 Lisa M. Shook
Lisa M. Shook Deidra Haygood
Deidra Haygood Charles T. Quinn
Charles T. Quinn- 1Division of Hematology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
- 2Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 3Erythrocyte Diagnostic Laboratory, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
Sickle cell disease (SCD) is a group of related yet genetically complex hemoglobinopathies. Universal newborn screening (NBS) for SCD is performed in the United States and many other nations. Classical, protein-based laboratory methods are often adequate for the diagnosis of SCD but have specific limitations in the context of NBS. A particular challenge is the differentiation of sickle cell anemia (SCA) from the benign condition, compound heterozygosity for HbS and gene-deletion hereditary persistence of fetal hemoglobin (HbS/HPFH). We describe a sequential cohort of 44 newborns identified over 4.5 years who had molecular genetic testing incorporated into NBS for presumed SCA (an “FS” pattern). The final diagnosis was something other than SCA in six newborns (12%). Three (7%) had HbS/HPFH. All had a final, correct diagnosis at the time of their first scheduled clinic visit in our center (median 8 weeks of age). None received initial counseling for an incorrect diagnosis. In summary, genetic testing as a component of NBS for SCD is necessary to provide correct genetic counseling and education for all newborns' families at their first visit to a sickle cell center. Genetic testing also permits the use of early, pre-symptomatic hydroxyurea therapy by preventing infants with HbS/HPFH from receiving unnecessary therapy. We argue that genetic testing should be incorporated into contemporary NBS for SCD.
Introduction
Sickle cell disease (SCD) is the name for a group of related hemoglobinopathies that affects numerous populations worldwide. Although it is a monogenic disease caused by mutations of the β-globin gene (HBB), there are a number of distinct genotypes of SCD that require differentiation to inform clinical care. The most common and severe form of SCD is the homozygous state for the sickle hemoglobin (HbS) mutation, called sickle cell anemia (SCA) or HbSS. There are also compound heterozygous forms of SCD, such as sickle-hemoglobin C disease (HbSC), sickle-β+-thalassemia (HbSβ+), and sickle-β0-thalassemia (HbSβ0). In contrast, compound heterozygosity for the HbS mutation and gene-deletion hereditary persistence of fetal hemoglobin (HbS/HPFH) is essentially benign.
Accurate NBS for SCD is needed to permit the prompt initiation of penicillin prophylaxis and referral to a specialized SCD center for ongoing comprehensive care, which has been shown to reduce morbidity and mortality (1–4). Most NBS programs use protein-based, Hb separation techniques that include electrophoresis (gel- or liquid-based), isoelectric focusing (IEF), and high-performance liquid chromatography (5). Although protein-based methods are the mainstay of Hb diagnostics (5), they may be inadequate in the context of NBS to detect β-thalassemia mutations (6) and unable to differentiate between HbSS and HbS/HPFH (7).
Without parental studies, the clinical differentiation of HbSS from HbS/HPFH can take several years to be correctly realized, given the prolonged postnatal decline in HbF concentration in children SCD (8). In the past, a watch-and-wait approach was reasonable given the limited and usually delayed initiation of treatment options. The hazards of this approach include unnecessary venipunctures, clinic visits, and prophylactic antibiotics for the children with HbS/HPFH and anxiety and fear about a serious disease, incorrectly diagnosed, for their parents and family. Now, the increasing use of early, pre-symptomatic disease-modifying therapy (e.g., initiation of hydroxyurea by 6 months of age for SCA) has made this critical diagnostic distinction time-sensitive (9). Treating children who have HbS/HPFH with hydroxyurea is not indicated. Here we describe a sequential cohort of newborns who had a presumed diagnosis of SCA by NBS (an “FS” pattern), several of whom actually had HbS/HPFH or other forms of SCD that were correctly and rapidly diagnosed by genetic testing before the child's first visit to our center, thereby preventing incorrect counseling and unnecessary medical interventions.
