 Qi Li
Qi Li Delma Veron
Delma Veron Alda Tufro
Alda Tufro
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med., 14 July 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.679518
This article is part of the Research TopicCell Cross-talk in Diabetic Kidney DiseasesView all 10 articles
The molecular pathogenesis of diabetic kidney disease progression is complex and remains unresolved. Rho-GAP MYO9A was recently identified as a novel podocyte protein and a candidate gene for monogenic FSGS. Myo9A involvement in diabetic kidney disease has been suggested. Here, we examined the effect of diabetic milieu on Myo9A expression in vivo and in vitro. We determined that Myo9A undergoes S-nitrosylation, a post-translational modification dependent on nitric oxide (NO) availability. Diabetic mice with nodular glomerulosclerosis and severe proteinuria associated with doxycycline-induced, podocyte-specific VEGF164 gain-of-function showed markedly decreased glomerular Myo9A expression and S-nitrosylation, as compared to uninduced diabetic mice. Immortalized mouse podocytes exposed to high glucose revealed decreased Myo9A expression, assessed by qPCR, immunoblot and immunocytochemistry, and reduced Myo9A S-nitrosylation (SNO-Myo9A), assessed by proximity link assay and biotin switch test, functionally resulting in abnormal podocyte migration. These defects were abrogated by exposure to a NO donor and were not due to hyperosmolarity. Our data demonstrate that high-glucose induced decrease of both Myo9A expression and SNO-Myo9A is regulated by NO availability. We detected S-nitrosylation of Myo9A interacting proteins RhoA and actin, which was also altered by high glucose and NO dependent. RhoA activity inversely related to SNO-RhoA. Collectively, data suggest that dysregulation of SNO-Myo9A, SNO-RhoA and SNO-actin may contribute to the pathogenesis of advanced diabetic kidney disease and may be amenable to therapeutic targeting.
Diabetic kidney disease (DKD) is a major complication of both type 1 and type 2 diabetes that leads to renal failure, and the single most frequent cause of end-stage renal disease (ESRD) worldwide (1). In the last few years novel therapies led to remarkable improvement of metabolic control in diabetic patients (2, 3). However, the prevalence and progression of DKD have not decreased as yet (2, 3). Incomplete understanding of the molecular mechanisms involved in DKD progression has precluded the development of effective treatments to prevent, halt or reverse progression to ESRD (4–6). In an effort to address this, we investigated the role of a novel podocyte protein, Myosin 9A, in progression to advanced DKD.
MYO9A was recently identified as a novel candidate gene for monogenic FSGS (7). This study also suggested dysregulation of Myo9A expression in other experimental proteinuric diseases, such as nephrotic syndrome and diabetic nephropathy (7). The relevance of Myo9A involvement in diabetic kidney disease is presently unknown. Myosins are a super family of actin binding molecular motors that regulate cell shape and motility, organelle trafficking and signaling (8–10). Several non-muscle myosins regulate foot process actin dynamics in podocytes (11). The 40 gene members of the myosin family share a common structure consisting of head, neck and tail domains and have been grouped in 18 classes based on their distinctive features. Class 2 non-muscle myosin MYH9 has been implicated in the pathogenesis of DKD (12) and MYO1E mutations cause monogenic FSGS (13). Class 9 myosins' unique features are their RhoGTPase-activating protein (Rho-GAP) tail domain and a loop insert in their head domain (14). Myosin 9A (Myo9A) crosslinks and bundles actin, inactivates RhoA and controls epithelial cell junction assembly (9, 10, 14). Myo9A is expressed by epithelial cells in brain, kidney, testis and lung (15). In the kidney Myo9A localizes to podocytes and proximal tubular cells (7). Myo9A loss-of-function increases kidney RhoA activity and alters podocyte function (7).
In diabetes, hyperglycemia induces uncoupling of nitric oxide synthase homodimers and overproduction of reactive oxygen species (ROS) relative to antioxidant molecules, resulting in low nitric oxide (NO) availability (16–18). NO signals through two distinct pathways: activation of guanylyl cyclase to produce cyclic GMP (cGMP) and protein S-nitrosylation. S-nitrosylation is the reversible, oxidative addition of NO to Cys residues to form S-nitrosothiols (SNOs) that modifies myriad proteins, providing a redox-based cellular signaling mechanism that conveys the ubiquitous influence of NO on cellular function (19). S-nitrosylation regulates protein activity of multiple proteins that play important roles in DKD, including all nitric oxide synthase (NOS) isoforms, guanylyl cyclase (GC), hypoxia-inducible factor1α (HiF1α), thioredoxin (20–23), as well as in cytoskeletal dynamics, such as actin and RhoA (24–26).
Since Myo9A directly interacts with both actin and RhoA (27, 28), we hypothesized that Myo9A might undergo S-nitrosylation and thereby participate in a transnitrosylation cascade to serve NO signaling. The goals of this study were to determine whether Myo9A dysregulation is involved in the severity of DKD and to assess whether the molecular mechanism involves Myo9A S-nitrosylation. We documented that Myo9A is S-nitrosylated in vivo and in cultured podocytes in control conditions. Diabetic mice with advanced DKD revealed downregulation of Myo9A expression and S-nitrosylation. Cultured podocytes showed that high glucose-induced Myo9A dysregulation is NO dependent and involves actin and RhoA S-nitrosylation. These findings uncover Myo9A relevance in advanced DKD and identify a targetable pathway that might influence DKD progression involving cross-talk among multiple nephron cell types.
Experiments were performed using kidney tissue from podocin-rtTA:tet-O-VEGF164 (iVEGF) diabetic mice, herein called DM-iVEGF mice, previously reported (29). Podocin-rtTA:tet-O-VEGF164 are podocyte-specific inducible transgenic mice that overexpress VEGF164 in podocytes upon induction with doxycycline, as described (30). Mice were crossbred on FVB/N background. Diabetes was induced using streptozotocin (50 mg/kg body weight i.p. for 5 consecutive days) following the Animal Models of Diabetic Complications Consortium (www.AMDCC.org) short protocol in 5.0 ± 0.6 week old iVEGF mice (n = 15). Diabetic iVEGF mice (DM-iVEGF) were fed doxycycline containing chow (0.625 mg/g chow; Harlan-Teklad) (DM-iVEGF +dox, n = 8), or fed standard chow (DM-iVEGF-dox, n = 7) for 12 weeks to induce VEGF164 expression or serve as diabetic controls, respectively (29). At the end of 12 weeks, mice were anesthetized and kidneys were perfused with sterile PBS and excised prior to euthanasia. All experimental protocols were approved by the Institutional Animal Care and Use Committee at Yale University School of Medicine.
Immortalized mouse podocytes were cultured in RPMI-1640 medium (11875-093, Life Technologies), 1% Insulin-Transferrin-Selenium (41400-045, Life Technologies), 10% heat inactivated FBS (10438-026, Life Technologies), 1% Pen/Strep at 33°C with 5% CO2. Podocyte differentiation was induced by incubation at 37°C for 7 days. Podocytes incubated in control medium (11 mM D-glucose), medium + 25 mM glucose, medium + 25 mM mannitol, or medium + 25 mM glucose + 10μM DETA NONOate (#82120,Cayman Chemical) for 24 h. For immunocytochemistry and proximity link assays, podocytes were cultured in 4-chamber slides; for cell migration assays, podocytes (1 × 105cell/ml) were cultured in 35 mm dishes.
