
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Med. , 28 June 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.684889
 Ying Tan1,2,3,4,5†
Ying Tan1,2,3,4,5† Yan Qin1,2,3,4,5,6†
Yan Qin1,2,3,4,5,6† Xiao-juan Yu1,2,3,4,5Rong Xu1,2,3,4,5
Xiao-juan Yu1,2,3,4,5Rong Xu1,2,3,4,5 Su-xia Wang1,2,3,4,5,7Fu-de Zhou1,2,3,4,5
Su-xia Wang1,2,3,4,5,7Fu-de Zhou1,2,3,4,5 Ming-hui Zhao1,2,3,4,5,8*
Ming-hui Zhao1,2,3,4,5,8*Isolated or dominant tubulointerstitial lupus nephritis is rare. Here, we reported a 67-year-old man diagnosed with systemic lupus erythematosus (SLE) based on clinical and laboratory criteria, who was showing impaired renal function and non-nephrotic range proteinuria in the past 2 years. Renal biopsy showed almost normal glomeruli, but the tubulointerstitium showed “storiform” pattern with interstitial infiltration of IgG3 predominant plasma cells. Immunofluorescence showed linear and granular staining of IgG and C1q along TBM and interstitium. He started on medium dose of oral steroids and mycophenolate mofetil, which were gradually tapered. As a result, his renal function improved over a few days. Now, he continued on low dose steroids and mycophenolate mofetil with no evidence of relapse.
Systemic lupus erythematosus (SLE) is a multisystemic immunological disorder with various manifestations. Lupus nephritis is the most common manifestation of SLE and contributes significantly toward morbidity and mortality in this disease. Tubulointerstitial involvement has been observed in ~90% of all patients with lupus nephritis1. However, the predominant or isolated presence of tubulointerstitial changes in the setting of minimal glomerular abnormalities in patients with SLE is rare. Here, we describe a case of predominant tubulointerstitial lupus nephritis, which was diagnosed with SLE based on clinical and laboratory criteria. The patient presented with impaired renal function, sub-nephrotic proteinuria, and evidence of tubular dysfunction.
In 2018, a 67-year-old Chinese man was admitted to our hospital with dryness of mouth for 6 years and elevated serum creatinine for 2 years. Six years ago, he felt dryness of mouth. Enlargement of salivary gland were discovered, and resection of bilateral submandibular gland was performed. The pathologic examination revealed severe chronic inflammation. Two years ago, the patient felt fatigue. The serum creatinine was 164 μmol/L (corresponding to estimated glomerular filtration rate of 40.2 ml/min/1.73 m2 as calculated by the CKD-EPI equation) with benign urinary analysis. Half a year later, his serum creatinine elevated to 175 μmol/L (corresponding to estimated glomerular filtration rate of 37.5 ml/min/1.73 m2 as calculated by the CKD-EPI equation). Positive antinuclear antibody (ANA) with titer of 1:3,200 was discovered. Oral methyprednisolone was prescribed as 32 mg per day and tapered quickly because of deteriorating dry mouth. After that, his serum creatinine suddenly elevated to 517 μmol/L within 3 months. And he had intermittent facial rash when exposed to the sun.
The patient had tuberculous pleurisy 50 years ago and fully recovered. Family history was of no significance.
On admission, physical examination revealed blood pressure of 125/70 mmHg, temperature of 36.3°C, pulse of 75 beats/min, and respiratory rate of 15/min. His tongue was dry, and dental caries could be found.
