 Irene Mattioli
Irene Mattioli Alessandra Bettiol
Alessandra Bettiol Güher Saruhan-Direskeneli2
Güher Saruhan-Direskeneli2 Haner Direskeneli
Haner Direskeneli Giacomo Emmi
Giacomo Emmi- 1Department of Experimental and Clinical Medicine, University of Firenze, Firenze, Italy
- 2Department of Physiology, Istanbul Medical Faculty, Istanbul University, Istanbul, Turkey
- 3Department of Internal Medicine, Division of Rheumatology, School of Medicine, Marmara University, Istanbul, Turkey
Behçet's syndrome (BS) is a rare systemic vasculitis, characterized by a wide range of different clinical involvements and unpredictable phases of recurrence and remission. BS can be described as a multifactorial disease with an incompletely known etiopathogenesis; in fact, though presenting some peculiar features, such as its typical geographic distribution and the strong association with the well-known genetic predisposing factor HLA-B*51, the cause behind the onset and progression of the disease remains currently not fully understood. Besides genetic HLA and non-HLA predisposing associations and epigenetic influence, environmental factors also play an important role in the pathogenesis of the disease, and among these, infectious agents (both bacterial and viral) and specific microbiome alterations are considered of particular relevance in BS pathogenesis. BS has been included for decades among autoimmune diseases, in light of evidence showing T- and B-cell aberrant responses. However, because of recurrent mucocutaneous lesions and episodes of inflammation without antigen-specific T-cell or autoantibody responses, BS has also been classified among autoinflammatory disorders. Nevertheless, differently from autoinflammatory diseases, BS mildly responds to therapies targeting IL-1, its onset is not usually in childhood, and has high neutrophilic vasculitic involvement. Finally, given the association with HLA class I alleles, similar to spondyloarthropathies, the concept of BS as a major histocompatibility complex (MHC) I -opathy has been introduced. Understanding the complex etiopathogenesis of BS is essential to identify modifiable risk factors of BS occurrence or exacerbation and to develop targeted therapies. This review summarizes current evidence on the main genetic, environmental and immunological factors contributing to BS development.
Introduction
Behçet syndrome (BS) is a rare systemic vasculitis, characterized by a wide range of different clinical involvements, which have unpredictable phases of recurrence and remission (1). Moreover, the different clinical manifestations may present alone, or co-exist in the same patient (2, 3).
BS can be described as a multifactorial disease with an incompletely known etiopathogenesis, and unique geographic distribution suggests that both genetic and environmental susceptibility factors might be involved. Indeed, the global pool prevalence of BS has been estimated around 10.3 (95% CI: 6.1–7.7)/100.000 people, but it is considerably higher in the countries along the ancient Silk Route (e.g., Turkey and Iran) (4).
BS has been included for decades among autoimmune diseases, in light of evidence showing T- and B-cell responses to heat-shock proteins (HSP), endothelial cells, enolase and retinal S antigen (5). However, there are some features that do not support the autoimmune nature of the disease, such as lack of anti-nuclear antibodies, female prevalence or increased risk of autoimmunity (6).
Because of its symptomatology including recurrent non-scarring mucocutaneous lesions and non-deforming arthritis, and episodes of inflammation without antigen-specific T-cell or autoantibody response, elevated neutrophil activation, and increased levels of certain proinflammatory cytokines, such as interleukin (IL)-1, BS has also been classified among autoinflammatory disorders (7, 8).
However, unlike other autoinflammatory diseases, BS mildly responds to therapies that specifically target IL-1, the first symptoms usually begin after puberty rather than in childhood, and there is generally high vasculitic involvement, which is quite rare in autoinflammatory diseases. According to the Chapel Hill classification revised in 2012, it is now counted among systemic vasculitis with variable caliber vessel involvement (9).
In recent years, clinical studies have shed light on a possible similarity with spondyloarthropathies (SpA), introducing the concept of BS as a human major histocompatibility complex (MHC)-I-opathy (10). This similarity is supported by the association of specific human leukocyte antigen (HLA) class I groups, namely HLA-B*51 and HLA-B*27, and the development of BS and SpA, respectively (11).
Although to date BS cannot be unambiguously classified as an autoimmune or an autoinflammatory disorder, a systemic vasculitis or a disease belonging to the MHC-I-opathies, but possesses common features with all these groups (Figure 1), it is increasingly important to categorize the disease because it helps understanding the underlying pathogenetic mechanisms, thus choosing the most appropriate therapeutic strategy.
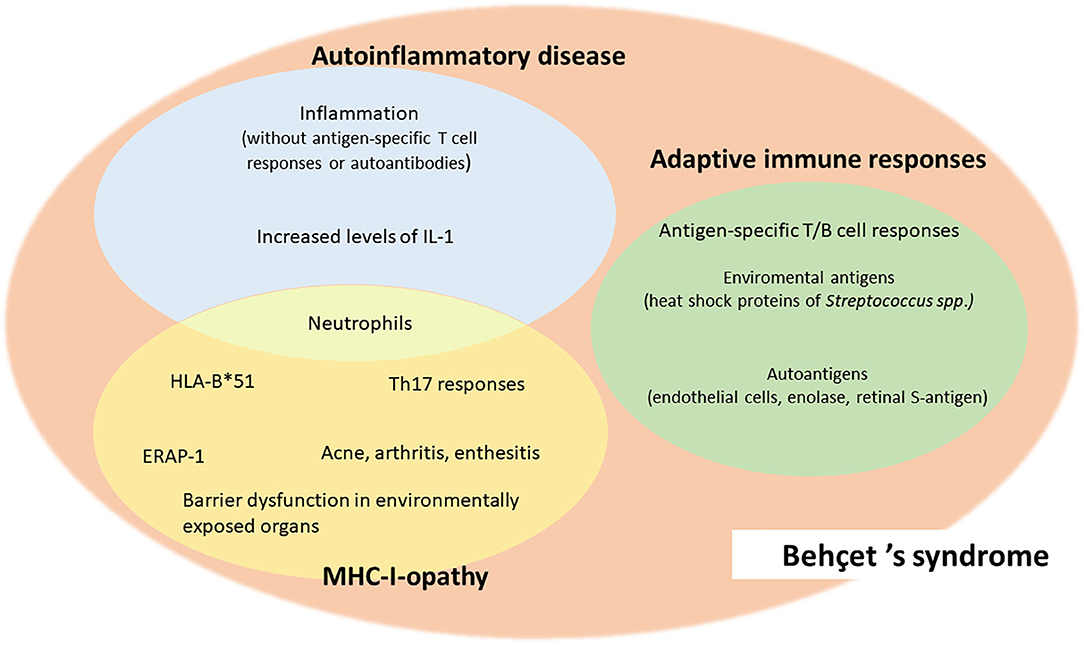
Figure 1. Common features of Behçet's syndrome and autoimmune diseases, autoinflammatory diseases and MHC-I-opathies.