Kidneys were snap frozen in liquid nitrogen at the time of euthanasia, and podocytes were pelleted by centrifugation at the end of culture experiments. Both tissues and cells were lyzed in lysis buffer (1% NP-40, 1% Triton X, 50 mM Hepes, 150 mM NaCl, 0.1 mM EDTA, 0.1 mM Neocuproine, complete protease inhibitor, Roche) for immunoblot and co-immunoprecipitation analysis, as previously described (7, 31). Proteins were resolved by SDS-PAGE in 10% or 4–20% SDS–polyacrylamide gels (BioRad), transferred to polyvinylidene difluoride membranes, blocked with 5% dry-milk or 5% BSA in TBST and incubated with primary antibodies: actin (A2066, Sigma), Myo9A (Abnova, clone 4C11) and RhoA (67B9,Cell Signaling), followed by appropriate species specific HRP-conjugated secondary antibodies (Jackson Immuno Research Laboratories Inc.). Immunoblotted proteins were visualized with ECL. Co-immunoprecipitation was performed using podocyte lysates, as previously described (7). Briefly, following pre-clearing with prewashed protein A agarose beads, lysate supernatants were incubated anti-MYO9A rabbit polyclonal antibody (A305-702A-M, Bethyl) at 4°C, pre-washed agarose beads were added and incubated overnight. Agarose beads were washed with PBS+protease inhibitors (Roche), spun and resuspended in Laemmli sample buffer for western blot analysis as described above.
Kidneys were perfused with sterile PBS for morphologic studies prior to euthanasia, incubated in 18% sucrose, embedded in optimal cutting temperature medium (OCT, Sakura Finetek USA), frozen in isopentane/dry ice and kept at −80°C for immunohistochemistry (IHC), as described (29) or processed for light microscopy. Histology was evaluated by periodic acid–Schiff's reagent (PAS) stain. Kidney frozen sections and podocytes were fixed in 4% PFA, permeabilized with 0.3% triton-X, blocked with 10% donkey serum, 5% BSA in PBST at room temperature, and incubated overnight at 4°C in primary antibodies: S-nitrosocysteine mouse monoclonal antibody (AG Scientific,1:100) and rabbit anti Myo9A (NBP1-92160, Novus, 1:50). Sections were washed, incubated with fluorescent-tagged secondary antibodies: goat anti-mouse Alexa Fluor 594 or goat anti-rabbit Alexa Fluor 488 (Life Technologies, 1:150) at room temperature. Coverslips were mounted with Vectashield + DAPI (Vector Labs). Stained sections and cells were examined using an Olympus IX 71 inverted fluorescence/phase and bright field microscope (Olympus, Tokyo, Japan) equipped with an Optronics (Goleta, CA) Microfire camera and Pictureframe version 3.00.30 software. Images were processed with Adobe Photoshop CC 2018 (Adobe Systems).
Myo9A S-nitrosylation was detected and localized using an in situ proximity link assay, as previously described (32, 33). Here, we used Myo9A rabbit polyclonal antibody (Novus) and S-nitrosocysteine mouse monoclonal antibody (AG Scientific), Duolink PLA probes and fluorescent labeled oligonucleotides to visualize the amplified reaction product attached to the antibody protein complex, following the Duolink® PLA fluorescence protocol (Sigma). Briefly, kidney frozen sections or podocytes were fixed, permeabilized and blocked as described above + 0.3% hydrogen peroxide in PBS and incubated overnight with primary antibodies Myo9A and S-nitrosocysteine at 4°C. Secondary antibodies (PLA probes) donkey anti-rabbit and anti-mouse conjugated with oligonucleotides were added and incubated at 37°C for 60 min. Sections were washed with PBS, incubated with ligation solution containing oligonucleotides for 30 min at 37°C. Ligation of oligonucleotides generates a circular DNA strand that serves as a template only if the probes are in close proximity. Then, sections were incubated at 37°C with DNA polymerase and fluorescently labeled oligonucleotides for 100 min. The amplification reaction product attaches to the antibody protein complex and is visualized as a fluorescent signal resulting from the hybridization of fluorescently labeled oligonucleotides. Kidney sections and podocytes were washed, and coverslips placed using mounting medium with DAPI (Vector). Cy3 and DAPI fluorescence signals were detected by inverted fluorescence microscopy at × 400 magnification and processed as described above.
S-nitrosylation of Myo9A, RhoA and actin was measured using a biotin switch assay (34) (S-nitrosylated protein detection kit, Cayman Chemical Co.), following the manufacturer's instructions. Briefly, podocyte lysates (1,000 μg) were re-suspended in blocking buffer to block free thiols, acetone precipitated, S-NO bonds were reduced, and the resulting free thiols were labeled with maleimide-biotin. Proteins were acetone-precipitated, the pellets were re-suspended in equal volumes of HENS/10 + 1% SDS buffer. To pull-down the biotinylated proteins we added streptavidin-agarose beads (Fluka). Beads were washed 5 times and bound proteins were eluted in 2X sample buffer. Myo9A, RhoA and actin presence in the eluates was detected by immunoblotting.
Active RhoA was measured with Rho-activation pulldown assay (Millipore) following manufacturer's instructions. Active RhoA was detected by immunoblotting using RhoA antibody (67B9, Cell Signaling), as described (7).
Data are analyzed with GraphPad-Prism-8 software (San Diego, CA) using unpaired Student's-t-test with Welch's correction, Welch's or Brown-Forsythe ANOVA, as appropriate. Non-parametric Kriskall-Wallis and Mann-Whitney test were used to analyze RhoA activity data. P < 0.05 was deemed statistically significant. Data are expressed as mean ± SD, unless otherwise indicated.
To begin to understand the role of Myo9A in DKD progression we compared Myo9A expression in diabetic mice with mild vs. advanced diabetic kidney disease (DKD). We examined Myo9A expression and distribution of S-nitrosylated proteins in kidneys from mice with streptozotocin-mediated diabetes and doxycycline-inducible, podocyte VEGF164 overexpression (DM- iVEGF164) (29, 30). As previously reported (29), uninduced diabetic mice (− dox) show discrete glomerular changes (Figure 1A) and mild albuminuria (ACR: 212 ± 18 μg/mg creatinine, Figure 1C), whereas doxycycline-induced diabetic mice overexpressing VEGF164 (DM- iVEGF164+dox) develop severe diabetic nodular glomerulosclerosis (Figure 1B) and nephrotic range proteinuria (ACR:1947 ± 708 μg/mg creatinine, Figure 1C), herein referred to as advanced DKD. Induced and uninduced diabetic mice developed similar hyperglycemia (29). Using immunoblotting we determined that kidney Myo9A expression is significantly decreased in mice with advanced DKD (+ dox) as compared to mice with mild DKD (- dox) (Figure 1D) and non-diabetic mice (7).