Patients and Methods
Newborn screening in the state of Ohio begins with an initial dried blood spot that is analyzed at a central laboratory using a combination of HPLC and IEF. Newborns with Hb variants detected by this initial phase of testing have a second, confirmatory panel of testing performed using new blood specimens obtained by venipuncture in the first 2–4 weeks of life at a Regional Sickle Cell Services Program (RSCP). The Ohio Department of Health Region 1 RSCP is located at the Cincinnati Comprehensive Sickle Cell Center at Cincinnati Children's Hospital Medical Center (CCHMC). Region 1 comprises eight counties in southwestern Ohio in which approximately 500 newborns undergo confirmatory testing for suspected hemoglobinopathies each year. For newborns with suspected SCD, protein-based confirmatory testing includes capillary zone electrophoresis, acid hemoglobin electrophoresis, and IEF. Additionally, beginning in July 2015, newborns with an “FS” pattern indicating suspected SCA (here defined to comprise HbSS and HbSβ0), also had HBB sequence analysis and copy number variation analysis of the α-globin and β-globin gene clusters performed simultaneously using the same blood specimen. The overall goal of this NBS-based genetic testing is to differentiate HbSS from HbS/HPFH before affected newborns have their first visits in our comprehensive sickle cell center. No a priori sample sized was calculated, rather all consecutive patients were included in this arbitrary 4.5-year sample (Figure 1). Medians were compared by the 2-tailed Mann Whitney test. P < 0.05 was considered statistically significant.
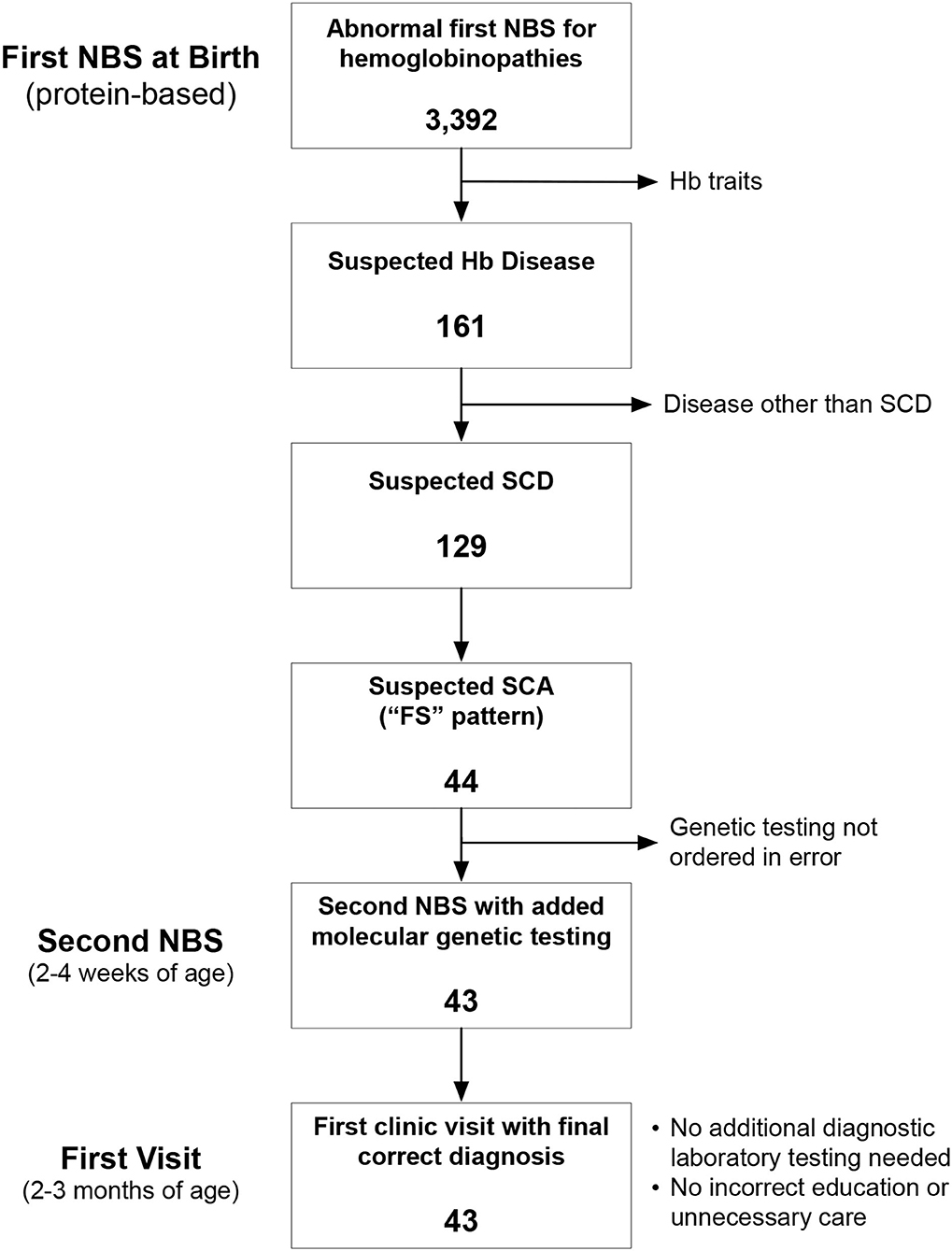
Figure 1. Identification of the study cohort. Between July 2015 and December 2020, there were 3,392 abnormal newborn screening (NBS) tests for hemoglobinopathies. We excluded newborns with hemoglobin (Hb) trait states and hemoglobinopathies other than sickle cell disease (SCD). We identified 44 newborns during this time with an “FS” pattern, indicating suspected sickle cell anemia (SCA), which we define here to include both homozygous sickle cell anemia (HbSS) and sickle-β0-thalassemia (HbSβ0).