It is now known that the complex etiopathogenesis of BS involves more than one pathogenetic pathway, namely (12):
(1) Genetic and epigenetic factors, including geographic distribution, the association with HLA and non-HLA genes, and micro-RNA (miRNA) polymorphisms.
(2) Environmental etiology, including infections, microbiome, and additional triggering factors.
(3) Immunological pathways, including neutrophils and other immune-mediated mechanisms of damage.
Genetic and Epigenetic Etiology
Genetic Factors
Different environmental factors can lead to epigenetic changes that may account for an increased incidence of the disease in some geographic areas. Similarly, the geographic distribution of BS has always implicated a genetic predisposition to the disease. The HLA class I antigen, HLA-B*51, has been identified as the predominant genetic susceptibility factor underlying BS in many populations, especially along the ancient Silk Road that ran from East Asia to the Middle East and Mediterranean basin (2, 13).
However, the frequency of HLA-B*51 in some ethnic groups (e.g., Italians and Portuguese) is similar to that in Silk Road areas, despite a much lower prevalence of BS (14), proposing that unknown environmental factors may interact with HLA-B*51 alleles, driving BS development.
A meta-analysis of data from 78 independent studies has shown that subjects presenting the HLA-B*51/B*5 had a 5-fold increased risk of BS compared with non-carriers (15). Although HLA-B*51 is the known genetic factor most closely associated with BS, it accounts for <20% of the genetic risk (16). Some other genes in HLA region such as Cw*1602 and A*26 are also found to be associated with BS (17).
In addition to the MHC class I region, several non-HLA genes are associated with BS. Common variants in IL10, IL23R-IL12RB2 loci and signal traducer and activator of transcription (STAT)-4 predispose individuals to BS (18). Expression studies have shown that disease-associated IL10 variants are correlated with reduced expression of this anti-inflammatory cytokine, which can lead to a susceptible inflammatory state, increasing BS susceptibility (16).
Other susceptibility genes recently identified by GWAS are ERAP1 (encoding for endoplasmic reticulum aminopeptidase 1, implicated in antigen presentation) and CCR1-CCR3, encoding for the chemokine receptor family, which play an important role in inflammatory cell recruitment and activation (19).
Another study has recently shown that non-synonymous variants (NSVs) of a gene involved in innate immune response (i.e., Toll-like receptor 4, TLR4) are associated with BS; also, the familial Mediterranean fever (MEFV) gene Met694Val mutation seems to confer BS risk in the Turkish population. Thus, the disease-associated NSVs in TLR4 and MEFV genes support the hypothesis of the correlation between innate immune response and bacterial sensing mechanisms in the pathogenesis of BS (19).
The NF-κB pathway is also increasingly implicated in the pathogenesis of BS (20). In this regard, it has been shown that the inflammatory features of BS might be associated with NF-κB hyperactivation in immune cells. Specifically, mutations in the TNFAIP3 gene encoding for the regulatory protein A20, a potent inhibitor of the NF-κB signaling pathway, have been shown to increase the risk of BS development. Indeed, A20 is a 790 amino acid protein encoded by the TNFAIP3 gene on chromosome 6 (20), which negatively regulate inflammation and immunity. Loss-of-function mutation in the TNFAIP3 gene leads to haploinsufficiency of A20 a monogenic auto-inflammatory/auto-immune disease, with Behçet-like features (21–23).
Epigenetic Factors
Epigenetics, namely the study of inheritable variations in gene expression, which do not involve alterations in the DNA sequence, is becoming increasingly available in BS (24). Differential methylation of genes associated with cytoskeleton remodeling and cell adhesion in CD4+ T cells and CD14+ monocytes has been identified in BS patients (25). Significantly, these alterations return to normal after the use of colchicine, an alkaloid with a well-known anti-inflammatory and anti-neutrophilic activity, widely used for different BS manifestations. Colchicine inhibits mitosis, also by interfering with genes implicated in microtubule structure (KIFA2 and TPPP), suggesting that methylation patterns could be useful as biomarkers or therapeutic targets which need further validation (26).
In addition, some evidence suggests a role of specific miRNA polymorphisms, another major form of epigenetic influence that may be involved in the pathogenesis of BS. In particular, altered expression of miRNA-155, miRNA-638, and miRNA-4488 has been described in BS (27, 28). In addition, the homozygous CC genotype and the C allele of the pre-miRNA-146a rs2910164 polymorphism were found to be protective against BS, whereas the rs3746444 and rs28362491 polymorphisms in miRNA-499 and the NF?B1 promoter were found to be involved in the genetic susceptibility of BS (29). Ubiquitination is another epigenetic factor that might influence the susceptibility to BS (30).
To sum up, epigenetic influences deserve attention in BS, as they may be instrumental in explaining the geographic and environmental differences that are prominent in this condition.