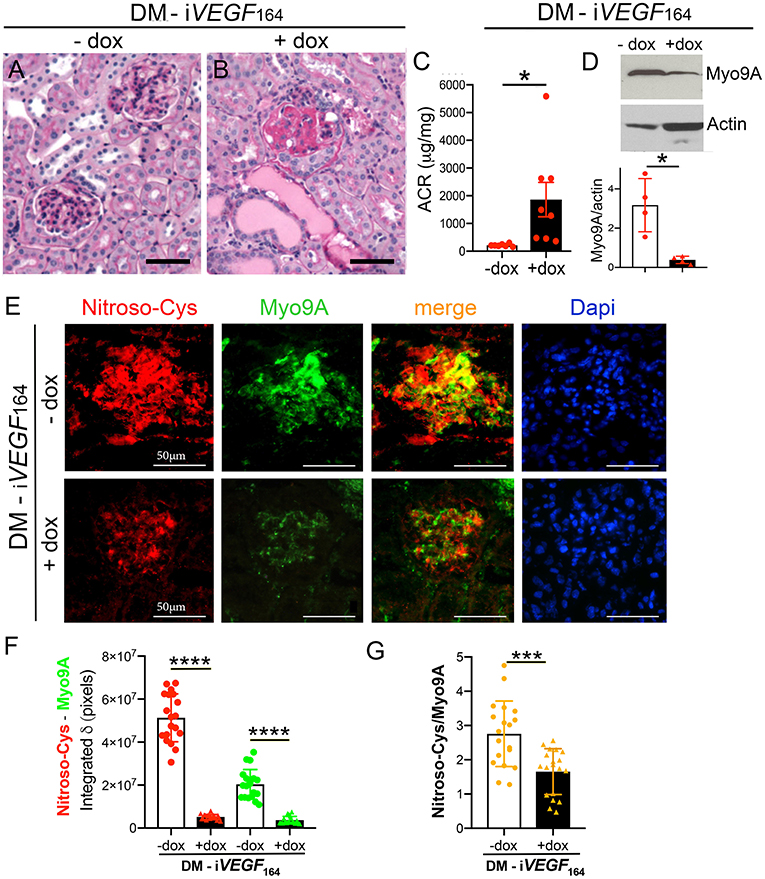
Figure 1. Myo9A is downregulated in advanced diabetic kidney disease. (A) Kidney PAS stain from uninduced diabetic mouse (DM-iVEGF164, - dox) showing mild mesangial proliferation; (B) kidney PAS stain from induced diabetic mouse (DM-iVEGF164, + dox) showing nodular glomerulosclerosis and large protein casts; (C) Urine ACR (albumin:creatinine ratio, mg/mg) shows mild albuminuria in uninduced diabetic mice (DM-iVEGF164, - dox, n = 7) and nephrotic range proteinuria in induced diabetic mice (DM-iVEGF164, + dox, n = 8), unpaired t-test with Welch's correction, P = 0.033; (D) representative immunoblot shows decreased kidney Myo9A expression in mice with advanced DKD (DM-iVEGF164, + dox); quantitation of Myo9A expression normalized to actin confirms significant Myo9A downregulation in n = 4 immunoblots (kidney lysates pooled from 4 to 6 mice/experimental group), mean ± SD, P < 0.05; (E) Fluorescence IHC shows S-nitrosylated proteins (red) and Myo9A (green) partially co-localized (merge) in glomeruli from uninduced diabetic kidneys (DM-iVEGF164, - dox), both Myo9A and nitroso-Cys IF signals are reduced in glomeruli from kidneys with advanced DKD (DM-iVEGF164, + dox); (F) quantitation of Myo9A and nitroso-Cys IF signals confirm a dramatic decrease in glomerular Myo9A expression and S-nitrosylated proteins in kidneys with advanced DKD (DM-iVEGF164, + dox), mean ± SD, n = 19 glomeruli/experimental group (each from 3 to 5 mice), unpaired t-test with Welch's correction, p < 0.0001; (G) quantitation of the IF signals' ratio Nitroso-Cys/Myo9A shows significant decrease in kidneys with advanced DKD, mean ± SD, n = 19 glomeruli/experimental group, unpaired t-test with Welch's correction, p = 0.0002. Scale bars = 50μm. *p < 0.05, ***p < 0.005, ****p < 0.0001.
Dual immunofluorescence labeling (IF) revealed that Myo9A and S-nitrosylated proteins localize to glomeruli from all diabetic mice (Figure 1E). S-nitrosylated proteins partially co-localize with Myo9A. We observed a significant decrease of glomerular Myo9A and S-nitrosylated proteins in induced diabetic mice (+ dox) as compared to non-induced diabetic mice (- dox). Quantitation of Myo9A and nitroso-Cys IF signals is shown in Figure 1F and the ratio of Nitroso-Cys/Myo9A IF signals is shown in Figure 1G. Together, IF data indicate that glomerular Myo9A and S-nitrosylated proteins partially co-localize and are decreased in the setting of advanced diabetic glomerulosclerosis, raising the possibility that Myo9A could be a S-nitrosylated protein.
To determine in situ whether glomerular Myo9A is S-nitrosylated we utilized a proximity link assay (PLA) (32, 33). Immunofluorescent PLA signals shown in Figure 2A demonstrate the presence of nitroso-Cys residues linked to Myo9A in uninduced glomeruli (- dox) and dramatically reduced SNO-Myo9A signal in glomeruli from induced (+ dox) diabetic mice. Quantitation of PLA signals is shown in Figure 2B. PLA data indicate that glomerular Myo9A is S-nitrosylated and that this post-translational modification is significantly downregulated in mouse kidneys with advanced DKD.
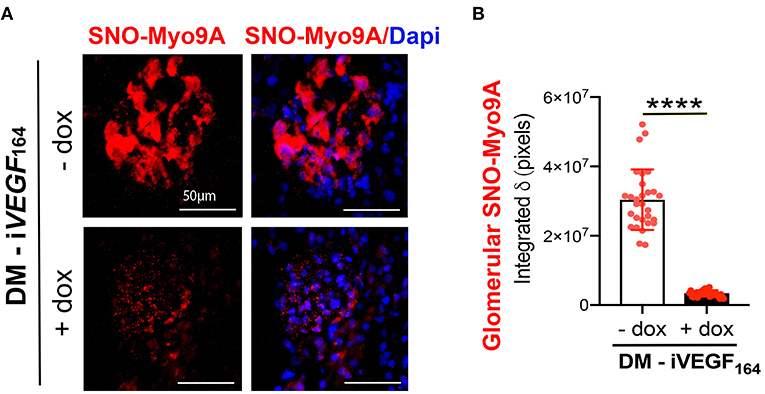
Figure 2. Glomerular Myo9A is S-nitrosylated in diabetic mice. (A) Proximity link assay IF signal (red) identifies abundant S-nitrosylated Myo9A (SNO-Myo9a) in glomeruli from uninduced diabetic mice with mild DKD (DM-iVEGF164 - dox), whereas SNO-Myo9A is clearly reduced in glomeruli from induced diabetic mice with advanced DKD (DM-iVEGF164, + dox). Dapi (blue) identifies cell nuclei. Scale bars = 50 μm. (B) Quantification of PLA IF signals, mean ± SD, n = 29–31/experimental group, unpaired t-test with Welch's correction, ****p < 0.0001.
A previous study demonstrated Myo9A expression in glomerular podocytes in vivo and in immortalized mouse and human podocytes (7). Therefore, we examined the effect of hyperglycemia on podocyte Myo9A expression and on S-nitrosylation. Immortalized mouse podocytes were exposed to normal glucose, mannitol, or high glucose, as described in the methods section. Mannitol was used as a control for hyperosmolarity-induced changes. Using IF dual labeling, we determined that Myo9A co-localizes with nitroso-Cys (SNO-Cys) signals in undifferentiated podocytes on normal glucose medium (Figure 3A, control, top panels) and differentiated podocytes exposed to mannitol (Figure 3A, middle panels), while differentiated podocytes exposed to high glucose showed reduced Myo9A and nitroso-Cys (SNO-Cys) proteins (Figure 3A, bottom panels). Quantification of IF signals demonstrating these highly significant changes are shown in Figure 3B.
