
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 24 August 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.708453
This article is part of the Research TopicClinical Application of Proteomics in Kidney DiseasesView all 7 articles
 Yunlong Li1,2†
Yunlong Li1,2† Kuai Ma3†
Kuai Ma3† Zhongyu Han2†
Zhongyu Han2† Mingxuan Chi2
Mingxuan Chi2 Xiyalatu Sai1Ping Zhu1Zhaolun Ding4
Xiyalatu Sai1Ping Zhu1Zhaolun Ding4 Linjiang Song2*
Linjiang Song2* Chi Liu1,5*
Chi Liu1,5*Kidney disease is a general term for heterogeneous damage that affects the function and the structure of the kidneys. The rising incidence of kidney diseases represents a considerable burden on the healthcare system, so the development of new drugs and the identification of novel therapeutic targets are urgently needed. The pathophysiology of kidney diseases is complex and involves multiple processes, including inflammation, autophagy, cell-cycle progression, and oxidative stress. Heme oxygenase-1 (HO-1), an enzyme involved in the process of heme degradation, has attracted widespread attention in recent years due to its cytoprotective properties. As an enzyme with known anti-oxidative functions, HO-1 plays an indispensable role in the regulation of oxidative stress and is involved in the pathogenesis of several kidney diseases. Moreover, current studies have revealed that HO-1 can affect cell proliferation, cell maturation, and other metabolic processes, thereby altering the function of immune cells. Many strategies, such as the administration of HO-1-overexpressing macrophages, use of phytochemicals, and carbon monoxide-based therapies, have been developed to target HO-1 in a variety of nephropathological animal models, indicating that HO-1 is a promising protein for the treatment of kidney diseases. Here, we briefly review the effects of HO-1 induction on specific immune cell populations with the aim of exploring the potential therapeutic roles of HO-1 and designing HO-1-based therapeutic strategies for the treatment of kidney diseases.
Kidney disease is an umbrella term for a number of diseased states characterized by impaired kidney function and/or structure. Due to the occult onset and the difficulty in early diagnosis, a large number of patients with kidney diseases end up with kidney failure and complete loss of kidney function (1). Currently, kidney diseases are still treated by surgery, chronic dialysis, renal transplantation, and other means, but these methods are suboptimal in the complete treatment of these diseases (1). Thus, the development of new treatments and drugs to change this status remains a pressing priority (1). Several potential renal protective therapies are currently being investigated, of which enhancing the heme oxygenase (HO) system to protect renal morphology is an area of great interest.
Heme oxygenase is a type of microsomal enzyme with anti-oxidant functions, and it is a member of the heat shock protein family (2). As the rate-limiting enzyme of heme catabolism, heme oxygenase decomposes heme to yield carbon monoxide (CO), catalytic iron, and biliverdin (3–5). Biliverdin is subsequently converted to unconjugated bilirubin by biliverdin reductase (3, 4, 6). HO can be subdivided into two subtypes: inducible heme oxygenase-1 (HO-1) and constitutive isoform heme oxygenase-2 (HO-2) (7). Different from HO-2, which is stably expressed in most organs (8), HO-1 can be highly up-regulated by a variety of oxidative stress stimuli to protect organs from the damage of oxidative stress (9). In other words, the induction of HO-1 is considered as an adaptive cellular response against the toxicity of oxidative stress (10). More recently, HO-1 has also been recognized as having significant immunomodulatory properties and anti-inflammatory functions (11). The immunomodulatory effects of HO-1 activity have been found in many immune cells (Figure 1). For example, HO-1 expression is particularly up-regulated in macrophages, where they inhibit the production of inflammatory mediators (12–15). Furthermore, HO-1 modulates the production of interferon-β (IFN) in macrophages and dendritic cells (DCs) through direct HO-1 binding to IFN regulatory factor 3 (IRF3) (16). On the other hand, the induction of HO-1 inhibits pro-inflammatory functions and maintains DCs in an immature-like phenotype to promote tolerogenic DCs (tolDCs), with a consequent reduction in effector T cell responses and a promotion of regulatory T cell (Treg cell) responses (17–22). Subsequent studies have reported that the up-regulation of HO-1 in mast cell (MC) can stabilize MC membranes to reduce the production of inflammatory cytokines, suggesting that HO-1 can suppress DC maturation induced by MC degranulation (23, 24). Moreover, HO-1 inhibits the phosphorylation of signal transducer and activator of transcription 3 (STAT3) in naive CD4+ T cells, thereby inhibiting T cell proliferation and T helper 17 cell (Th17 cell) differentiation (25), and decreasing T helper 2 cell (Th2 cell)-related cytokine production (26). Furthermore, HO-1 is a critical transcriptional suppressor for B cell development and growth (27), as well as an inhibitor for natural killer cell (NK cell) effector functions (28). These results demonstrate that HO-1 has anti-inflammatory, anti-oxidant, and immunomodulatory properties, and they also imply the therapeutic potential of HO-1 in inflammatory diseases.
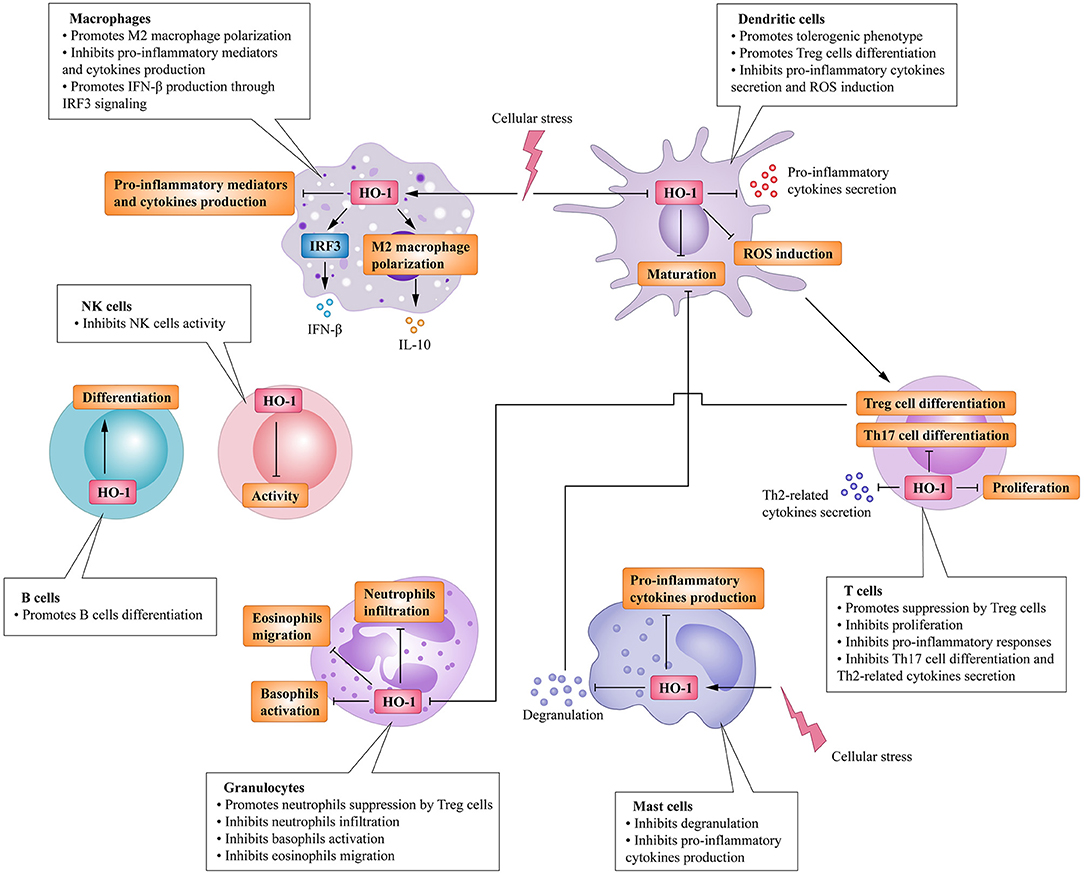
Figure 1. Immunomodulatory activity of heme oxygenase 1 (HO-1) in immune cells. HO-1 is stress-inducible by macrophages and promotes the phenotypic shift to M2 macrophages, associated anti-inflammatory activities. HO-1 also modulates the production of interferon (IFN)-β in macrophages through direct HO-1 binding to interferon regulatory factor 3 (IRF3) in response to pro-inflammatory stimuli. HO-1 induction in dendritic cells (DCs) inhibits their maturation, secretion of pro-inflammatory cytokines, and induction of ROS. Mast cells (MCs) can sense local stressor insults to induce HO-1, thereby suppressing their degranulation and production of inflammatory cytokines. Moreover, inhibiting MCs' degranulation can also inhibit DCs' maturation. These effects all result in the promotion of DCs into a tolerogenic phenotype, thus promoting regulatory T cell (Treg cell) differentiation. HO-1 expression by T cells can additionally inhibit T helper 17 cells (TH17 cells) differentiation, Th2-related cytokines secretion, as well as limit proliferation. Activated Treg cells can initiate HO-1 expression in neutrophils shifting them to a suppressive phenotype, which inhibits their infiltration, basophil activation as well as migration of eosinophils. HO-1 expression by B cells can additionally promote their development and growth. HO-1 induction can also inhibit NK cells effector functions and activity.
The beneficial effects of HO-1 induction in inflammation have also been associated with the degradation of the heme group (29). Under pathogenic conditions, hemoproteins release the heme group, which is a universal danger-associated molecular pattern (DAMP) (30). Free heme subsequently binds to toll-like receptor 4 (TLR4) (31), which triggers the production of tumor necrosis factor α (TNFα) (32). Moreover, free heme readily promotes lipid peroxidation and induces oxidative stress through the generation of reactive oxygen species (ROS), which damage cellular structures and tissues (33). HO-1 in macrophages has the ability to catalyze the decomposition of heme, thereby reducing TLR4 activation and ROS formation (34). Meanwhile, HO-1 prevents heme-induced pro-inflammatory M1 macrophages from undergoing polarization and drives the phenotypic shift to anti-inflammatory M2 macrophages (35, 36). Moreover, the products of heme degradation, namely, CO and biliverdin, exert anti-inflammatory and anti-oxidant properties, respectively (37). Biliverdin and its metabolite, bilirubin, exhibit suppressive properties on the activation of CD4+ T cells (11, 38, 39). On the other hand, CO can selectively decrease the expression of several pro-inflammatory genes and increase the expression of anti-inflammatory interleukins-10 (IL-10) in macrophages (40). In rodent models of inflammation, HO-1 and its metabolites (CO and biliverdin) have been shown to reduce leukocyte rolling, adhesion, and neutrophil infiltration as well as migration of eosinophils to inflammatory sites (41–45).
Taken collectively, HO-1 activity has been confirmed to affect both innate and adaptive immune responses (Table 1), leading to the reduction of the early inflammatory response and limiting the subsequent tissue damage (5, 55). As a response to inflammatory stimuli and oxidative stress, the expression of HO-1 could activate or inhibit cell-intrinsic pathways in most cell types (20). Therefore, HO-1 is considered as one of the key regulators of the immune system. Here, we focus this review on the influence of HO-1 on kidney diseases in different immune cell populations and kidney resident cells, with a comprehensive understanding of HO-1 and its therapeutic potential in these diseases. Figure 2 summarizes the beneficial effects of HO-1 in these kidney diseases.
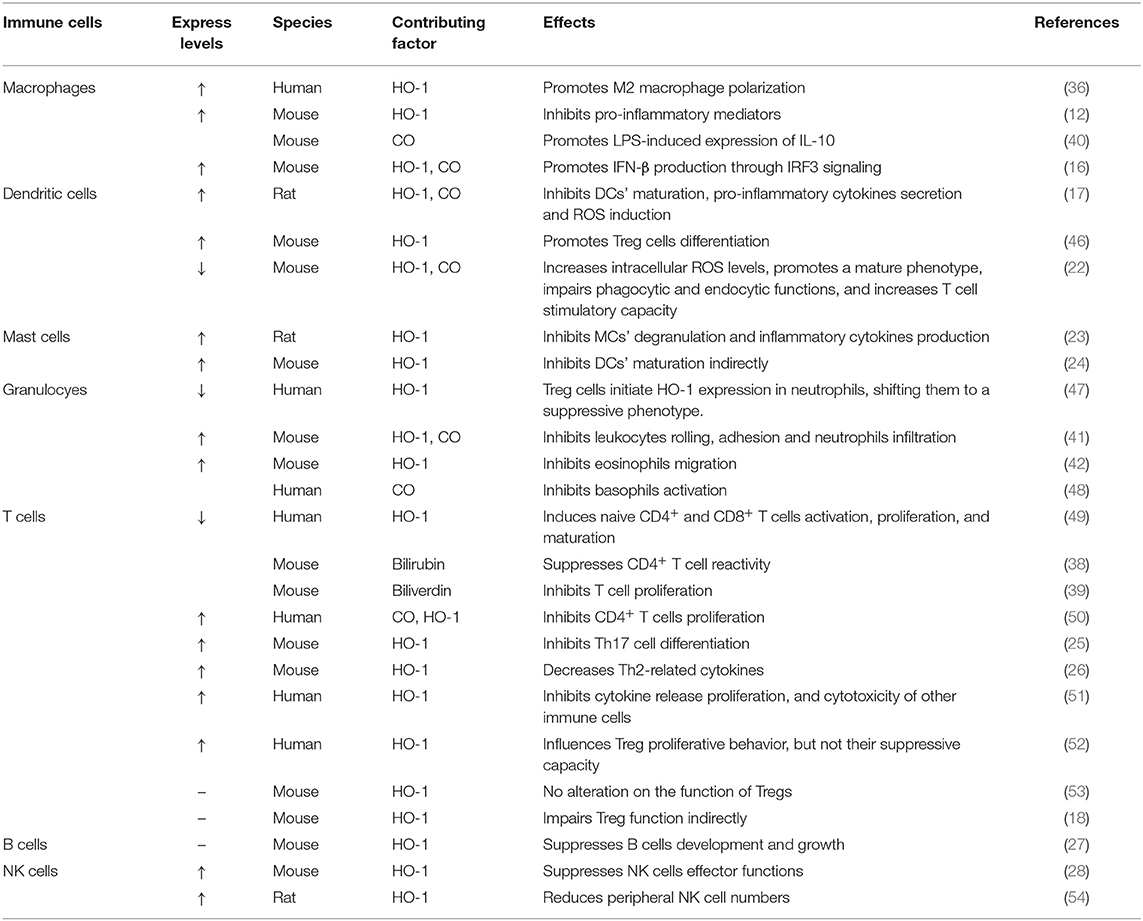
Table 1. Effects of heme oxygenase-1 on innate and adaptive immune responses based on experiments.
