
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 08 June 2021
Sec. Nuclear Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.675209
This article is part of the Research Topic Nuclear Medicine in Cancer Diagnosis View all 11 articles
 Caroline Dagallier1,2
Caroline Dagallier1,2 François Avry2
François Avry2 Yann Touchefeu3,4Frédéric Buron5Sylvain Routier5
Yann Touchefeu3,4Frédéric Buron5Sylvain Routier5 Michel Chérel3
Michel Chérel3 Nicolas Arlicot1,2,6*
Nicolas Arlicot1,2,6*Colorectal cancer (CRC) is the second most common cause of cancer death, making early diagnosis a major public health challenge. The role of inflammation in tumorigenesis has been extensively explored, and among the identified markers of inflammation, cyclooxygenase-2 (COX-2) expression seems to be linked to lesions with a poor prognosis. Until now, COX-2 expression could only be accessed by invasive methods, mainly by biopsy. Imaging techniques such as functional Positron Emission Tomography (PET) could give access to in vivo COX-2 expression. This could make the staging of the disease more accurate and would be of particular interest in the exploration of the first metastatic stages. In this paper, we review recent progress in the development of COX-2 specific PET tracers by comparing the radioligands' characteristics and highlighting the obstacles that remain to be overcome in order to achieve the clinical development of such a radiotracer, and its evaluation in the management of CRC.
Colorectal cancer (CRC) is one of the most frequent types of cancer, ranking second in most developed countries, and has the second highest mortality rate (1, 2). This rate is especially high for late diagnosis and advanced stage disease. Thanks to an increase in CRC screening, the incidence rate has been decreasing for the last two decades (2). However, this high mortality rate draws early detection of CRC and prediction of recurrences and metastases as two major public health challenges, in order to initiate the appropriate treatment as early as possible.
Inflammation seems to play a key role in CRC physiopathology. Among the many known markers of inflammation, cyclooxygenase-2 (COX-2) was identified as having a crucial role from the first stages of tumorigenesis (3, 4). Reviews of over 30 epidemiologic studies showed that regular consumption of non-steroidal anti-inflammatory drugs (NSAID) was associated with a 30–50% reduced incidence of several cancer types, including CRC (5, 6). Interestingly, patients with familial adenomatous polyposis receiving COX-2 inhibitors as a preventive treatment developed fewer adenomas than patients receiving a placebo (7).
COX-2 is an enzyme that intervenes in the first steps of prostaglandin E2 synthesis from arachidonic acid and can be induced by various pro-inflammatory signals. COX-2 overexpression has been identified in different malignant neoplastic tissues, especially in up to 85% of adenocarcinomas (8–10) and is also associated with the potential for progression and recurrences of colorectal tumors. Molecular biology studies on CRC tissue samples concluded that COX-2 expression is significantly correlated with invasive (11) and metastatic phenotypes (12, 13). Moreover, high COX-2 expression in patients treated with radical surgery is a prognostic factor for recurrences, mainly because of undetected metastasis (13, 14). The COX-2 expression level is high in CRC cells. On the contrary, COX-2 has a low basal expression in healthy colon epithelial cells (15), making COX-2 a relevant biomarker for molecular imaging, especially for Positron Emission Tomography (PET).
The correlation of elevated COX-2 expression with the first stages of tumorigenesis and cancer progression suggests that COX-2 could be a target for the early imaging of pre-cancerous colorectal lesions. Since inflammation seems to be a hallmark of malignant CRC, PET imaging could allow the early detection of CRC dissemination and thus spot metastasis. In addition, in other cancerous pathologies (breast, prostate), COX-2 expression increases with the stage of the disease (16, 17). PET-scanning could therefore be a tool for CRC staging. Furthermore, positive therapeutic results in cohorts of patients with different types of solid tumors receiving COX-2 inhibitors in addition to their respective standard chemotherapy (18–20) suggest that COX-2 molecular imaging could provide stratification of the patients, enabling individualized treatment approaches. Notably, it could help to decide whether neoadjuvant chemotherapy is needed or determine whether patients would respond to COX-2 inhibitors therapy.
Up to now, numerous radioligands targeting COX-2 have been developed for SPECT or PET applications. Reviews by Laube et al., de Vries et al., and Pacelli et al., summarized their structures and synthesis methods and highlighted the challenges encountered in the development of COX-2 radiotracers (21–23). In the present review, we explore the recent progress of COX-2 molecular imaging in CRC, by comparing the radioligands' characteristics and highlighting the obstacles that remain to be overcome in order to achieve the clinical development of such a radiotracer.
The PubMed database was screened using pre-defined search dates (January 1995–January 2021). The search terms used were as follows: “colorectal cancer” and “COX-2” or “cyclooxygenase 2” and “PET.” It yielded 78 results. We screened preclinical results performed only on colorectal cancer cell lines. Title-, abstract- or full text-reading led to the exclusion of 70 papers because they did not focus on CRC or did not include in vivo PET radioligand evaluation or did not target COX-2. Eight original papers fulfilled these criteria and were thus included in the scope of the present review. Chemical structures of the PET radioligands are presented. The main conditions and results of these PET imaging preclinical studies assessing COX-2 radiotracers in CRC xenograft models are summarized in Table 1.

Table 1. Summary of characterization studies assessing COX-2 PET radiotracers in CRC xenograft models.
Celecoxib 1 (Figure 1) is a well-known COX-2 specific inhibitor used as an anti-inflammatory drug. Its specificity and selectivity for COX-2 over COX-1 makes it a natural candidate for PET applications. In 2005, Prabhakaran et al. synthesized 11C-Celecoxib 2 (Figure 1) in 8 efficient steps including a palladium catalyzed radiomethylation (32), but this radioligand has not yet been tested in a CRC preclinical model. A biodistribution study in baboons showed unsatisfactory pharmacokinetic properties. 11C-2 underwent a fast metabolism (80% after 30 min in plasma samples) and was quickly cleared out of the organism by urinary excretion (33). Therefore, several series of celecoxib derivatives were proposed to work around the limitations of celecoxib. In 2011, Uddin et al. developed a series of celecoxib [18F]-fluorine derivatives (24). Synthesis in 7 steps involving an aliphatic nucleophilic substitution led to the best candidate. Diethylaminosulfurtrifluoride (DAST) mediated fluoration of its alcohol precursor gave the fluoromethyl derivative 18F-3 (Figure 2). In inhibition assays using purified COX-2, 18F-3 exhibited an IC50 of 160 vs. 30 nM for 1. In vivo evaluation was conducted in a Nu-fox1nu mice model bearing human head and neck squamous cell carcinoma cells HNSCC 1483 (COX-2+) or human colorectal carcinoma cells HCT-116 (COX-2–). A biodistribution study gave promising results, with an uptake ratio in COX-2 positive/COX-2 negative tumors of 3, similar to the COX-2+ tumor to muscle ratio (T/M). Blocking experiments with cold celecoxib produced a clear decrease of the radiotracer uptake in the COX-2+ tumor (T/M = 1). However, only the COX-2 negative tumors were CRC cells (HCT-116); the COX-2 positive model was a xenograft of human head and neck squamous cell carcinoma.
