 Emaan Madany
Emaan Madany Derick Okwan-Duodu1
Derick Okwan-Duodu1 Raisa Balbuena-Merle
Raisa Balbuena-Merle Jeanne E. Hendrickson
Jeanne E. Hendrickson David R. Gibb
David R. Gibb- 1Department of Pathology and Laboratory Medicine, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 2Department of Laboratory Medicine, Yale New Haven Hospital, New Haven, CT, United States
At the onset of the corona virus disease 19 (COVID-19) pandemic, there were concerns that patients with sickle cell disease (SCD) might be especially vulnerable to severe sequelae of SARS-CoV-2 infection. While two reports support this conclusion, multiple studies have reported unexpectedly favorable outcomes in patients with SCD. However, mechanisms explaining these disparate conclusions are lacking. Here, we review recent studies indicating that the majority of patients with SCD express elevated levels of anti-viral type 1 interferons (IFNα/β) and interferon stimulated genes, independent of COVID-19, during their baseline state of health. We also present our data from the pre-COVID-19 era, illustrating elevated expression of a well-characterized interferon stimulated gene in a cohort of patients with SCD, compared to race-matched controls. These type 1 interferons and interferon stimulated genes have the potential to contribute to the variable progression of COVID-19 and other viral infections in patients with SCD. While the majority of evidence supports a protective role, the role of IFNα/β in COVID-19 severity in the general population remains an area of current investigation. We conclude that type 1 interferon responses in patients with SCD may contribute to the variable COVID-19 responses reported in prior studies. Additional studies investigating the mechanisms underlying IFNα/β production and other clinical consequences of IFNα/β-mediated inflammation in SCD disease are warranted.
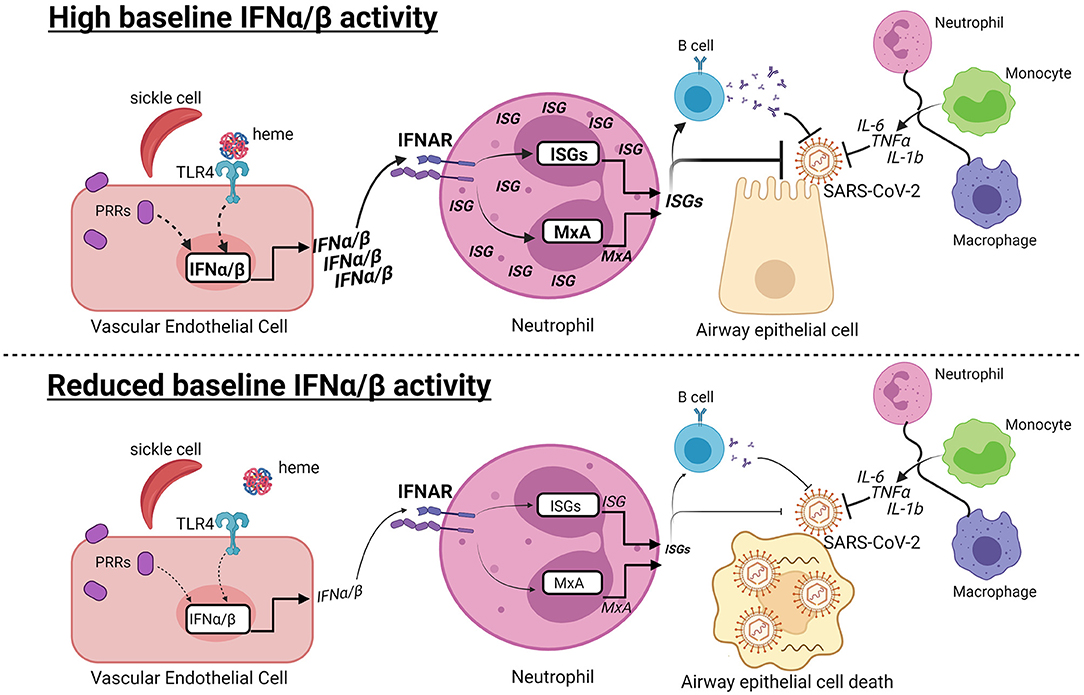
Graphical Abstract. Hypothesis: Baseline type I interferon activity may contribute to variable COVID-19 progression in SCD. (Top) At early stages of SARS-CoV-2 infection, high baseline IFNα/β activity may contribute to the anti-viral response in patients with SCD. Recognition of damage-associated molecular patterns by pattern recognition receptors (PRRs) induces IFNα/β production. Heme released from hemolyzed sickle cells binds Toll-like receptor 4 (TLR4), which may induce IFNα/β in vascular endothelial cells. IFNα/β bind to the IFNα/β receptor (IFNAR) in neutrophils and other cells types, leading to production of MxA and other interferon-stimulated genes (ISGs). ISGs can directly inhibit viral replication and promote B cell production of neutralizing antibodies. The IFNα/β response is one of multiple responses, including production of IL-6, TNFα, and IL-1b, by innate and adaptive immune cells that have the potential to limit COVID-19 progression. (Bottom) In contrast, reduced or absent IFNα/β activity may increase susceptibility to viral infection, leading to airway epithelial cell death and COVID-19. Dashed lines indicate potentially connected pathways, while solid lines are supported by prior studies.