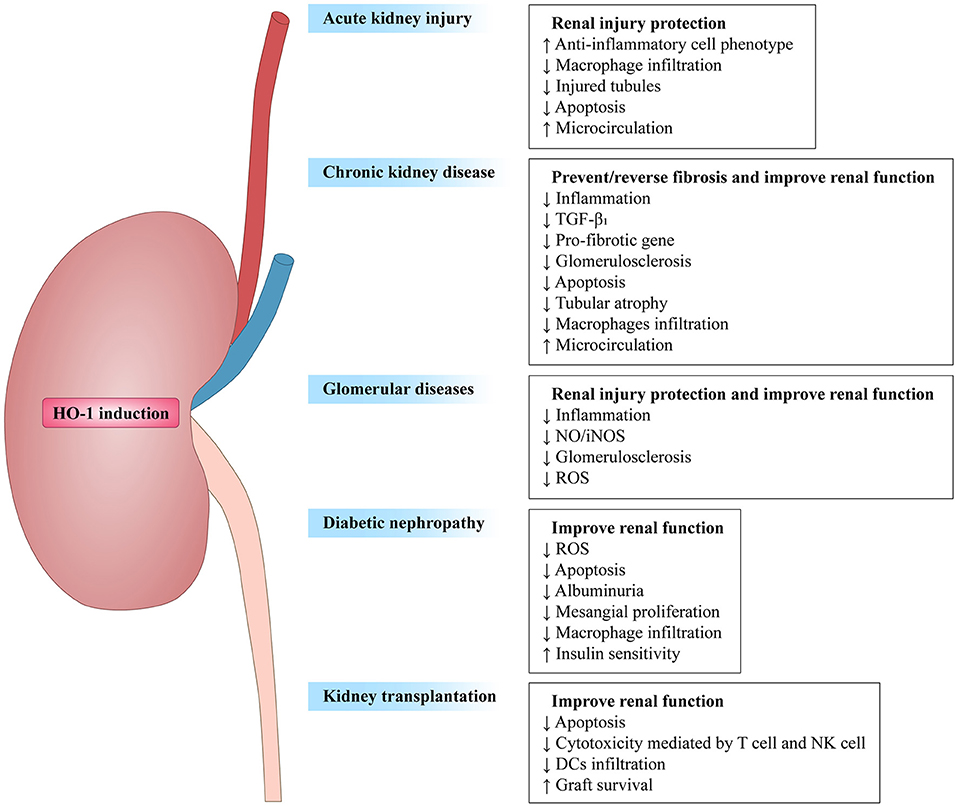
Figure 2. Overview of heme oxygenase-1 in renal diseases. HO-1 induction prevents renal damage in diverse renal diseases, such as acute kidney injury (AKI), chronic kidney disease (CKD), glomerular diseases, diabetic nephropathy (DN), and kidney transplantion. Arrows followed by text indicate increase (↑) or decrease (↓).
Acute kidney injury (AKI), characterized by a rapid increase in serum creatinine and/or a decrease in urine output, is associated with high rates of morbidity and mortality in hospital and outpatient settings (56, 57). Despite the heterogeneity of causes, the response following an acute insult involves similar pathways, including apoptosis, necrosis, and infiltration of inflammatory cells (58). These processes lead to an exaggerated inflammatory response, further aggravating acute tubular necrosis (ATN) and functional derangement in the form of AKI (59). Certain diseases that result in AKI may cause heme release such as intravascular hemolysis or rhabdomyolysis (60). Due to its small molecular weight and hydrophobicity, heme can translocate across the glomerular vascular endothelial cell membrane, subsequently activating TLR4 signaling and other pathways, and mediating the activation of endothelial cells and the production of inflammatory cytokines (61, 62). Furthermore, excessive proteins, such as hemoglobin or myoglobin, can be released into the circulation (63). These processes may lead to severe adenosine triphosphate (ATP) depletion due to excess protein reabsorption, subsequently causing depletion of energy stores and excessive production of ROS (64). High levels of ROS can induce oxidative stress, followed by impaired cellular homeostasis and cell death (65). Under these conditions, inflammatory cells can be locally activated or depleted from hematopoietic organs, thereby leading to an exaggerated inflammatory response (66).
At early stages of AKI, excessive oxidative stress ultimately results in cell death, which accelerates inflammation directly and indirectly, and further increases oxidative stress (67). In this context, HO-1 can be induced in both renal parenchymal cells and tissue resident leukocytes (68, 69). Innate immune cells, such as macrophages, DCs, and neutrophils, are chief participants in the acute insult and recruited to the injured kidneys (70). HO-1 expression in monocytes/macrophages is beneficial in alleviating the inflammatory response of AKI. HO-1-expressing macrophages tend to be polarized toward the M2 phenotype, up-regulate the expression of anti-inflammatory cytokines (IL-10), suppress pro-inflammatory cytokine (TNFα) secretion, and express reparative genes that are beneficial for tissue recovery after AKI (68, 71). A recent study by Hull et al. demonstrated that HO-1 expression by renal DCs could regulate their migration, allowing them to reside in the kidneys where they enhanced recovery and decreased renal fibrosis after ischemia-reperfusion injury (IRI) (68). Furthermore, the authors also found that HO-1 expression in monocytes/macrophages not only accelerates the exit of these cells from the ischemic kidney tissue, but also their migration to extra-renal sites, in turn attenuating their involvement in renal ischemic injury (68). Additionally, HO-1 expression in antigen-presenting cells (e.g., DCs) is required for optimal Treg cell function, which has been suggested to facilitate recovery following AKI (72, 73).
It is worth mentioning that the cytoprotective effects of HO-1 are associated with the by-products of heme degradation. CO exhibits potent anti-proliferative effects on T cells via the down-regulation of IL-2 and caspase activity, which mitigates inflammation (74). Moreover, CO also exerts inhibitory effects on the migration of DCs and promotes immune tolerance (75, 76). CO has significant effects on the circulation by inhibiting platelet aggregation and inducing potent vasodilatory effects (77). Dual-treatment studies with biliverdin and CO in a rat renal transplantation model demonstrated synergistic effects on both blood flow rates and graft survival (78). Furthermore, biliverdin can protect cells from apoptosis induced by cisplatin (CP), a very effective anti-cancer drug, through its anti-oxidative effects (79). CO inhalation therapy can also protect the kidneys from nephrotoxicity induced by CP by limiting renal tubule cell apoptosis (80). Moreover, HO-1 induction is related to the increased availability of ferritin, leading to prompt conjugation and removal of free iron, thereby removing another source of potential oxidative stress (81). Thus, the mechanism of HO-1 may be multifaceted and diversified in terms of the maintenance of renal blood flow and the promotion of cell survival (Figure 3).
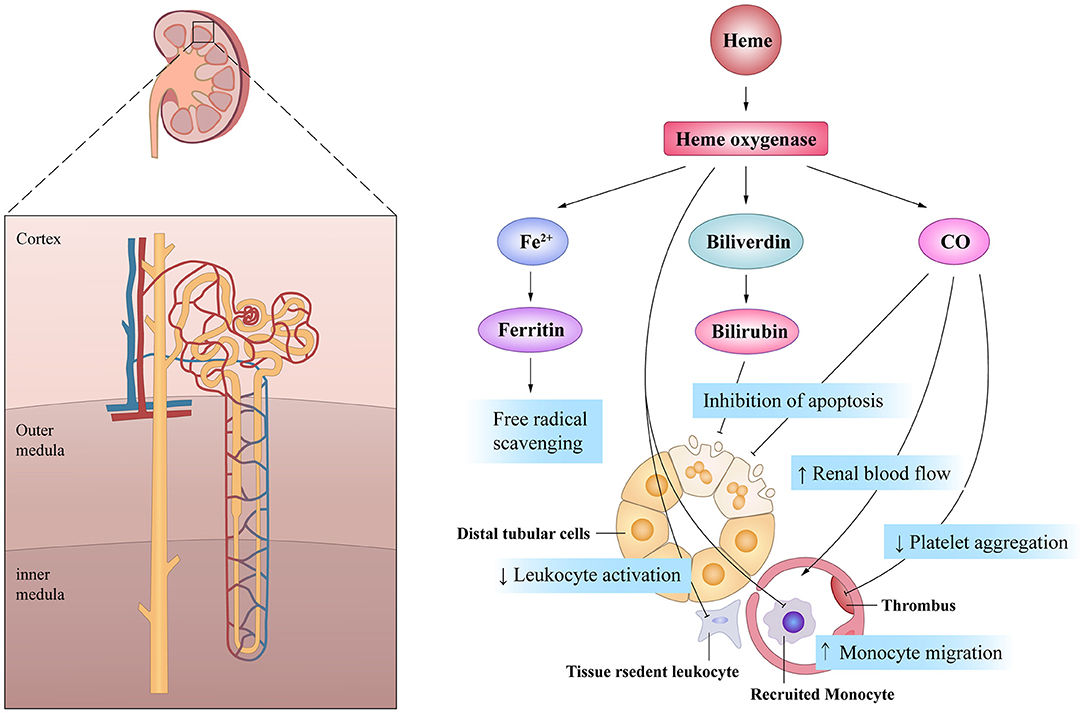
Figure 3. The beneficial actions of HO-1 on renal tubule in acute kidney injury. HO-1 and its products of heme exhibit pleiotropic effects on the cell survival, the inflammatory response and the microcirculation, all of which are of potential importance in mediating its potential protective effects after AKI.
The preventive role of HO-1, as well as the induction of other cytoprotective stress proteins, is collectively referred in the literature as “preconditioning treatment” (56). Numerous subsequent studies have confirmed the potential of pharmacologic or genetic HO-1 induction in immune modulation against AKI (71, 82, 83). For instance, statins are capable of exaggerating HO-1 induction post ischemia in vascular smooth muscle cells and macrophages, the administration of which can reduce the severity of IRI-induced AKI, and this protection can be blocked by HO inhibition (83). Moreover, intravenous injection of HO-1-overexpressing macrophages preferentially homes these cells to the ischemic kidney to ameliorate AKI (71). Besides the induction of HO-1 expression under macroscopic conditions, recent studies also have focused on the molecular mechanisms in the induction of HO-1 expression and the regulation of its activity, which includes transcription factors and upstream signaling molecules. For instance, the nuclear factor erythroid 2-related factor 2 (Nrf2) is one of the key transcription factors involved in the regulation of HO-1 (84). During injury, Nrf2 hyperactivation induces the expression of anti-oxidant and detoxification enzymes and downstream proteins, including HO-1 (85). This Nrf2/HO-1 signaling axis can trigger a variety of immunological processes against AKI. Firstly, as one of the catalytic products of HO-1, CO can decrease the activity of nuclear histone acetyltransferase, preventing high-mobility group box 1 (HMGB1) acetylation and release (86), thereby inhibiting the secretion of HMGB1 (87). Recently, studies have shown that HMGB1 activates inflammatory responses by stimulating receptors such as TLR4, and reduces the survival of tubular epithelial cells (86), which promotes renal IR injury (88). Secondly, activation of the Nrf2/HO-1 pathway can reduce the activity of the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome, which is activated by ROS that subsequently decrease the secretion of pro-inflammatory IL-1β and IL-18 (89). Furthermore, T cell-specific augmentation of Nrf2 affects phenotypic diversity, activation, and recruitment of immune cells, which includes increasing anti-inflammatory Tregs, decreasing pro-inflammatory M1 macrophages, and reducing intracellular cytokine production by T cells in ischemic AKI (90). These studies suggest that HO-1, which is induced by the activation of its transcription factor Nrf2, can protect kidneys from the exaggerated inflammatory response during AKI. Given that the multifactorial mechanisms of HO-1 induction are adverse toward the pathophysiological processes of AKI, preventive HO-1 induction may provide new insights that can improve the treatment of AKI.
Chronic kidney disease (CKD) is a clinical syndrome secondary to the definitive dysfunction and/or the structure of the kidneys and is a public health problem that causes substantial morbidity and mortality (91). In CKD, various abnormally filtered urinary proteins stimulate tubular epithelial cells to synthesize pro-inflammatory mediators, including ROS and chemokines (92). Proteinuria is well-recognized as a prognostic index of the severity of progressive kidney disease and the degree of decline in renal function in CKD (93). Reabsorption of urinary proteins induces ROS production within tubular epithelial cells, which results in the high up-regulation of HO-1 (94). The overexpression of HO-1 in proximal tubular epithelial cells reduces the albumin-stimulated production of cytokines such as monocyte chemoattractant protein-1 (MCP-1) (95). In addition, MCP-1 is a potent chemoattractant, affecting monocytes/macrophages, and increased MCP-1 levels can stimulate transforming growth factor (TGF)-β induction in resident glomerular cells (96). Moreover, MCP-1 induces inflammation by activating nuclear factor-kappaB (NF-κB), thereby directly producing pro-inflammatory effects on the proximal tubules (97, 98). HO-1 knockout mice exhibit drastic interstitial cellular inflammation accompanied by the striking up-regulation of MCP-1 and the activation of NF-κB, which is consistent with previous findings (99).