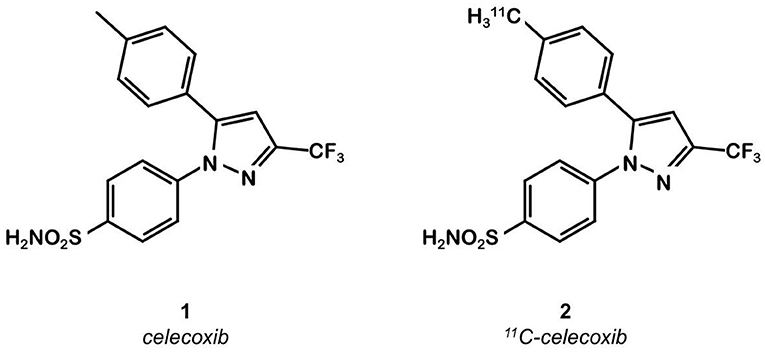
Figure 1. Celecoxib and 11C-celecoxib structures.
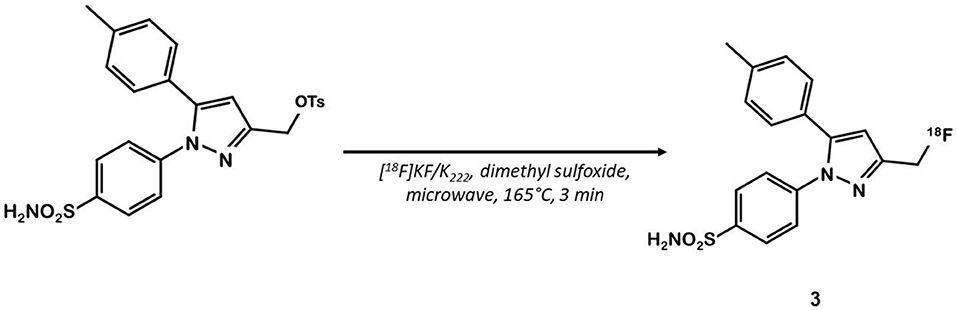
Figure 2. Radiosynthesis of pyrazole derivative 18F-3.
In 2015, Kaur et al. synthesized a radiolabeled derivative 18F-4 from a key sulfonylchorine and its amination by the prosthetic 4-[18F]fluorobenzylamine scaffold (25) (Figure 3). Derivative 4 exhibited interesting in vitro properties on human recombinant COX-2 (log P = 3.18; IC50 = 360 vs. 40 nM for 1) but suffered from a lack of specificity. Indeed, cellular uptake experiments in human CRC cells expressing COX-2 (HCA-7) showed an accumulation of the radioligand that could not be inhibited by a celecoxib or rofecoxib pre-treatment. In vivo, NIH-III mice xenografted with HCA-7 cells exhibited a maximum tumor to muscle ratio 10 min post injection (p.i.) of 1.4, slowly decreasing over time. Dynamic acquisitions showed a rapid elimination of the radioactivity through the intestinal tract. In vitro results were not convincing, and specificity was not investigated in vivo.
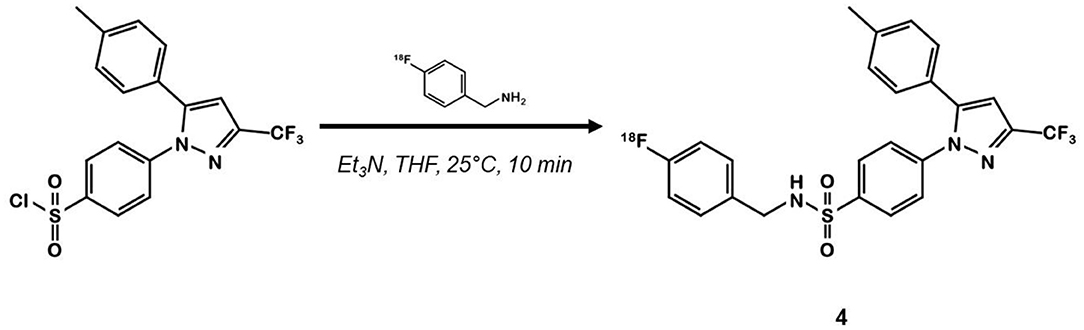
Figure 3. Radiosynthesis of pyrazole derivative 18F-4.
Other teams focused on different coxib-like derivatives. Derivative 5 was first studied by Li et al. and exhibited a very promising COX-2 affinity, with an IC50 in the range of the nanomolar concentration (IC50 = 5 nM) and a high in vitro selectivity for COX-2 over COX-1 (COX-1/COX-2 = 2,000) (34). Wuest et al. synthetized the [11C]methoxy-1,2-diarylcyclopentene 11C-5 in 4 steps from 2-dibromocyclopentene using a double Suzuki cross coupling and as final step a Williamson type O-methylation with [11C]CH3I (26) (Figure 4). The authors confirmed the specificity of compound 5 for COX-2 with cellular uptake studies (especially in the COX-2+ human colorectal adenocarcinoma cell line HT-29 vs. COX-2- RAW264.7). Its lipophilicity of 4.2 enabled it to cross the cell membranes but might be responsible for a lack of specificity in vivo. In fact, in vivo assays on a preclinical rodent xenograft model of human CRC cells (HT-29 cells, COX-2+) exhibited an accumulation of the tracer in the tumor, that could not be inhibited in blocking experiments with an excess of non-radioactive competitor. Moreover, this radiotracer was quickly metabolized, with only 17% remaining intact in plasma samples 10 min p.i.