Laboratory data were as follows. Urinary analysis showed proteinuria+, glucose+ with normal blood glucose. Urine sediment analysis revealed red blood cell 0–2 cells per high power field (HPF) with white blood cell 0–1/HPF. Urinary protein excretion was 1.00 g/24 h (urine volume 2,700 ml). The urinary alpha 1 microglobulin was 343 mg/L (normal range: 0.00–12.00 mg/L) and N-Acetyl-D-Glucosaminidase 22 IU/L (normal range: 0–21 IU/L). Urine osmotic pressure was 314 mOsm/kg with normal blood osmotic pressure. Urine protein electrophoresis showed 58.90% of small molecule protein, 32.9% of albumin, and 8.20% of large molecule protein. Blood routine test showed mild anemia (hemoglobin 95 g/L). Blood chemistry tests showed increased levels of blood urea nitrogen and serum creatinine (16.91 mmol/L and 456.00 μmol/L, respectively). Serological studies revealed a high titer of ANA (1:10,000) and an increased level of anti-dsDNA antibody (>800 IU/ml) with negative ENA. Decreased serum levels of C3 (0.240 g/L) and C4 (<0.017 g/L) were detected. Total IgG was elevated to 33.80 g/L, and IgG subtypes were IgG1 19.8 g/L, IgG2 3.84 g/L, IgG3 2.24 g/L, and IgG4 2.43 g/L. Coombs test was positive.
Tear break-up time (BUT) indicated dryness of eye (<10 s). Labial gland biopsy did not indicate the diagnosis of Sjogren's Syndrome. Submandibular gland pathology revealed severe chronic inflammation of the left submandibular gland. Immunochemistry staining of immunoglobulin showed IgG3 positive.
Percutaneous renal biopsy was performed. Light microscopy examination showed that 3/14 glomeruli were globally sclerosed and 6/14 glomeruli showed ischemic sclerosis. Other glomeruli showed minor shrinkage of glomerular capillary wall, but no significant glomerular change. Tubular epithelial cells exhibited focal vacuolization and eosinophilic granules in the cytoplasm and focal loss of brush border. There were profound interstitial infiltration with lymphocytes, plasma cells, and a few eosinophils. “Storiform” fibrosis with tubular atrophy can be seen in the tubulointerstitium (Figures 1A–C). Immunofluorescence microscopy revealed one out of seven glomeruli with crescent and granular mesangial staining for IgG and C3, and granular staining for IgG, κ, λ, IgG1, IgG2, IgG3, IgG4, and C1q along TBM and interstitium and linear staining for λ also along TBM (Figure 1D). Electron microscopy revealed one glomerulus with numerous electronic dense deposits along tubular basement membrane with focal mesangial deposits (Figure 1E). Immunohistochemistry revealed that IgG1-positive plasma cells were 35–45/HPF, IgG2-positive plasma cells 40–50/HPF, IgG3-positive plasma cells 90–100/HPF, and IgG4-positive plasma cells 10–15/HP (Figures 1F–I). Taken together with the diagnosis of SLE, these findings indicated the renal lesion as predominant acute and chronic tubulointerstitial nephritis with dominant IgG3 positive plasma cell infiltration.
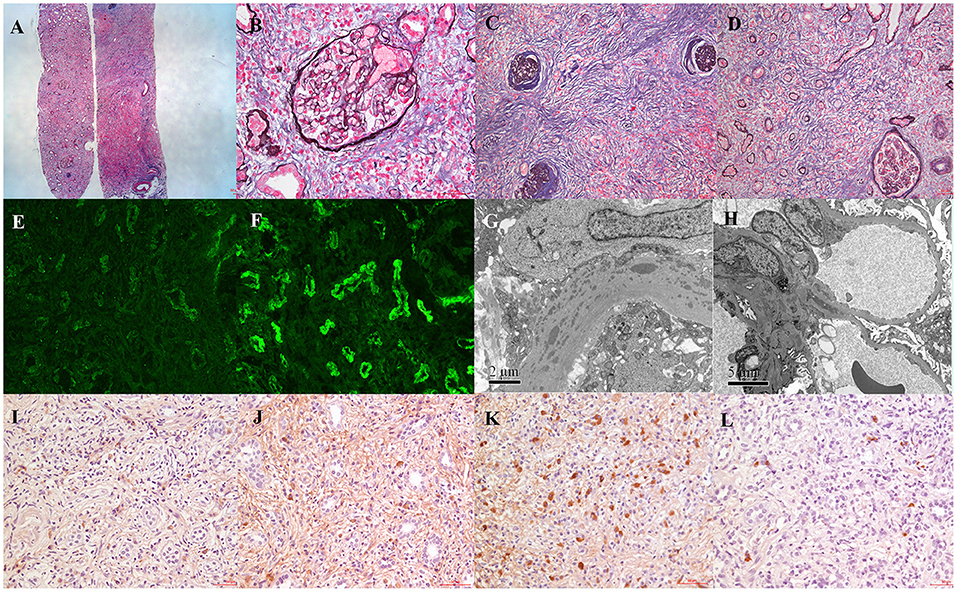