Theoretically, any etiology that can cause progressive and permanent death of renal tissue and the subsequent replacement of functional nephrons has the potential to lead the fibrosis characteristics of CKD (100). Despite the heterogeneity of etiologies, TGF-β has a critical role in initiating and modulating tissue repair, and its aberrant expression is involved in the pathogenesis of progressive CKD (101). However, this factor can activate the expression of HO-1 to stabilize and attenuate tissue injury (102). Interestingly, a therapeutic approach for HO-1 induction has been proposed in mitigating TGF-β-mediated renal disease (103). In mice with unilateral ureteral obstruction (UUO), a progressive interstitial fibrosis model, the preventive enhancement of HO-1 expression 48 h before UUO attenuated fibrosis by down-regulating inflammatory pro-fibrotic genes as well as pro-apoptotic pathways (caspase-3 activation), which decreased proteinuria and renal dysfunction (104, 105). Zinc protoporphyrin IX (ZnPP), an HO-1 inhibitor, can block these protective effects, thereby inducing increased fibrosis, up-regulating tubular TGF-β1 expression and inflammation, and enhancing the epithelial-to-mesenchymal transition with the increased infiltration of macrophages (106). In transgenic mice, HO-1 overexpression has been reported to limit the tubule-interstitial infiltration of macrophages and to regulate the secretion of inflammatory cytokines, significantly reducing renal interstitial fibrosis in the UUO model (107). Recent evidence indicates that HO-1 can act as a protective agent against renal fibrosis through the regulation of microRNAs, such as the p53-regulated miR-34a and the pro-fibrotic miR-21, but the underlying regulatory mechanisms remain unclear (108). Moreover, administration of low-dose CO to mice has protective effects via the MAPK kinase 3 (MKK3) pathway, thereby inhibiting the development of renal fibrosis in obstructive nephropathy (109). These results suggest that the augmentation of HO-1 levels may be a therapeutic strategy against renal interstitial fibrosis.
Interestingly, the level of HO-1 expression is variable across individuals due to the high degree of polymorphism in the number of the guanosine thymidine (GT)n fragment in the promoter (110). This is clinically meaningful, as shown in patients with coronary artery disease in whom a greater number of GT dinucleotide repeats in the HO-1 gene promoter was found to be associated with an increased risk of CKD (111). Thus, dysregulated pathways of inflammation and repair, such as the up-regulated failure of HO-1, may increase oxidative stress and inflammation, which in turn can contribute to this self-injury state (112). Despite the reduced oxygen supply in CKD, hypoxia inducible factors (HIF) are down-regulated and associated with decreased HO-1 expression, although HIF induction was found to restore HO-1 expression in a mouse model of CKD, together with other target genes and angiogenesis (113). Furthermore, Nrf2 activation (and downstream HO-1) may lessen maladaptive repair after repeated acute injuries (114). Thus, the up-regulation of HO-1 expression by Nrf2 or HIF seems to be a potential target for delaying the progression of CKD.
Hemodialysis is one of three renal replacement therapies, and the arteriovenous fistula (AVF) is the preferred hemodialysis vascular access, although it is complicated by high failure rates and attendant morbidity (115). AVF blood flow is markedly reduced in the setting of CKD, and the venous wall is significantly thickened (116). The risk of venous thrombus formation is also increased due to the up-regulation of genes with procoagulant properties (117). However, venous HO-1 induction can improve AVF blood flow and decrease venous wall thickness of the AVF in a murine model of CKD (118). Moreover, the authors found that the administration of carbon monoxide-releasing molecules-3 (CORM-3) (40 mg/kg ip) after nephrectomy and before AVF surgery acutely increases AVF blood flow (118). This study indicates that induction of HO-1 and/or administration of its products may confer beneficial effects in terms of improving certain classical therapies. To conclude, up-regulation of HO-1 during CKD may interrupt the progressive loss of renal function by inhibiting the progression of renal fibrosis. As such, aberrant expression of HO-1 may favor fibrosis by decreasing blood flow, thereby establishing and maintaining a chronic pro-inflammatory state.
Glomerular diseases are a leading cause of chronic and end stage renal disease (ESRD) worldwide (119). The unique anatomical location and highly specialized structure of the glomerulus makes the glomerular microvasculature particularly vulnerable, exposes it to a variety of HO-1 inducers. In line with this, up-regulation or induction of HO-1 has been observed in diverse glomerular diseases, including sickle cell nephropathy (120), minimal change disease (121), and IgA nephropathy (121). Interestingly, although the glomerular microvasculature is commonly attacked in these diseases, the above studies demonstrate dominant HO-1 induction primarily in renal tubular rather than glomerular cells (122, 123). This indicates that injury in glomeruli can induce HO-1 expression in renal tubules, which establishes the potential association between glomeruli and tubules.
Glomerulonephritis (GN) refers to a group of renal diseases that attack glomeruli due to damage mediated by immunological mechanisms (124). To study HO-1 expression in acute GN, a model of nephrotoxic nephritis (NTN) has mostly been used (125). In this model, the location of HO-1 expression was also predominantly found in tubular cells and not in glomeruli (126). Under certain circumstances, such as the administration of HO-1 inducer in this model, HO-1 expression can also be induced by glomerulus cells, showing reduced glomerular neutrophils and macrophage infiltration, and decreased glomerular thrombosis (127). In kidney injury, HO-1 expression can be induced in renal parenchymal cells and tissue resident leukocytes (68, 69). These renal tissue resident leukocytes can contribute to lymphocyte differentiation and activation, thereby linking innate and adaptive immune systems (128). HO-1 can also exhibit inhibitory effects on autophagy, which is a highly regulated mechanism to eliminate damaged organelles and proteins from cells and to maintain homeostasis (129).
Inflammatory responses are also regulated by nitric oxide (NO), which is synthesized by nitric oxide synthase (NOS) (130). In GN, intraglomerular inducible nitric oxide synthase (iNOS) activation leads to high levels of NO generation, which results in supraphysiologic amounts of NO within glomeruli (131). The excessive NO can move freely in and out of cells and bind to the heme-iron present in iNOS itself and other hemoproteins (132, 133). Thus, excessive NO can inhibit the subsequent production of NO catalyzed by iNOS (134). Additionally, the binding of NO to heme-containing enzymes can promote destabilization, fragmentation, and proteolysis by proteasomes (135). Thus, NO promotes the release of heme from iNOS, and HO-1 activity is enhanced by the released heme, which can undergo degradation and removal by HO-1 (135). Consistent with these findings, iNOS and HO-1 are co-induced in an NTN model (136). NO production derived from iNOS stimulates HO-1 expression in glomerular mesangial cells, whereas HO-1 activation reduces iNOS expression/activity, which provides evidence for a link between iNOS and HO-1 in NTN rats (136). This may be partly attributable to the expression products of HO-1, all of which can suppress the effects of iNOS expression (137, 138). For instance, CO can bind heme within iNOS and influence the enzymatic activity of iNOS and the production of NO (139). The released iron can suppress iNOS transcription, which down-regulates iNOS expression (140). Another possible reason may be that the heme prosthetic group required by iNOS is degraded by HO-1 (141). Increased HO-1 activity can decrease the availability of heme needed for incorporation into newly synthesized apo-iNOS protein, thereby impairing the synthesis of functional iNOS (142). Moreover, iNOS activity is inhibited when excessive NO binds to heme within the iNOS protein (134). As a result, the activity of iNOS is suppressed and the additional production of NO is blocked, allowing the cells to survive after being exposed to both oxidative and nitrosative stresses. This negative feedback seems to be an excellent mechanism in that glomerular cells can rapidly defend against NO-mediated oxidative injury.
It has also been reported that oxidative stress constitutes a key and a common event in the pathogenesis of IgA nephropathy (143, 144), which is the most common form of primary GN worldwide (145). Nakamura et al. reported that oxidative DNA damage and oxidative stress were increased in patients with IgA nephropathy compared with healthy controls (146). Chen et al. observed that polymorphonuclear leukocyte infiltration, which has a high potential to produce ROS, increased in patients with IgA nephropathy (144). These data suggest that the production of activated ROS is associated with renal dysfunction in IgA nephropathy. ROS are considered to be activated in IgA nephropathy due to the lower mRNA expression of superoxide dismutase (SOD) in moderately or severely damaged tissues from patients with IgA nephropathy and non-IgA mesangial proliferative glomerulonephritis compared with normal or mildly damaged tissues (147). Furthermore, decreased SOD levels may suppress the superoxide-scavenging reaction, thereby rendering the tissues more vulnerable to oxidative stress (147). Accordingly, HO-1 immunoreactivity in the kidneys of patients with IgA nephropathy was significantly higher than that in the kidneys of controls (144). Activated HO-1 can reduce oxidative stress-mediated cellular injury through at least two possible mechanisms, namely, the degradation of cellular heme and the elevation of biliverdin concentrations (148, 149). However, the underlying mechanisms of HO-1 in IgA nephropathy remain unclear, with research pointing to the presence of HO-1 promoter polymorphisms, which predispose individuals to the development of IgA nephropathy (150).
In addition, inflammatory reactions and oxidative stress are involved in the pathogenesis of focal segmental glomerulosclerosis (FSGS), a very common type of chronic kidney disease with clinical features of excessive proteinuria or nephrotic syndrome (151). HO-1-deficient rats exhibit FSGS-type lesions that associate with proteinuria, which implicates HO-1 in the pathobiology of FSGS (152). Aggravated oxidative stress resulting from the absence of HO-1 may be an underlying mechanism that explains the presence of FSGS-type lesions and proteinuria observed in the glomeruli of rats lacking HO-1. Furthermore, several studies have already reported that targeting the Nrf2 anti-oxidant pathway may hold promise as a renoprotective therapy for FSGS (153, 154). Yang et al. observed that targeting the Nrf2-mediated anti-oxidant pathway significantly prevented the development of FSGS in treated mice (153). This delayed effect may involve mechanistic pathways, such as the binding of Nrf2 to anti-oxidant-response elements in the promoter region of several Nrf2-downstream genes encoding anti-oxidant enzymes, including HO-1, glutathione peroxidase (GPx), catalase, and SOD (155, 156). However, it is not entirely clear to what extent HO-1 plays a role in the protection of FSGS.
Taken collectively, inflammatory responses and oxidative stress appear to be a major part of the pathophysiologic process in glomerular diseases. Under these conditions, HO-1 can be induced in tubules, and glomerular injury may initiate HO-1 expression in renal tubules via a potential mechanism. Among them, excessive NO production stimulates HO-1 expression in glomerular mesangial cells, and activation of HO-1 may mitigate NO-mediated toxicity by negatively modulating iNOS expression or activity. HO-1 expression is also upregulated in tissue resident leukocytes. Consequently, glomerular neutrophils and macrophage infiltration are significantly reduced. As an adaptive response to oxidative stress, increased HO-1 expression is also needed to protect cells from oxidative stress, and it may be an emerging therapy in clinical studies. Further studies are required to understand the role of HO-1 in these glomerular diseases.
Systemic lupus erythematosus (SLE) is a typical systemic autoimmune disease of unknown etiology that predominantly affects women of child-bearing age (157). It is characterized by chronic inflammation and immunological abnormalities (158). Although inflammation may impact multiple organ systems in SLE, lupus nephritis (LN) remains the representative manifestation and the major contributor to mortality caused by SLE (159). The pro-inflammatory role of monocytes/macrophages in the pathogenesis of SLE has been established. Patients with SLE exhibit a lower level of HO-1 expression in monocytes, suggesting a potential connection between HO-1 expression by myeloid cells and lupus nephritis (160). Cuitino et al. found that the activated neutrophils of patients with LN showed low HO-1 expression, and the baseline ROS level in patients' monocytes was increased, and cobalt protoporphyrin (Co-PP) restored the HO-1 level to the baseline level (161). Thus, the pro-inflammatory environment of LN patients may be associated with decreased HO-1 expression in circulating and infiltrating monocytes/macrophages and neutrophils. However, further studies are needed to determine whether these alterations in immune cells are a cause or a consequence of the disease.
In LN, renal damage is initiated by the production of anti-nuclear antibodies (ANA) and the glomerular deposition of immune complexes (IC) (162). These results are attributed to the failure of T cell and B cell suppression, which is mediated by defects in cell signaling, immune tolerance, and apoptotic mechanisms that promote autoimmunity (163). Accordingly, the administration of tolDCs has been suggested as a potential strategy in the treatment of SLE (164, 165). In a mouse model of SLE, DCs treated in vitro with HO-1 inducer showed a stable tolerogenic profile, and treatment with these DCs alleviated SLE symptoms, including decreased ANA and reduced skin lesion severity (166). Likewise, after the administration of hemin as an HO-1 inducer, these mice showed decreased proteinuria, reduced glomerular immune complex deposition, and increased expression of iNOS in the kidneys (167). They also showed decreased circulating levels of anti-dsDNA IgG (a group of ANA) and IFNγ (167). Moreover, CO treatment can also ameliorate proteinuria and renal inflammation in FcγRIIb-deficient mice, a model for SLE (165). CO exposure also significantly decreases the number of activated T cells in the kidneys and lungs, as well as ANA levels in lupus-prone mice (168).
Based on the adverse effects of low HO-1 expression in immune cells, it has become possible to use HO-1 inducers to delay the progression of LN and even ameliorate the systemic conditions of SLE patients (Figure 4). For instance, baicalein can alleviate the symptoms of pristane-induced LN by regulating the balance between Nrf2/HO-1 signaling and NLRP3 expression (169). Furthermore, dietary extra virgin olive oil could attenuate renal damage in a mice SLE model via the activation of the Nrf-2/HO-1 pathway and the reduction of pro-inflammatory cytokines (170). However, further studies are required to provide greater insight into the effects of HO-1 induction and its byproducts.