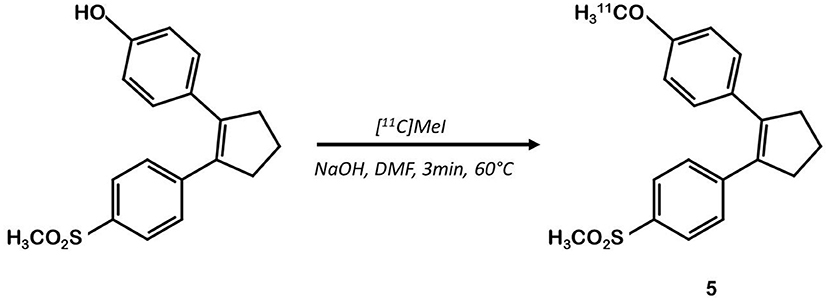
Figure 4. Radiosynthesis of [11C]diarylcyclopentene 5.
In 2013, Tietz et al. developed 18F-Pyricoxib 6, designed by replacing the celecoxib pyrazole ring by a pyrazine isoster (35). In 2016, Tietz et al. proved that derivative 6 showed better in vitro affinity and specificity than its parent Celecoxib 1 (IC50 = 7 vs. 40 nM for 1) (27). In vitro blocking experiments in HCA-7 and HCT-116 cells were promising. Radiotracer uptake in COX-2 positive cells (HCA-7) was significantly higher than in COX-2 negative cells (HCT-116), and pre-treatment with diverse non-radiolabeled COX-2 inhibitors led to a maximum uptake reduction of 65%. In vivo experiments were then conducted on NIH-III mice xenografted with either HCA-7 or HCT-116 cells, showing a satisfactory uptake in HCA-7 tumors (T/M ratio of 2.25 after 4 h p.i.). Pre-treatment with 2 mg of Celecoxib 1 (intraperitoneal administration, i.p.) led to a marked decrease in radiotracer uptake in the tumors. Biodistribution results showed a %ID/g of 2.12% in HCA-7 tumors, scaled down to 1.04% after a celecoxib pretreatment. However, radiotracer uptake was similar in HCA-7 and HCT-116 tumors. Expression of COX-2 in HCT-116 tumors was confirmed by immunohistochemistry, highlighting a cell line drift and questioning the relevance of the HCT-116 cell line as a COX-2 negative model. They concluded that 18F-pyricoxib 6 was a very promising candidate for a “first in human” study (27). In 2018, Tietz et al. deepened their understanding of this chemical entity by performing pharmacomodulation experiments (28). For the O-analog 18F-7 the radiolabeling was carried out on a preoxidized iodyl precursor. The authors started from 4-chloro-2-(methylsulphonyl)-6-(trifluoromethyl)pyrimidine after 5 steps including radiolabeling with 4-[18F]fluorobenzylamine. 18F-8 radiosynthesis was similar to that of 18F-pyricoxib 6, using sulfonyl precursors and a 4-[18F]fluorobenzylamine (FBA) as building block (Figure 5). The three tested probes 18F-6-8 exhibited similar in vitro properties (lipophilicity, affinity, specificity for COX-2). However, even slight modifications of the structure led to radically different pharmacokinetic, biodistribution and uptake profiles (Table 1). One of the radiotracers was slightly metabolized and excreted via the urinary tract but did not accumulate in the tumors, while the other was rapidly eliminated but had a better T/M ratio.
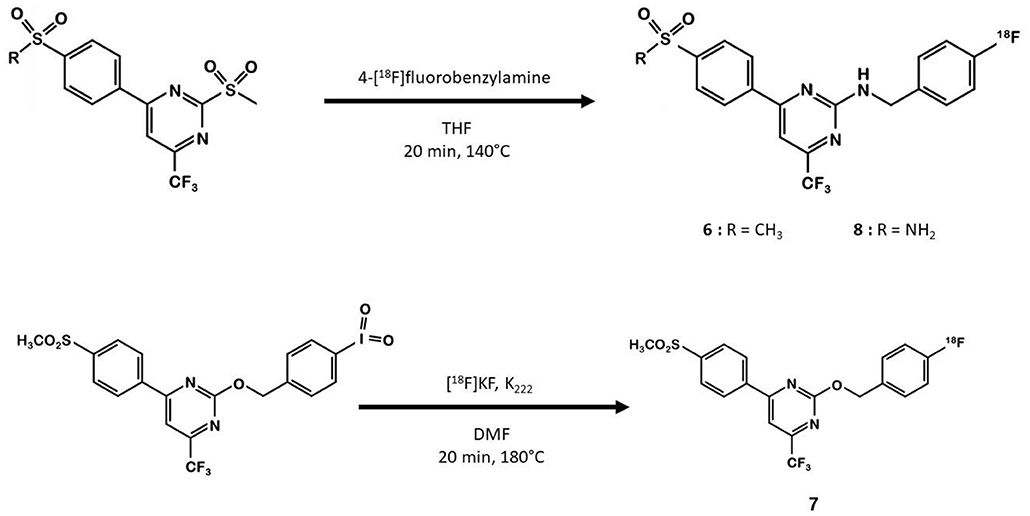
Figure 5. Radiosynthesis of 18F-6-8 probes.
The same team developed a pyricoxib derivative to reduce this unspecific binding previously observed with 18F-pyricoxib 6 (29). They designed 18F-triacoxib 9 which was obtained from the Celecoxib 1 structure by replacing the CF3 by a CH3 group and the acidic sulfonylamide by a simple methylsulfonyl. Nevertheless, the best innovation in this series consisted in the insertion of a triazole between the pyrazole and the phenyl ring, which required building an azidopyrazole as key intermediate. Noteworthy, 18F-fluorination was achieved using a pinacol boronic ester (in the presence of a copper catalyst [Cu(OTf)2(py)4]) as leaving group (Figure 6). Compound 9 displayed a satisfactory in vitro affinity and selectivity for COX-2 (IC50 = 90 vs. 70 nM for 1; IC50 COX-1 > 100 μM for both compounds, HCA-7 cells). In vitro binding experiments with increasing doses of non-radiolabeled compounds (celecoxib or triacoxib) resulted in an inhibition of the radiotracer uptake by 47 to 63%. In vivo experiments on BALB/c mice bearing HCA-7 xenografts suggested an improved stability of 18F-triacoxib 9 compared to 18F-pyricoxib 6 (respectively, 90 vs. 75% intact radiotracer in plasma samples 60 min p.i.). PET acquisitions confirmed a higher uptake in the tumors than in the muscle, within a similar range as 18F-pyricoxib 6 (tumor-to-muscle ratio of 1.49 vs. 1.47, respectively, 60 min p.i.). In Celecoxib 1 pretreated mice, a partial inhibition of 18F-triacoxib 9 uptake was observed (20%). Unspecific binding was also observed in lipid rich tissues. 18F-triacoxib 9 did not seem to be a better candidate than 18F-pyricoxib 6 as a diagnostic tool for CRC.
