 Celien Kuiper-Makris
Celien Kuiper-Makris Jaco Selle
Jaco Selle Eva Nüsken
Eva Nüsken Jörg Dötsch2
Jörg Dötsch2 Miguel A. Alejandre Alcazar
Miguel A. Alejandre Alcazar- 1Department of Pediatric and Adolescent Medicine, Translational Experimental Pediatrics—Experimental Pulmonology, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany
- 2Department of Pediatric and Adolescent Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany
- 3Center for Molecular Medicine Cologne (CMMC), Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany
- 4Excellence Cluster on Stress Responses in Aging-associated Diseases (CECAD), Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany
- 5Member of the German Centre for Lung Research (DZL), Institute for Lung Health, University of Giessen and Marburg Lung Centre (UGMLC), Gießen, Germany
Lung development is not completed at birth, but expands beyond infancy, rendering the lung highly susceptible to injury. Exposure to various influences during a critical window of organ growth can interfere with the finely-tuned process of development and induce pathological processes with aberrant alveolarization and long-term structural and functional sequelae. This concept of developmental origins of chronic disease has been coined as perinatal programming. Some adverse perinatal factors, including prematurity along with respiratory support, are well-recognized to induce bronchopulmonary dysplasia (BPD), a neonatal chronic lung disease that is characterized by arrest of alveolar and microvascular formation as well as lung matrix remodeling. While the pathogenesis of various experimental models focus on oxygen toxicity, mechanical ventilation and inflammation, the role of nutrition before and after birth remain poorly investigated. There is accumulating clinical and experimental evidence that intrauterine growth restriction (IUGR) as a consequence of limited nutritive supply due to placental insufficiency or maternal malnutrition is a major risk factor for BPD and impaired lung function later in life. In contrast, a surplus of nutrition with perinatal maternal obesity, accelerated postnatal weight gain and early childhood obesity is associated with wheezing and adverse clinical course of chronic lung diseases, such as asthma. While the link between perinatal nutrition and lung health has been described, the underlying mechanisms remain poorly understood. There are initial data showing that inflammatory and nutrient sensing processes are involved in programming of alveolarization, pulmonary angiogenesis, and composition of extracellular matrix. Here, we provide a comprehensive overview of the current knowledge regarding the impact of perinatal metabolism and nutrition on the lung and beyond the cardiopulmonary system as well as possible mechanisms determining the individual susceptibility to CLD early in life. We aim to emphasize the importance of unraveling the mechanisms of perinatal metabolic programming to develop novel preventive and therapeutic avenues.
Introduction
Chronic lung diseases (CLD) such as asthma, chronic obstructive pulmonary disease (COPD) and pulmonary arterial hypertension (PAH) have a major impact on global health, with COPD being the third leading cause of death worldwide (WHO Global Health Estimates, 2020). CLDs do not only have an enormous impact on the patient's quality of life, but also on health care costs (e.g., an average of $4147 per COPD patient per year) (1, 2). While the pathology of adult lung diseases and the influence of environmental factors such as smoking have been extensively studied, the mechanisms determining the individual susceptibility to CLD early in life remain elusive. This review will provide insights in the current knowledge on how perinatal nutritional and metabolic conditions adversely affect lung development and contribute to the origin of CLDs.