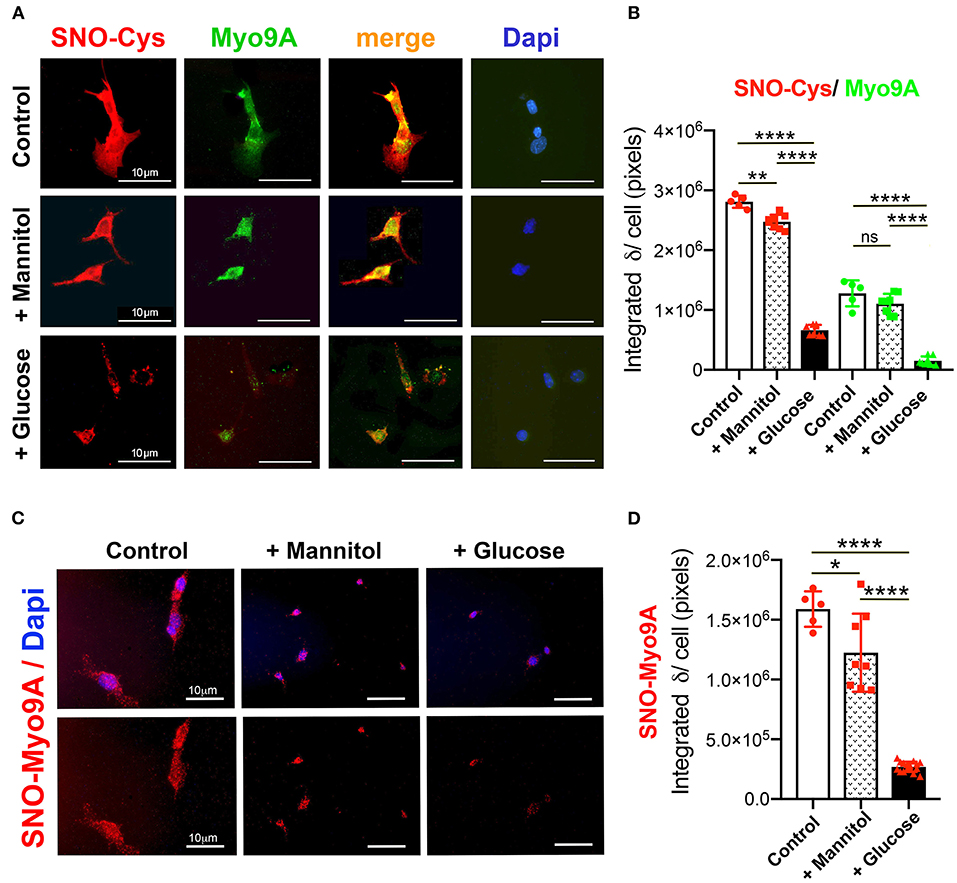
Figure 3. Podocyte Myo9A expression and S-nitrosylation are downregulated by high glucose. (A) IHC shows abundant SNO-Cys proteins (red) and Myo9A (green) partially co-localized in normal podocytes (top panels) and in podocytes exposed to mannitol (middle panels), whereas both SNO-Cys and Myo9A signals are clearly reduced in podocytes exposed to high glucose (bottom panels). Scale bars= 10μm. (B) Quantitation of IHC IF signals demonstrate highly significant decrease in Myo9A and SNO-Cys proteins in podocytes exposed to high glucose, data expressed as mean ± SD, n = 24–32 cells/experimental group, Welch's ANOVA p < 0.0001, unpaired t-test with Welch's correction p < 0.027 or n.s. control vs. mannitol, p < 0.0001 mannitol vs. high glucose. (C) Proximity link assay IF signal (red) identifies SNO-Myo9A in control podocytes, a mild decrease in podocytes exposed to mannitol and barely detected SNO-Myo9A in podocytes exposed to high glucose; Dapi (blue) identifies cell nuclei. Scale bars = 10μm. (D) Quantification of PLA IF signals, mean ± SD, n = 20–31 cells/experimental group, Welch's ANOVA p < 0.0001, unpaired t-test with Welch's correction p < 0.02 control vs. mannitol, p < 0.0001 mannitol vs. high glucose. *p < 0.05, **p < 0.01, ****p < 0.0001.
Proximity linked assay (PLA) revealed in situ S-nitrosylated Myo9A (SNO-Myo9A) fluorescent signals in podocytes grown in normal glucose and in podocytes exposed to mannitol, whereas SNO-MyoA signals were barely detected in podocytes exposed to high glucose (Figure 3C). Quantitation of PLA IF signals confirmed that exposure to high glucose significantly decreases SNO-Myo9A in podocytes (Figure 3D), demonstrating that SNO-Myo9A is regulated by glucose in podocytes.
Since IF revealed decreased podocyte Myo9A protein expression upon exposure to high glucose, we performed qPCR and immunoblotting to quantitate this effect at both mRNA and protein levels. Immortalized differentiated mouse podocytes were exposed to normal glucose, mannitol, high glucose or high glucose + nitric oxide donor (DETA). Podocyte Myo9A mRNA and protein decreased significantly upon exposure to high glucose as compared to normal glucose or mannitol (Figures 4A,B). The long acting NO donor DETA abrogated the high glucose-induced defect in Myo9A expression (Figures 4A,B).
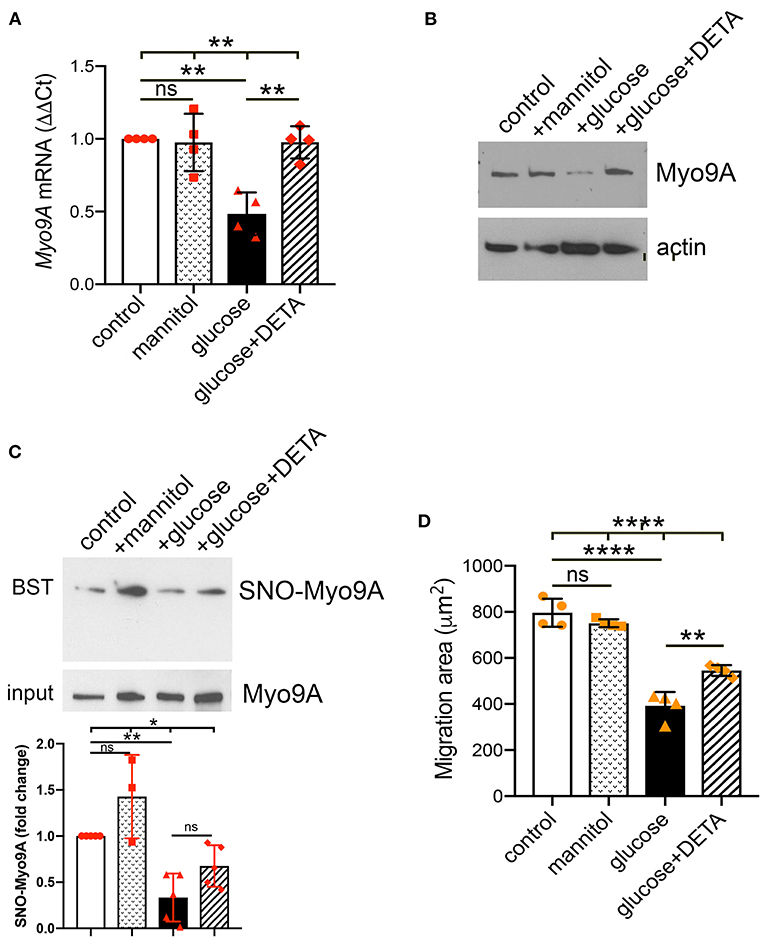
Figure 4. Podocyte Myo9A expression and SNO-Myo9A are regulated by glucose and NO. (A) qPCR shows that Myo9A mRNA is not affected by mannitol, decreases ~50% in podocytes exposed to high glucose and addition of NO donor prevents Myo9A mRNA downregulation, mean ± SD, n = 4 independent experiments; Welch's ANOVA p < 0.02, unpaired t-test with Welch's correction: n.s. control vs. mannitol, p < 0.02 control vs. high glucose, p < 0.02 high glucose vs. high glucose + DETA. (B) Immunoblots show that Myo9A protein expression is not altered by mannitol, decreases ≥50% in podocytes exposed to high glucose and addition of NO donor prevents Myo9A downregulation. (C) BST shows SNO-Myo9A in control podocytes, SNO-Myo9A ~50% decrease in podocytes exposed to high glucose, addition of NO donor partially prevents Myo9A de-nitrosylation. Input shows total Myo9A loading, mean ± SD, n = 3–5 independent experiments, Brown-Forsythe ANOVA test, p = 0.022, unpaired t-test with Welch's correction non-significant (n.s.) control vs. mannitol, **p = 0.0046 control vs. high glucose, p = 0.0575 (n.s.) high glucose vs. high glucose + DETA. (D) Migration ‘wound' assay shows that podocyte migration is not affected by mannitol, whereas high glucose clearly reduces podocyte migration and addition of NO donor partially prevents this defect, mean ± SD, n = 4 independent experiments; Welch's ANOVA p < 0.0001, unpaired t-test with Welch's correction non-significant (n.s.) control vs. mannitol, p < 0.005 control vs. high glucose, p < 0.02 high glucose vs. high glucose + DETA. *p < 0.05, **p < 0.01, ****p < 0.0001.
