal K, Sharma M, Parnell SC, Zelenchuk L, Charlesworth MC, et al. Analysis of the Polycystin Complex (PCC) in human urinary exosome-like vesicles (ELVs). Sci Rep. (2020) 10:1500. doi: 10.1038/s41598-020-58087-3
52. Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, et al. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol. (2007) 27:3241–52. doi: 10.1128/MCB.00072-07
53. O'Meara CC, Hoffman M, Sweeney WE, Tsaih S-W, Xiao B, Jacob HJ, et al. Role of genetic modifiers in an orthologous rat model of ARPKD. Physiol Genomics. (2012) 44:741–53. doi: 10.1152/physiolgenomics.00187.2011
54. Cabezas OR, Flanagan SE, Stanescu H, García-Martínez E, Caswell R, Lango-Allen H, et al. Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. J Am Soc Nephrol. (2017) 28:2529–39. doi: 10.1681/ASN.2016121312
55. Guay-Woodford LM. Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol. (2003) 285:F1034–1049. doi: 10.1152/ajprenal.00195.2003
56. Watnick T, Germino GG. mTOR inhibitors in polycystic kidney disease. N Engl J Med. (2010) 363:879–81. doi: 10.1056/NEJMe1006925
57. Li Q-W, Lu X-Y, You Y, Sun H, Liu X-Y, Ai J-Z, et al. Comparative proteomic analysis suggests that mitochondria are involved in autosomal recessive polycystic kidney disease. Proteomics. (2012) 12:2556–70. doi: 10.1002/pmic.201100590
58. Burgmaier K, Kunzmann K, Ariceta G, Bergmann C, Buescher AK, Burgmaier M, et al. Risk factors for early dialysis dependency in autosomal recessive polycystic kidney disease. J Pediatr. (2018) 199:22-8.e6. doi: 10.1016/j.jpeds.2018.03.052
59. Kraus A, Peters DJM, Klanke B, Weidemann A, Willam C, Schley G, et al. HIF-1α promotes cyst progression in a mouse model of autosomal dominant polycystic kidney disease. Kidney Int. (2018) 94:887–99. doi: 10.1016/j.kint.2018.06.008
Keywords: PKD, fibrocystin, PKHD1, PKD1, PKD2, polycystin
Citation: Liebau MC (2021) Is There a Functional Role of Mitochondrial Dysfunction in the Pathogenesis of ARPKD? Front. Med. 8:739534. doi: 10.3389/fmed.2021.739534
Received: 11 July 2021; Accepted: 03 September 2021;
Published: 05 October 2021.
Edited by:
Alessandra Boletta, San Raffaele Hospital (IRCCS), ItalyReviewed by:
John Andrew Sayer, Newcastle University, United KingdomCopyright © 2021 Liebau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Max Christoph Liebau, bWF4LmxpZWJhdUB1ay1rb2Vsbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.