Environmental Etiology
Infectious Factors and Microbiome
Infectious agents have long been proposed as triggering factors in BS development (31). Antigens from viruses such as herpes simplex virus (HSV)-1 or bacteria belonging to Streptococcus species such as Streptococcus sanguinis have been suspected to have high homologies with human proteins such as human HSP, leading to a cross-reactive immune response in genetically predisposed individuals (32).
Considering that BS usually starts from the oral mucosa and that oral ulcers are almost pathognomonic in BS, especially after dental procedures, it has been hypothesized that oral microbial flora may be implicated in the pathogenesis of the disease (33). Indeed, higher levels of various strains of streptococci were found in the oral mucosa of BS patients (29).
Moreover, HSP-specific T cells are known to mediate tissue damage either through the production of Th1 cytokines, such as IFN-γ and TNF-α, or through activation of cell-mediated cytotoxicity (34, 35). Some studies have also demonstrated the association between chronic Chlamydia pneumoniae infection and the development of BS (36).
In addition to the aforementioned role of bacterial infections in the development and course of BS, viral infections, in particular HSV, have also been identified as possible triggers. Studies in Behçet-like mouse models and in BS patients demonstrated elevated levels of serum IgA and IgG anti-HSV antibodies against the UL48 protein of HSV (37). Moreover, the presence of HSV-1 DNA has been demonstrated in peripheral blood polymorphonuclear cells and in oral and genital ulcers (38).
Beyond infectious agents, the endogenous microbiota has also been suggested to be potentially involved in BS pathogenesis. Namely, a poorer and dysbiotic salivary and fecal microbial community has been described in BS compared to healthy controls (39, 40). Indeed, oral flora had less diversity, with Haemophilus parainfluenzae as the most overabundant species, while Alloprevotella rava and genus Leptotrichia species are depleted (40). Similarly, fecal samples from BS patients have a lower bacterial diversity in terms of Roseburia, Subdoligranulum, Megamonas, Prevotella, and butyrate-producing bacteria Clostridum spp. and methanogens (41), whereas a higher abundance of Bifidobacterium, Eggerthella, Bilophilia spp. and opportunistic pathogens has been described (42). Reduced microbial diversity has been associated with lower butyrate production, one of the most representative short-chain fatty acids (SCFA), able to mediate T regulatory cell (Tregs)and activation of immunopathological T-effector responses (39, 43).
Supporting the key role of butyrate in BS pathogenesis, it has recently been shown that two different butyrate-enriched diets are able to improve disease activity, while also modulating the blood redox and prothrombotic status, which is impaired in BS (44).
Additional Triggering Factors
Beside infectious and microbiome, a wide spectrum of additional triggering factors may contribute to the etiopathogenesis of BS (31). Increasing evidence suggests a role for environmental trigger factors in the development of BS in patients with genetic susceptibility.
An association between oral ulcer recurrences and an external event, such as stress, fatigue, and food intake are reported in 78.3% of patients with BS (45). The results of these and other studies on psychological factors related to BS suggest that these factors may be considered as main triggers of oral ulcer recurrence in these patients (46, 47).
Next to psychological factors, some foods have been identified as triggers for the development of BS-related oral ulcers. Specifically, nuts, cheese, citrus fruits, pineapple, and strawberries have been reported as triggers for the development of oral ulcers (48), whereas eggplant, walnut and tomato are implicated in another series (45). Regarding patient-related factors, hormone levels have been linked to disease activity. In particular, menstruation has been described as a trigger of skin and mucosal lesions in women with BS (49), whereas testosterone has been associated with neutrophil activation in men with BS (50).
In contrast, although smoking habit reduces oral mucocutaneous inflammation in BS, it is responsible for increased atherosclerosis and increased risk of vascular events, particularly in men with BS carrying glutathione S-transferase gene polymorphisms (51).
Finally, the presence of atopic disorders, such as respiratory, skin, or food allergies, have also been associated with a lower risk of BS (31). This is probably due to the balance between pro-inflammatory Th1- and Th17-associated responses typical of BS and Th2-associated IL-4 and IL-13 responses typical of allergies (52).
Immunological Etiology
Neutrophil Mediated Mechanism of Damage
Neutrophils are known to play a key role in the etiopathogenesis of BS, which has traditionally been considered a neutrophilic vasculitis (53). Hyperactivated neutrophils in perivascular infiltration and tissue injury have been described in BS patients (54). These granulocytes have shown increased phagocytosis and superoxide production, potentially contributing to clot formation by fibrinogen oxidation (55). Indeed, it has been recently shown that neutrophil activation promotes fibrinogen oxidation and thrombosis formation in BS. Specifically, these findings suggest that altered fibrinogen structure and fibrinogen function are related to neutrophil activation and production of enhanced ROS, which are mainly derived from neutrophil NADPH oxidases (55). In addition to the role of ROS, other mechanisms implicated in typical thrombo-inflammation are also documented, including the release of neutrophil extracellular traps (NETs) (56). Under inflammatory or infectious conditions, neutrophils are able to generate NETs through a distinct cell death process called NETosis (57). NETs consist of extruded cell-free DNA (CfDNA) decorated with histones and granular components that include antimicrobial peptides and proteases. Molecular pathways leading to NETosis include calcium mobilization, generation of ROS, nuclear delobulation involving the enzymatic activities of myeloperoxidase (MPO) and neutrophil elastase, and chromatin modification through citrullination of histones by peptidyl arginine deiminase (PAD)-4 (58, 59).
Notably, among the major producers of NETs is a specific subtype of neutrophils with proinflammatory activity, namely low-density granulocytes (LDGs) that are widely involved in the pathogenesis of cardiovascular manifestations associated with various autoimmune and autoinflammatory disorders (60, 61). Moreover, NETs are now recognized as a key factor in the initiation and progression of thrombosis in pathological conditions such as deep vein thrombosis in mice and humans, but also in arterial diseases such as stroke and myocardial infarction (62, 63).