To examine further the regulation of Myo9A S-nitrosylation we performed biotin-switch test (BST). Consistent with the PLA results (Figures 3C,D), biotin-switch tests (BST) demonstrated that Myo9A S-nitrosylation (SNO-Myo9A) decreases ~50% upon podocyte exposure to high glucose (Figure 4C). Addition of NO donor DETA partially improves the Myo9A S-nitrosylation defect induced by podocyte exposure to high glucose (Figure 4C). We evaluated the effect of high glucose on podocyte function using a migration assay. Upon exposure to high glucose podocyte migration was significantly reduced, as compared to normal glucose or mannitol (Figure 4D). The migration defect was partially abrogated by addition of DETA (Figure 4D).
We assessed whether high glucose and NO regulate S-nitrosylation of Myo9A interacting proteins, RhoA and actin (7, 14). We determined that Myo9A interacts with RhoA in podocytes using immunoprecipitation (Figure 5A). Then, we performed BST to evaluate SNO-RhoA and SNO-actin under the conditions described above. These experiments revealed that both RhoA and actin are S-nitrosylated in control podocytes (Figures 5B,C). Remarkably, high glucose decreased SNO-RhoA, while addition of NO donor DETA abrogated podocyte RhoA de-nitrosylation (Figure 5B). High glucose also induced >50% SNO-actin decrease in podocytes and exposure to NO donor partially prevented this defect (Figure 5C). We measured RhoA activity using a pull down assay (7) and determined that high glucose induces an increase in RhoA activity, which is partially abrogated by NO donor (Figure 5D).
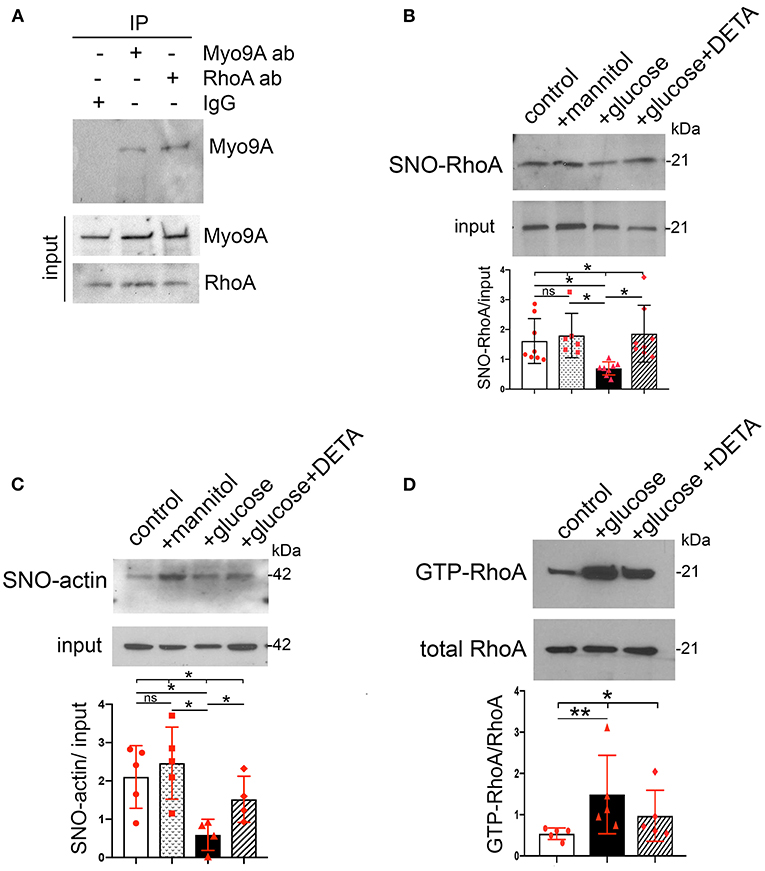
Figure 5. Podocyte SNO-RhoA, SNO-actin and RhoA activity regulation by glucose and NO. (A) Immunoprecipitation (IP): Myo9A and RhoA, WB: RhoA and Myo9A demonstrate Myo9A-RhoA interaction in podocytes. (B) BST shows SNO-RhoA in normal podocytes, ~50% SNO-RhoA decrease in podocytes exposed to high glucose, addition of NO donor prevents RhoA de-nitrosylation. Input shows total RhoA, mean ± SD, n = 6–8 independent experiments, Welch's ANOVA test, p = 0.002, unpaired t-test with Welch's correction non-significant (n.s.) control vs. mannitol, p < 0.01 control vs. high glucose, p < 0.01 high glucose vs. high glucose + DETA. (C) BST shows SNO-actin >50% decrease induced by high glucose, partially prevented by the NO donor DETA. Input shows actin loading, mean ± SD, n = 4–6 independent experiments, Welch's ANOVA test, p = 0.013; unpaired t-test with Welch's correction n.s. control vs. mannitol, p = 0.0011 control vs. high glucose, p < 0.05 high glucose vs. high glucose + DETA. (D) RhoA activity assay shows that exposure to high glucose increases active GTP-RhoA and addition of NO donor partially prevents activation of RhoA. Total RhoA shows equal input, mean ± SD, n = 5 independent experiments, Kruskall-Wallis test, p = 0.013; Mann-Whitney test p = 0.0079 control vs. high glucose. *p < 0.05 and **p < 0.01.
Taken together, our findings indicate that in podocytes high glucose-induced downregulation of SNO-Myo9A is associated with similar decreases in SNO-actin and SNO-RhoA, as well as with increased RhoA activity, all of which are regulated by NO availability.
This study demonstrates that the unconventional myosin Myo9A is S-nitrosylated in normal podocytes and that diabetic milieu downregulates Myo9A expression and S-nitrosylation in vivo. Our findings revealed that Myo9A S-nitrosylation is regulated by glucose and nitric oxide availability in cultured podocytes, consistent with in vivo findings. Data uncover S-nitrosylation as an integrated signaling between Myo9A and its interacting proteins RhoA and actin that transduces metabolic cues (high glucose + low NO), modifies cytoskeletal effectors (RhoA) function and impacts podocyte behavior.