Introduction
Certain comorbidities are associated with an increased severity of corona virus disease 19 (COVID-19) resulting from SARS-CoV-2 infection (1). Given that patients with sickle cell disease (SCD) have underlying chronic inflammation, significant cardiopulmonary dysfunction, and immune deficiency due in part to hyposplenism, concern exists regarding the severity of COVID-19 in patients with SCD (2). In March of 2020, the National Haemoglobinopathy Panel in the United Kingdom deemed that patients with SCD and other rare hemoglobinopathies are exceptionally vulnerable populations requiring strict self-isolation (3). In November, the Center for Disease Control added SCD to the list of comorbidities that may increase the severity of COVID-19 (https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html).
Multiple case series and reports have described the disease course of patients with SCD and COVID-19. One common finding is that COVID-19, like H1N1 influenza and other respiratory infections, can trigger acute chest syndrome and vaso-occlusive crises (4, 5). However, it is unclear whether patients with SCD have a more or less severe COVID-19 disease course than those without SCD. Minniti et al. reported that pre-existing co-morbidities, including renal and cardiopulmonary disease, are associated with poor COVID-19 outcomes in patients with SCD (6). In addition, Singh et al. found an increase in hospitalizations and pneumonia, but no increase in mortality, when comparing patients with SCD to race-matched controls with comorbidities (7). In contrast, smaller studies have reported favorable outcomes in patients with SCD (4, 8–11). As reviewed by Sahu et al., most SARS-CoV-2 infected patients with SCD experience mild disease with few patients requiring respiratory support (12). While these outcomes may be due to early diagnosis and treatment in an at-risk population, underlying inflammatory mechanisms in SCD may also influence COVID-19 progression.
Following SARS-CoV-2 infection, multiple cell types produce cytokines, including IL-1b, TNFα, and IL-6, which contribute to the anti-viral response and inflammation (13). Patients with SCD have chronic inflammation at baseline, characterized by leukocytosis, endothelial damage, oxidative stress, and production of pro-inflammatory cytokines. Interleukin-6 is induced during COVID-19, and there are reports of improved COVID-19 outcomes following anti-IL-6 therapy in patients with SCD (14, 15). In addition, there is particular interest in the role of anti-viral type 1 interferons (IFNα/β) produced following SARS-CoV-2 infection. IFNα/β, including IFNβ, IFNω and 12 subtypes of IFNα, were discovered in 1957 for their critical role in anti-viral immunity (16). During infection, viral nucleic acids bind pattern recognition receptors and induce IFNα/β, which in turn activate signaling via the IFNα/β receptor to produce interferon-stimulated genes (ISGs) that interfere with viral replication and promote the production of neutralizing antibodies. Independent of COVID-19, Hounkpe et al. performed a meta-analysis of gene expression studies and identified a cluster of ISGs enriched in patients with SCD (17). In addition, Hermand et al. recently reported that serum IFNα and ISGs produced by neutrophils are elevated in children with SCD, compared to healthy blood donors (18). Here, we assessed the baseline expression of a well-characterized ISG, Myxovirus Resistance Protein 1 (MxA), in adults with SCD compared to race-matched controls, and describe SARS-CoV-2 test results and COVID-19 hospitalization outcomes in one patient cohort.
Methods and Results
Discovered in 1962 for its role in resistance to orthomyxoviruses, including influenza, MxA is a GTPase that targets nucleocapsid proteins and inhibits viral transcription and replication (19, 20). MxA has since been reported to be a clinically applicable biomarker of IFNα/β activation (21). In the pre-COVID era, we utilized an ELISA-based assay (BioVendor, Czech Republic) to quantify MxA levels in the whole blood of patients with and without SCD, according to manufacturer instructions. Initially, 13 SCD and 37 control de-identified remnant type and screen samples were randomly selected from the blood bank. All samples were analyzed within 72 h of the blood draw. In this initial analysis, patients with SCD expressed a significantly elevated level of MxA, compared to controls (Figure 1A).