BS patients have been shown to be more prone to NETosis than healthy controls (64, 65), and the role of NETs in BS has been mainly implicated in the pathogenesis of vascular manifestations (58, 66, 67).
However, considering that in BS a dense neutrophilic infiltration can be found not only at the vascular level, but also at the cutaneous, articular, ocular, intestinal, and neurological locations, it is possible to assume that neutrophils and neutrophil-derived products might also be involved in the pathogenesis of non-vascular BS involvement (68).
Other Immunological Mechanisms
In addition to neutrophils, natural killer (NK) cells are also known to play a role in BS. NK cells are the main components of innate immunity and not only play a cytotoxic role in infected cells, but also regulate the function of other immune cells, including dendritic cells (DCs) and T lymphocytes (69). Among their abilities, NK cells mediate the release of cytokines implicated in the pacing of CD4+T cells toward differentiation into Th1 cells (70), the presence of which is associated with BS activity. Therefore, overproduction of inflammatory cytokines by innate immune cells such as NK or other immune-cells (e.g., macrophages and DC) may cause a higher production of adaptive Th1- and Th17-related cytokines. Additionally, a decreased regulation by Tregs and a Th1/Th17 cytokine polarization of CD4+ T cells is a consistent finding in disease lesions and peripheral blood from BS patients, with IFN-γ, TNF-α, IL-8 and IL-17 levels correlating with BS activity (71).
Specifically, at the level of antigen-processing cells (APCs), a series of molecules such as ERAP1 assists to the exposure of microbial antigens on the surface of the same cells via MHC class I and class II molecules. The linking of activated CD4+ T lymphocytes to APCs presenting these antigens on MHC molecules is responsible for up-regulation of Th17 and Th1 lymphocytes and down-regulation of Treg lymphocytes (72).
Cytotoxic Th1 and Th17 cells are directly involved in mucosal damage in the early stages of intestinal BS involvement (73). In addition, increased levels of IL-17, IL-23 and IFN-γ have been described in patients with active ocular, mucocutaneous, and joint involvement (74). In turn, IL-17 and IFN-γ secreted by lymphocytes from BS patients have been shown to induce innate responses, late adaptive immunity, and neutrophil infiltration (75). These immune features are shared between different spondyloarthropathies and support the concept of MHC-I-opathies (10).
Upregulation of different Th subtypes may also be linked to upregulated Janus kinase (JAK)/STAT pathway in both CD4+ T cells and CD14+ monocytes in BS (76). Recently IFN-γ receptor-1 (IFNGR1) gene has been shown as a susceptibility locus for BS in a large multi-ethnic GWAS study (77). IFNGR1 encodes the binding subunit, α chain, of the IFN-γ receptor and the binding of IFN-γ stimulates the activation of JAK-STAT signaling pathway. BS-associated variant leads to higher levels of expression of the receptor after stimulation in vitro, suggesting a functional effect in BS pathogenesis.
Furthermore, CD8 + T cells also play a significant role in BS pathogenesis. Indeed, some studies showed a significant association between CD8+ T cells and disease activity in BS patients, thus suggesting their potential critical role in BS recurrence (78, 79).
Finally, along with CD4+ and CD8+ T cells, circulating γδ T cells in BS patients have also been correlated with disease status. γδT lymphocytes also play an important role in the regulation of the autoimmune response. γδT cells are a minor population of T cells present in ~0.5–5% of total blood, which express the γ and δ chains of the T cell receptor (TCR), providing an essential contribution to many types of immune response. Among their pleiotropic effector functions, they are able to recognize qualitatively distinct antigens, protect multiple defined anatomical sites, and are capable of mediating and modulating responses to specific pathogens (80).
γδT cells have been identified in BS inflammatory lesions and have been suggested to be contributory to the induction and/or maintenance of the proinflammatory environment characteristic of the disease (81).
Discussion
Understanding the pathogenesis of BS is a critical step in the development of novel and effective therapies. A genetic predisposition (HLA and non-HLA-driven), with an abnormal innate hyperinflammatory response and neutrophil hyperactivity are well-known hallmarks of the disorder.
On this susceptible background, environmental and microbial risk factors can trigger inflammation of innate origin, which can subsequently be sustained by adaptive immune responses.
However, to date, the etiopathogenesis of the disease remains poorly understood. Recent findings of monogenic diseases sharing features with BS strongly suggest an important role for dysregulated innate immune activation due to mutations in autoinflammatory cascades such as NF- kB pathway in familial and polygenic BS cases. Activation of JAK/STAT pathway associated with IFNGR1 polymorphism and pro-inflammatory cytokine milieu (IL-6, IL-17) leading to Th1/Th17 activation is also implicated.
Moreover, T cells also play a crucial role, especially CD8+ T cells, as they seem to be correlated with disease status and therefore representing a potential therapeutic target for BS.
In addition, there is evidence suggesting a crucial role also of γδT cells as responsible for the induction and/or maintenance of the proinflammatory environment characteristic of the disease (Figure 2).