Maternal obesity and intrauterine growth restriction (IUGR) represent alterations of the antenatal, perinatal and postnatal nutritional and metabolic status with adverse consequences for the fetus and newborn. (1) First, both maternal obesity and IUGR increase the risk of pregnancy complications and prematurity of the offspring. Epidemiological studies have shown that not only the risk of pregnancy complications for overweight and obese mothers is higher; it is also associated with an early pregnancy loss, congenital malformations, premature birth and stillbirth (3). In addition, the offspring has an increased risk of being either macrosome or IUGR, both introducing their own risk of comorbidity. IUGR is diagnosed in 5–10% of all pregnancies, characterized as a rate of fetal growth less than the growth potential that is appropriate for the gestational age, and well–recognized as an additional risk factor for prematurity (4, 5). (2) Second, fetal and postnatal nutritional supply as well as maternal weight and metabolism can adversely affect the long-term health of the child. This is referred to as perinatal or metabolic programming (6, 7). This concept was initially coined by Barker as the fetal origins hypothesis, also known as fetal programming. Barker et al. proposed that the developing fetus adapts its growth rate and metabolism as a response to variations in the supply of nutrients (and oxygen), which may lead to permanent changes of organs' structure and physiology in the newborn (8). Over the past two decades, the developmental origins of health and disease have gained increasing scientific interest. There has been an enormous effort and an accumulation of studies devoted to elucidating the underlying mechanisms of perinatal (metabolic) programming of diseases as well as its prevention and therapy. (3) Lastly, maternal obesity and IUGR are associated with long-term alterations of lung function and lung structure. For example, clinical reports showed a positive linear trend between birth weight, adjusted for maternal factors, and lung function in adulthood (9). Furthermore, children that were exposed to maternal obesity during pregnancy or gestational diabetes mellitus (GDM) have an increased risk of developing asthma in childhood (10–12). These findings indicate the significant impact of body weight, nutrition, and metabolism during critical phases of pregnancy and the early postnatal period on the lung development and later pulmonary function of a child (13).
In addition to the adverse nutritive and metabolic influences, the time of exposure is of great importance with regard to the resulting lung pathology. There are different critical windows of lung development with diverse developmental biological processes. The lung develops in five stages, with the last (alveolarization) starting shortly before birth and continuing beyond infancy (14). The window and the nature of exposure to adverse influences render not only the prenatal, but also postnatal lung development highly susceptible to injury and CLDs (15). This basic principle of timing emphasizes the far-reaching complex consequences of antenatal, perinatal and postnatal nutrition. Here, we provide an overview of the impact and mechanisms of nutritive surplus with metabolic disorder (maternal obesity) as well as nutritive deprivation (e.g., IUGR) on the child's lung health (schematic representation in Figure 1).
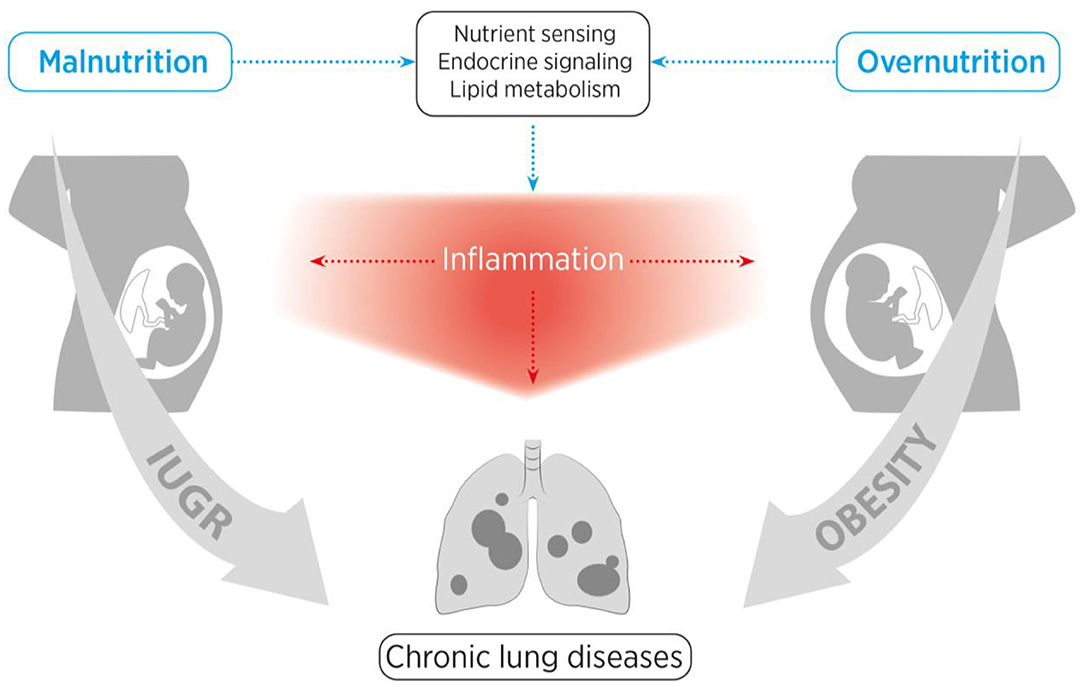
Figure 1. Schematic representation of the structure of this review. We aim to provide a comprehensive overview of the influences of maternal obesity, early childhood obesity and intrauterine growth restriction (IUGR) on nutrient sensing as well as endocrine and inflammatory pathways, and how these adverse perinatal effects contribute to the early origins of chronic lung diseases.