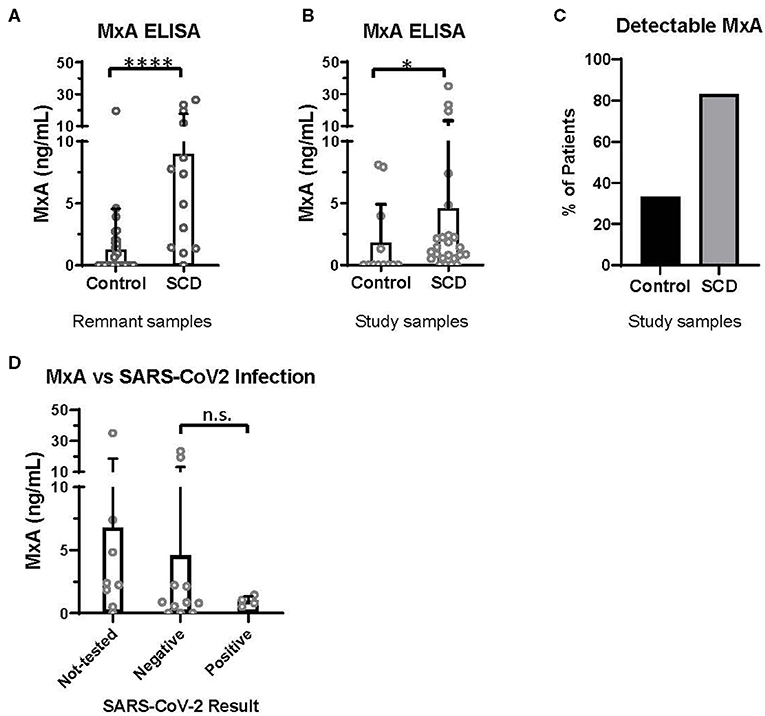
Figure 1. MxA expression is increased in patients with SCD, compared to controls. MxA expression in whole blood measured by MxA Protein Human ELISA (BioVendor). (A) MxA expression in de-identified remnant samples from patients with (n = 13) or without SCD (n = 37). (B) MxA expression in patients with SCD (n = 24) and race-matched controls (n = 12) summarized in Table 1. (C) Percentage of subjects summarized in Table 1 with detectable levels of MxA. (D) MxA expression of patients with SCD (n = 24) who were not tested or tested positive (n = 4) or negative (n = 11) for SARS-CoV-2 by PCR. n.s., not significant by Kruskal Wallis test with a Dunn's post-test. *p < 0.05, ****p < 0.0001 by Mann-Whitney U-test. Bars represent the mean. Circles represent values from individual patient or control samples, and error bars represent the standard deviation.
Given that these initial samples were de-identified and the health status of patients and controls was unknown, a second study comparing MxA levels in patients with SCD seen in out-patients clinics for routine care and race-matched controls was initiated. Blood was drawn from 24 patients and 12 controls in their baseline state of health. 22 patients had Hgb SS, 1 had Hgb SC, and 1 had Sβ0 disease (Table 1). Exclusion criteria included acute illness or crisis, pregnancy, and lack of competency to provide informed consent. The study was approved by the Yale Institutional Review Board.
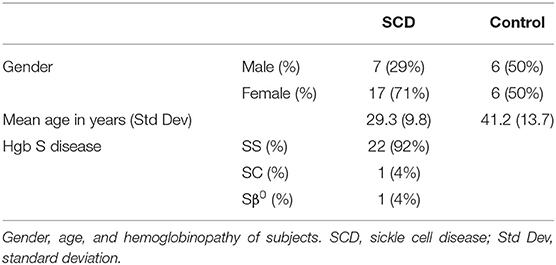
Table 1. Demographics of subjects enrolled in second study.
In this second study, MxA was also significantly elevated in patients with SCD compared to control subjects (Figure 1B). 83% of patients with SCD had detectable MxA, compared to 33% of controls (Figure 1C). It is noteworthy that expression of MxA, as well as IFNα and other ISGs (18), is highly variable amongst patients with SCD, which may contribute to differences in anti-viral immunity. In addition, MxA was detected in four of 12 controls, which may reflect the role of IFNα/β and MxA in other diseases, including autoimmunity and viral infections (19, 21).
Finally, we retrospectively examined SARS-CoV-2 testing results and COVID-19 severity of patients with SCD enrolled in the second study. Fifteen of the 24 patients had test results of SARS-CoV-2 PCR documented in the electronic medical record. Eleven patients had only negative test results and four patients tested positive. Three of the four with positive PCR results were admitted to the hospital: two were diagnosed with acute chest syndrome and received supplemental oxygen but neither required intubation; all patients survived. MxA levels of these patients, according to SARS-CoV-2 test results, are shown in Figure 1D. There were no significant differences in MxA levels between untested patients, patients with negative results, and patients with positive results.