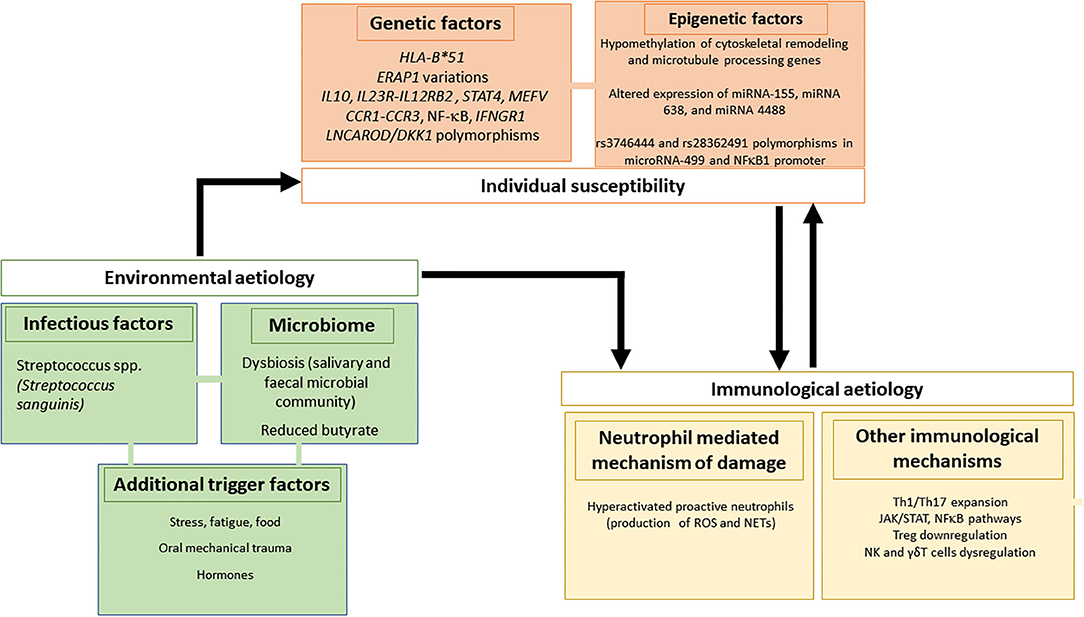
Figure 2. Genetic/epigenetic, infectious and immunological factors involved in the pathogenesis of Behçet's syndrome.
In the meanwhile, a growing interest is focusing on the introduction of new validated biomarkers for disease diagnosis. In this context, in the absence of validated laboratory parameters for BS diagnosis, an emerging role is being attributed to specific miRNA signature associated with BS. Moreover, the identification of miRNA profiles connected with specific BS features could also provide valuable insights to reveal key pathogenetic mechanism, and also representing future targets for tailored therapeutic interventions.
Considering that in BS common inflammatory biomarkers poorly reflect disease course, NETs are also emerging as promising biomarkers for monitoring disease activity and could represent a significant step forward in the clinical approach, as well as in the knowledge of BS pathogenesis.
Finally, a specific gut microbiota (GM) characterized by changes in SCFA profiles (among which especially butyrate) is now known. Intriguingly, a growing interest in nutritional interventions aimed at restoring this GM imbalance to ameliorate both pathogenic and clinical features in BS is recently arising (43, 44) (NCT03962335).
Further studies are needed to understand the role of these processes to better clarify BS etiopathogenesis.
Author Contributions
IM, AB, and GE conceived and drafted the manuscript. GS-D and HD critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bettiol A, Prisco D, Emmi G. Behçet: the syndrome. Rheumatol. (2020) 59:iii101–7. doi: 10.1093/rheumatology/kez626
2. Emmi G, Silvestri E, Squatrito D, D'Elios MM, Ciucciarelli L, Prisco D, et al. Behçet's syndrome pathophysiology and potential therapeutic targets. Intern Emerg Med. (2014) 9:257–65. doi: 10.1007/s11739-013-1036-5
3. Bettiol A, Hatemi G, Vannozzi L, Barilaro A, Prisco D, Emmi G. Treating the different phenotypes of behçet's syndrome. Front Immunol. (2019) 10:2830. doi: 10.3389/fimmu.2019.02830
4. Davatchi F, Chams-Davatchi C, Shams H, Shahram F, Nadji A, Akhlaghi M, et al. Behcet's disease: epidemiology, clinical manifestations, and diagnosis. Expert Rev Clin Immunol. (2017) 13:57–65. doi: 10.1080/1744666X.2016.1205486
5. Direskeneli H. Autoimmunity vs autoinflammation in Behcet's disease: do we oversimplify a complex disorder? Rheumatology. (2006) 45:1461–5. doi: 10.1093/rheumatology/kel329
6. Yazici H. The place of Behcet's syndrome among the autoimmune diseases. Int Rev Immunol. (1997) 14:1–10. doi: 10.3109/08830189709116840
7. Stojanov S, Kastner DL. Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol. (2005) 17:586–99. doi: 10.1097/bor.0000174210.78449.6b
8. Franks WA, Limb GA, Stanford MR, Ogilvie J, Wolstencroft RA, Chignell AH, et al. Cytokines in human intraocular inflammation. Curr Eye Res. (1992) 11:187–91. doi: 10.3109/02713689208999531
9. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. Revised International Chapel Hill consensus conference nomenclature of vasculitides. Arthr Rheum. (2013) 65:1–11. doi: 10.1002/art.37715
10. McGonagle D, Aydin SZ, Gül A, Mahr A, Direskeneli H. 'MHC-I-opathy'-unified concept for spondyloarthritis and Behçet disease. Nat Rev Rheumatol. (2015) 11:731–40. doi: 10.1038/nrrheum.2015.147
11. Direskeneli H. Behçet's disease: infectious aetiology, new autoantigens, and HLA-B51. Ann Rheum Dis. (2001) 60:996–1002. doi: 10.1136/ard.60.11.996
12. Leccese P, Alpsoy E. Behçet's disease: an overview of etiopathogenesis. Front Immunol. (2019) 10:1067. doi: 10.3389/fimmu.2019.01067
13. Gul A, Ohno S. HLA-B*51 and behçet disease. Ocul Immunol Inflamm. (2012) 20:37–43. doi: 10.3109/09273948.2011.634978
14. Maldini C, Druce K, Basu N, LaValley MP, Mahr A. Exploring the variability in Behçet's disease prevalence: a meta-analytical approach. Rheumatology. (2018) 57:185–95. doi: 10.1093/rheumatology/kew486