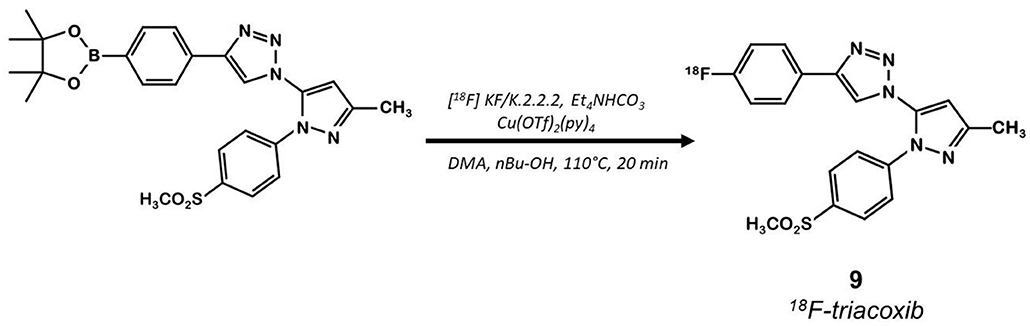
Figure 6. Radiosynthesis of 18F-Triacoxib 9.
In 2003, Hu et al. synthesized a large series of original 2,3-diaryl indoles (36). Structure-Activity Relationships (SAR) were established from about thirty derivatives obtained from anthranilic acids in 5 or 6 steps including indole formation by McMurry cyclization (Figure 7). Derivative 10 exhibited high affinity and selectivity for COX-2 (IC50 = 20 vs. 520 nM for 1) over COX-1 (IC50 >10 μM for both compounds), and better anti-inflammatory properties than celecoxib in a rat carrageenan-induced foot pad edema assay. In 2012, Kniess et al. radiolabeled this promising compound using the nucleophilic substitution of a trimethylammonium salt by 18F-fluorine (30). They investigated the determination of PGE2 levels in cell culture supernatants, which is therefore a direct measure of the COX activity. The cellular inhibition assay results demonstrated 10 to be a potent cyclooxygenase inhibitor with only low COX-1/COX-2 selectivity. The authors evaluated the selectivity of 10 using an enzymatic competitive inhibition assay with celecoxib as reference. IC50 = 6.6 μM for COX-1 and 1.2 μM for COX-2 were determined (IC50 COX-1 = 115 and 0.06 μM for 1). Pre-incubation of HT-29 cells with cold compound reduced the cellular uptake by almost 80% compared to incubation with the radioactive compound alone. In vivo PET imaging of HT-29 xenografted mice showed no accumulation of the tracer in the tumor. Despite a low lipophilicity (log D = 1.2), the tracer was rapidly eliminated through the intestinal tract (half-life: 8 min).
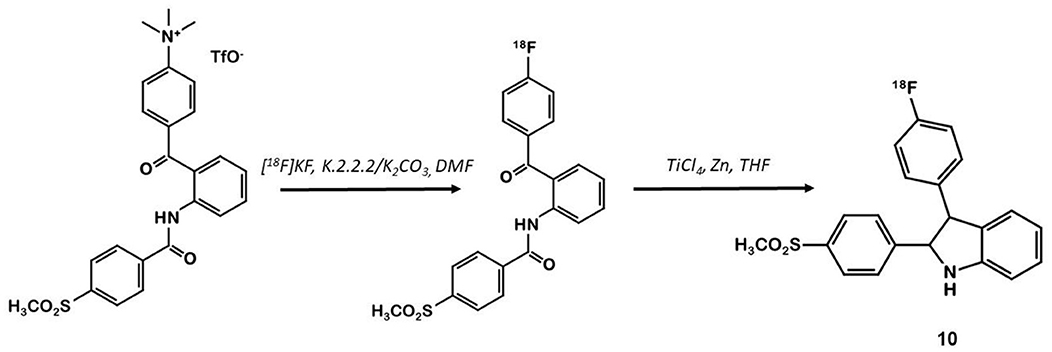
Figure 7. Radiosynthesis of 18F-10 via McMurry cyclisation.
Indomethacin 11 (Figure 8) is a COX ligand that binds to both COX-1 and COX-2. Kalgutar et al. modified its chemical structure by adding an amide group (37). After SAR analysis and the building of nearly twenty analogs, they clearly improved the selectivity of the derivatives for COX-2, which was inhibited in the nanomolar range. Following this work, Morgenroth et al. synthetized radio iodinated analogs from stannyl precursors (31). Derivative 125I-12 (Figure 9) was found to be the most suitable tracer. In vitro studies on CRC cell cultures HT-29 and HCT-116 showed a significantly higher concentration of 12 in COX-2 positive (HT-29) cells (COX-2+/COX-2– ratio of 2.5 and 7.6 for the two tested radiotracers). Blocking experiments were performed on human embryonic kidney (HEK) and human umbilical vein endothelial cell (HUVEC) cell lines, showing a partial decrease of the uptake in cells pre-treated with celecoxib 1. These affinities and selectivities were confirmed using both in vivo microPET and bio-distribution studies. The compound with the highest uptake was radiolabeled with 124I and used for PET imaging of HT-29 and HCT-116 xenografted mice. Due to the lipophilicity of this compound (log D = 4.41), the radioactivity was mainly localized in the liver and the gastrointestinal tract. No tracer uptake was observed in HCT-116 tumors whereas HT-29 tumors displayed a significant uptake. Measurement of the remaining activity in each organ with a gamma counter confirmed a COX-2+/COX-2– ratio of 5. The T/M ratio was over 50 times higher in HT-29 tumors than in HCT-116 tumors.
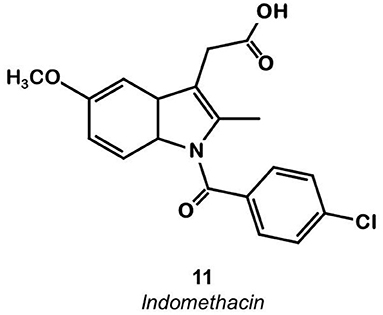
Figure 8. Indomethacin.