Using an experimental type 1 diabetes (T1D) mouse model we determined that Myo9A expression in the kidney is decreased in diabetic mice with advanced DKD, while in diabetic mice with mild DKD Myo9A expression is not different from non-diabetic mice (7). Glomerular Myo9A is S-nitrosylated in mice with mild DKD whereas SNO-Myo9A is significantly reduced in mice with advanced DKD. These findings suggest (but do not prove) that downregulation of Myo9A expression and S-nitrosylation are mechanistically involved in the progression or severity of DKD. In this experimental model the development of diabetic nodular glomerulosclerosis is driven by inducible podocyte VEGF164 overexpression (29). Thus, the observed changes in Myo9A expression and SNO-Myo9A in induced mice with severe DKD could be a direct effect of excess glomerular VEGF-A and not mechanistically contributing to DKD progression. A previous study showed that VEGF-A cell autonomously decreases laminin S-nitrosylation in podocytes (33). Alternatively, hyperglycemia and VEGF164-induced NOS uncoupling reduce NO availability (17), which in turn could influence Myo9A expression and SNO-Myo9A in diabetic glomeruli and thereby contribute to DKD progression. Results from a DNA array showing >2-fold decrease of Myo9A expression in diabetic Zucker rats, a model of type 2 diabetes (T2D) are consistent with the latter possibility (35). Further studies assessing Myo9A role in DKD progression in other experimental mouse models of advanced DKD, e.g., T1D or T2D + eNOS KO, are warranted. Here we used podocytes to examine how the diabetic milieu influences Myo9A at the cellular level.
A key finding of this study is that Myo9A is S-nitrosylated in normal podocyte culture conditions and de-nitrosylates in diabetic milieu. Data indicate that this is not due to hyperosmolarity associated with high glucose and it is abrogated by addition of NO donor, demonstrating that SNO-Myo9A is glucose and NO dependent. Myo9A expression is also glucose and NO dependent, raising the intriguing possibility that Myo9A regulation is both transcriptional and post-translational in podocytes. High glucose-induced Myo9A downregulation and de-nitrosylation were associated with decreased podocyte migration, which was partially abrogated by a NO donor. Although this abnormal podocyte behavior in the diabetic milieu could be mediated via multiple pathways, it is remarkably similar to that reported in Myo9A knockdown podocytes (7), suggesting that Myo9A dysregulation is involved.
Myo9A binds actin at one of the two actin-binding sites in loop 2 of the catalytic domain forming crosslinks that bridge across actin filaments in parallel polarity at 36 nm regular intervals matching the actin helical repeat, thereby bundling actin filaments to form ordered networks (36). Experimental conditions such as calcium-calmodulin, ATP and redox status influence Myo9A actin crosslinking activity in vitro (36). However, it is presently unknown whether SNO-Myo9A is required for actin crosslinking in vivo.
We report for the first time SNO-actin in normal podocytes, which is regulated by high glucose and NO dependent alike SNO-Myo9A. In physiological conditions all actin isoforms are S-nitrosylated on Cys374 and probably on additional Cys residues (37). Because actin is abundantly expressed and largely S-nitrosylated in most cells, it has been proposed that actin serves as a cell SNO-thiol reservoir that trans-nitrosylates with GSH-nitroso-glutathione (GSNO) (24, 26). S-nitrosylation affects actin polymerization and its interaction with proteins that are relevant for actin dynamics, including VASP, cofilin1, profilin and α-actinin (37).
An important finding of this work is that SNO-RhoA occurs in normal podocytes and inversely relates with RhoA activity. Myo9A interacts directly with RhoA through its tail RhoGAP domain (14, 28). Upon binding, Myo9A dephosphorylates RhoA GTPase rendering it inactive (14). We recently reported that Myo9A haploinsufficiency increases RhoA activity in kidneys and podocytes, consistent with loss of RhoGTPase function (7). Here we show that SNO-RhoA is regulated by high glucose and NO dependent, i.e., inversely related to high glucose and positively related to NO availability, and that RhoA activity is inversely related to SNO-RhoA in podocytes. Our results are consistent with a report showing that endothelial cell RhoA S-nitrosylation occurs in physiological conditions, is NO dependent and inhibited by increased intracellular Ca+2, while RhoA activity is inversely correlated to SNO-RhoA (38). It is well-established that RhoA activity is elevated in T1D and T2D experimental models and that high glucose increases RhoA activity in endothelial cells (39), mesangial cells (40) and podocytes (41). Collectively, these findings suggest that RhoA de-nitrosylation induced by the diabetic milieu may mediate RhoA activation in all three glomerular cell types.
Our novel findings of podocyte Myo9A, actin and RhoA S-nitrosylation, which are regulated similarly, suggest that these post-translational modifications are linked. SNO transnitrosylation, i.e., the transfer of NO− between Cys residues, occurs between interacting proteins or those closely adjacent within a cell compartment or microdomain, and its specificity is spatially determined (19, 42, 43). We have previously described Myo9A-actin interaction in podocytes (7). Here we report for the first time Myo9A-RhoA interaction in podocytes. We propose a model of transnitrosylation cascade involving Myo9A, RhoA and actin, three interacting proteins that are critical for podocyte cytoskeleton homeostasis (Figure 6). In this model, the three proteins are S-nitrosylated in control conditions (A) and Myo9A interacts physically and functionally with actin through its catalytic domain that hydrolyses ATP, crosslinks and bundles actin, as well as with RhoA via the tail RhoGAP domain that de-phosphorylates and inactivates the RhoA GTPase (14, 36). In the diabetic milieu (B), high glucose and low NO decrease SNO-Myo9A, SNO-actin and SNO-RhoA leading to increased RhoA activity and abnormal actin dynamics, which alter podocyte function, as assessed by decreased migration. Remarkably, these changes are at least partially reversible. SNO-actin changes upon high-glucose exposure are consistent with the hypothesis that it acts as a SNO reservoir (26). Alternatively, S-nitrosylation of actin isoforms may be regulated differently (24, 44). We speculate that SNO-RhoA may also be regulated by TRPC6-mediated increases in iCa+2, known to be stimulated by AngII and VEGF-A and to mediate RhoA activation in the diabetic milieu (45–47). Akin to SNO inhibition of PKM2 (48–50), the reversible inhibitory S-nitrosylation of RhoA described herein may provide a novel mode of regulation amenable to therapeutic intervention in DKD.
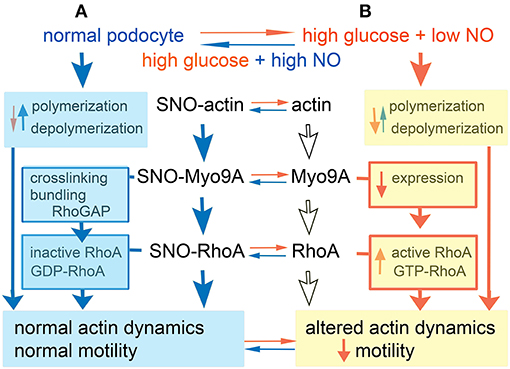
Figure 6. Model. (A) In normal podocyte culture conditions actin, Myo9A and RhoA are S-nitrosylated (SNO), whereby F actin polymerization predominates, Myo9A crosslinks and bundles actin and inactivates RhoA leading to normal actin dynamics and podocyte motility. (B) In high glucose + low NO conditions podocyte SNO-Myo9A, SNO-RhoA and SNO-actin decrease significantly (de-nitrosylate), actin depolymerization may predominate, Myo9A expression decreases and RhoA activity increases, together resulting in altered actin dynamics and reduced podocyte motility. Most of these changes are partially reversible by addition of NO donor.