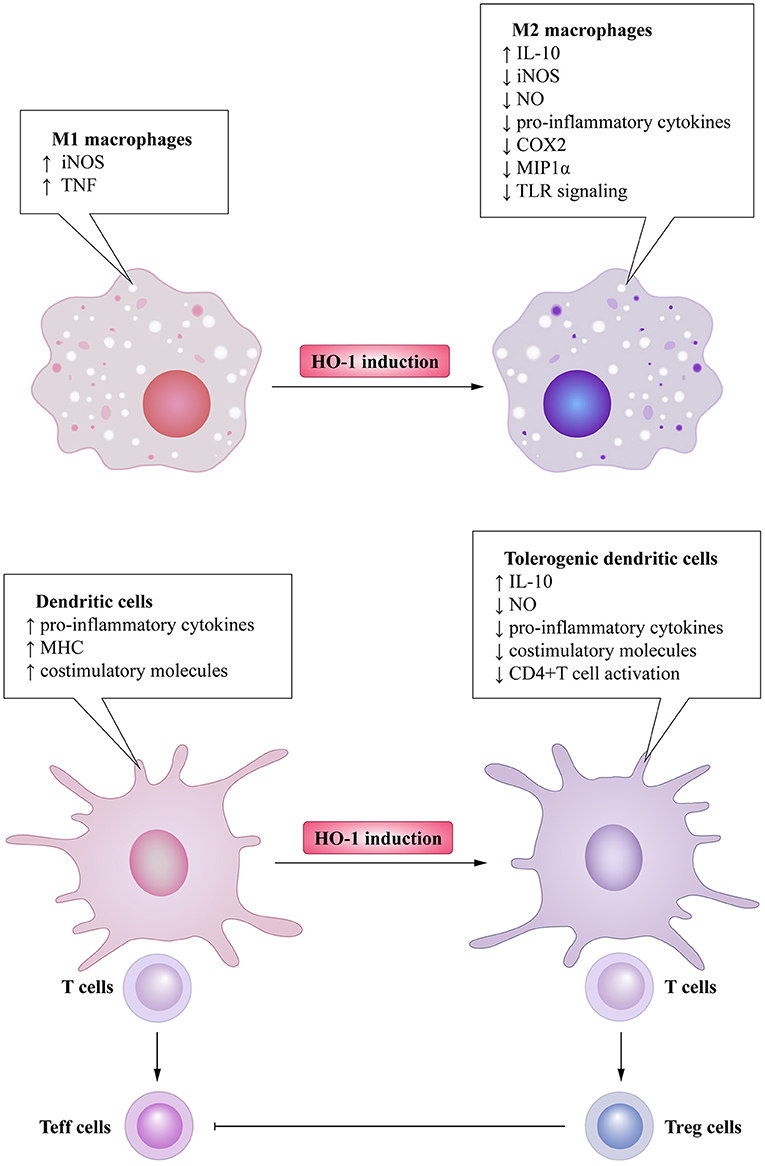
Figure 4. The beneficial effect of the use of HO-1 inducers on the SLE patient. HO-1 induction in immune cells promotes polarization of macrophages into the M2 profile with anti-inflammatory property. They are capable of increasing secretion of IL-10 and decreasing secretion of pro-inflammatory cytokines and mediators. These favor the obtainment of a tolerogenic profile in DCs, thus generating regulatory T (Treg) cells differentiation, which prevents autoimmunity by inhibiting the activation and proliferation of effector T (Teff) cells.
Diabetic nephropathy (DN) is one of the most common microvascular complications coinciding with both type I and II diabetes (171). It is characterized by glomerular hypertrophy, proteinuria, decreased glomerular filtration, and renal fibrosis with progressive renal injury caused by high glucose levels (172). About 45% diabetics are affected by this microvascular complication, which ultimately leads to ESRD (173). Chronic hyperglycemia is the major cause of increased ROS production and the leading cause of CKD through the increase of oxidative stress and the induction of cellular damage in patients with diabetes, and thus, it is strongly implicated in the pathogenesis of diabetic-related complications (174). High glucose levels suppress the activation of the HO-1 gene, and reduced HO-1 activity not only increases the levels of heme and superoxide anion (O2−) but also decreases the levels of CO and bilirubin, thereby aggravating glucose-mediated oxidative stress (175–178). These observations have been confirmed by experiments with TNF, heme/hemoglobin, and H2O2 exposure, in which decreased HO-1 activity resulted in reduced cell viability (179–182).
The diabetic db/db mouse model shows increased glomerular HO-1 expression (183). This particular phenomenon was also observed for other animal models of diabetes such as streptozotocin (STZ)-induced diabetes mellitus (184). In vitro experiments, high glucose-treated podocytes showed increased HO-1 expression and apoptosis, and the inhibition of HO-1 accentuated podocytes apoptosis (185). Additionally, Nrf2 overexpression in mouse mesangial cells up-regulated the expression of HO-1, as well as reduced high glucose-induced ROS and cell proliferation (186). The reverse effects were observed in cells with Nrf2 knockdown, suggesting a protective role for HO-1 in both podocytes and mesangial cells. Furthermore, HO-1 overexpression prevents apoptosis and cell death mediated by hyperglycemia due to the increased levels of the anti-apoptotic protein B-cell leukemia-lymphoma-xL (Bcl-xL) and several growth factors, as well as the decreased level of MCP-1 (187), again verifying the beneficial effects of HO-1.
Among the HO-1 inducers, hemin can drastically increase HO-1 expression, especially in tubules (188). After chronic administration of hemin in STZ DN rats, these animals showed increased creatinine clearance, decreased iNOS, and decreased urea levels (189). However, hemin can selectively stimulate anti-inflammatory M2 macrophages and IL-10 production, thereby reducing interstitial macrophage infiltration in STZ DN rats (190). Pro-inflammatory M1 macrophages, as well as the suppressive extracellular matrix/profibrotic factors, are also concomitantly abated (190). A recent study has reported that hemin can improve insulin sensitivity by up-regulating HO-1, presumably due to plasma insulin and potentiated agents implicated in insulin sensitization and insulin signaling (191). On the other hand, chromium mesoporphyrin (CrMP), an HO-1 inhibitor, can nullify the hemin-dependent anti-diabetic and insulin-sensitizing effects (191). Several other HO-1 inducers, such as sitagliptin, can mitigate renal injury in STZ-treated rats by activating phosphatidylinositol 3-kinase (PI3K) and Nrf2 (192). Additionally, inducers of HO-1 can improve insulin sensitivity (193), which supports their protective effects and offers the possibility of new therapeutic approaches. Finally, the relevant role of HO-2 in protecting individuals from DN cannot be ignored. STZ-induced diabetes in HO-2-deficient mice stimulates superoxide anion production and provokes prominent tubulointerstitial injury, thereby resulting in enhanced renal dysfunction. Conversely, these negative effects are attenuated when HO-1 is upregulated in these mice (194).
To conclude, chronic hyperglycemia in DN has the capacity to increase oxidative stress and cell apoptosis. In this context, HO-1 can be induced in several types of cells, including podocytes and mesangial cells, thereby limiting podocyte apoptosis and mesangial proliferation. HO-1 also plays an unexpected role in DN under the action of HO-1 inducers by reducing interstitial macrophage infiltration and improving insulin sensitivity.
Kidney transplantation is considered to be the mainstay of treatment and the preferred replacement therapy for patients with ESRD (195). The failure of transplanted organs is usually attributed to the early complications of IRI, namely, acute and chronic rejection (196). Although most research has focused on preventing T-cell responses that lead to acute rejection in a more specific and less toxic way, none of these problems have been completely overcome. Transplantation is a state of ischemia-reperfusion, and the main factors affecting organ function after transplantation are nutrient deprivation and hypoxia, with reperfusion aggravating organ damage initially caused by ischemia (197). Oxidative stress, apoptosis, and a non-specific innate immune response that subsequently activates the specific immune system are the leading causes of deteriorated early graft function (198). Accordingly, HO-1 can potentially prevent oxidative stress due to its anti-oxidant and anti-apoptotic properties and suppress the immune response through its immunomodulatory effects.
During organ transplantation, grafts are successively subjected to global cold ischemia, warm ischemia, and blood reperfusion. These steps are thought to compromise graft function and to aggravate both acute and chronic rejection (199). The length of warm ischemia is associated with the extent of tissue damage in renal IRI. The prolonged duration of warm ischemia results in inflammation, local tissue injury, higher free heme levels, and upregulated levels of HO-1, C5a receptor (C5aR), IL-6, and TNF-α (200). However, during the cold ischemia period, high HO-1 expression is related to an inferior outcome, although previous studies cannot identify a direct association between the longer ischemia time and the higher HO-1 expression (201). In a rat model of kidney transplantation, hyperthermic preconditioning or Co-PP induces HO-1 expression. Furthermore, HO-1-induced animals show decreased expression of apoptosis markers and signs of injury, indicating that graft survival and function are improved (202). HO-1 induction by hemin or fenoldopam in donors has also yielded similar beneficial effects that associate with preserved kidney graft function and prevention of apoptosis after reperfusion (203, 204).
T cell and B cell activation by the graft is recognized as the acute rejection episode after kidney transplantation (205). During acute renal allograft rejection, HO-1 is mainly induced in infiltrating macrophages (206). HIF-1 is also significantly up-regulated in both tubules and infiltrating cells, indicating that the rejected grafts are hypoxic (207). This effect may result in the up-regulation of HO-1 in animal models, which may inhibit the cytotoxicity mediated by T cells and NK cells and may reduce the number of DCs in the graft and lymph nodes derived by the donor, and, thus, improve the graft survival (28). Moreover, administration of low doses of cyclosporine A, immune-modulatory peptides, can induce HO-1 to reduce allograft injury and to improve graft function (208). However, recent observations indicate that HO-1 may also be involved in B cell differentiation, which may potentially increase the risk of acute rejection (27).
In a rat chronic renal transplantation model, the induction of HO-1 in the donor kidney by Co-PP improves survival, kidney function, and the morphologic characteristics of grafts (209). The kidneys from Co-PP-treated donor animals exhibit longer preservation under ischemic conditions, and thus, better graft survival (209). Similar findings have also been reported in brain-dead donors, with the up-regulation of HO-1 by Co-PP ameliorating survival of the kidney graft (210). HO-1 overexpression in the recipient can also reduce chronic kidney allograft injury (211). In this regard, there is evidence to suggest that HO-1 can engender cell death induced by the activation of alloreactive T cells, which facilitates graft tolerance (212).
In addition, some of the effects of HO-1 may be associated with the expression of the byproducts of heme degradation such as CO and biliverdin (4, 213, 214). For instance, CO can reduce graft immunogenicity after engraftment and improve allograft function, thereby slowing the progression of chronic allograft nephropathy (76). Despite high doses of CO and their clinical limitations, low doses of CO have been shown to attenuate IRI (207), which was probably due to the stabilization of various enzymes, thereby reducing their degradation and release of heme (215). In a swine model of a kidney allograft, CO exposure reduced acute tubular necrosis and apoptosis, as well as the expression of tissue factor and P-selectin, and enhanced cell proliferative repair (216). Moreover, CO induction before organ procurement may avoid chronic rejection of the kidney in rats (217, 218), and this effect may be more significant with co-administration of CO and biliverdin (78). Furthermore, administration of biliverdin reduced the CD4+ T cell response by suppressing immune transcription factor activation, inhibiting co-stimulatory activity, and down-regulating major histocompatibility complex (MHC) class II expression (38). The beneficial effects of bilirubin administration have been proven in patients with islet allografts (219). Thus, HO-1 induction and administration of its products may confer beneficial effects to the transplanted kidney.
During the past few decades, important roles for HO-1 and its byproducts in the pathophysiology of kidney diseases have been reported and supported by an abundance of evidence. As mentioned above, HO-1 has the capacity to affect the development and the function of a variety of immune cells and kidney resident cells. Under physiological conditions, HO-1 expression in these cells contributes directly or indirectly to the protection of renal function by eliminating free heme and preventing heme-induced inflammation. Under certain pathological conditions, such as kidney transplantation, autoimmune disease, and autoinflammatory disease, HO-1 can regulate and/or inhibit excessive immune responses to cellular stress. HO-1-based immunotherapy may represent a promising strategy to circumvent kidney diseases. These approaches will likely include dietary and herbal medicines or gene therapy-mediated induction of HO-1 as well as the administration of its byproducts. However, it is crucial to consider the pharmacological properties of these compounds as well as the reported discrepancies between ex vivo/in vivo simulations and the actual clinical situations. The administration of heme and HO-1 byproducts for prolonged periods or at high concentrations exhibits toxic properties. Thus, the enhancement of HO-1 and its byproducts and the down-regulation of free heme should be maintained at acceptable non-toxic levels. Furthermore, despite the amelioration of the underlying disease, chronic HO-1-induced immune suppression remains a significant challenge.
YL, KM, and ZH were involved in the conception of the study. YL and KM were involved in writing the article. XS, PZ, ZD, LS, and CL critically revised the manuscript. All authors read and approved the final manuscript.