Figure 1. Patient renal biopsy findings. (A) Light microscopy showed minor shrinkage of glomerular capillary wall with dense interstitial lymphoplasmacytic cell infiltrates and variable degree of interstitial fibrosis (periodic methenamine silver and Masson trichrome staining, ×40). (B) Almost normal glomeruli (periodic methenamine silver and Masson trichrome staining, ×400). (C,D) “Storiform” fibrosis with tubular atrophy in the tubulointerstitium (periodic methenamine silver and Masson trichrome staining, ×200). (E) Immunofluorescence staining showed IgG granular staining along TBM and interstitium (×400). (F) Immunofluorescence staining showed λ linear staining along TBM (×400). (G) Electronic dense deposits along tubular basement membrane (×12,000). (H) Electronic dense deposits in the mesangium (×12,000). (I–L) Immunohistochemical staining of IgG subclass (IgG1–IgG4) (×200).
Oral prednisone 30 mg was prescribed immediately. Hydroxychloroquine 0.2 g and mycophenolate mofetil 1.0 g were given later. The serum creatinine was gradually reduced from 456 μmol/L to 210 μmol/L. The urinary protein was reduced to 0.17 g/24 h in 8 months. The patient was stable for the last 31 months during follow-up.
The 2003 International Society of Nephrology/Renal Pathology Society (ISN/RPS) system for classifying patients with lupus nephritis was based on glomerular lesions. However, previous studies have indicated that severe tubulointerstitial involvement has been observed in approximately more than half of all patients with lupus nephritis and helpful in predicting renal outcome in patients with lupus nephritis (1). Major or isolated tubulointerstitial damage is rare.
Here, we presented a patient with SLE who presented with acute onset of chronic disease attributable to predominant tubulointerstitial lupus nephritis. The diagnosis of lupus was based on the presence of 5 of the 11 criteria for the diagnosis of SLE (photosensitivity, autoimmune hemolytic anemia, a positive ANA, a positive anti-dsDNA antibody, and the presence of renal disease). In addition, the patient was noted to have hypocomplementemia and hyperglobulinemia.
The chief complaint of the patient was dryness of mouth. Taking the enlargement of salivary gland, we needed to exclude primary Sjögren's syndrome. The specific antibody of Sjögren's syndrome and anti-SSA and anti-SSB antibodies were negative. Though the dryness of eyes was diagnosed by BUT, the labial gland biopsy did not meet the criteria of primary Sjogren's Syndrome (2). Moreover, we also needed to exclude IgG4 disease since the elevated IgG4 (≥135 mg/dl), enlargement of salivary gland, and the kidney biopsy with storiform fibrosis and a lymphoplasmacytic infiltration. Thus, the immunohistochemistry of IgG subtype on tubulointerstitial plasma cells and submandibular gland was performed with all IgG3 predominant, and the ratio of IgG4/IgG <40%, which did not support IgG4-related disease (IgG4-RD) (3–5). However, there's still possibility that there might be an overlap of IgG4-RD and SLE since “storiform fibrosis” was more prevalent in IgG4-RD and more research work needs to be done to further differentiate the new disease entity (6, 7).
Meanwhile, a detailed history did not reveal the consumption of any potentially nephrotoxic medications or exposure to radiation or environmental toxin (e.g., cadmium or lead). Furthermore, though the renal biopsy revealed a few eosinophils in the interstitial infiltrate, it was not suggestive of an allergic interstitial nephritis. Similarly, there was no evidence of an infection, a neoplastic process, or a metabolic factor, which could have accounted for the current presentation.