Discussion
Data presented here, in combination with prior studies (17, 18), indicate that patients with SCD express an IFNα/β gene signature. As IFNα/β and ISGs are upregulated as a result of SARS-CoV-2 infection and vaccination (22), it is worth noting that these laboratory studies were completed prior to the COVID-19 pandemic. This allowed for baseline measurement of MxA without the potential variable of SARS-CoV-2 infection or vaccination. While SARS-CoV-2 test results and COVID-19 hospitalization outcomes were examined, based on the sample size, no definitive conclusions can be drawn about baseline MxA levels and the risk of SARS-CoV-2 infection or disease severity. While it is plausible that elevated baseline anti-viral IFNα/β and ISGs, including MxA, contribute to the variable progression of COVID-19 and other viral infections in patients with SCD, it is certain that other inflammatory responses, including production of IL-1b, TNFα, and IL-6, also significantly contribute to the anti-viral response (23).
Multiple groups have investigated the role of IFNα/β in COVID-19 disease progression, independent of SCD. Utilizing mass cytometry and Nanostring technology, Hadjadj et al. performed immune profiling of 50 COVID-19 patients and observed an impaired IFNα/β response in severe and critically ill patients, compared to those with mild to moderate disease (24). Also in support of a protective role of IFNα/β, another group found inborn errors of IFNα/β immunity and an increased prevalence of autoantibodies against IFNα/β, including IFNα and IFNω, in critically ill COVID-19 patients, compared to those with mild disease or asymptomatic infection (25, 26). These results are consistent with recent reports of reduced COVID-19 associated mortality in patients treated with IFNβ (27, 28), and reduced duration of detectable virus and inflammation in a cohort treated with IFN-α2b (29).
Conversely, another group concluded that the IFNα/β response can also exacerbate deleterious COVID-19 induced inflammation. Utilizing single cell RNA sequencing, Lee et al. observed that an IFNα/β response was absent in mild disease but co-existed with Tumor Necrosis Factor α (TNFα) and IL-1b production in severe COVID-19 disease (13). However, it is unclear whether the correlation between IFNα/β and severe disease in this study is causal or responsive. In addition, Ziegler et al. demonstrated that angiotensin converting enzyme 2 (ACE2), a receptor for the SARS-CoV-2 spike protein, is an ISG expressed by multiple pulmonary cell types, suggesting that the virus may exploit the IFNα/β response to gain viral entry (30).
Although evidence supporting a protective role of IFNα/β outweighs that supporting a deleterious role in COVID-19 disease progression, it remains an area of current investigation. Ongoing clinical trials have shown conflicting results. A phase 2 trial of inhaled IFNβ showed clinical improvement (31), while data from the World Health Organization Solidarity trial indicate that IFNβ is not effective in improving mortality or other endpoints (32). Others have postulated that the timing of IFNα/β responses may impact the outcomes of the aforementioned studies (33). Early robust responses and early treatment with inhaled IFNβ may lead to reduced viral load; whereas delayed and dysregulated IFNα/β responses or IFNα/β-based treatments may exacerbate deleterious hyperinflammation at later stages of disease (34, 35).
It is noteworthy that IFNα/β has also been implicated in the pathogenesis of autoimmune diseases, including rheumatoid arthritis, myositis, Sjögren's syndrome (SS), systemic sclerosis and systemic lupus erythematosus (SLE) (36–40). Approximately two-thirds of adult patients and nearly all children with SLE express an IFNα/β gene signature (41, 42), and more than 50% of SLE-associated genetic variants have been linked to the IFNα/β pathway (43). In addition, approximately 25% of patients with SLE produce anti-IFNα/β autoantibodies, which are associated with decreased disease activity (44, 45). Due to the IFNα/β gene signature and the use of hydroxychloroquine in patients with SLE, many groups have investigated COVID-19 disease progression in patients with SLE and have reported variable results (46, 47). Unfortunately, it has been difficult to separate the effects of baseline IFNα/β activity and the use of immunosuppressive therapies, which result in an increased incidence of multiple viral infections in patients with SLE (48).