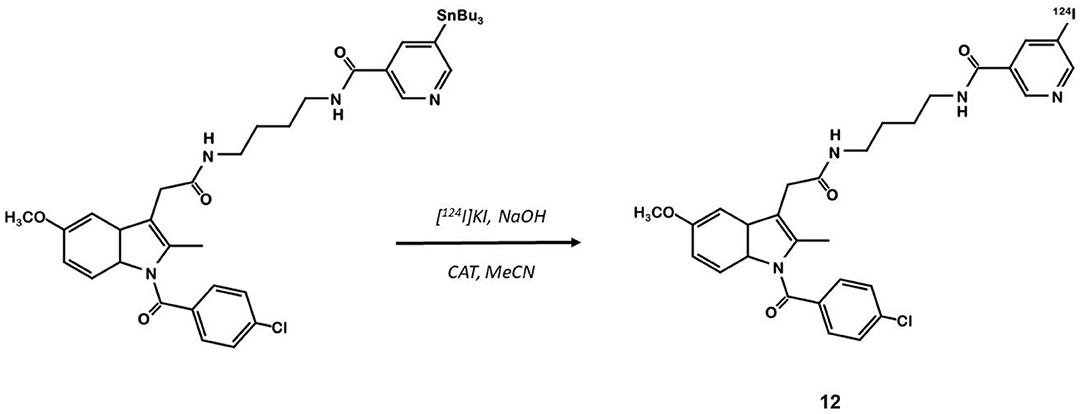
Figure 9. Radiosynthesis of indomethacin derivative 124I-12.
In 2011, Uddin et al. also synthesized a series of fluorine-containing indomethacin derivatives. These probes were satisfactory COX-2 inhibitors with an IC50 in the range of the nanomolar both in inhibition assays using purified COX-2 and intact cells. However, they could not be successfully radiolabeled due to a lack of stability in the severe reaction conditions required (24).
This review focuses on COX-2 radiotracers tested in CRC models. Other COX-2 radiotracers have also been developed for other oncologic indications as well as applications in the field of neuroinflammation (22).
These eight studies on CRC xenografts display heterogeneous results. Kaur et al. (25), Wuest et al. (26), and Kniess et al. (30) concluded that their radiotracer was not appropriate for further clinical investigation, whereas Uddin et al. (24), Tietz et al. (27, 28), Litchfield et al. (29), and Morgenroth et al. (31) judged their radiotracer to be promising for clinical applications (satisfactory T/M ratio). However, for all the tested compounds, most of the radioactivity uptake was located in the digestive tract. The T/M ratios were deemed satisfactory, ranging from 1.4 to 5.0, but the tumor-to-intestine ratio was not always mentioned in these studies, and, when available, was well above the T/M ratio. The tumors were detectable as xenografts in mice flanks, but these T/M ratios would probably not be sufficient to detect in situ CRC tumors (Figure 10). Thus, the challenge to develop the ideal COX-2 radiotracer for CRC staging remains to date. Two areas of work can be explored to achieve this radiotracer development for CRC imaging.
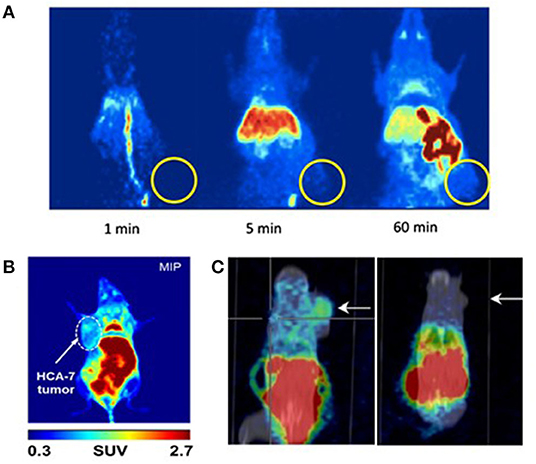
Figure 10. microPET images of mice xenografted with human colorectal cancer cell lines after injection of a COX-2 specific radiotracer. (A) Kniess et al., 2012 (30): Maximum intensity projection (MIP) images at 1, 5, and 60 min p.i. after a single IV injection of 18F-3 into HT-29 tumor-bearing (right flank) NMRI nu/nu mice. Authors concluded that 18F-3 was not promising. Reprinted from Bioorg Med Chem. 2012 Jun 1;20(11):3410–21, Radiosynthesis of a 18F-labeled 2,3-diarylsubstituted indole via McMurry coupling for functional characterization of cyclooxygenase-2 (COX-2) in vitro and in vivo. Kniess T, Laube M, Bergmann R, Sehn F, Graf F, Steinbach J, et al. Copyright (2012), with permission from Elsevier, license number 5067540791584. (B) Litchfield et al. (29): Maximum intensity projection (MIP) images at 60 min p.i. of 18F-9 into HCA-7 tumor-bearing (left flank) BALB/c mice. Authors concluded that 18F-9 was promising. (C) Morgenroth et al. (31): μPET/CT molecular imaging of COX-2 with 124I-12 in HT29 (left panel) and HCT-116 (right panel) xenografted SCID mice at 4 h p.i. Arrows indicate tumor. Authors concluded that 124I-12 was promising.
All the tested compounds display a high lipophilicity (log P or log D ϵ [1.7–4.4]), required to reach their intracellular target COX-2, but also responsible for hepatobiliary excretion that could mask CRC tumors. This assumption questions the relevance of COX-2 as a CRC imaging target and could be tested using in situ CRC preclinical models. 11C-celecoxib was deemed unfit for clinical applications but benefited from a renal excretion that would be a serious advantage in CRC imaging. To increase the affinity for COX-2, pharmacomodulations on this parent compound led to more lipophilic molecules. There is still a need to balance affinity and lipophilicity with ideal pharmacokinetic properties. The use of innovative vectors could prove to be the solution to selectively deliver radiolabeled coxib derivatives into cancerous cells. In particular, the use of the RGD sequence (a tripeptide composed of arginine, glycine and aspartic acid) (38), dendrimers (39) or dehydropeptides (40) proved their potential to carry COX inhibitors into inflammatory or cancerous cells. Other pharmacokinetic properties should be taken into account, especially the metabolization rate and the radiotracer half-life. To be used routinely, the ideal radiotracer should be more than 50% intact 1 h p.i. and the half-life of the vector should not be shorter than 30 min. Other well-known PET tracers, such as 18F-FDG or 18F-FDOPA comply with these requirements (respective renal excretion of 20% 2 h p.i.1 and 50% 45 min p.i.2).
The second parameter to consider is the radioisotope. Due to its 110 min half-life, the use of 18F is widespread in PET imaging, and most of the COX-2 binders were labeled with 18F. Morgenroth et al. obtained the maximum T/M with a radio-iodinate derivative (31). However, due to both the higher energy of emitted beta particles and the longer half-life of 124I (4.2 days), radio-iodinate derivatives exhibit a poorer dosimetry profile compared to 18F-compounds. On the contrary, the shorter half-life of 11C makes it less accessible for routine use (on-site cyclotron required) but would endow it with a better dosimetry profile.