Results
We analyzed 3,392 consecutive newborns with abnormal NBS results between July 2015 and December 2020 and identified 44 who had a presumed diagnosis of SCA (Figure 1). Genetic testing was not performed for 1 newborn at the time of the second, confirmatory NBS test in error (only protein-based testing was done); genetic testing later confirmed that she had HbSS. Of the remaining 43 newborns who had genetic testing as part of NBS (Table 1), 37 (86%) had a final diagnosis of HbSS. The other six newborns (14%) had different SCD genotypes. Three (7%) had HbS/HPFH, two (5%) had sickle-β+-thalassemia (HbSβ+), and one (2%) had sickle-β0-thalassemia (Hb Sβ0).

Table 1. Results of molecular genetic testing as a component of newborn screening (NBS) for suspected sickle cell anemia (SCA).
Of the newborns 3 who had HbS/HPFH, 2 had the HPFH-2 (Ghanaian) deletion and 1 had the HPFH-1 (Black) deletion. These three newborns had a final genetic diagnosis of HbS/HPFH by the time of their first clinic visits in our center (ages at first visits: 6, 8, and 11 weeks). None was started on hydroxyurea. If prophylactic penicillin was initiated before the first clinic visit it was discontinued, and each is now followed only yearly in our center. The two with HbSβ+ also had a final genetic diagnosis of by the time of their first clinic visit.
Of the patients with SCA, 37/38 (97.3%) were prescribed hydroxyurea starting at a median of 8.0 months of age (interquartile range: 7.1–10.0). The single patient with SCA not yet prescribed hydroxyurea was last seen in our clinic at 4 months of age, and he has a scheduled visit at 7 months of age to start this medication. Comparing the first and second halves of this cohort, the median age at prescription of hydroxyurea decreased from 9.6 to 7.8 months (P = 0.023).