15. De Menthon M, LaValley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the risk of Behçet's disease: a systematic review and meta-analysis of case-control genetic association studies. Arthr Care Res. (2009) 61:1287–96. doi: 10.1002/art.24642
16. Remmers EF, Cosan F, Kirino Y, Ombrello MJ, Abaci N, Satorius C, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet's disease. Nat Genet. (2010) 42:698–702. doi: 10.1038/ng.625
17. Hughes T, Coit P, Adler A, Yilmaz V, Aksu K, Düzgün N, et al. Identification of multiple independent susceptibility loci in the HLA region in Behçet's disease. Nat Genet. (2013) 45:319–24. doi: 10.1038/ng.2551
18. Hou S, Yang Z, Du L, Jiang Z, Shu Q, Chen Y, et al. Identification of a susceptibility locus in STAT4 for Behçet's disease in Han chinese in a genome-wide association study. Arthritis Rheum. (2012) 64:4104–13. doi: 10.1002/art.37708
19. Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Genome-wide association analysis identifies new susceptibility loci for Behçet's disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. (2013) 45:202–7. doi: 10.1038/ng.2520
20. Aksentijevich I, Zhou Q. NF-κB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol. (2017) 8:399. doi: 10.3389/fimmu.2017.00399
21. Berteau F, Rouviere B, Delluc A, Nau A, Le Berre R, Sarrabay G, et al. Autosomic dominant familial Behçet disease and haploinsufficiency A20: a review of the literature. Autoimmun Rev. (2018) 17:809–15. doi: 10.1016/j.autrev.2018.02.012
22. Tsuchida N, Kirino Y, Soejima Y, Onodera M, Arai K, Tamura E, et al. Haploinsufficiency of A20 caused by a novel nonsense variant or entire deletion of TNFAIP3 is clinically distinct from Behçet's disease. Arthritis Res Ther. (2019) 21:137. doi: 10.1186/s13075-019-1928-5
23. Zhou Q, Wang H, Schwartz DM, Stoffels M, Hwan Park Y, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. (2015) 48:67–73. doi: 10.1038/ng.3459
24. Alipour S, Nouri M, Sakhinia E, Samadi N, Roshanravan N, Ghavami A, et al. Epigenetic alterations in chronic disease focusing on Behçet's disease: Review. Biomed Pharmacother. (2017) 91:526–33. doi: 10.1016/j.biopha.2017.04.106
25. Hughes T, Ture-Ozdemir F, Alibaz-Oner F, Coit P, Direskeneli H, Sawalha AH. Epigenome-wide scan identifies a treatment-responsive pattern of altered dna methylation among cytoskeletal remodeling genes in monocytes and cd4+ t cells from patients with behçet's disease. Arthritis Rheumatol. (2014) 66:1648–58. doi: 10.1002/art.38409
26. Coit P, Direskeneli H, Sawalha AH. An update on the role of epigenetics in systemic vasculitis. Curr Opin Rheumatol. (2018) 30:4–15. doi: 10.1097/BOR.0000000000000451
27. Yu H, Liu Y, Bai L, Kijlstra A, Yang P. Predisposition to Behçet's disease and VKH syndrome by genetic variants of miR-182. J Mol Med. (2014) 92:961–7. doi: 10.1007/s00109-014-1159-9
28. Woo MY, Yun SJ, Cho O, Kim K, Lee ES, Park S. MicroRNAs differentially expressed in Behçet disease are involved in interleukin-6 production. J Inflamm. (2016) 13:22. doi: 10.1186/s12950-016-0130-7
29. Oner T, Yenmis G, Tombulturk K, Cam C, Kucuk OS, Yakicier MC, et al. Association of Pre-miRNA-499 rs3746444 and Pre-miRNA-146a rs2910164 polymorphisms and susceptibility to Behcet's disease. Genet Test Mol Biomarkers. (2015) 19:424–30. doi: 10.1089/gtmb.2015.0016
30. Sawalha AH, Hughes T, Nadig A, Yilmaz V, Aksu K, Keser G, et al. A putative functional variant within the UBAC2 gene is associated with increased risk of Behçet's disease. Arthritis Rheum. (2011) 63:3607–12. doi: 10.1002/art.30604
31. Mumcu G, Direskeneli H. Triggering agents and microbiome as environmental factors on Behçet's syndrome. Intern Emerg Med. (2019) 14:653–60. doi: 10.1007/s11739-018-2000-1
32. Direskeneli H, Saruhan-Direskeneli G. The role of heat shock proteins in Behçet's disease. Clin Exp Rheumatol. (2003) 21:S44–8.