Finally, pharmacomodulations were performed to increase the specificity for COX-2 compared to COX-1, but other targets are known for coxibs. For instance, celecoxib or valdecoxib bind significantly (IC50 in the order of nM) to carbonic anhydrase isoenzymes (CA) (41). In addition, celecoxib is able to bind to PDK1 (42, 43), a cell survival regulation enzyme via the Akt/PKB pathway, with an IC50 in the range of μM (44). Likewise, coxibs bind to the transmembrane protein SERCA (45–47), a pump that can induce cellular apoptosis by increasing the intracellular calcium concentration. These different pro-apoptotic and anti-oncogenic targets have been confirmed by an increasing number of reports indicating that celecoxib does not require the presence of COX-2 to exert its anti-tumoral activity (44, 48, 49). Even more striking, it has been shown that structural analogs close to celecoxib, devoid of any COX-2 inhibitory activity, were able to mimic the anti-tumor properties of celecoxib studied so far, not only in vitro but also in various in vivo xenograft models (43, 50, 51). These data question the relevance of radiolabeled coxibs as specific COX-2 binders.
Xenografts are known to lead to derangement of the normal tumor architecture and nearby healthy tissues and vasculature, and cause altered drug-sensitivity (52, 53). To address these possible biases, the use of orthotopic xenograft or genetically engineered mice expressing human colorectal cancer genes would mimic in situ CRC more accurately (54), easing the evaluation of the impact of unspecific intestinal uptake. Imaging the xenografts with 18F-FDG prior to the radiolabeled COX-2 inhibitor would also inform about the accessibility of tumors for PET radioligands, notably regarding their perfusion.
Ultimately, most of these radioligands exhibit promising affinity and specificity in vitro but fail to prove their efficiency in vivo on xenograft models. Chemical screening and pharmacomodulations have yet to work out the ideal COX-2 radiotracer. The use of innovative vectors should be considered to selectively deliver radiotracers in the tumors. Given the hepatobiliary excretion of most of the known coxib derivatives, in situ CRC models should be considered for future explorations. In vivo preclinical studies on in situ CRC models would be decisive to conclude whether COX-2 is a relevant target in CRC imaging. More conclusive in vivo results are required before conducting a “first-in-human” study.
CD wrote the present paper and contributed to the design of the review. FA contributed to the redaction of the manuscript and to the prior bibliographic study. YT contributed to the clinical rational of the work. FB and SR wrote chemical sections of the manuscript. MC contributed to manuscript revision and improvement. NA designed the objectives of the review and supervised its preparation and redaction. All authors have substantially contributed to this work. The manuscript has been re-read and approved by all authors, and they have all contributed to its scientific improvement.
This work was supported by the French National Agency for Research (Investissements d'Avenir No. ANR-11-LABX-0018-01), IRON, by the Site de Recherche Intégrée sur le Cancer Imaging and Longitudinal Investigations to Ameliorate Decision-making SIRIC-ILIAD, and by the Région Centre Val de Loire RTR Motivhealth.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ^Résumé des Caractéristiques du Produit FDG. Available online at: http://agence-prd.ansm.sante.fr/php/ecodex/rcp/R0187540.htm (accessed June 9, 2020).
2. ^Résumé des Caractéristiques du Produit FDOPA. Available online at: http://agence-prd.ansm.sante.fr/php/ecodex/rcp/R0157439.htm (accessed October 6, 2020).
1. Lambert R. Épidémiologie du cancer colorectal (CCR). Cancéro Dig. (2009) 1:2–6. doi: 10.4267/2042/25061
2. Siegel RL, Miller KD, Sauer AG, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. (2020) 70:145–64. doi: 10.3322/caac.21601
3. Wang D, DuBois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. (2010) 29:781–8. doi: 10.1038/onc.2009.421
4. Liu Y, Sun H, Hu M, Zhang Y, Chen S, Tighe S, et al. The role of cyclooxygenase-2 in colorectal carcinogenesis. Clin Colorectal Cancer. (2017) 16:165–72. doi: 10.1016/j.clcc.2016.09.012
5. Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. (2002) 94:252–66. doi: 10.1093/jnci/94.4.252
6. Chan AT, Arber N, Burn J, Chia JW-K, Elwood P, Hull MA, et al. Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prev Res. (2012) 5:164–78. doi: 10.1158/1940-6207.CAPR-11-0391
7. Phillips RKS, Wallace MH, Lynch PM, Hawk E, Gordon GB, Saunders BP, et al. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut. (2002) 50:857–60. doi: 10.1136/gut.50.6.857
8. Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. (1994) 107:1183–8. doi: 10.1016/0016-5085(94)90246-1
9. Gupta RA, DuBois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. (2001) 1:11–21. doi: 10.1038/35094017
10. Marnett LJ, DuBois RN. COX-2: a target for colon cancer prevention. Annu Rev Pharmacol Toxicol. (2002) 42:55–80. doi: 10.1146/annurev.pharmtox.42.082301.164620
11. Fujita T, Matsui M, Takaku K, Uetake H, Ichikawa W, Taketo MM, et al. Size- and invasion-dependent increase in cyclooxygenase 2 levels in human colorectal carcinomas. Cancer Res. (1998) 58:4823–6.