Our patient was a predominant tubulointerstitial lupus nephritis with normal glomeruli. Fifteen similar previously reported cases are summarized in Table 1: acute kidney injury or tubular acidosis with trace proteinuria as onset. Renal pathology showed no or only slight glomerular lesions, but interstitial fibrosis, inflammatory cell infiltration, and renal tubular atrophy. Immunofluorescence showed IgG, C1q, and C3 deposition along TBM. Electron microscopy showed electron dense deposits on TBM. Most patients had a good prognosis with steroids just like our patient (8–21) (Table 1).
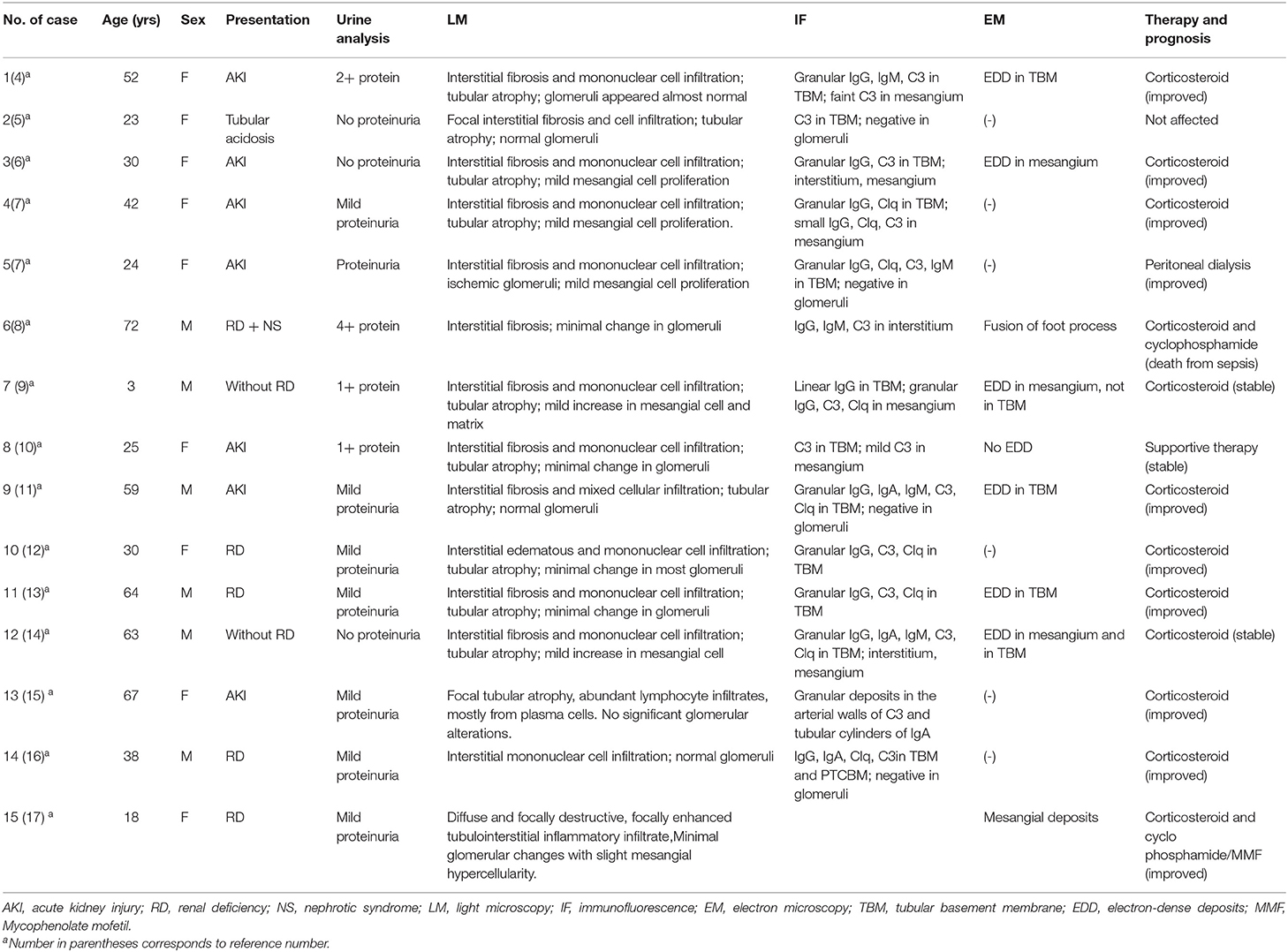
Table 1. Reported cases of predominant tubulointerstitial lupus nephritis.