This research was funded by National Key Research and Development Program of China (2018YFA0108700, 2017YFA0105602), NSFC Projects of International Cooperation and Exchanges (81720108004), National Natural Science Foundation of China (81974019), and The Research Team Project of Natural Science Foundation of Guangdong Province of China (2017A030312007). The key program of guangzhou science research plan (201904020047). The Special Project of Dengfeng Program of Guangdong Provincial People's Hospital (DFJH201812; KJ012019119; KJ012019423). Overseas Famous Teacher Project of Guangdong Provincial Department of Science and Technology (2020A1414010311).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Luyckx VA, Al-Aly Z, Bello AK, Bellorin-Font E, Carlini RG, Fabian J, et al. Sustainable development goals relevant to kidney health: an update on progress. Nat Rev Nephrol. (2021) 17:15–32. doi: 10.1038/s41581-020-00363-6
2. Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. (1997) 37:517–54. doi: 10.1146/annurev.pharmtox.37.1.517
3. Li B-Z, Guo B, Zhang H-Y, Liu J, Tao S-S, Pan H-F, et al. Therapeutic potential of HO-1 in autoimmune diseases. Inflammation. (2014) 37:1779–88. doi: 10.1007/s10753-014-9908-z
4. Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. (2006) 70:432–43. doi: 10.1038/sj.ki.5001565
5. Funes SC, Rios M, Fernandez-Fierro A, Covian C, Bueno SM, Riedel CA, et al. Naturally derived heme-oxygenase 1 inducers and their therapeutic application to immune-mediated diseases. Front Immunol. (2020) 11:1467. doi: 10.3389/fimmu.2020.01467
6. Tenhunen R, Ross ME, Marver HS, Schmid R. Reduced nicotinamide-adenine dinucleotide phosphate dependent biliverdin reductase: partial purification and characterization. Biochemistry. (1970) 9:298–303. doi: 10.1021/bi00804a016
7. Spencer AL, Bagai I, Becker DF, Zuiderweg ER, Ragsdale SW. Protein/protein interactions in the mammalian heme degradation pathway: heme oxygenase-2, cytochrome P450 reductase, and biliverdin reductase. J Biol Chem. (2014) 289:29836–58. doi: 10.1074/jbc.M114.582783
8. Munoz-Sanchez J, Chanez-Cardenas ME. A review on hemeoxygenase-2: focus on cellular protection and oxygen response. Oxid Med Cell Longev. (2014) 2014:604981. doi: 10.1155/2014/604981
9. Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem. (1986) 261:411–9. doi: 10.1016/S0021-9258(17)42488-4
10. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. (2010) 50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600
11. Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. (2010) 80:1895–903. doi: 10.1016/j.bcp.2010.07.014
12. Wang Y, Yu C, Pan Y, Li J, Zhang Y, Ye F, et al. A novel compound C12 inhibits inflammatory cytokine production and protects from inflammatory injury in vivo. PLoS ONE. (2011) 6:e24377. doi: 10.1371/journal.pone.0024377
13. Borges MC, Vinolo MA, Crisma AR, Fock RA, Borelli P, Tirapegui J, et al. High-fat diet blunts activation of the nuclear factor-kappaB signaling pathway in lipopolysaccharide-stimulated peritoneal macrophages of Wistar rats. Nutrition. (2013) 29:443–9. doi: 10.1016/j.nut.2012.06.008
14. Jeong YH, Oh YC, Cho WK, Lee B, Ma JY. Anti-inflammatory effects of melandrii herba ethanol extract via inhibition of NF-kappaB and MAPK signaling pathways and induction of HO-1 in RAW 264.7 cells and mouse primary macrophages. Molecules. (2016) 21:818. doi: 10.3390/molecules21060818
15. Vijayan V, Wagener F, Immenschuh S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem Pharmacol. (2018) 153:159–67. doi: 10.1016/j.bcp.2018.02.010
16. Tzima S, Victoratos P, Kranidioti K, Alexiou M, Kollias G. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J Exp Med. (2009) 206:1167–79. doi: 10.1084/jem.20081582
17. Chauveau C, Bouchet D, Roussel JC, Mathieu P, Braudeau C, Renaudin K, et al. Gene transfer of heme oxygenase-1 and carbon monoxide delivery inhibit chronic rejection. Am J Transplant. (2002) 2:581–92. doi: 10.1034/j.1600-6143.2002.20702.x
18. George JF, Braun A, Brusko TM, Joseph R, Bolisetty S, Wasserfall CH, et al. Suppression by CD4+CD25+ regulatory T cells is dependent on expression of heme oxygenase-1 in antigen-presenting cells. Am J Pathol. (2008) 173:154–60. doi: 10.2353/ajpath.2008.070963
19. Moreau A, Hill M, Thebault P, Deschamps JY, Chiffoleau E, Chauveau C, et al. Tolerogenic dendritic cells actively inhibit T cells through heme oxygenase-1 in rodents and in nonhuman primates. FASEB J. (2009) 23:3070–7. doi: 10.1096/fj.08-128173
20. Chauveau C, Rémy S, Royer PJ, Hill M, Tanguy-Royer S, Hubert FX, et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood. (2005) 106:1694–702. doi: 10.1182/blood-2005-02-0494
21. Schumacher A, Wafula PO, Teles A, El-Mousleh T, Linzke N, Zenclussen ML, et al. Blockage of heme oxygenase-1 abrogates the protective effect of regulatory T cells on murine pregnancy and promotes the maturation of dendritic cells. PLoS ONE. (2012) 7:e42301. doi: 10.1371/journal.pone.0042301
22. Al-Huseini LM, Aw Yeang HX, Hamdam JM, Sethu S, Alhumeed N, Wong W, et al. Heme oxygenase-1 regulates dendritic cell function through modulation of p38 MAPK-CREB/ATF1 signaling. J Biol Chem. (2014) 289:16442–51. doi: 10.1074/jbc.M113.532069
23. Takamiya R, Murakami M, Kajimura M, Goda N, Makino N, Takamiya Y, et al. Stabilization of mast cells by heme oxygenase-1: an anti-inflammatory role. Am J Physiol Heart Circ Physiol. (2002) 283:H861–70. doi: 10.1152/ajpheart.00740.2001
24. Ma YY, Yang MQ, Wang CF, Ding J, Li JY. Inhibiting mast cell degranulation by HO-1 affects dendritic cell maturation in vitro. Inflamm Res. (2014) 63:527–37. doi: 10.1007/s00011-014-0722-8
25. Lin XL, Lv JJ, Lv J, Di CX, Zhang YJ, Zhou T, et al. Heme oxygenase-1 directly binds STAT3 to control the generation of pathogenic Th17 cells during neutrophilic airway inflammation. Allergy. (2017) 72:1972–87. doi: 10.1111/all.13216
26. Lin X, Lv J, Ge D, Bai H, Yang Y, Wu J. Heme oxygenase-1 alleviates eosinophilic inflammation by inhibiting STAT3-SOCS3 signaling. Pediatr Pulmonol. (2020) 55:1440–7. doi: 10.1002/ppul.24759
27. Zhou Z, Ma D, Liu P, Wang P, Wei D, Yu K, et al. Deletion of HO-1 blocks development of B lymphocytes in mice. Cell Signal. (2019) 63:109378. doi: 10.1016/j.cellsig.2019.109378
28. Woo J, Iyer S, Cornejo MC, Mori N, Gao L, Sipos I, et al. Stress protein-induced immunosuppression: inhibition of cellular immune effector functions following overexpression of haem oxygenase (HSP 32). Transpl Immunol. (1998) 6:84–93. doi: 10.1016/S0966-3274(98)80022-1
29. Kim KS, Zhang DL, Kovtunovych G, Ghosh MC, Ollivierre H, Eckhaus MA, et al. Infused wild-type macrophages reside and self-renew in the liver to rescue the hemolysis and anemia of Hmox1-deficient mice. Blood Adv. (2018) 2:2732–43. doi: 10.1182/bloodadvances.2018019737
30. Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol. (2014) 5:115. doi: 10.3389/fphar.2014.00115
31. Canesin G, Hejazi SM, Swanson KD, Wegiel B. Heme-derived metabolic signals dictate immune responses. Front Immunol. (2020) 11:66. doi: 10.3389/fimmu.2020.00066
32. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. (2007) 282:20221–9. doi: 10.1074/jbc.M610737200
33. Preiser JC. Oxidative stress. JPEN J Parenter Enteral Nutr. (2012) 36:147–54. doi: 10.1177/0148607111434963
34. Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. (2016) 127:473–86. doi: 10.1182/blood-2015-08-663245
35. Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys. (2014) 564:83–8. doi: 10.1016/j.abb.2014.09.005
36. Weis N, Weigert A, von Knethen A, Brune B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol Biol Cell. (2009) 20:1280–8. doi: 10.1091/mbc.e08-10-1005
37. Bozza MT, Jeney V. Pro-inflammatory actions of heme and other hemoglobin-derived DAMPs. Front Immunol. (2020) 11:1323. doi: 10.3389/fimmu.2020.01323
38. Liu Y, Li P, Lu J, Xiong W, Oger J, Tetzlaff W, et al. Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J Immunol. (2008) 181:1887–97. doi: 10.4049/jimmunol.181.3.1887
39. Yamashita K, McDaid J, Ollinger R, Tsui TY, Berberat PO, Usheva A, et al. Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J. (2004) 18:765–7. doi: 10.1096/fj.03-0839fje
40. Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. (2000) 6:422–8. doi: 10.1038/74680
41. Freitas A, Alves-Filho JC, Secco DD, Neto AF, Ferreira SH, Barja-Fidalgo C, et al. Heme oxygenase/carbon monoxide-biliverdin pathway down regulates neutrophil rolling, adhesion and migration in acute inflammation. Br J Pharmacol. (2006) 149:345–54. doi: 10.1038/sj.bjp.0706882
42. Xia ZW, Xu LQ, Zhong WW, Wei JJ, Li NL, Shao J, et al. Heme oxygenase-1 attenuates ovalbumin-induced airway inflammation by up-regulation of foxp3 T-regulatory cells, interleukin-10, and membrane-bound transforming growth factor- 1. Am J Pathol. (2007) 171:1904–14. doi: 10.2353/ajpath.2007.070096
43. Lin YT, Chen YH, Yang YH, Jao HC, Abiko Y, Yokoyama K, et al. Heme oxygenase-1 suppresses the infiltration of neutrophils in rat liver during sepsis through inactivation of p38 MAPK. Shock. (2010) 34:615–21. doi: 10.1097/SHK.0b013e3181e46ee0
44. Chiang N, Shinohara M, Dalli J, Mirakaj V, Kibi M, Choi AM, et al. Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J Immunol. (2013) 190:6378–88. doi: 10.4049/jimmunol.1202969
45. Zhang Y, Zhang L, Wu J, Di C, Xia Z. Heme oxygenase-1 exerts a protective role in ovalbumin-induced neutrophilic airway inflammation by inhibiting Th17 cell-mediated immune response. J Biol Chem. (2013) 288:34612–26. doi: 10.1074/jbc.M113.494369
46. Wong TH, Chen HA, Gau RJ, Yen JH, Suen JL. Heme oxygenase-1-expressing dendritic cells promote Foxp3+ regulatory T Cell differentiation and induce less severe airway inflammation in murine models. PLoS ONE. (2016) 11:e0168919. doi: 10.1371/journal.pone.0168919
47. Lewkowicz N, Klink M, Mycko MP, Lewkowicz P. Neutrophil–CD4+CD25+ T regulatory cell interactions: a possible new mechanism of infectious tolerance. Immunobiology. (2013) 218:455–64. doi: 10.1016/j.imbio.2012.05.029
48. Vannacci A, Baronti R, Zagli G, Marzocca C, Pierpaoli S, Bani D, et al. Carbon monoxide modulates the response of human basophils to FcepsilonRI stimulation through the heme oxygenase pathway. Eur J Pharmacol. (2003) 465:289–97. doi: 10.1016/S0014-2999(03)01489-4
49. Burt TD, Seu L, Mold JE, Kappas A, McCune JM. Naive human T cells are activated and proliferate in response to the heme oxygenase-1 inhibitor tin mesoporphyrin. J Immunol. (2010) 185:5279–88. doi: 10.4049/jimmunol.0903127
50. Pae HO, Oh GS, Choi BM, Chae SC, Kim YM, Chung KR, et al. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. J Immunol. (2004) 172:4744–51. doi: 10.4049/jimmunol.172.8.4744
51. Andersen MH, Sorensen RB, Brimnes MK, Svane IM, Becker JC, Thor Straten P. Identification of heme oxygenase-1-specific regulatory CD8+ T cells in cancer patients. J Clin Invest. (2009) 119:2245–56. doi: 10.1172/JCI38739
52. Biburger M, Theiner G, Schadle M, Schuler G, Tiegs G. Pivotal advance: heme oxygenase 1 expression by human CD4+ T cells is not sufficient for their development of immunoregulatory capacity. J Leukoc Biol. (2010) 87:193–202. doi: 10.1189/jlb.0508280
53. Zelenay S, Chora A, Soares MP, Demengeot J. Heme oxygenase-1 is not required for mouse regulatory T cell development and function. Int Immunol. (2007) 19:11–8. doi: 10.1093/intimm/dxl116
54. Shen Z, Teng X, Qian X, He M, Hu Y, Ye W, et al. Immunoregulation effect by overexpression of heme oxygenase-1 on cardiac xenotransplantation. Transplant Proc. (2011) 43:1994–7. doi: 10.1016/j.transproceed.2011.03.037
55. Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, et al. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. (2004) 165:1045–53. doi: 10.1016/S0002-9440(10)63365-2
56. Nath M, Agarwal A. New insights into the role of heme oxygenase-1 in acute kidney injury. Kidney Res Clin Pract. (2020) 39:387–401. doi: 10.23876/j.krcp.20.091
57. Ronco C, Bellomo R, Kellum JA. Acute kidney injury. Lancet. (2019) 394:1949–64. doi: 10.1016/S0140-6736(19)32563-2
58. Price PM, Safirstein RL, Megyesi J. The cell cycle and acute kidney injury. Kidney Int. (2009) 76:604–13. doi: 10.1038/ki.2009.224
59. Kumar S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int. (2018) 93:27–40. doi: 10.1016/j.kint.2017.07.030
60. Nath KA, Croatt AJ, Haggard JJ, Grande JP. Renal response to repetitive exposure to heme proteins: chronic injury induced by an acute insult. Kidney Int. (2000) 57:2423–33. doi: 10.1046/j.1523-1755.2000.00101.x
61. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
62. Schaer CA, Laczko E, Schoedon G, Schaer DJ, Vallelian F. Chloroquine interference with hemoglobin endocytic trafficking suppresses adaptive heme and iron homeostasis in macrophages: the paradox of an antimalarial agent. Oxid Med Cell Longev. (2013) 2013:870472. doi: 10.1155/2013/870472
63. Bagley WH, Yang H, Shah KH. Rhabdomyolysis. Intern Emerg Med. (2007) 2:210–8. doi: 10.1007/s11739-007-0060-8
64. Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. (2011) 7:189–200. doi: 10.1038/nrneph.2011.16
65. Ratliff BB, Abdulmahdi W, Pawar R, Wolin MS. Oxidant mechanisms in renal injury and disease. Antioxid Redox Signal. (2016) 25:119–46. doi: 10.1089/ars.2016.6665
66. Tuttolomondo A, Pecoraro R, Pinto A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: a review of the evidence to date. Drug Des Devel Ther. (2014) 8:2221–38. doi: 10.2147/DDDT.S67655
67. Chen Y, Qiu J, Chen B, Lin Y, Chen Y, Xie G, et al. Long non-coding RNA NEAT1 plays an important role in sepsis-induced acute kidney injury by targeting miR-204 and modulating the NF-κB pathway. Int Immunopharmacol. (2018) 59:252–60. doi: 10.1016/j.intimp.2018.03.023
68. Hull TD, Kamal AI, Boddu R, Bolisetty S, Guo L, Tisher CC, et al. Heme oxygenase-1 regulates myeloid cell trafficking in AKI. J Am Soc Nephrol. (2015) 26:2139–51. doi: 10.1681/ASN.2014080770
69. Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, et al. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest. (1992) 90:267–70. doi: 10.1172/JCI115847
70. Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm. (2009) 2009:137072. doi: 10.1155/2009/137072
71. Ferenbach DA, Ramdas V, Spencer N, Marson L, Anegon I, Hughes J, et al. Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Mol Ther. (2010) 18:1706–13. doi: 10.1038/mt.2010.100
72. Hull TD, Agarwal A, George JF. The mononuclear phagocyte system in homeostasis and disease: a role for heme oxygenase-1. Antioxid Redox Signal. (2014) 20:1770–88. doi: 10.1089/ars.2013.5673
73. Brusko TM, Wasserfall CH, Agarwal A, Kapturczak MH, Atkinson MA. An integral role for heme oxygenase-1 and carbon monoxide in maintaining peripheral tolerance by CD4+CD25+ regulatory T cells. J Immunol. (2005) 174:5181–6. doi: 10.4049/jimmunol.174.9.5181
74. Song R, Mahidhara RS, Zhou Z, Hoffman RA, Seol DW, Flavell RA, et al. Carbon monoxide inhibits T lymphocyte proliferation via caspase-dependent pathway. J Immunol. (2004) 172:1220–6. doi: 10.4049/jimmunol.172.2.1220
75. Kotsch K, Martins PN, Klemz R, Janssen U, Gerstmayer B, Dernier A, et al. Heme oxygenase-1 ameliorates ischemia/reperfusion injury by targeting dendritic cell maturation and migration. Antioxid Redox Signal. (2007) 9:2049–63. doi: 10.1089/ars.2007.1801
76. Martins PN, Reutzel-Selke A, Jurisch A, Denecke C, Attrot K, Pascher A, et al. Induction of carbon monoxide in donor animals prior to organ procurement reduces graft immunogenicity and inhibits chronic allograft dysfunction. Transplantation. (2006) 82:938–44. doi: 10.1097/01.tp.0000232716.91887.c5
77. Chlopicki S, Olszanecki R, Marcinkiewicz E, Lomnicka M, Motterlini R. Carbon monoxide released by CORM-3 inhibits human platelets by a mechanism independent of soluble guanylate cyclase. Cardiovasc Res. (2006) 71:393–401. doi: 10.1016/j.cardiores.2006.03.011
78. Nakao A, Neto JS, Kanno S, Stolz DB, Kimizuka K, Liu F, et al. Protection against ischemia/reperfusion injury in cardiac and renal transplantation with carbon monoxide, biliverdin and both. Am J Transplant. (2005) 5:282–91. doi: 10.1111/j.1600-6143.2004.00695.x
79. Lv Q, Yao Y, Wang W, Xiong W, Liao WH. Biliverdin protects against cisplatin-induced apoptosis of renal tubular epithelial cells. J Huazhong Univ Sci Technolog Med Sci. (2016) 36:48–52. doi: 10.1007/s11596-016-1540-8
80. Tayem Y, Johnson TR, Mann BE, Green CJ, Motterlini R. Protection against cisplatin-induced nephrotoxicity by a carbon monoxide-releasing molecule. Am J Physiol Renal Physiol. (2006) 290:F789–94. doi: 10.1152/ajprenal.00363.2005
81. Gozzelino R, Soares MP. Coupling heme and iron metabolism via ferritin H chain. Antioxid Redox Signal. (2014) 20:1754–69. doi: 10.1089/ars.2013.5666
82. Rossi M, Thierry A, Delbauve S, Preyat N, Soares MP, Roumeguere T, et al. Specific expression of heme oxygenase-1 by myeloid cells modulates renal ischemia-reperfusion injury. Sci Rep. (2017) 7:197. doi: 10.1038/s41598-017-00220-w
83. Gueler F, Park JK, Rong S, Kirsch T, Lindschau C, Zheng W, et al. Statins attenuate ischemia-reperfusion injury by inducing heme oxygenase-1 in infiltrating macrophages. Am J Pathol. (2007) 170:1192–9. doi: 10.2353/ajpath.2007.060782
84. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. (2009) 284:13291–5. doi: 10.1074/jbc.R900010200
85. Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. (2009) 47:1304–9. doi: 10.1016/j.freeradbiomed.2009.07.035
86. Ruan Y, Wang L, Zhao Y, Yao Y, Chen S, Li J, et al. Carbon monoxide potently prevents ischemia-induced high-mobility group box 1 translocation and release and protects against lethal renal ischemia-reperfusion injury. Kidney Int. (2014) 86:525–37. doi: 10.1038/ki.2014.80
87. Seo MS, Kim HJ, Kim H, Park SW. Ethyl pyruvate directly attenuates active secretion of HMGB1 in proximal tubular cells via induction of heme oxygenase-1. J Clin Med. (2019) 8:629. doi: 10.3390/jcm8050629
88. Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. (2010) 21:1878–90. doi: 10.1681/ASN.2009101048
89. Chen Z, Zhong H, Wei J, Lin S, Zong Z, Gong F, et al. Inhibition of Nrf2/HO-1 signaling leads to increased activation of the NLRP3 inflammasome in osteoarthritis. Arthritis Res Ther. (2019) 21:300. doi: 10.1186/s13075-019-2085-6
90. Noel S, Martina MN, Bandapalle S, Racusen LC, Potteti HR, Hamad AR, et al. T Lymphocyte-specific activation of Nrf2 protects from AKI. J Am Soc Nephrol. (2015) 26:2989–3000. doi: 10.1681/ASN.2014100978
91. Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet. (2017) 389:1238–52. doi: 10.1016/S0140-6736(16)32064-5
92. Hagmann H, Brinkkoetter PT. ROS and oxidative stress in CKD patients: is it the mitochondria that keeps CKD patients in bed? Nephrol Dial Transplant. (2015) 30:867–8. doi: 10.1093/ndt/gfv052
93. Hemmelgarn BR, Manns BJ, Lloyd A, James MT, Klarenbach S, Quinn RR, et al. Relation between kidney function, proteinuria, and adverse outcomes. JAMA. (2010) 303:423–9. doi: 10.1001/jama.2010.39
94. Shimizu M, Ohta K, Yang Y, Nakai A, Toma T, Saikawa Y, et al. Glomerular proteinuria induces heme oxygenase-1 gene expression within renal epithelial cells. Pediatr Res. (2005) 58:666–71. doi: 10.1203/01.PDR.0000180557.68222.5A
95. Murali NS, Ackerman AW, Croatt AJ, Cheng J, Grande JP, Sutor SL, et al. Renal upregulation of HO-1 reduces albumin-driven MCP-1 production: implications for chronic kidney disease. Am J Physiol Renal Physiol. (2007) 292:F837–44. doi: 10.1152/ajprenal.00254.2006
96. Wolf G, Jocks T, Zahner G, Panzer U, Stahl RA. Existence of a regulatory loop between MCP-1 and TGF-beta in glomerular immune injury. Am J Physiol Renal Physiol. (2002) 283:F1075–84. doi: 10.1152/ajprenal.00349.2001
97. Viedt C, Dechend R, Fei J, Hansch GM, Kreuzer J, Orth SR. MCP-1 induces inflammatory activation of human tubular epithelial cells: involvement of the transcription factors, nuclear factor-kappaB and activating protein-1. J Am Soc Nephrol. (2002) 13:1534–47. doi: 10.1097/01.ASN.0000015609.31253.7F
98. Viedt C, Orth SR. Monocyte chemoattractant protein-1 (MCP-1) in the kidney: does it more than simply attract monocytes? Nephrol Dial Transplant. (2002) 17:2043–7. doi: 10.1093/ndt/17.12.2043
99. Nath KA, Vercellotti GM, Grande JP, Miyoshi H, Paya CV, Manivel JC, et al. Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int. (2001) 59:106–17. doi: 10.1046/j.1523-1755.2001.00471.x
100. Eddy AA, Neilson EG. Chronic kidney disease progression. J Am Soc Nephrol. (2006) 17:2964–6. doi: 10.1681/ASN.2006070704
101. Bottinger EP. TGF-beta in renal injury and disease. Semin Nephrol. (2007) 27:309–20. doi: 10.1016/j.semnephrol.2007.02.009
102. Hill-Kapturczak N, Jarmi T, Agarwal A. Growth factors and heme oxygenase-1: perspectives in physiology and pathophysiology. Antioxid Redox Signal. (2007) 9:2197–207. doi: 10.1089/ars.2007.1798
103. Zarjou A, Agarwal A. Heme oxygenase-1 as a target for TGF-beta in kidney disease. Semin Nephrol. (2012) 32:277–86. doi: 10.1016/j.semnephrol.2012.04.007
104. Kim JH, Yang JI, Jung MH, Hwa JS, Kang KR, Park DJ, et al. Heme oxygenase-1 protects rat kidney from ureteral obstruction via an antiapoptotic pathway. J Am Soc Nephrol. (2006) 17:1373–81. doi: 10.1681/ASN.2005091001
105. Correa-Costa M, Semedo P, Monteiro AP, Silva RC, Pereira RL, Goncalves GM, et al. Induction of heme oxygenase-1 can halt and even reverse renal tubule-interstitial fibrosis. PLoS ONE. (2010) 5:e14298. doi: 10.1371/journal.pone.0014298
106. Kie JH, Kapturczak MH, Traylor A, Agarwal A, Hill-Kapturczak N. Heme oxygenase-1 deficiency promotes epithelial-mesenchymal transition and renal fibrosis. J Am Soc Nephrol. (2008) 19:1681–91. doi: 10.1681/ASN.2007101099
107. Chen X, Wei SY, Li JS, Zhang QF, Wang YX, Zhao SL, et al. Overexpression of Heme Oxygenase-1 prevents renal interstitial inflammation and fibrosis induced by unilateral ureter obstruction. PLoS ONE. (2016) 11:e0147084. doi: 10.1371/journal.pone.0147084
108. Loboda A, Stachurska A, Podkalicka P, Sobczak M, Mucha O, Witalisz-Siepracka A, et al. Effect of heme oxygenase-1 on ochratoxin A-induced nephrotoxicity in mice. Int J Biochem Cell Biol. (2017) 84:46–57. doi: 10.1016/j.biocel.2017.01.003
109. Wang L, Lee JY, Kwak JH, He Y, Kim SI, Choi ME. Protective effects of low-dose carbon monoxide against renal fibrosis induced by unilateral ureteral obstruction. Am J Physiol Renal Physiol. (2008) 294:F508–17. doi: 10.1152/ajprenal.00306.2007
110. Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet. (2000) 66:187–95. doi: 10.1086/302729
111. Chen YH, Kuo KL, Hung SC, Hsu CC, Chen YH, Tarng DC. Length polymorphism in heme oxygenase-1 and risk of CKD among patients with coronary artery disease. J Am Soc Nephrol. (2014) 25:2669–77. doi: 10.1681/ASN.2013111205
112. Zager RA, Johnson AC, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am J Physiol Renal Physiol. (2011) 301:F1334–45. doi: 10.1152/ajprenal.00431.2011
113. Schellinger IN, Cordasic N, Panesar J, Buchholz B, Jacobi J, Hartner A, et al. Hypoxia inducible factor stabilization improves defective ischemia-induced angiogenesis in a rodent model of chronic kidney disease. Kidney Int. (2017) 91:616–27. doi: 10.1016/j.kint.2016.09.028
114. Strausser SA, Nakano D, Souma T. Acute kidney injury to chronic kidney disease transition: insufficient cellular stress response. Curr Opin Nephrol Hypertens. (2018) 27:314–22. doi: 10.1097/MNH.0000000000000424
115. Al-Jaishi AA, Oliver MJ, Thomas SM, Lok CE, Zhang JC, Garg AX, et al. Patency rates of the arteriovenous fistula for hemodialysis: a systematic review and meta-analysis. Am J Kidney Dis. (2014) 63:464–78. doi: 10.1053/j.ajkd.2013.08.023
117. Ocak G, Verduijn M, Vossen CY, Lijfering WM, Dekker FW, Rosendaal FR, et al. Chronic kidney disease stages 1-3 increase the risk of venous thrombosis. J Thromb Haemost. (2010) 8:2428–35. doi: 10.1111/j.1538-7836.2010.04048.x
118. Kang L, Grande JP, Hillestad ML, Croatt AJ, Barry MA, Katusic ZS, et al. A new model of an arteriovenous fistula in chronic kidney disease in the mouse: beneficial effects of upregulated heme oxygenase-1. Am J Physiol Renal Physiol. (2016) 310:F466–76. doi: 10.1152/ajprenal.00288.2015
119. Marshall CB, Shankland SJ. Cell cycle and glomerular disease: a minireview. Nephron Exp Nephrol. (2006) 102:e39–48. doi: 10.1159/000088400
120. Nath KA, Grande JP, Haggard JJ, Croatt AJ, Katusic ZS, Solovey A, et al. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol. (2001) 158:893–903. doi: 10.1016/S0002-9440(10)64037-0
121. Yokoyama T, Shimizu M, Ohta K, Yuno T, Okajima M, Wada T, et al. Urinary heme oxygenase-1 as a sensitive indicator of tubulointerstitial inflammatory damage in various renal diseases. Am J Nephrol. (2011) 33:414–20. doi: 10.1159/000327020
122. Shepard M, Dhulipala P, Kabaria S, Abraham NG, Lianos EA. Heme oxygenase-1 localization in the rat nephron. Nephron. (2002) 92:660–4. doi: 10.1159/000064113
123. Datta PK, Reddy S, Sharma M, Lianos EA. Differential nephron HO-1 expression following glomerular epithelial cell injury. Nephron Exp Nephrol. (2006) 103:e131–8. doi: 10.1159/000092544
124. Chadban SJ, Atkins RC. Glomerulonephritis. Lancet. (2005) 365:1797–806. doi: 10.1016/S0140-6736(05)66583-X
125. Wenderfer SE, Dubinsky WP, Hernandez-Sanabria M, Braun MC. Urine proteome analysis in murine nephrotoxic serum nephritis. Am J Nephrol. (2009) 30:450–8. doi: 10.1159/000242430
126. Vogt BA, Shanley TP, Croatt A, Alam J, Johnson KJ, Nath KA. Glomerular inflammation induces resistance to tubular injury in the rat. A novel form of acquired, heme oxygenase-dependent resistance to renal injury. J Clin Invest. (1996) 98:2139–45. doi: 10.1172/JCI119020
127. Mosley K, Wembridge DE, Cattell V, Cook HT. Heme oxygenase is induced in nephrotoxic nephritis and hemin, a stimulator of heme oxygenase synthesis, ameliorates disease. Kidney Int. (1998) 53:672–8. doi: 10.1046/j.1523-1755.1998.00798.x
128. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. (2015) 135:626–35. doi: 10.1016/j.jaci.2014.11.001
129. Bolisetty S, Traylor AM, Kim J, Joseph R, Ricart K, Landar A, et al. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J Am Soc Nephrol. (2010) 21:1702–12. doi: 10.1681/ASN.2010030238
130. Moncada S, Bolaños JP. Nitric oxide, cell bioenergetics and neurodegeneration. J Neurochem. (2006) 97:1676–89. doi: 10.1111/j.1471-4159.2006.03988.x
131. Nathan C, Xie QW. Regulation of biosynthesis of nitric oxide. J Biol Chem. (1994) 269:13725–8. doi: 10.1016/S0021-9258(17)36703-0
132. Sharma VS, Traylor TG, Gardiner R, Mizukami H. Reaction of nitric oxide with heme proteins and model compounds of hemoglobin. Biochemistry. (1987) 26:3837–43. doi: 10.1021/bi00387a015
133. Cooper CE. Nitric oxide and iron proteins. Biochim Biophys Acta. (1999) 1411:290–309. doi: 10.1016/S0005-2728(99)00021-3
134. Karupiah G, Harris N. Inhibition of viral replication by nitric oxide and its reversal by ferrous sulfate and tricarboxylic acid cycle metabolites. J Exp Med. (1995) 181:2171–9. doi: 10.1084/jem.181.6.2171
135. Kim YM, Bergonia HA, Muller C, Pitt BR, Watkins WD, Lancaster JR, Jr. Loss and degradation of enzyme-bound heme induced by cellular nitric oxide synthesis. J Biol Chem. (1995) 270:5710–3. doi: 10.1074/jbc.270.11.5710
136. Datta PK, Gross EJ, Lianos EA. Interactions between inducible nitric oxide synthase and heme oxygenase-1 in glomerulonephritis. Kidney Int. (2002) 61:847–50. doi: 10.1046/j.1523-1755.2002.00231.x
137. Dlaska M, Weiss G. Central role of transcription factor NF-IL6 for cytokine and iron-mediated regulation of murine inducible nitric oxide synthase expression. J Immunol. (1999) 162:6171–7.