12. Cai J, Huang L, Huang J, Kang L, Lin H, Huang P, et al. Associations between the cyclooxygenase-2 expression in circulating tumor cells and the clinicopathological features of patients with colorectal cancer. J Cell Biochem. (2019) 120:4935–41. doi: 10.1002/jcb.27768
13. Tomozawa S, Tsuno NH, Sunami E, Hatano K, Kitayama J, Osada T, et al. Cyclooxygenase-2 overexpression correlates with tumour recurrence, especially haematogenous metastasis, of colorectal cancer. Br J Cancer. (2000) 83:324–8. doi: 10.1054/bjoc.2000.1270
14. Ogino S, Kirkner GJ, Nosho K, Irahara N, Kure S, Shima K, et al. Cyclooxygenase-2 expression is an independent predictor of poor prognosis in colon cancer. Clin Cancer Res. (2008) 14:8221–7. doi: 10.1158/1078-0432.CCR-08-1841
15. Wasilewicz MP, Kołodziej B, Bojułko T, Kaczmarczyk M, Sulzyc-Bielicka V, Bielicki D. Expression of cyclooxygenase-2 in colonic polyps. Pol Arch Med Wewn. (2010) 120:313–20. doi: 10.20452/pamw.955
16. Kerlikowske K, Molinaro AM, Gauthier ML, Berman HK, Waldman F, Bennington J, et al. Biomarker expression and risk of subsequent tumors after initial ductal carcinoma in situ diagnosis. J Natl Cancer Inst. (2010) 102:627–37. doi: 10.1093/jnci/djq101
17. Shao N, Feng N, Wang Y, Mi Y, Li T, Hua L. Systematic review and meta-analysis of COX-2 expression and polymorphisms in prostate cancer. Mol Biol Rep. (2012) 39:10997–1004. doi: 10.1007/s11033-012-2001-5
18. Edelman MJ, Watson D, Wang X, Morrison C, Kratzke RA, Jewell S, et al. Eicosanoid modulation in advanced lung cancer: cyclooxygenase-2 expression is a positive predictive factor for celecoxib + chemotherapy—cancer and leukemia group B trial 30203. JCO. (2008) 26:848–55. doi: 10.1200/JCO.2007.13.8081
19. Fabi A, Metro G, Papaldo P, Mottolese M, Melucci E, Carlini P, et al. Impact of celecoxib on capecitabine tolerability and activity in pretreated metastatic breast cancer: results of a phase II study with biomarker evaluation. Cancer Chemother Pharmacol. (2008) 62:717–25. doi: 10.1007/s00280-007-0650-1
20. Rini BI, Weinberg V, Dunlap S, Elchinoff A, Yu N, Bok R, et al. Maximal COX-2 immunostaining and clinical response to celecoxib and interferon alpha therapy in metastatic renal cell carcinoma. Cancer. (2006) 106:566–75. doi: 10.1002/cncr.21661
21. Laube M, Kniess T, Pietzsch J. Radiolabeled COX-2 inhibitors for non-invasive visualization of COX-2 expression and activity — a critical update. Molecules. (2013) 18:6311–55. doi: 10.3390/molecules18066311
22. de Vries EFJ. Imaging of cyclooxygenase-2 (COX-2) expression: potential use in diagnosis and drug evaluation. Curr Pharm Des. (2006) 12:3847–56. doi: 10.2174/138161206778559650
23. Pacelli A, Greenman J, Cawthorne C, Smith G. Imaging COX-2 expression in cancer using PET/SPECT radioligands: current status and future directions. J Label Comp Radiopharm. (2014) 57:317–22. doi: 10.1002/jlcr.3160
24. Uddin MdJ, Crews BC, Ghebreselasie K, Huda I, Kingsley PJ, Ansari MS, et al. Fluorinated cyclooxygenase-2 inhibitors as agents in PET imaging of inflammation and cancer. Cancer Prev Res. (2011) 4:1536–45. doi: 10.1158/1940-6207.CAPR-11-0120
25. Kaur J, Tietz O, Bhardwaj A, Marshall A, Way J, Wuest M, et al. Design, synthesis, and evaluation of an (18)F-labeled radiotracer based on celecoxib-NBD for positron emission tomography (PET) imaging of cyclooxygenase-2 (COX-2). ChemMedChem. (2015) 10:1635–40. doi: 10.1002/cmdc.201500287
26. Wuest F, Kniess T, Bergmann R, Pietzsch J. Synthesis and evaluation in vitro and in vivo of a 11C-labeled cyclooxygenase-2 (COX-2) inhibitor. Bioorg Med Chem. (2008) 16:7662–70. doi: 10.1016/j.bmc.2008.07.016
27. Tietz O, Wuest M, Marshall A, Glubrecht D, Hamann I, Wang M, et al. PET imaging of cyclooxygenase-2 (COX-2) in a pre-clinical colorectal cancer model. EJNMMI Res. (2016) 6:37. doi: 10.1186/s13550-016-0192-9
28. Tietz O, Marshall A, Bergman C, Wuest M, Wuest F. Impact of structural alterations on the radiopharmacological profile of 18F-labeled pyrimidines as cyclooxygenase-2 (COX-2) imaging agents. Nucl Med Biol. (2018) 62–63:9–17. doi: 10.1016/j.nucmedbio.2018.05.001
29. Litchfield M, Wuest M, Glubrecht D, Wuest F. Radiosynthesis and biological evaluation of [18F]Triacoxib: a new radiotracer for PET imaging of COX-2. Mol Pharm. (2020) 17:251–61. doi: 10.1021/acs.molpharmaceut.9b00986
30. Kniess T, Laube M, Bergmann R, Sehn F, Graf F, Steinbach J, et al. Radiosynthesis of a 18F-labeled 2,3-diarylsubstituted indole via McMurry coupling for functional characterization of cyclooxygenase-2 (COX-2) in vitro and in vivo. Bioorg Med Chem. (2012) 20:3410–21. doi: 10.1016/j.bmc.2012.04.022
31. Morgenroth A, Vogg ATJ, Neumaier B, Mottaghy FM, Zlatopolskiy BD. Radioiodinated indomethacin amide for molecular imaging of cyclooxygenase-2 expressing tumors. Oncotarget. (2017) 8:18059–69. doi: 10.18632/oncotarget.15437
32. Prabhakaran J, Majo VJ, Simpson NR, Heertum RLV, Mann JJ, Kumar JSD. Synthesis of [11C]celecoxib: a potential PET probe for imaging COX-2 expression. J Label Comp Radiopharm. (2005) 48:887–95. doi: 10.1002/jlcr.1002
33. Kumar JSD, Bai B, Zanderigo F, DeLorenzo C, Prabhakaran J, Parsey RV, et al. In vivo brain imaging, biodistribution, and radiation dosimetry estimation of [11C]Celecoxib, a COX-2 PET ligand, in nonhuman primates. Molecules. (2018) 23:1929. doi: 10.3390/molecules23081929
34. Li JJ, Anderson GD, Burton EG, Cogburn JN, Collins JT, Garland DJ, et al. 1,2-diarylcyclopentenes as selective cyclooxygenase-2 inhibitors and orally active anti-inflammatory agents. J Med Chem. (1995) 38:4570–8. doi: 10.1021/jm00022a023