Discussion
In this consecutive cohort of newborns with suspected SCA, the final diagnosis was something other than HbSS or HbSβ0 in about 12%. Three (7%) had HbS/HPFH and 2 (5%) had HbSβ+. The distinction between HbSS and HbSβ0 by NBS is not critical, because both diseases are clinically similar and managed identically, and these two genotypes are often both called sickle cell anemia (10, 11). Nevertheless, this genetic distinction can be clinically helpful to explain microcytosis, provide accurate genetic counseling, and determine eligibility for genetic therapies.
A diagnosis of HbSβ+ will become clear within a few months of age by the detection of a small amount of HbA by Hb analysis, but a diagnosis of HbS/HPFH might take several years to be correctly realized. In children with SCA, the postnatal decline in HbF concentration may take 5–6 years to be completed (8). In the past, a “watch-and-wait” approach to distinguish HbSS from HbS/HPFH with serial measurements of HbF and blood counts was reasonable given the formerly limited and delayed initiation of disease-modifying therapy. Now, the increasing use of early, pre-symptomatic disease-modifying therapy (e.g., hydroxyurea initiation at 6 months of age) has made this critical diagnostic distinction time-sensitive (9, 12). For infants with SCA, hydroxyurea should be started to prevent and reverse this early decline in HbF rather than waiting for the HbF level to decline to some arbitrary level (13). However, treating children who have HbS/HPFH with hydroxyurea is not indicated and exposes them to unnecessary risks, expenses, laboratory monitoring, and inconvenience. Genetic testing incorporated into NBS can facilitate this new standard (6).
We also determine α-globin gene copy number (data not shown), because it provides supplemental prognostic information in SCA, and copy number variation analysis of the β-globin gene cluster is already being performed. We do not test for common non-gene-deletion HPFH determinants at the time of NBS, because these polymorphisms are not likely to inform early clinical management, but practice may change with the decreasing costs of broader genetic testing over time (e.g., next generation sequencing panels). Currently, the cost of genetic testing is offset by the elimination of both parental testing and additional diagnostic laboratory testing in the infant.
Molecular genetic testing is not required to determine if an infant has HbS/HPFH if parental studies can be performed. However, both parents may not be available or willing to be tested. The time needed to perform this testing may also take longer than needed to permit the earliest initiation of hydroxyurea. In this 4.5-year study period, the median age of initiation of hydroxyurea decreased from 9.6 to 7.8 months of age, highlighting the need for a timely correct diagnosis. Genetic testing added to NBS overcomes these obstacles and can be performed on a blood sample that had already been obtained for protein-based testing. A recent plea has been published for the diagnosis of HbS/HPFH in newborns (7), which we have answered here. Larger studies are needed to demonstrate the generalizability and cost-effectiveness of our approach.
In summary, molecular genetic testing incorporated into NBS for SCD allows correct education and counseling for the newborn's family to be given at the first visit in a sickle cell center and eliminates additional diagnostic testing (no additional venipuncture). It prevents unnecessary grief and anxiety from incorrectly diagnosing a newborn with SCA, only later to “revise” the diagnosis to a benign condition. Moreover, a timely correct diagnosis can prevent infants with HbS/HPFH from receiving unnecessary medical interventions and treatments.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author Contributions