Specific mechanisms leading to IFNα/β activation during viral infection or SLE are fairly well-characterized. However, IFNα/β-inducing stimuli and pathways in SCD have not been identified. Heme-induced inflammation is one of many candidates yet to be investigated. Heme, released from hemolyzed RBCs, binds to Toll-like receptor 4 and induces NFκb-mediated production of pro-inflammatory cytokines, including IL-6, IL-12, and TNFα (49). However, its role in IFNα/β activation has not been reported, and many other ligands and pathways are worthy of investigation. Moreover, while anti-IFNα/β antibodies that prevent IFNα/β-induced inflammation have been described in patients with autoimmune polyendocrinopathy syndrome, SLE and COVID-19 (25, 50, 51), it is unclear whether they are produced by patients with SCD and whether they contribute to the variable disease progression during viral infection.
Finally, unlike in viral infection and autoimmunity, the contribution of IFNα/β activation to chronic inflammation and the numerous sequelae of SCD are unknown and warrant investigation. For example, endothelial damage is a hallmark of SCD associated with vaso-occlusive crises, including acute chest syndrome and cerebrovascular accidents, and mortality (52, 53). IFNα/β has been shown to induce endothelial damage in patients with anti-phospholipid syndrome, thrombotic microangiopathy, and SLE, and recipients of IFNα/β therapy (54–57). However, the role of IFNα/β in endothelial damage, and its associated adverse events, in SCD has not been investigated. In addition, prior studies reported that IFNα/β is a critical regulator of RBC alloimmunization in transfusion mouse models (58–61). Alloantibody production against minor RBC antigens can lead to significant hemolytic events and severely limit availability of blood products for alloimmunized patients. As patients with SCD have the highest incidence of RBC alloimmunization (62) compared with any other disease population, the role of IFNα/β in RBC alloimmunization in patients with SCD warrants further study.
In conclusion, data presented here, in combination with other studies, indicate that the majority of patients with SCD express an IFNα/β gene signature. While the impact of IFNα/β and ISGs on SARS-CoV-2 infection risk or COVID-19 disease severity remains to be fully determined, baseline IFNα/β activity may contribute to the variable disease progression reported in prior studies. Additional studies investigating mechanisms underlying IFNα/β production and clinical consequences of IFNα/β-mediated inflammation in SCD are needed. Future studies elucidating the role of IFNα/β in chronic inflammation, RBC alloimmunization, and vaso-occlusive events could lead to targeted therapies to mitigate severe sequelae in patients with SCD.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Yale University Institutional Review Board. Written informed consent to participate in this study was provided by the participants or the participants' legal guardian/next of kin.
Author Contributions
Project design and data analysis were completed by EM, DO-D, JEH, and DRG. Experiments were completed by DRG, EM, and RB-M. DRG wrote the initial draft of the manuscript. All authors contributed to edits of the manuscript.
Funding
This work was funded by the NIH/NHLBI, R01 HL132951 (to JEH), T32 HL007974-14 (to Brian Smith for RB-M), K08 HL141446 to DRG, the American Society of Hematology (5283), and the National Blood Foundation (R13672) to DRG.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the volunteers and patients at Yale New Haven Hospital for their participation in this study.
References
1. Shevel E. Conditions favoring increased COVID-19 morbidity and mortality: their common denominator and its early treatment. Mo Med. (2021) 118:113–5.
2. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. (1994) 330:1639–44. doi: 10.1056/NEJM199406093302303
3. Telfer P, De la Fuente J, Sohal M, Brown R, Eleftheriou P, Roy N, et al. Real-time national survey of COVID-19 in hemoglobinopathy and rare inherited anemia patients. Haematologica. (2020) 105:2651–4. doi: 10.3324/haematol.2020.259440
4. McCloskey KA, Meenan J, Hall R, Tsitsikas DA. COVID-19 infection and sickle cell disease: a UK centre experience. Br J Haematol. (2020) 190:e57–e8. doi: 10.1111/bjh.16779
5. Nur E, Gaartman AE, van Tuijn CFJ, Tang MW, Biemond BJ. Vaso-occlusive crisis and acute chest syndrome in sickle cell disease due to 2019 novel coronavirus disease (COVID-19). Am J Hematol. (2020) 95:725–6. doi: 10.1002/ajh.25821