Further studies are needed to provide detailed insight on the proposed model. For example, it is critical to elucidate whether SNO-MyoA is required for Myo9A's actin crosslinking activity and RhoGAP function and to determine how does SNO-actin influence the balance of actin polymerization-depolymerization dynamics in podocytes. Actin is also oxidized on Met44 and Met47 by MICAL, a flavo-oxygenase expressed in the kidney and in podocytes (51, 52) that leads to F-actin disassembly (52, 53). It is not known if oxidation of actin Met residues is regulated by glucose or NO dependent. Characterization of Myo9A post-translational modifications is limited (14). Identification of Myo9A Cys residues that undergo S-nitrosylation has been elusive as yet, precluding definitive experiments testing our model. Limitations of this study include not examining S-nitrosylation of Myo9A and its interacting partners in biopsy samples from DKD patients, in proximal tubular cells, glomerular endothelial and mesangial cells, known to be involved in DKD progression, nor in additional experimental T1D and T2D models. Further in vivo studies are needed to ascertain whether decreased SNO-RhoA and SNO-actin contribute to DKD progression and to evaluate the effect of NO donors on S-nitrosylation of Myo9A, RhoA and actin.
In summary, this work shows that Myo9A, RhoA and actin are S-nitrosylated in normal podocytes and that diabetic milieu induces Myo9A, actin and RhoA de-nitrosylation, resulting in increased RhoA activity and impaired podocyte migration, which proved to be partially reversible, and therefore potentially targetable. Collectively, our findings uncover S-nitrosylation of Myo9A, actin and RhoA as an integrated signaling crosstalk that reversibly transduces metabolic cues to regulate actin dynamics and podocyte motility.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee at the Yale University School of Medicine.
QL and DV performed experiments, analyzed data, and contributed to manuscript writing. AT designed and supervised experiments, analyzed data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Institutes of Health (NIH) RO1DK109434 grant to AT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Diane Dynia for technical support on this project.
1. Tuttle KR, Bakris GL, Bilous RW, Chiang JL, de Boer IH, Goldstein-Fuchs J, et al. Diabetic kidney disease: a report from an ADA consensus conference. Am J Kidney Dis. (2014) 64:510–33. doi: 10.1053/j.ajkd.2014.08.001
2. Warren AM, Knudsen ST, Cooper ME. Diabetic nephropathy: an insight into molecular mechanisms and emerging therapies. Exp Opin Ther Targ. (2019) 23:579–91. doi: 10.1080/14728222.2019.1624721
3. Ingelfinger JR, Rosen CJ. Clinical credence - SGLT2 inhibitors, diabetes, and chronic kidney disease. N Engl J Med. (2019) 380:2371–73. doi: 10.1056/NEJMe1904740
4. Tonneijck L, Muskiet MH, Smits MM, Van Bommel EJ, Heerspink HJ, Van Raalte DH, et al. Glomerular hyperfiltration in diabetes: mechanisms, clinical significance, and treatment. J Am Soc Nephrol. (2017) 28:1023–39. doi: 10.1681/ASN.2016060666
5. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. (2014) 124:2333–40. doi: 10.1172/JCI72271
6. Tufro A, Veron D. VEGF and podocytes in diabetic nephropathy. Semin Nephrol. (2012) 32:385–93. doi: 10.1016/j.semnephrol.2012.06.010
7. Li Q, Gulati A, Lemaire M, Nottoli T, Bale A, Tufro A. Rho-GTPase activating protein myosin MYO9A identified as a novel candidate gene for monogenic FSGS. Kidney Int. (2021) 99:1102–17. doi: 10.1016/j.kint.2020.12.022
8. Masters TA, Kendrick-Jones J, Buss F. Myosins: domain organization, motor properties, physiological roles and cellular functions. Handb Exp Pharmacol. (2017) 235:77–122. doi: 10.1007/164_2016_29
9. Liu KC, Cheney RE. Myosins in cell junctions. Bioarchitecture. (2012) 2:158–70. doi: 10.4161/bioa.21791
10. Omelchenko T, Hall A. Myosin-IXA regulates collective epithelial cell migration by targeting RhoGAP activity to cell-cell junctions. Curr Biol. (2012) 22:278–88. doi: 10.1016/j.cub.2012.01.014
11. Noris M, Remuzzi G. Non-muscle myosins and the podocyte. Clin Kidney J. (2012) 5:94–101. doi: 10.1093/ckj/sfs032
12. Zhao H, Ma L, Yan M, Wang Y, Zhao T, Zhang H, et al. Association between MYH9 and APOL1 gene polymorphisms and the risk of diabetic kidney disease in patients with type 2 diabetes in a chinese han population. J Diabetes Res. (2018) 9:5068578. doi: 10.1155/2018/5068578
13. Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med. (2011) 365:295–306. doi: 10.1056/NEJMoa1101273
14. Hanley PJ, Vollmer V, Bähler M. Class IX myosins: motorized RhoGAP signaling molecules. In: Coluccio L. editors. Myosins. Advances in Experimental Medicine and Biology. Cham: Springer (2020).
15. Gorman SW, Haider NB, Grieshammer U, Swiderski RE, Kim E, Welch JW, et al. The cloning and developmental expression of unconventional myosin IXA (MYO9A) a gene in the Bardet-Biedl syndrome (BBS4) region at chromosome 15q22-q23. Genomics. (1999) 59:150–60. doi: 10.1006/geno.1999.5867
16. Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. (2001) 108:1341–48. doi: 10.1172/JCI11235
17. Nakagawa T. Uncoupling of VEGF with NO as a mechanism for diabetic nephropathy. Diabetes Res Clin Pract. (2008) 82(Suppl. 1):S67–9. doi: 10.1016/j.diabres.2008.09.030
18. Yuen DA, Stead BE, Zhang Y, White KE, Kabir MG, Thai K, et al. eNOS deficiency predisposes podocytes to injury in diabetes. J Am Soc Nephrol. (2012) 23:1810–23. doi: 10.1681/ASN.2011121170
19. Stomberski CT, Hess DT, Stamler JS. Protein S-Nitrosylation: Determinants of specificity and enzymatic regulation of S-Nitrosothiol-based signaling. Antioxid Redox Signal. (2019) 30:1331–51. doi: 10.1089/ars.2017.7403
20. Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. (2005) 280:19888–94. doi: 10.1074/jbc.M413058200
21. Sayed N, Baskaran P, Ma X, van den Akker F, Beuve A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc Natl Acad Sci USA. (2007) 104:12312–17. doi: 10.1073/pnas.0703944104
22. Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell. (2007) 26:63–74. doi: 10.1016/j.molcel.2007.02.024
23. Sengupta R, Holmgren A. The role of thioredoxin in the regulation of cellular processes by S-nitrosylation. Biochim Biophys Acta. (2012) 1820:689–700. doi: 10.1016/j.bbagen.2011.08.012
24. Dalle-Donne I, Milzani A, Giustarini D, Di Simplicio P, Colombo R, Rossi R. S-NO-actin: S-nitrosylation kinetics and the effect on isolated vascular smooth muscle. J Muscle Res Cell Motil. (2000) 21:171–81. doi: 10.1023/A:1005671319604