LS and CQ: conceptualization, visualization, and funding acquisition. CQ: formal analysis. DH: investigation and data curation. LS: writing—original draft preparation. DH and CQ: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by Ohio Department of Health, grant number 0313011SK1320 (to LS) and by NIH-NHLBI, grant number U01HL117709 (to CQ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee A, Thomas P, Cupidore L, Serjeant B, Serjeant G. Improved survival in homozygous sickle cell disease: lessons from a cohort study. BMJ. (1995) 311:1600–2. doi: 10.1136/bmj.311.7020.1600
2. Gill FM, Sleeper LA, Weiner SJ, Brown AK, Bellevue R, Grover R, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood. (1995) 86:776–83. doi: 10.1182/blood.V86.2.776.bloodjournal862776
3. Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. (2010) 115:3447–52. doi: 10.1182/blood-2009-07-233700
4. Quinn CT, Rogers ZR, Buchanan GR. Survival of children with sickle cell disease. Blood. (2004) 103:4023–7. doi: 10.1182/blood-2003-11-3758
5. Therrell BL, Lloyd-Puryear MA, Eckman JR, Mann MY. Newborn screening for sickle cell diseases in the United States: a review of data spanning 2 decades. Semin Perinatol. (2015) 39:238–51. doi: 10.1053/j.semperi.2015.03.008
6. Shook LM, Haygood D, Quinn CT. Clinical utility of confirmatory genetic testing to differentiate sickle cell trait from sickle-β+-thalassemia by newborn screening. Int J Neonatal Screen. (2020) 6:7. doi: 10.3390/ijns6010007
7. Serjeant GR, Serjeant BE, Hambleton IR, Oakley M, Thein SL, Clark B. A plea for the newborn diagnosis of Hb S-hereditary persistence of fetal hemoglobin. Hemoglobin. (2017) 41:216–7. doi: 10.1080/03630269.2017.1360336
8. Mason KP, Grandison Y, Hayes RJ, Serjeant BE, Serjeant GR, Vaidya S, et al. Post-natal decline of fetal haemoglobin in homozygous sickle cell disease: relationship to parenteral Hb F levels. Br J Haematol. (1982) 52:455–63. doi: 10.1111/j.1365-2141.1982.tb03915.x
9. Ware RE, McGann PT, Quinn CT. Hydroxyurea for children with sickle cell anemia: prescribe it early and often. Pediatr Blood Cancer. (2019) 66:e27778. doi: 10.1002/pbc.27778
10. Evidence-Based Management of Sickle Cell Disease: Expert Panel Report 2014. National Heart, Lung, and Blood Institute (2014). Available online at: https://www.nhlbi.nih.gov/health-topics/evidence-based-management-sickle-cell-disease (accessed October 27, 2021).
11. Quinn CT. Sickle cell disease in childhood: from newborn screening through transition to adult medical care. Pediatr Clin N Am. (2013) 60:1363–81. doi: 10.1016/j.pcl.2013.09.006
12. McGann PT, Niss O, Dong M, Marahatta A, Howard TA, Mizuno T, et al. Robust clinical and laboratory response to hydroxyurea using pharmacokinetically guided dosing for young children with sickle cell anemia. Am J Hematol. (2019) 94:871–9. doi: 10.1002/ajh.25510
13. Quinn CT, Niss O, Dong M, Pfeiffer A, Korpik J, Reynaud M, et al. Early initiation of hydroxyurea (hydroxycarbamide) using individualised, pharmacokinetics-guided dosing can produce sustained and nearly pancellular expression of fetal haemoglobin in children with sickle cell anaemia. Brit J Haematol. (2021) 194:617–25. doi: 10.1111/bjh.17663
Keywords: hemoglobinopathies, newborn screening, sickle cell anemia, electrophoresis, genetic testing, HPFH, hydroxyurea
Citation: Shook LM, Haygood D and Quinn CT (2021) Clinical Utility of the Addition of Molecular Genetic Testing to Newborn Screening for Sickle Cell Anemia. Front. Med. 8:734305. doi: 10.3389/fmed.2021.734305
Received: 30 June 2021; Accepted: 18 October 2021;
Published: 10 November 2021.
Edited by:
Dipty Jain, Government Medical College, Nagpur, IndiaReviewed by:
Mervat Mattar, Cairo University, EgyptGraham Serjeant, Sickle Cell Support Foundation of Jamaica (SCSFJ), Jamaica
Copyright © 2021 Shook, Haygood and Quinn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa M. Shook, TGlzYS5TaG9va0BjY2htYy5vcmc=