138. Minamoto K, Harada H, Lama VN, Fedarau MA, Pinsky DJ. Reciprocal regulation of airway rejection by the inducible gas-forming enzymes heme oxygenase and nitric oxide synthase. J Exp Med. (2005) 202:283–94. doi: 10.1084/jem.20050377
139. McMillan K, Bredt DS, Hirsch DJ, Snyder SH, Clark JE, Masters BS. Cloned, expressed rat cerebellar nitric oxide synthase contains stoichiometric amounts of heme, which binds carbon monoxide. Proc Natl Acad Sci USA. (1992) 89:11141–5. doi: 10.1073/pnas.89.23.11141
140. Weiss G, Werner-Felmayer G, Werner ER, Grunewald K, Wachter H, Hentze MW. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med. (1994) 180:969–76. doi: 10.1084/jem.180.3.969
141. White KA, Marletta MA. Nitric oxide synthase is a cytochrome P-450 type hemoprotein. Biochemistry. (1992) 31:6627–31. doi: 10.1021/bi00144a001
142. Xie QW, Leung M, Fuortes M, Sassa S, Nathan C. Complementation analysis of mutants of nitric oxide synthase reveals that the active site requires two hemes. Proc Natl Acad Sci USA. (1996) 93:4891–6. doi: 10.1073/pnas.93.10.4891
143. Descamps-Latscha B, Witko-Sarsat V, Nguyen-Khoa T, Nguyen AT, Gausson V, Mothu N, et al. Early prediction of IgA nephropathy progression: proteinuria and AOPP are strong prognostic markers. Kidney Int. (2004) 66:1606–12. doi: 10.1111/j.1523-1755.2004.00926.x
144. Chen HC, Tomino Y, Yaguchi Y, Fukui M, Yokoyama K, Watanabe A, et al. Oxidative metabolism of polymorphonuclear leukocytes (PMN) in patients with IgA nephropathy. J Clin Lab Anal. (1992) 6:35–9. doi: 10.1002/jcla.1860060108
145. Barratt J, Feehally J. IgA nephropathy. J Am Soc Nephrol. (2005) 16:2088–97. doi: 10.1681/ASN.2005020134
146. Nakamura T, Inoue T, Sugaya T, Kawagoe Y, Suzuki T, Ueda Y, et al. Beneficial effects of olmesartan and temocapril on urinary liver-type fatty acid-binding protein levels in normotensive patients with immunoglobin A nephropathy. Am J Hypertens. (2007) 20:1195–201. doi: 10.1016/j.amjhyper.2007.06.003
147. Kashem A, Endoh M, Yamauchi F, Yano N, Nomoto Y, Sakai H, et al. Superoxide dismutase activity in human glomerulonephritis. Am J Kidney Dis. (1996) 28:14–22. doi: 10.1016/S0272-6386(96)90125-0
148. Kobori H, Katsurada A, Ozawa Y, Satou R, Miyata K, Hase N, et al. Enhanced intrarenal oxidative stress and angiotensinogen in IgA nephropathy patients. Biochem Biophys Res Commun. (2007) 358:156–63. doi: 10.1016/j.bbrc.2007.04.105
149. Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. (1987) 235:1043–6. doi: 10.1126/science.3029864
150. Chin HJ, Cho HJ, Lee TW, Na KY, Yoon HJ, Chae DW, et al. The heme oxygenase-1 genotype is a risk factor to renal impairment of IgA nephropathy at diagnosis, which is a strong predictor of mortality. J Korean Med Sci. (2009) 24 Suppl(Suppl 1):S30–7. doi: 10.3346/jkms.2009.24.S1.S30
151. D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. (2011) 365:2398–411. doi: 10.1056/NEJMra1106556
152. Atsaves V, Detsika MG, Poulaki E, Gakiopoulou H, Lianos EA. Phenotypic characterization of a novel HO-1 depletion model in the rat. Transgenic Res. (2017) 26:51–64. doi: 10.1007/s11248-016-9986-9
153. Yang SM, Chan YL, Hua KF, Chang JM, Chen HL, Tsai YJ, et al. Osthole improves an accelerated focal segmental glomerulosclerosis model in the early stage by activating the Nrf2 antioxidant pathway and subsequently inhibiting NF-kappaB-mediated COX-2 expression and apoptosis. Free Radic Biol Med. (2014) 73:260–9. doi: 10.1016/j.freeradbiomed.2014.05.009
154. Lin EY, Bayarsengee U, Wang CC, Chiang YH, Cheng CW. The natural compound 2,3,5,4'-tetrahydroxystilbene-2-O-β-d glucoside protects against adriamycin-induced nephropathy through activating the Nrf2-Keap1 antioxidant pathway. Environ Toxicol. (2018) 33:72–82. doi: 10.1002/tox.22496
155. Guerrero-Beltran CE, Calderon-Oliver M, Pedraza-Chaverri J, Chirino YI. Protective effect of sulforaphane against oxidative stress: recent advances. Exp Toxicol Pathol. (2012) 64:503–8. doi: 10.1016/j.etp.2010.11.005
156. Pedruzzi LM, Stockler-Pinto MB, Leite M Jr., Mafra D. Nrf2-keap1 system versus NF-kappaB: the good and the evil in chronic kidney disease? Biochimie. (2012) 94:2461–6. doi: 10.1016/j.biochi.2012.07.015
157. Tsokos GC. Systemic lupus erythematosus. N Engl J Med. (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
158. Rekvig OP. Systemic lupus erythematosus: definitions, contexts, conflicts, enigmas. Front Immunol. (2018) 9:387. doi: 10.3389/fimmu.2018.00387
159. Ocampo-Piraquive V, Nieto-Aristizábal I, Cañas CA, Tobón GJ. Mortality in systemic lupus erythematosus: causes, predictors and interventions. Expert Rev Clin Immunol. (2018) 14:1043–53. doi: 10.1080/1744666X.2018.1538789
160. Herrada AA, Llanos C, Mackern-Oberti JP, Carreño LJ, Henriquez C, Gómez RS, et al. Haem oxygenase 1 expression is altered in monocytes from patients with systemic lupus erythematosus. Immunology. (2012) 136:414–24. doi: 10.1111/j.1365-2567.2012.03598.x
161. Cuitino L, Obreque J, Gajardo-Meneses P, Villarroel A, Crisostomo N, San Francisco IF, et al. Heme-Oxygenase-1 Is decreased in circulating monocytes and is associated with impaired phagocytosis and ROS production in lupus nephritis. Front Immunol. (2019) 10:2868. doi: 10.3389/fimmu.2019.02868
162. Ren J, Catalina MD, Eden K, Liao X, Read KA, Luo X, et al. Selective Histone Deacetylase 6 inhibition normalizes B Cell activation and germinal center formation in a model of systemic lupus erythematosus. Front Immunol. (2019) 10:2512. doi: 10.3389/fimmu.2019.02512
163. Lech M, Anders HJ. The pathogenesis of lupus nephritis. J Am Soc Nephrol. (2013) 24:1357–66. doi: 10.1681/ASN.2013010026
164. Funes SC, Manrique de Lara A, Altamirano-Lagos MJ, Mackern-Oberti JP, Escobar-Vera J, Kalergis AM. Immune checkpoints and the regulation of tolerogenicity in dendritic cells: Implications for autoimmunity and immunotherapy. Autoimmun Rev. (2019) 18:359–68. doi: 10.1016/j.autrev.2019.02.006
165. Mackern-Oberti JP, Llanos C, Carreno LJ, Riquelme SA, Jacobelli SH, Anegon I, et al. Carbon monoxide exposure improves immune function in lupus-prone mice. Immunology. (2013) 140:123–32. doi: 10.1111/imm.12124
166. Funes SC, Rios M, Gomez-Santander F, Fernandez-Fierro A, Altamirano-Lagos MJ, Rivera-Perez D, et al. Tolerogenic dendritic cell transfer ameliorates systemic lupus erythematosus in mice. Immunology. (2019) 158:322–39. doi: 10.1111/imm.13119
167. Takeda Y, Takeno M, Iwasaki M, Kobayashi H, Kirino Y, Ueda A, et al. Chemical induction of HO-1 suppresses lupus nephritis by reducing local iNOS expression and synthesis of anti-dsDNA antibody. Clin Exp Immunol. (2004) 138:237–44. doi: 10.1111/j.1365-2249.2004.02594.x
168. Mackern-Oberti JP, Obreque J, Mendez GP, Llanos C, Kalergis AM. Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus. Clin Exp Immunol. (2015) 182:1–13. doi: 10.1111/cei.12657
169. Li D, Shi G, Wang J, Zhang D, Pan Y, Dou H, et al. Baicalein ameliorates pristane-induced lupus nephritis via activating Nrf2/HO-1 in myeloid-derived suppressor cells. Arthritis Res Ther. (2019) 21:105. doi: 10.1186/s13075-019-1876-0
170. Aparicio-Soto M, Sanchez-Hidalgo M, Cardeno A, Rosillo MA, Sanchez-Fidalgo S, Utrilla J, et al. Dietary extra virgin olive oil attenuates kidney injury in pristane-induced SLE model via activation of HO-1/Nrf-2 antioxidant pathway and suppression of JAK/STAT, NF-kappaB and MAPK activation. J Nutr Biochem. (2016) 27:278–88. doi: 10.1016/j.jnutbio.2015.09.017
171. Kong LL, Wu H, Cui WP, Zhou WH, Luo P, Sun J, et al. Advances in murine models of diabetic nephropathy. J Diabetes Res. (2013) 2013:797548. doi: 10.1155/2013/797548
172. Lu Z, Liu N, Wang F. Epigenetic regulations in diabetic nephropathy. J Diabetes Res. (2017) 2017:7805058. doi: 10.1155/2017/7805058
173. Sagoo MK, Gnudi L. Diabetic nephropathy: an overview. Methods Mol Biol. (2020) 2067:3–7. doi: 10.1007/978-1-4939-9841-8_1
174. Aghadavod E, Khodadadi S, Baradaran A, Nasri P, Bahmani M, Rafieian-Kopaei M. Role of oxidative stress and inflammatory factors in diabetic kidney disease. Iran J Kidney Dis. (2016) 10:337–43. Available online at: https://pubmed.ncbi.nlm.nih.gov/27903991/
175. Appleton SD, Lash GE, Marks GS, Nakatsu K, Brien JF, Smith GN, et al. Effect of glucose and oxygen deprivation on heme oxygenase expression in human chorionic villi explants and immortalized trophoblast cells. Am J Physiol Regul Integr Comp Physiol. (2003) 285:R1453–60. doi: 10.1152/ajpregu.00234.2003
176. Chang SH, Barbosa-Tessmann I, Chen C, Kilberg MS, Agarwal A. Glucose deprivation induces heme oxygenase-1 gene expression by a pathway independent of the unfolded protein response. J Biol Chem. (2002) 277:1933–40. doi: 10.1074/jbc.M108921200
177. Chang SH, Garcia J, Melendez JA, Kilberg MS, Agarwal A. Haem oxygenase 1 gene induction by glucose deprivation is mediated by reactive oxygen species via the mitochondrial electron-transport chain. Biochem J. (2003) 371:877–85. doi: 10.1042/bj20021731
178. Quan S, Kaminski PM, Yang L, Morita T, Inaba M, Ikehara S, et al. Heme oxygenase-1 prevents superoxide anion-associated endothelial cell sloughing in diabetic rats. Biochem Biophys Res Commun. (2004) 315:509–16. doi: 10.1016/j.bbrc.2004.01.086
179. Abraham NG, Da Silva JL, Dunn MW, Kigasawa K, Shibahara S. Retinal pigment epithelial cell-based gene therapy against hemoglobin toxicity. Int J Mol Med. (1998) 1:657–63. doi: 10.3892/ijmm.1.4.657
180. Colombrita C, Lombardo G, Scapagnini G, Abraham NG. Heme oxygenase-1 expression levels are cell cycle dependent. Biochem Biophys Res Commun. (2003) 308:1001–8. doi: 10.1016/S0006-291X(03)01509-2
181. Kushida T, Li Volti G, Quan S, Goodman A, Abraham NG. Role of human heme oxygenase-1 in attenuating TNF-alpha-mediated inflammation injury in endothelial cells. J Cell Biochem. (2002) 87:377–85. doi: 10.1002/jcb.10316
182. Yang L, Quan S, Abraham NG. Retrovirus-mediated HO gene transfer into endothelial cells protects against oxidant-induced injury. Am J Physiol. (1999) 277:L127–33. doi: 10.1152/ajplung.1999.277.1.L127
183. Liu J, Wang L, Tian XY, Liu L, Wong WT, Zhang Y, et al. Unconjugated bilirubin mediates heme oxygenase-1-induced vascular benefits in diabetic mice. Diabetes. (2015) 64:1564–75. doi: 10.2337/db14-1391
184. Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 GeneAblation and expression. J Am Soc Nephrol. (2000) 11:965–73. doi: 10.1681/ASN.V115965
185. Lee SC, Han SH, Li JJ, Lee SH, Jung DS, Kwak SJ, et al. Induction of heme oxygenase-1 protects against podocyte apoptosis under diabetic conditions. Kidney Int. (2009) 76:838–48. doi: 10.1038/ki.2009.286