35. Tietz O, Sharma SK, Kaur J, Way J, Marshall A, Wuest M, et al. Synthesis of three 18F-labelled cyclooxygenase-2 (COX-2) inhibitors based on a pyrimidine scaffold. Org Biomol Chem. (2013) 11:8052–64. doi: 10.1039/c3ob41935e
36. Hu W, Guo Z, Chu F, Bai A, Yi X, Cheng G, et al. Synthesis and biological evaluation of substituted 2-sulfonyl-phenyl-3-phenyl-indoles: a new series of selective COX-2 inhibitors. Bioorg Med Chem. (2003) 11:1153–60. doi: 10.1016/S0968-0896(03)00046-4
37. Kalgutkar AS, Crews BC, Rowlinson SW, Marnett AB, Kozak KR, Remmel RP, et al. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors. Proc Natl Acad Sci USA. (2000) 97:925–30. doi: 10.1073/pnas.97.2.925
38. Mohammadi R, Shokri B, Shamshirian D, Zarghi A, Shahhosseini S. Synthesis and biological evaluation of RGD conjugated with Ketoprofen/Naproxen and radiolabeled with [99mTc] via N4(GGAG) for αVβ3 integrin-targeted drug delivery. Daru. (2020) 28:87–96. doi: 10.1007/s40199-019-00318-8
39. Ficker M, Theeuwen MJM, Janaszewska A, Gorzkiewicz M, Svenningsen SW, Klajnert-Maculewicz B, et al. Complexes of indomethacin with 4-carbomethoxy-pyrrolidone PAMAM dendrimers show improved anti-inflammatory properties and temperature-dependent binding and release profile. Mol Pharm. (2018) 15:3573–82. doi: 10.1021/acs.molpharmaceut.8b00567
40. Moreira R, Jervis PJ, Carvalho A, Ferreira PMT, Martins JA, Valentão P, et al. Biological evaluation of naproxen–dehydrodipeptide conjugates with self-hydrogelation capacity as dual LOX/COX inhibitors. Pharmaceutics. (2020) 12:122. doi: 10.3390/pharmaceutics12020122
41. Di Fiore A, Pedone C, D'Ambrosio K, Scozzafava A, De Simone G, Supuran CT. Carbonic anhydrase inhibitors: valdecoxib binds to a different active site region of the human isoform II as compared to the structurally related cyclooxygenase II ‘selective' inhibitor celecoxib. Bioorg Med Chem Lett. (2006) 16:437–42. doi: 10.1016/j.bmcl.2005.09.040
42. Arico S, Pattingre S, Bauvy C, Gane P, Barbat A, Codogno P, et al. Celecoxib induces apoptosis by inhibiting 3-phosphoinositide-dependent protein kinase-1 activity in the human colon cancer HT-29 cell line. J Biol Chem. (2002) 277:27613–21. doi: 10.1074/jbc.M201119200
43. Kulp SK, Yang Y-T, Hung C-C, Chen K-F, Lai J-P, Tseng P-H, et al. 3-phosphoinositide-dependent protein kinase-1/Akt Signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Res. (2004) 64:1444–51. doi: 10.1158/0008-5472.CAN-03-2396
44. Grösch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)–independent anticarcinogenic effects of selective COX-2 inhibitors. J Natl Cancer Inst. (2006) 98:736–47. doi: 10.1093/jnci/djj206
45. Johnson AJ, Hsu A-L, Lin H-P, Song X, Chen C-S. The cyclo-oxygenase-2 inhibitor celecoxib perturbs intracellular calcium by inhibiting endoplasmic reticulum Ca2+-ATPases: a plausible link with its anti-tumour effect and cardiovascular risks. Biochem J. (2002) 366:831–7. doi: 10.1042/bj20020279
46. Tanaka K, Tomisato W, Hoshino T, Ishihara T, Namba T, Aburaya M, et al. Involvement of intracellular Ca2+ levels in nonsteroidal anti-inflammatory drug-induced apoptosis. J Biol Chem. (2005) 280:31059–67. doi: 10.1074/jbc.M502956200
47. Pyrko P, Kardosh A, Liu Y-T, Soriano N, Xiong W, Chow RH, et al. Calcium-activated endoplasmic reticulum stress as a major component of tumor cell death induced by 2,5-dimethyl-celecoxib, a non-coxib analogue of celecoxib. Mol Cancer Ther. (2007) 6:1262–75. doi: 10.1158/1535-7163.MCT-06-0629
48. Kashfi K, Rigas B. Non-COX-2 targets and cancer: expanding the molecular target repertoire of chemoprevention. Biochem Pharmacol. (2005) 70:969–86. doi: 10.1016/j.bcp.2005.05.004
49. Schönthal AH. Induction of apoptosis by celecoxib in cell culture: an uncertain role for cyclooxygenase-2. Cancer Res. (2007) 67:5575–6. doi: 10.1158/0008-5472.CAN-06-3414
50. Song X, Lin H-P, Johnson AJ, Tseng P-H, Yang Y-T, Kulp SK, et al. Cyclooxygenase-2, player or spectator in cyclooxygenase-2 inhibitor-induced apoptosis in prostate cancer cells. J Natl Cancer Inst. (2002) 94:585–91. doi: 10.1093/jnci/94.8.585
51. Schönthal AH. Antitumor properties of dimethyl-celecoxib, a derivative of celecoxib that does not inhibit cyclooxygenase-2: implications for glioma therapy. Neurosurg Focus. (2006) 20:E21. doi: 10.3171/foc.2006.20.4.14
52. Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. (2007) 7:654–8. doi: 10.1038/nrc2192
53. Sikder H, Huso DL, Zhang H, Wang B, Ryu B, Hwang ST, et al. Disruption of Id1 reveals major differences in angiogenesis between transplanted and autochthonous tumors. Cancer Cell. (2003) 4:291–9. doi: 10.1016/S1535-6108(03)00245-9
Keywords: COX-2, PET, colorectal cancer, radioligands, preclinical model
Citation: Dagallier C, Avry F, Touchefeu Y, Buron F, Routier S, Chérel M and Arlicot N (2021) Development of PET Radioligands Targeting COX-2 for Colorectal Cancer Staging, a Review of in vitro and Preclinical Imaging Studies. Front. Med. 8:675209. doi: 10.3389/fmed.2021.675209
Received: 02 March 2021; Accepted: 03 May 2021;
Published: 08 June 2021.
Edited by:
Anil Kumar Mishra, Institute of Nuclear Medicine & Allied Sciences (DRDO), IndiaReviewed by:
Linjing Mu, ETH Zürich, SwitzerlandCopyright © 2021 Dagallier, Avry, Touchefeu, Buron, Routier, Chérel and Arlicot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolas Arlicot, bmljb2xhcy5hcmxpY290QHVuaXYtdG91cnMuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.