33. Mumcu G, Ergun T, Inanc N, Fresko I, Atalay T, Hayran O, et al. Oral health is impaired in Behçet's disease and is associated with disease severity. Rheumatology. (2004) 43:1028–33. doi: 10.1093/rheumatology/keh236
34. Alpsoy E. Behçet's disease: a comprehensive review with a focus on epidemiology, etiology and clinical features, and management of mucocutaneous lesions. J Dermatol. (2016) 43:620–32. doi: 10.1111/1346-8138.13381
35. Lehner T. The role of heat shock protein, microbial and autoimmune agents in the aetiology of Behcet's disease. Int Rev Immunol. (1997) 14:21–32. doi: 10.3109/08830189709116842
36. Ayaşlioglu E, Düzgün N, Erkek E, Inal A. Evidence of chronic chlamydia pneumoniae infection in patients with Behçet's disease. Scand J Infect Dis. (2004) 36:428–30. doi: 10.1080/00365540410020730
37. Kim DY, Cho S, Choi MJ, Sohn S, Lee E-S, Bang D. Immunopathogenic role of herpes simplex virus in Behçet's disease. Genet Res Int. (2013) 2013:1–6. doi: 10.1155/2013/638273
38. Zheng Z, Sohn S, Ahn K, Bang D, Cho S. Serum reactivity against herpes simplex virus type 1 UL48 protein in Behçet's disease patients and a Behçet's disease-like mouse model. Acta Derm Venereol. (2015) 95:952–8. doi: 10.2340/00015555-2127
39. Consolandi C, Turroni S, Emmi G, Severgnini M, Fiori J, Peano C, et al. Behçet's syndrome patients exhibit specific microbiome signature. Autoimmun Rev. (2015) 14:269–76. doi: 10.1016/j.autrev.2014.11.009
40. Coit P, Mumcu G, Ture-Ozdemir F, Unal AU, Alpar U, Bostanci N, et al. Sequencing of 16S rRNA reveals a distinct salivary microbiome signature in Behçet's disease. Clin Immunol. (2016) 169:28–35. doi: 10.1016/j.clim.2016.06.002
41. van der Houwen TB, van Laar JAM, Kappen JH, van Hagen PM, de Zoete MR, van Muijlwijk GH, et al. Behçet's disease under microbiotic surveillance? A combined analysis of two cohorts of Behçet's disease patients. Front Immunol. (2020) 11:1192. doi: 10.3389/fimmu.2020.01192
42. Shimizu J, Kubota T, Takada E, Takai K, Fujiwara N, Arimitsu N, et al. Bifidobacteria abundance-featured gut microbiota compositional change in patients with behcet's disease. PLoS ONE. (2016) 11:e0153746. doi: 10.1371/journal.pone.0153746
43. Pagliai G, Dinu M, Fiorillo C, Becatti M, Turroni S, Emmi G, et al. Modulation of gut microbiota through nutritional interventions in Behçet's syndrome patients (the MAMBA study): study protocol for a randomized controlled trial. Trials. (2020) 21:511. doi: 10.1186/s13063-020-04444-6
44. Emmi G, Bettiol A, Niccolai E, Ramazzotti M, Amedei A, Pagliai G, et al. Butyrate-rich diets improve redox status and fibrin lysis in Behçet's syndrome. Circ Res. (2021) 128:278–80. doi: 10.1161/CIRCRESAHA.120.317789
45. Iris M, Ozcikmak E, Aksoy A, Alibaz-Oner F, Inanc N, Ergun T, et al. The assessment of contributing factors to oral ulcer presence in Behçet's disease: dietary and non-dietary factors. Eur J Rheumatol. (2018) 5:240–3. doi: 10.5152/eurjrheum.2018.18094
46. Shenavandeh S, Asis M, Eftekhari MH, Aflaki E, Abdollahifard GR, Abnavi MA, et al. The patients' beliefs regarding the role of food, mucosal trauma, menstruation, and psychological stress in the recurrence of Behçet's disease symptoms. J Med Life. (2020) 13:164–169. doi: 10.25122/jml-2019-0153
47. Ilhan B, Can M, Alibaz-Oner F, Yilmaz-Oner S, Polat-Korkmaz O, Ozen G, et al. Fatigue in patients with Behçet's syndrome: relationship with quality of life, depression, anxiety, disability and disease activity. Int J Rheum Dis. (2018) 21:2139–45. doi: 10.1111/1756-185X.12839
48. Volle G, Fraison JB, Gobert D, Goulenok T, Dhote R, Fain O, et al. Dietary and nondietary triggers of oral ulcer recurrences in behçet's disease. Arthritis Care Res. (2017) 69:1429–36. doi: 10.1002/acr.23155
49. Gul Guzelant 1, Yesim Ozguler 1, Sinem Nihal Esatoglu 1, Guzin Karatemiz 1, Huri Ozdogan 1, Sebahattin Yurdakul 1, Hasan Yazici 1, ES 2; Exacerbation of Behçet's Syndrome and Familial Mediterranean Fever With Menstruation - PubMed. Clin Exp Rheumatol. Available online at: https://pubmed.ncbi.nlm.nih.gov/29148415/ (accessed April 7, 2021).
50. Yavuz S, Ozilhan G, Elbir Y, Tolunay A, Eksioglu-Demiralp E DH. Activation of neutrophils by testosterone in Behçet's disease - PubMed. Clin Exp Rheumatol. (2007) 25:S46–51.
51. Özer HT, Günesaçar R, Dinkçi S, Özbalkan Z, Yildiz F EE. The impact of smoking on clinical features of Behçet's disease patients with glutathione S-transferase polymorphisms - PubMed. Clin Exp Rheumatol. (2012) 30:s14–7.
52. Tong B, Liu X, Xiao J, Su G. Immunopathogenesis of Behcet's disease. Front Immunol. (2019) 10:665. doi: 10.3389/fimmu.2019.00665
53. Emmi G, Becatti M, Bettiol A, Hatemi G, Prisco D, Fiorillo C. Behçet's syndrome as a model of thrombo-inflammation: The role of neutrophils. Front Immunol. (2019) 10:1085. doi: 10.3389/fimmu.2019.01085
54. Becatti M, Emmi G, Silvestri E, Bruschi G, Ciucciarelli L, Squatrito D, et al. Neutrophil activation promotes fibrinogen oxidation and thrombus formation in behçet disease. Circulation. (2016) 133:302–11. doi: 10.1161/CIRCULATIONAHA.115.017738
55. Batu ED. Neutrophil-mediated thrombosis and NETosis in behcet's disease: a hypothesis. J Korean Med Sci. (2020) 35:e213. doi: 10.3346/jkms.2020.35.e213
56. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. (2009) 15:623–5. doi: 10.1038/nm.1959
57. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular Traps Kill Bacteria. Science (80-). (2004) 303:1532–5. doi: 10.1126/science.1092385