6. Minniti CP, Zaidi AU, Nouraie M, Manwani D, Crouch GD, Crouch AS, et al. Clinical predictors of poor outcomes in patients with sickle cell disease and COVID-19 infection. Blood Adv. (2021) 5:207–15. doi: 10.1182/bloodadvances.2020003456
7. Singh A, Brandow AM, Panepinto JA. COVID-19 in individuals with sickle cell disease/trait compared with other black individuals. Blood Adv. (2021) 5:1915–21. doi: 10.1182/bloodadvances.2020003741
8. Appiah-Kubi A, Acharya S, Fein Levy C, Vlachos A, Ostovar G, Murphy K, et al. Varying presentations and favourable outcomes of COVID-19 infection in children and young adults with sickle cell disease: an additional case series with comparisons to published cases. Br J Haematol. (2020) doi: 10.1111/bjh.17013
9. Azerad MA, Bayoudh F, Weber T, Minon JM, Ketelslegers O, Hoyoux M, et al. Sickle cell disease and COVID-19: atypical presentations and favorable outcomes. EJHaem. (2020) doi: 10.1002/jha2.74
10. Balanchivadze N, Kudirka AA, Askar S, Almadhoun K, Kuriakose P, Fadel R, et al. Impact of COVID-19 infection on 24 patients with sickle cell disease. One Center Urban Experience, Detroit, MI, USA. Hemoglobin. (2020) 44:284–9. doi: 10.1080/03630269.2020.1797775
11. Chakravorty S, Padmore-Payne G, Ike F, Tshibangu V, Graham C, Rees D, et al. COVID-19 in patients with sickle cell disease - a case series from a UK tertiary hospital. Haematologica. (2020) 105:2691–3. doi: 10.3324/haematol.2020.254250
12. Sahu KK, Siddiqui AD, Cerny J. Managing sickle cell patients with COVID-19 infection: the need to pool our collective experience. Br J Haematol. (2020) 190:e86–e89. doi: 10.1111/bjh.16880
13. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type i interferons in development of severe COVID-19. Sci Immunol. (2020) 5:1554. doi: 10.1126/sciimmunol.abd1554
14. De Luna G, Habibi A, Deux JF, Colard M, Pham Hung AL, Schlemmer F, Joher N, et al. Rapid and severe covid-19 pneumonia with severe acute chest syndrome in a sickle cell patient successfully treated with tocilizumab. Am J Hematol. (2020) 95:876–878. doi: 10.1002/ajh.25833
15. Odievre MH, de Marcellus C, Ducou Le Pointe H, Allali S, Romain AS, Youn J, et al. Dramatic improvement after tocilizumab of severe COVID-19 in a child with sickle cell disease and acute chest syndrome. Am J Hematol. (2020) 95:E192–E4. doi: 10.1002/ajh.25855
16. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
17. Hounkpe BW, Fiusa MM, Colella MP, da Costa LN, Benatti Rde O, Saad ST, et al. Role of innate immunity-triggered pathways in the pathogenesis of Sickle Cell Disease: a meta-analysis of gene expression studies. Sci Rep. (2015) 5:17822. doi: 10.1038/srep17822
18. Hermand P, Azouzi S, Gautier EF, Guillonneau F, Bondet V, Duffy D, et al. The proteome of neutrophils in sickle cell disease reveals an unexpected activation of interferon alpha signaling pathway. Haematologica. (2020) 105:2851–4. doi: 10.3324/haematol.2019.238295
19. Haller O, Staeheli P, Schwemmle M, Kochs G. Mx gTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol. (2015) 23:154–63. doi: 10.1016/j.tim.2014.12.003
20. Lindenmann J. Resistance of mice to mouse-adapted influenza A virus. Virology. (1962) 16:203–4. doi: 10.1016/0042-6822(62)90297-0
21. Maria NI, Brkic Z, Waris M, van helden-Meeuwsen CG, Heezen K, van de Merwe JP, et al. MxA as a clinically applicable biomarker for identifying systemic interferon type I in primary Sjogren's syndrome. Ann Rheum Dis. (2014) 73:1052–9. doi: 10.1136/annrheumdis-2012-202552
22. Teijaro JR, Farber DL. COVID-19 vaccines: modes of immune activation and future challenges. Nat Rev Immunol. (2021) 21:195–97. doi: 10.1038/s41577-021-00526-x
23. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-CoV-2 drives development of covid-19. Cell. (2020) 181:1036–45 e9 doi: 10.1016/j.cell.2020.04.026
24. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type i interferon activity and inflammatory responses in severe COVID-19 patients. Science. (2020) 369:718–24. doi: 10.1126/science.abc6027
25. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science. (2020) 370:eabd4585. doi: 10.1126/science.abd4585
26. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. (2020) 370:eabd4570. doi: 10.1126/science.abd4570
27. Nakhlband A, Fakhari A, Azizi H. Interferon-beta offers promising avenues to COVID-19 treatment: a systematic review and meta-analysis of clinical trial studies. Naunyn Schmiedebergs Arch Pharmacol. (2021) 15:1–10. doi: 10.1007/s00210-021-02061-x
28. Baghaei P, Dastan F, Marjani M, Moniri A, Abtahian Z, Ghadimi S, et al. Combination therapy of iFNbeta1 with lopinavir-ritonavir, increases oxygenation, survival and discharging of sever COVID-19 infected inpatients. Int Immunopharmacol. (2021) 92:107329. doi: 10.1016/j.intimp.2020.107329