186. Li H, Wang F, Zhang L, Cao Y, Liu W, Hao J, et al. Modulation of Nrf2 expression alters high glucose-induced oxidative stress and antioxidant gene expression in mouse mesangial cells. Cell Signal. (2011) 23:1625–32. doi: 10.1016/j.cellsig.2011.05.016
187. Sacerdoti D, Olszanecki R, Li Volti G, Colombrita C, Scapagnini G, Abraham NG. Heme oxygenase overexpression attenuates glucose-mediated oxidative stress in quiescent cell phase: linking heme to hyperglycemia complications. Curr Neurovasc Res. (2005) 2:103–11. doi: 10.2174/1567202053586802
188. Chebotareva N, Bobkova I, Shilov E. Heat shock proteins and kidney disease: perspectives of HSP therapy. Cell Stress Chaperones. (2017) 22:319–43. doi: 10.1007/s12192-017-0790-0
189. Ptilovanciv EO, Fernandes GS, Teixeira LC, Reis LA, Pessoa EA, Convento MB, et al. Heme oxygenase 1 improves glucoses metabolism and kidney histological alterations in diabetic rats. Diabetol Metab Syndr. (2013) 5:3. doi: 10.1186/1758-5996-5-3
190. Ndisang JF, Jadhav A. Hemin therapy improves kidney function in male streptozotocin-induced diabetic rats: role of the heme oxygenase/atrial natriuretic peptide/adiponectin axis. Endocrinology. (2014) 155:215–29. doi: 10.1210/en.2013-1050
191. Ndisang JF, Jadhav A. Heme oxygenase system enhances insulin sensitivity and glucose metabolism in streptozotocin-induced diabetes. Am J Physiol Endocrinol Metab. (2009) 296:E829–41. doi: 10.1152/ajpendo.90783.2008
192. Wang J, Hu L, Chen Y, Fu T, Jiang T, Jiang A, et al. Sitagliptin improves renal function in diabetic nephropathy in male Sprague Dawley rats through upregulating heme oxygenase-1 expression. Endocrine. (2019) 63:70–8. doi: 10.1007/s12020-018-1721-2
193. Nicolai A, Li M, Kim DH, Peterson SJ, Vanella L, Positano V, et al. Heme oxygenase-1 induction remodels adipose tissue and improves insulin sensitivity in obesity-induced diabetic rats. Hypertension. (2009) 53:508–15. doi: 10.1161/HYPERTENSIONAHA.108.124701
194. Goodman AI, Chander PN, Rezzani R, Schwartzman ML, Regan RF, Rodella L, et al. Heme oxygenase-2 deficiency contributes to diabetes-mediated increase in superoxide anion and renal dysfunction. J Am Soc Nephrol. (2006) 17:1073–81. doi: 10.1681/ASN.2004121082
195. Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med. (1999) 341:1725–30. doi: 10.1056/NEJM199912023412303
196. Petra H, Eva H, Irena B, Petra H, Ondrej V. Molecular profiling of acute and chronic rejections of renal allografts. Clin Dev Immunol. (2013) 2013:509259. doi: 10.1155/2013/509259
197. Faucher Q, Alarcan H, Marquet P, Barin-Le Guellec C. Effects of ischemia-reperfusion on tubular cell membrane transporters and consequences in kidney transplantation. J Clin Med. (2020) 9:2610. doi: 10.3390/jcm9082610
198. Boros P, Bromberg JS. New cellular and molecular immune pathways in ischemia/reperfusion injury. Am J Transplant. (2006) 6:652–8. doi: 10.1111/j.1600-6143.2005.01228.x
199. Land W, Schneeberger H, Schleibner S, Illner WD, Abendroth D, Rutili G, et al. The beneficial effect of human recombinant superoxide dismutase on acute and chronic rejection events in recipients of cadaveric renal transplants. Transplantation. (1994) 57:211–7. doi: 10.1097/00007890-199401001-00010
200. Wang L, Vijayan V, Jang MS, Thorenz A, Greite R, Rong S, et al. Labile heme aggravates renal inflammation and complement activation after ischemia reperfusion injury. Front Immunol. (2019) 10:2975. doi: 10.3389/fimmu.2019.02975
201. Corona D, Ekser B, Gioco R, Caruso M, Schipa C, Veroux P, et al. Heme-oxygenase and kidney transplantation: a potential for target therapy? Biomolecules. (2020) 10:840. doi: 10.3390/biom10060840
202. Wagner M, Cadetg P, Ruf R, Mazzucchelli L, Ferrari P, Redaelli CA. Heme oxygenase-1 attenuates ischemia/reperfusion-induced apoptosis and improves survival in rat renal allografts. Kidney Int. (2003) 63:1564–73. doi: 10.1046/j.1523-1755.2003.00897.x
203. Hölzen JP, August C, Bahde R, Minin E, Lang D, Heidenreich S, et al. Influence of heme oxygenase-1 on microcirculation after kidney transplantation. J Surg Res. (2008) 148:126–35. doi: 10.1016/j.jss.2007.10.007
204. Salahudeen AK, Yang M, Huang H, Dore S, Stec DE. Fenoldopam preconditioning: role of heme oxygenase-1 in protecting human tubular cells and rodent kidneys against cold-hypoxic injury. Transplantation. (2011) 91:176–82. doi: 10.1097/TP.0b013e3181fffff2
205. Goldberg RJ, Weng FL, Kandula P. Acute and chronic allograft dysfunction in kidney transplant recipients. Med Clin North Am. (2016) 100:487–503. doi: 10.1016/j.mcna.2016.01.002
206. Agarwal A, Kim Y, Matas AJ, Alam J, Nath KA. Gas-generating systems in acute renal allograft rejection in the rat. Co-induction of heme oxygenase and nitric oxide synthase. Transplantation. (1996) 61:93–8. doi: 10.1097/00007890-199601150-00019
207. Rosenberger C, Pratschke J, Rudolph B, Heyman SN, Schindler R, Babel N, et al. Immunohistochemical detection of hypoxia-inducible factor-1alpha in human renal allograft biopsies. J Am Soc Nephrol. (2007) 18:343–51. doi: 10.1681/ASN.2006070792
208. Magee CC, Azuma H, Knoflach A, Denton MD, Chandraker A, Iyer S, et al. In vitro and in vivo immunomodulatory effects of RDP1258, a novel synthetic peptide. J Am Soc Nephrol. (1999) 10:1997–2005. doi: 10.1681/ASN.V1091997
209. Tullius SG, Nieminen-Kelha M, Buelow R, Reutzel-Selke A, Martins PN, Pratschke J, et al. Inhibition of ischemia/reperfusion injury and chronic graft deterioration by a single-donor treatment with cobalt-protoporphyrin for the induction of heme oxygenase-1. Transplantation. (2002) 74:591–8. doi: 10.1097/00007890-200209150-00001
210. Kotsch K, Francuski M, Pascher A, Klemz R, Seifert M, Mittler J, et al. Improved long-term graft survival after HO-1 induction in brain-dead donors. Am J Transplant. (2006) 6:477–86. doi: 10.1111/j.1600-6143.2005.01208.x
211. Bédard EL, Jiang J, Parry N, Wang H, Liu W, Garcia B, et al. Peritransplant treatment with cobalt protoporphyrin attenuates chronic renal allograft rejection. Transpl Int. (2005) 18:341–9. doi: 10.1111/j.1432-2277.2004.00062.x
212. McDaid J, Yamashita K, Chora A, Ollinger R, Strom TB, Li XC, et al. Heme oxygenase-1 modulates the allo-immune response by promoting activation-induced cell death of T cells. FASEB J. (2005) 19:458–60. doi: 10.1096/fj.04-2217fje
213. Sikorski EM, Hock T, Hill-Kapturczak N, Agarwal A. The story so far: Molecular regulation of the heme oxygenase-1 gene in renal injury. Am J Physiol Renal Physiol. (2004) 286:F425–41. doi: 10.1152/ajprenal.00297.2003
214. Jarmi T, Agarwal A. Heme oxygenase and renal disease. Curr Hypertens Rep. (2009) 11:56–62. doi: 10.1007/s11906-009-0011-z
215. Ozaki KS, Kimura S, Murase N. Use of carbon monoxide in minimizing ischemia/reperfusion injury in transplantation. Transplant Rev. (2012) 26:125–39. doi: 10.1016/j.trre.2011.01.004
216. Hanto DW, Maki T, Yoon MH, Csizmadia E, Chin BY, Gallo D, et al. Intraoperative administration of inhaled carbon monoxide reduces delayed graft function in kidney allografts in Swine. Am J Transplant. (2010) 10:2421–30. doi: 10.1111/j.1600-6143.2010.03289.x
217. Martins PN, Reuzel-Selke A, Jurisch A, Atrott K, Pascher A, Pratschke J, et al. Induction of carbon monoxide in the donor reduces graft immunogenicity and chronic graft deterioration. Transplant Proc. (2005) 37:379–81. doi: 10.1016/j.transproceed.2004.11.079
218. Kirkby KA, Adin CA. Products of heme oxygenase and their potential therapeutic applications. Am J Physiol Renal Physiol. (2006) 290:F563–71. doi: 10.1152/ajprenal.00220.2005
219. Lee SS, Gao W, Mazzola S, Thomas MN, Csizmadia E, Otterbein LE, et al. Heme oxygenase-1, carbon monoxide, and bilirubin induce tolerance in recipients toward islet allografts by modulating T regulatory cells. FASEB J. (2007) 21:3450–7. doi: 10.1096/fj.07-8472com
HO, heme oxygenase; CO, carbon monoxide; HO-1, heme oxygenase-1; HO-2, heme oxygenase-2; IFN, interferon; DCs, dendritic cells; IRF3, IFN regulatory factor 3; tolDCs, tolerogenic dendritic cells; Treg cells, regulatory T cells; MC, mast cell; STAT3, signal transducer and activator of transcription 3; Th17 cells, T helper 17 cells; Th2 cells, T helper 2 cells; NK cells, natural killer cells; DAMP, danger-associated molecular pattern; TLR4, toll-like receptor 4; TNF, tumor necrosis factor; ROS, reactive oxygen species; IL, interleukins; AKI, acute kidney injury; ATN, acute tubular necrosis; IR, ischemia-reperfusion; ATP, adenosine triphosphate; IRI, ischemia-reperfusion injury; CP, cisplatin; Nrf2, nuclear factor erythroid 2-related factor 2; HMGB1, high-mobility group box 1; NLRP3, NACHT, LRR, and PYD domains-containing protein 3; CKD, chronic kidney disease; MCP-1, monocyte chemoattractant protein-1; TGF, transforming growth factors; NF-κB, nuclear factor-kappaB; UUO, unilateral ureteral obstruction; ZnPP, Zinc protoporphyrin IX; MKK3, MAPK kinase 3; GT, guanosine thymidine; HIF, hypoxia inducible factors; AVF, arteriovenous fistula; CORM-3, carbon monoxide-releasing molecules-3; ESRD, end stage renal disease; GN, glomerulonephritis; NTN, nephrotoxic nephritis; NO, nitric oxide; NOS, nitric oxide synthase; iNOS, inducible nitric oxide synthase; SOD, superoxide dismutase; FSGS, focal segmental glomerulosclerosis; NADPH, nicotinamide adenine dinucleotide phosphate; GPx, glutathione peroxidase; LN, lupus nephritis; SLE, systemic lupus erythematosus; Co-PP, Cobalt Protoporphyrin; ANA, anti-nuclear antibodies; IC, immune complexes; DN, diabetic nephropathy; O2–, superoxide anion; STZ, streptozotocin; CrMP, chromium mesoporphyrin; PI3K, phosphatidylinositol 3-kinase; Bcl-xL, B-cell leukemia-lymphoma-Xl; C5aR, C5a receptor; MHC, major histocompatibility complex.
Keywords: heme oxygenase-1, kidney diseases, immune regulation, oxidative stress, carbon monoxide
Citation: Li Y, Ma K, Han Z, Chi M, Sai X, Zhu P, Ding Z, Song L and Liu C (2021) Immunomodulatory Effects of Heme Oxygenase-1 in Kidney Disease. Front. Med. 8:708453. doi: 10.3389/fmed.2021.708453
Received: 01 June 2021; Accepted: 31 July 2021;
Published: 24 August 2021.
Edited by:
Alain Le Moine, Université Libre de Bruxelles, BelgiumReviewed by:
Stefan W. Ryter, Harvard Medical School, United StatesCopyright © 2021 Li, Ma, Han, Chi, Sai, Zhu, Ding, Song and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chi Liu, bGl1Y2hpMTk4NUAxNjMuY29t; Linjiang Song, bGluanNvbmdfc2N1QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.