58. Michailidou D, Mustelin T, Lood C. Role of neutrophils in systemic vasculitides. Front Immunol. (2020) 11:619705. doi: 10.3389/fimmu.2020.619705
59. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. (2012) 189:2689–95. doi: 10.4049/jimmunol.1201719
60. Liu Y, Xia C, Chen J, Fan C, He J. Elevated circulating pro-inflammatory low-density granulocytes in adult-onset Still's disease. Rheumatol. (2021) 60:297–303. doi: 10.1093/rheumatology/keaa324
61. Liu Y, Kaplan MJ. Neutrophils in the pathogenesis of rheumatic diseases: fueling the fire. Clin Rev Allergy Immunol. (2021) 60:1–16. doi: 10.1007/s12016-020-08816-3
62. Sørensen OE, Borregaard N. Neutrophil extracellular traps - the dark side of neutrophils. J Clin Invest. (2016) 126:1612–20. doi: 10.1172/JCI84538
63. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. (2017) 8:81. doi: 10.3389/fimmu.2017.00081
64. Le Joncour A, Martos R, Loyau S, Lelay N, Dossier A, Cazes A, et al. Critical role of neutrophil extracellular traps (NETs) in patients with Behcet's disease. Ann Rheum Dis. (2019) 78:1274–82. doi: 10.1136/annrheumdis-2018-214335
65. Safi R, Kallas R, Bardawil T, Mehanna CJ, Abbas O, Hamam R, et al. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in Behçet's disease. J Dermatol Sci. (2018) 92:143–50. doi: 10.1016/j.jdermsci.2018.08.010
66. Li L, Yu X, Liu J, Wang Z, Li C, Shi J, et al. Neutrophil extracellular traps promote aberrant macrophages activation in behçet's disease. Front Immunol. (2021) 11:590622. doi: 10.3389/fimmu.2020.590622
67. Chen J, Liu T, He J, Liu Y. Correspondence on â € Critical role of neutrophil extracellular traps (NETs) in patients with Behcet's disease'. Ann Rheum Dis. (2020). doi: 10.1136/annrheumdis-2020-219472
68. Greco A, De Virgilio A, Ralli M, Ciofalo A, Mancini P, Attanasio G, et al. Behçet's disease: new insights into pathophysiology, clinical features and treatment options. Autoimmun Rev. (2018) 17:567–75. doi: 10.1016/j.autrev.2017.12.006
69. Moretta A, Marcenaro E, Parolini S, Ferlazzo G, Moretta L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. (2008) 15:226–33. doi: 10.1038/sj.cdd.4402170
70. Salmaninejad A, Gowhari A, Hosseini S, Aslani S, Yousefi M, Bahrami T, et al. Genetics and immunodysfunction underlying Behçet's disease and immunomodulant treatment approaches. J Immunotoxicol. (2017) 14:137–51. doi: 10.1080/1547691X.2017.1346008
71. Shimizu J, Takai K, Fujiwara N, Arimitsu N, Ueda Y, Wakisaka S, et al. Excessive CD4 + T cells co-expressing interleukin-17 and interferon-γ in patients with Behçet's disease. Clin Exp Immunol. (2012) 168:68–74. doi: 10.1111/j.1365-2249.2011.04543.x
72. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
73. Emmi G, Silvestri E, Bella C Della, Grassi A, Benagiano M, Cianchi F, et al. Cytotoxic Th1 and Th17 cells infiltrate the intestinal mucosa of Behcet patients and exhibit high levels of TNF-α in early phases of the disease. Med. (2016) 95:e5516. doi: 10.1097/MD.0000000000005516
74. Chi W, Zhu X, Yang P, Liu X, Lin X, Zhou H, et al. Upregulated IL-23 and IL-17 in Behçet patients with active uveitis. Investig Ophthalmol Vis Sci. (2008) 49:3058–64. doi: 10.1167/iovs.07-1390
75. Deniz R, Tulunay-Virlan A, Ture Ozdemir F, Unal AU, Ozen G, Alibaz-Oner F, et al. Th17-inducing conditions lead to in vitro activation of both Th17 and Th1 responses in Behcet's disease. Immunol Invest. (2017) 46:518–25. doi: 10.1080/08820139.2017.1306865
76. Tulunay A, Dozmorov MG, Ture-Ozdemir F, Yilmaz V, Eksioglu-Demiralp E, Alibaz-Oner F, et al. Activation of the JAK/STAT pathway in Behcet's disease. Genes Immun. (2015) 16:170–5. doi: 10.1038/gene.2014.64
77. Ortiz Fernández L, Coit P, Yilmaz V, Yentür SP, Alibaz-Oner F, Aksu K, et al. Genetic association of a gain of function interferon gamma receptor 1 (IFNGR1) polymorphism and the intergenic region LNCAROD/DKK1 with Behçet's disease. Arthritis Rheumatol. (2021) 73:1244–1252. doi: 10.1002/art.41637
78. Yasuoka H, Yamaguchi Y, Mizuki N, Nishida T, Kawakami Y, Kuwana M. Preferential activation of circulating CD8+ and gammadelta T cells in patients with active Behçet's disease and HLA-B51. Clin Exp Rheumatol. (2008) 26:S59–63.
79. Kim SM, Park MJ, Park S, Cheng JY, Lee ES. Differential expression of novel genes and signalling pathways of senescent CD8+ T cell subsets in Behçet's disease. Clin Exp Rheumatol. (2020) 38(Suppl. 1):17–25.
80. Hasan MS, Bergmeier LA, Petrushkin H, Fortune F. Gamma Delta (γδ) T cells and their involvement in behçet's disease. J Immunol Res. (2015) 2015:705831. doi: 10.1155/2015/705831
Keywords: Behçet's syndrome, pathogenesis, HLA-B*51, neutrophils, epigenetic, microbiome
Citation: Mattioli I, Bettiol A, Saruhan-Direskeneli G, Direskeneli H and Emmi G (2021) Pathogenesis of Behçet's Syndrome: Genetic, Environmental and Immunological Factors. Front. Med. 8:713052. doi: 10.3389/fmed.2021.713052
Received: 21 May 2021; Accepted: 13 September 2021;
Published: 08 October 2021.
Edited by:
Tulin Ergun, Marmara University, TurkeyReviewed by:
Wei Cao, Baylor College of Medicine, United StatesRuth J. Napier, Oregon Health and Science University, United States
Copyright © 2021 Mattioli, Bettiol, Saruhan-Direskeneli, Direskeneli and Emmi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Bettiol, YWxlc3NhbmRyYS5iZXR0aW9sQHVuaWZpLml0
†These authors have contributed equally to this work
‡These authors share senior authorship