29. Zhou Q, Chen V, Shannon CP, Wei XS, Xiang X, Wang X, et al. Interferon-alpha2b treatment for COVID-19. Front Immunol. (2020) 11:1061. doi: 10.1101/2020.04.06.20042580
30. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 Receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. (2020) 181:1016−35 e19. doi: 10.1016/j.cell.2020.04.035
31. Monk PD, Marsden RJ, Tear VJ, Brookes J, Batten TN, Mankowski M, et al. Inhaled Interferon Beta. Safety and efficacy of inhaled nebulised interferon beta-1a (SNG001) for treatment of SARS-CoV-2 infection: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir Med. (2021) 9:196–206. doi: 10.1016/S2213-2600(20)30511-7
32. Consortium OST, Pan H, Peto R, Henao-Restrepo AM, Preziosi MP, Sathiyamoorthy V, et al. Repurposed antiviral drugs for Covid-19 - interim WHO solidarity trial results. N Engl J Med. (2021) 384:497–511. doi: 10.1056/NEJMoa2023184
33. Carvalho T, Krammer F, Iwasaki A. The first 12 months of COVID-19: a timeline of immunological insights. Nat Rev Immunol. (2021) 21:245–56. doi: 10.1038/s41577-021-00522-1
34. Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M, Meyerholz DK, Perlman S, et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. (2019) 129:3625–39. doi: 10.1172/JCI126363
35. Wang N, Zhan Y, Zhu L, Hou Z, Liu F, Song P, et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microbe. (2020) 28:455–64 e2. doi: 10.1016/j.chom.2020.07.005
36. Olsen N, Sokka T, Seehorn CL, Kraft B, Maas K, Moore J, et al. A gene expression signature for recent onset rheumatoid arthritis in peripheral blood mononuclear cells. Ann Rheum Dis. (2004) 63:1387–92. doi: 10.1136/ard.2003.017194
37. Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type i interferon pathway. Ann Rheum Dis. (2011) 70:2029–36. doi: 10.1136/ard.2011.150326
38. Bave U, Nordmark G, Lovgren T, Ronnelid J, Cajander S, Eloranta ML, et al. Activation of the type i interferon system in primary sjogren's syndrome: a possible etiopathogenic mechanism. Arthritis Rheum. (2005) 52:1185–95. doi: 10.1002/art.20998
39. Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. (2007) 13:59–68. doi: 10.2119/2006-00085.Baechler
40. Assassi S, Mayes MD, Arnett FC, Gourh P, Agarwal SK, McNearney TA, et al. Systemic sclerosis and lupus: points in an interferon-mediated continuum. Arthritis Rheum. (2010) 62:589–98. doi: 10.1002/art.27224
41. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. (2003) 100:2610–5. doi: 10.1073/pnas.0337679100
42. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. (2003) 197:711–23. doi: 10.1084/jem.20021553
43. Bronson PG, Chaivorapol C, Ortmann W, Behrens TW, Graham RR. The genetics of type i interferon in systemic lupus erythematosus. Curr Opin Immunol. (2012) 24:530–7. doi: 10.1016/j.coi.2012.07.008
44. Harris BD, Kuruganti S, Deshpande A, Goepfert PA, Chatham WW, Walter MR. Characterization of type-I IFN subtype autoantibodies and activity in SLE serum and urine. Lupus. (2020) 29:1095–105. doi: 10.1177/0961203320935976
45. Morimoto AM, Flesher DT, Yang J, Wolslegel K, Wang X, Brady A, et al. Association of endogenous anti-interferon-alpha autoantibodies with decreased interferon-pathway and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. (2011) 63:2407–15. doi: 10.1002/art.30399
46. Gianfrancesco M, Hyrich KL, Al-Adely S, Carmona L, Danila MI, Gossec L, et al. Characteristics associated with hospitalisation for COVID-19 in people with rheumatic disease. data from the COVID-19 Global Rheumatology Alliance physician-reported registry. Ann Rheum Dis. (2020) 79:859–66. doi: 10.1136/annrheumdis-2020-217871
47. Mathian A, Mahevas M, Rohmer J, Roumier M, Cohen-Aubart F, Amador-Borrero B, et al. Clinical course of coronavirus disease 2019 (COVID-19) in a series of 17 patients with systemic lupus erythematosus under long-term treatment with hydroxychloroquine. Ann Rheum Dis. (2020) 79:837–9. doi: 10.1136/annrheumdis-2020-218795
48. Danza A, Ruiz-Irastorza G. Infection risk in systemic lupus erythematosus patients: susceptibility factors and preventive strategies. Lupus. (2013) 22:1286–94. doi: 10.1177/0961203313493032
49. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers tLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
50. Meager A, Visvalingam K, Peterson P, Moll K, Murumagi A, Krohn K, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. (2006) 3:e289. doi: 10.1371/journal.pmed.0030289
51. Panem S, Check IJ, Henriksen D, Vilcek J. Antibodies to alpha-interferon in a patient with systemic lupus erythematosus. J Immunol. (1982) 129:1–3.
52. Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. (2004) 11:129–51. doi: 10.1080/mic.11.2.129.151
53. Kato GJ, Martyr S, Blackwelder WC, Nichols JS, Coles WA, Hunter LA, et al. Levels of soluble endothelium-derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. Br J Haematol. (2005) 130:943–53. doi: 10.1111/j.1365-2141.2005.05701.x
54. Grenn RC, Yalavarthi S, Gandhi AA, Kazzaz NM, Nunez-Alvarez C, Hernandez-Ramirez D, et al. Endothelial progenitor dysfunction associates with a type I interferon signature in primary antiphospholipid syndrome. Ann Rheum Dis. (2017) 76:450–7. doi: 10.1136/annrheumdis-2016-209442
55. Jia H, Thelwell C, Dilger P, Bird C, Daniels S, Wadhwa M. Endothelial cell functions impaired by interferon in vitro: insights into the molecular mechanism of thrombotic microangiopathy associated with interferon therapy. Thromb Res. (2018) 163:105–16. doi: 10.1016/j.thromres.2018.01.039
56. Jodele S, Medvedovic M, Luebbering N, Chen J, Dandoy CE, Laskin BL, et al. Interferon-complement loop in transplant-associated thrombotic microangiopathy. Blood Adv. (2020) 4:1166–77. doi: 10.1182/bloodadvances.2020001515
57. Tyden H, Lood C, Gullstrand B, Nielsen CT, Heegaard NHH, Kahn R, et al. Endothelial dysfunction is associated with activation of the type i interferon system and platelets in patients with systemic lupus erythematosus. RMD Open. (2017) 3:e000508. doi: 10.1136/rmdopen-2017-000508
58. Gibb DR, Liu J, Natarajan P, Santhanakrishnan M, Madrid DJ, Eisenbarth SC, et al. Type I IFN is necessary and sufficient for inflammation-Induced red blood cell alloimmunization in mice. J Immunol. (2017) 199:1041–50. doi: 10.4049/jimmunol.1700401
59. Gibb DR, Liu J, Santhanakrishnan M, Natarajan P, Madrid DJ, Patel S, et al. B cells require type 1 interferon to produce alloantibodies to transfused KEL-expressing red blood cells in mice. Transfusion. (2017) 57:2595–608. doi: 10.1111/trf.14288
60. M. E. Lee JY, El Kadi N, Pandya S, Ng K, Yamashita M, Jefferies CA, et al. Type 1 interferon gene signature promotes rBC alloimmunizaiton in a lupus mouse model. Front Immunol. (2020) 11:254. doi: 10.3389/fimmu.2020.584254
61. Liu D, Gibb DR, Escamilla-Rivera V, Liu J, Santhanakrishnan M, Shi Z, et al. Type 1 iFN signaling critically regulates influenza-induced alloimmunization to transfused KEL rbcs in a murine model. Transfusion. (2019) 59:3243–52. doi: 10.1111/trf.15482
Keywords: sickle cell disease, type 1 interferons, COVID-19, myxovirus resistance interferon stimulated genes, SARS-CoV-2
Citation: Madany E, Okwan-Duodu D, Balbuena-Merle R, Hendrickson JE and Gibb DR (2021) Potential Implications of a Type 1 Interferon Gene Signature on COVID-19 Severity and Chronic Inflammation in Sickle Cell Disease. Front. Med. 8:679030. doi: 10.3389/fmed.2021.679030
Received: 10 March 2021; Accepted: 25 June 2021;
Published: 22 July 2021.
Edited by:
Lakshmanan Krishnamurti, Emory University, United StatesReviewed by:
Manisha Rajan Madkaikar, Indian Council of Medical Research, IndiaEleonora Timperi, Institut Curie, France
Copyright © 2021 Madany, Okwan-Duodu, Balbuena-Merle, Hendrickson and Gibb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David R. Gibb, ZGF2aWQuZ2liYkBjc2hzLm9yZw==