The pathogenesis of predominant tubulointerstitial lupus nephritis is still unknown. The deposition of immune complex on TBM may reflect in situ formation of immune complexes after the binding of circulating autoantibodies to exogenous or natural antigens since no obvious glomerular lesions were discovered. Recent studies indicated that germinal center-like structure might be formed in the interstitial nephritis of SLE, which could secrete autoantibodies that form immune complex along TBM, activate complement system, and result in focal inflammatory lesions and fibrosis (22). However, this case showed no germinal center-like structure in the kidney biopsies.
In summary, we reported an old man who presented with acute kidney disease attributable to predominant tubulointerstitial lupus nephritis. Renal biopsy showed almost normal glomeruli but the tubulointerstitium showed “storiform” pattern with interstitial infiltration of IgG3 predominant plasma cells. Immunofluorescence showed IgG and C1q linear and granular staining along TBM and interstitium. He was responsive to medium dose steroids and mycophenolate mofetil. However, the underlying pathogenesis in the predominant tubulointerstitial lupus nephritis still needs to be addressed.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Peking University First Hospital, approval number: 2017[1333]. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
YT and YQ analyzed and interpreted the patient data and were major contributors in writing the manuscript. X-jY and S-xW performed interpretation of pathological data. RX, F-dZ, and M-hZ performed interpretation of the clinical data and substantively revised it. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (No. 81670639), Beijing Natural Science Foundation (No. 7192207), and CAMS Innovation Fund for Medical Sciences (2019-I2M-5-046).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.684889/full#supplementary-material
Supplementary Figure 1. (A) Immunohistochemical staining of CD3 (×100); (B) Immunohistochemical staining of CD20 (×100); (C) Immunohistochemical staining of CD138 (×100); (D) Immunohistochemical staining of total IgG (×200).
1. Yu F, Wu LH, Tan Y, Li LH, Wang CL, Wang WK, et al. Tubulointerstitial lesions of patients with lupus nephritis classified by the 2003 International Society of Nephrology and Renal Pathology Society system. Kidney Int. (2010) 77:820–9. doi: 10.1038/ki.2010.13
2. Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjogren's syndrome: a consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis. (2017) 76:9–16. doi: 10.1136/annrheumdis-2016-210571
3. Mann S, Seidman MA, Barbour SJ, Levin A, Carruthers M, Chen LY. Recognizing IgG4-related tubulointerstitial nephritis. Can J Kidney Health Dis. (2016) 3:34. doi: 10.1186/s40697-016-0126-5
4. Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. (2012) 22:21–30. doi: 10.3109/s10165-011-0571-z
5. Wallace ZS, Naden RP, Chari S, Choi HK, Della-Torre E, Dicaire JF, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis. (2020) 79:77–87. doi: 10.1136/annrheumdis-2019-216561
6. Naramala S, Biswas S, Adapa S, Gayam V, Konala VM, Bose S. An overlapping case of IgG4-related disease and systemic lupus erythematosus. J Investig Med High Impact Case Rep. (2019) 7:2324709619862297. doi: 10.1177/2324709619862297
7. Yoshita K, Kawano M, Mizushima I, Hara S, Ito Y, Imai N, et al. Light-microscopic characteristics of IgG4-related tubulointerstitial nephritis: distinction from non-IgG4-related tubulointerstitial nephritis. Nephrol Dial Transplant. (2012) 27:2755–61. doi: 10.1093/ndt/gfr761
8. Epstein WH, McCluskey RT. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 2-1976. N Engl J Med. (1976) 294:100–5. doi: 10.1056/NEJM197601082940208
9. Cunningham E, Provost T, Brentjens J, Reichlin M, Venuto RC. Acute renal failure secondary to interstitial lupus nephritis. Arch Intern Med. (1978) 138:1560–1. doi: 10.1001/archinte.138.10.1560
10. Disler PB, Lewin JR, Laidley L, Meyers AM. Systemic lupus erythematosus with pure interstitial disease: a case report (abstract). Kidney Int. (1978) 13:428.