25. Heo J, Raines KW, Mocanu V, Campbell SL. Redox regulation of RhoA. Biochemistry. (2006) 45:14481–89. doi: 10.1021/bi0610101
26. Horenberg AL, Houghton AM, Pandey S, Seshadri V, Guilford WH. S-nitrosylation of cytoskeletal proteins. Cytoskeleton. (2019) 76:243–53. doi: 10.1002/cm.21520
27. Liao W, Elfrink K, Bähler M. Head of myosin IX binds calmodulin and moves processively toward the plus-end of actin filaments. J Biol Chem. (2010) 285:24933–42. doi: 10.1074/jbc.M110.101105
28. Yi F, Kong R, Ren J, Zhu L, Lou J, Wu JY, et al. Noncanonical Myo9b-RhoGAP accelerates RhoA GTP hydrolysis by a dual-Arginine-finger mechanism. J Mol Biol. (2016) 428:3043–57. doi: 10.1016/j.jmb.2016.06.014
29. Veron D, Bertuccio CA, Marlier A, Reidy K, Garcia AM, Jimenez J, Velazquez H, et al. Podocyte vascular endothelial growth factor (Vegf 164) overexpression causes severe nodular glomerulosclerosis in a mouse model of type 1 diabetes. Diabetologia. (2011) 54:1227–41. doi: 10.1007/s00125-010-2034-z
30. Veron D, Reidy KJ, Bertuccio C, Teichman J, Villegas G, Jimenez J, et al. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. (2010) 77:989–99. doi: 10.1038/ki.2010.64
31. Bertuccio C, Veron D, Aggarwal PK, Holzman L, Tufro A. Vascular endothelial growth factor receptor 2 direct interaction with nephrin links VEGF-A signals to actin in kidney podocytes. J Biol Chem. (2011) 286:39933–44. doi: 10.1074/jbc.M111.241620
32. Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Meth. (2006) 3:995–1000. doi: 10.1038/nmeth947
33. Veron D, Aggarwal PK, Velazquez H, Kashgarian M, Moeckel G, Tufro A. Podocyte-specific VEGF-a gain of function induces nodular glomerulosclerosis in eNOS null mice. J Am Soc Nephrol. (2014) 25:1814–24. doi: 10.1681/ASN.2013070752
34. Jaffrey SR, Snyder SH. The biotin switch method for detection of S-nitrosylated proteins. Sci Stke. (2001) 86:pl1. doi: 10.1126/stke.2001.86.pl1
35. Sárközy M, Zvara A, Gyémánt N, Fekete V, Kocsis GF, Pipis J. Metabolic syndrome influences cardiac gene expression pattern at the transcript level in male ZDF rats. Cardiovasc Diabetol. (2013) 12:16. doi: 10.1186/1475-2840-12-16
36. Saczko-Brack D, Warchol E, Rogez B, Kröss M, Heissler SM, Sellers JR, et al. Self-organization of actin networks by a monomeric myosin. Proc Natl Acad Sci USA. (2016) 113:E8387–95. doi: 10.1073/pnas.1612719113
37. Xu Q, Huff LP, Fujii M, Griendling KK. Redox regulation of the actin cytoskeleton and its role in the vascular system. Free Radic Biol Med. (2017) 109:84–107. doi: 10.1016/j.freeradbiomed.2017.03.004
38. Chen F, Wang Y, Rafikov R, Haigh S, Zhi WB, Kumar S, et al. RhoA S-nitrosylation as a regulatory mechanism influencing endothelial barrier function in response to G +-bacterial toxins. Biochem Pharmacol. (2017) 127:34–45. doi: 10.1016/j.bcp.2016.12.014
39. Peng H, Luo P, Li Y, Wang C, Liu X, et al. Simvastatin alleviates hyperpermeability of glomerular endothelial cells in early-stage diabetic nephropathy by inhibition of RhoA/ROCK1. PLoS ONE. (2013) 8:e80009. doi: 10.1371/journal.pone.0080009
40. Zhang Y, Peng F, Gao B, Ingram AJ, Krepinsky JC. High glucose-induced RhoA activation requires caveolae and PKCbeta1-mediated ROS generation. Am J Physiol Renal Physiol. (2012) 302:F159–72. doi: 10.1152/ajprenal.00749.2010
41. Yang H, Zhao B, Liao C, Zhang R, Meng K, Xu J, et al. High glucose-induced apoptosis in cultured podocytes involves TRPC6-dependent calcium entry via the RhoA/ROCK pathway. J Biochem Biophys Res Commun. (2013) 434:394–400. doi: 10.1016/j.bbrc.2013.03.087
42. Evangelista AM, Kohr MJ, Murphy E. S-nitrosylation: specificity, occupancy, and interaction with other post-translational modifications. Antioxid Redox Signal. (2013) 19:1209–19. doi: 10.1089/ars.2012.5056
43. Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, et al. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci USA. (2006) 103:19777–82. doi: 10.1073/pnas.0605907103
44. Thom SR, Bhopale VM, Mancini DJ, Milovanova TN. Actin S-nitrosylation inhibits neutrophil beta2 integrin function. J Biol Chem. (2008) 283:10822–34. doi: 10.1074/jbc.M709200200
45. Sonneveld R, van der Vlag J, Baltissen MP, Verkaart SA, Wetzels JF, Berden JH, et al. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII dependent manner. Am J Pathol. (2014) 184:1715–26. doi: 10.1016/j.ajpath.2014.02.008
46. Ilatovskaya DV, Blass G, Palygin O, Levchenko V, Pavlov TS, Grzybowski MN, et al. A NOX4/TRPC6 pathway in podocyte calcium regulation and renal damage in diabetic kidney disease. J Am Soc Nephrol. (2018) 29:1917–27. doi: 10.1681/ASN.2018030280
47. Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, et al. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal. (2010) 3:ra77. doi: 10.1126/scisignal.2001200
48. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. (2017) 23:753–62. doi: 10.1038/nm.4328
49. Zhou HL, Zhang R, Anand P, Stomberski CT, Qian Z, Hausladen A, et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature. (2019) 565:96–100. doi: 10.1038/s41586-018-0749-z
50. Mitchell AR, Yuan M, Morgan HP, McNae IW, Blackburn EA, Le Bihan T, et al. Redox regulation of pyruvate kinase M2 by cysteine oxidation and S-nitrosation. Biochem J. (2018) 475:3275–91. doi: 10.1042/BCJ20180556
51. Tufro A. Podocyte shape regulation by semaphorin 3A and MICAL-1. Methods Mol Biol. (2017) 1493:393–99. doi: 10.1007/978-1-4939-6448-2_28
52. Aggarwal PK, Veron D, Thomas DB, Siegel D, Moeckel G, Kashgarian M, et al. Semaphorin3a promotes advanced diabetic nephropathy. Diabetes. (2015) 64:1743–59. doi: 10.2337/db14-0719
Keywords: diabetic kidney disease, cell cross-talk, RhoA, S-nitrosylation, MYO9A, actin
Citation: Li Q, Veron D and Tufro A (2021) S-Nitrosylation of RhoGAP Myosin9A Is Altered in Advanced Diabetic Kidney Disease. Front. Med. 8:679518. doi: 10.3389/fmed.2021.679518
Received: 11 March 2021; Accepted: 18 June 2021;
Published: 14 July 2021.
Edited by:
Quan Hong, Chinese PLA General Hospital, ChinaReviewed by:
Rui Zeng, Huazhong University of Science and Technology, ChinaCopyright © 2021 Li, Veron and Tufro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alda Tufro, YWxkYS50dWZyb0B5YWxlLmVkdQ==
†Present address: Delma Veron, Facultad de Ciencias de la Salud, Universidad Estatal de Milagro, Milagro, Ecuador
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.