11. Tron F, Ganeval D, Droz D. Immunologically-mediated acute renal failure of nonglomerular origin in the course of systemic lupus erythematosus [SLE]. Report of two cases. Am J Med. (1979) 67:529–32. doi: 10.1016/0002-9343(79)90806-4
12. Klahr S, Lynch R. Clinicopathological conference: interstitial nephritis in a patient with systemic lupus erythematosus. Am J Med. (1980) 69:775–81. doi: 10.1016/0002-9343(80)90451-9
13. Makker SP. Tubular basement membrane antibody-induced interstitial nephritis in systemic lupus erythematosus. Am J Med. (1980) 69:949–52. doi: 10.1016/S0002-9343(80)80025-8
14. Gur H, Kopolovic Y, Gross DJ. Chronic predominant interstitial nephritis in a patient with systemic lupus erythematosus: a follow up of 3 years and review of the literature. Ann Rheum Dis. (1987) 46:617–23. doi: 10.1136/ard.46.8.617
15. Singh AK, Ucci A, Madias NE. Predominant tubulointerstitial lupus nephritis. Am J Kidney Dis. (1996) 27:273–8. doi: 10.1016/S0272-6386(96)90553-3
16. Michail S, Stathakis C, Marinaki S, Revenas C, Nakopoulou L, Vaiopoulos G. Relapse of predominant tubulointerstitial lupus nephritis. Lupus. (2003) 12:728–9. doi: 10.1191/0961203303lu434xx
17. Mori Y, Kishimoto N, Yamahara H, Kijima Y, Nose A, Uchiyama-Tanaka Y, et al. Predominant tubulointerstitial nephritis in a patient with systemic lupus nephritis. Clin Exp Nephrol. (2005) 9:79–84. doi: 10.1007/s10157-004-0338-3
18. Omokawa A, Wakui H, Okuyama S, Togashi M, Ohtani H, Komatsuda A, et al. Predominant tubulointerstitial nephritis in a patient with systemic lupus erythematosus: phenotype of infiltrating cells. Clin Nephrol. (2008) 69:436–44. doi: 10.5414/CNP69436
19. Moyano Franco MJ, Amor Sanchez J, Ortega Ruano R, Armas Padron JR. [Isolated tubular interstitial nephritis in a patient with systemic lupus erythematosus]. Nefrologia. (2009) 29:501–2. doi: 10.3265/nefrologia.2009.29.5.5540.en.full
20. Ali A, Al-Windawi S. Tubulointerstitial lupus nephritis. J Nephropathol. (2013) 2:75–80. doi: 10.5812/nephropathol.9000
21. Odler B, Pollheimer MJ, Kirsch AH, Moazedi-Fuerst F, Rosenkranz AR, Eller K. A patient with predominant interstitial nephritis as a renal manifestation of systemic lupus erythematosus. Rheumatology. (2020) 59:685–7. doi: 10.1093/rheumatology/kez401
Keywords: systemic lupus erythematosus, lupus nephritis, predominant interstitial nephritis, IgG3, storiform pattern
Citation: Tan Y, Qin Y, Yu X-j, Xu R, Wang S-x, Zhou F-d and Zhao M-h (2021) Case Report: Predominant Tubulointerstitial Lupus Nephritis or the Combination With IgG4-Related Disease? Front. Med. 8:684889. doi: 10.3389/fmed.2021.684889
Received: 24 March 2021; Accepted: 27 May 2021;
Published: 28 June 2021.
Edited by:
Sergey Brodsky, Ohio State University Hospital, United StatesReviewed by:
Beom Jin Lim, Yonsei University College of Medicine, South KoreaCopyright © 2021 Tan, Qin, Yu, Xu, Wang, Zhou and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-hui Zhao, bWh6aGFvQGJqbXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.