The Impact of Perinatal Nutritive Surplus on the Origins of Chronic Lung Disease
Obesity and overweight result from an imbalance of energy consumption and energy intake, causing fat accumulation in adipose tissue (16). The origin of obesity is multifactorial and comprises a complex interaction of genetic and life style factors (17, 18). It is widely accepted that each individual has a certain level of predisposition for obesity due to genetic and epigenetic adaptations along with modifying environmental factors that can in part contribute to familiar obesity (17). Two central endocrine pathways in obesity are those of insulin and leptin. Insulin is a critical regulator of adipocyte biology that promotes the uptake of glucose and fatty acids and stimulates lipogenesis while inhibiting lipolysis (19). In obesity, the glucose transport and adipocyte metabolism are decreased despite high circulating levels of insulin, also known as insulin resistance (20). Leptin is produced by adipose tissue and acts as a regulator of appetite and energy expenditure (21). Obesity is associated with high levels of circulating leptin combined with leptin resistance (22). Leptin and insulin directly interact with each other and in addition, leptin influences insulin sensitivity through the regulation of glucose metabolism (23). Interestingly, targeting the energy balance to favor weight loss might induce compensatory behavioral and metabolic actions that favor the maintenance of bodyweight (24). This is one of the explanations for the further increasing numbers of obesity, despite multiple broad scale attempts on lifestyle and dietary interventions. Instead, obesity has grown into a worldwide pandemic. Surveys conducted by the WHO in 2008 showed that around 1.5 billion adults worldwide suffer from overweight, which corresponds to a body mass index (BMI) of over 25. Of far greater concern are the ~200 million men and 300 million women with a BMI of more than 30, therefore considered to be obese (25). It is alarming that the prevalence of overweight and obesity is not only increasing dramatically among adults, but also children (26).
Linking Maternal to Childhood Obesity: a Transgenerational Vicious Circle
Early childhood obesity has been proposed as a strong predictor of overweight in early adulthood (27). It has also been reported that maternal obesity and GDM can cause early-onset childhood obesity, which is associated with a higher prevalence of overweight or obesity in adulthood (28). In the USA, 17% of children are already considered obese (26, 29). Studies show that 1 to 2-year-old overweight children are more likely to be obese in their teenage and middle age years, and are prone to develop early-onset metabolic syndrome (30–33). Metabolic syndrome is characterized by obesity, type 2 diabetes, cardiovascular diseases, dyslipidemia and hypertension. As the prevalence of obesity amongst young adults continues to rise, the number of overweight/obese pregnant women is also steadily increasing. Interestingly, there is accumulating evidence of a transgenerational effect of obesity that adversely affects child health throughout life (34, 35). Specifically, maternal BMI shows a significant correlation with high offspring's birth weight and children's overweight (30, 36, 37). Thus, children of overweight mothers are at high risk of developing overweight later in life and tend to suffer from overweight-associated diseases (38). These findings are supported by experimental studies that show higher tendency for obesity and impaired insulin response in offspring of obese dams (39–41).
These transgenerational effects can be attributed, in part, to epigenetic changes in the offspring of obese mothers. The DNA is hypomethylated at the start of embryonic development, therefore, the developing embryo is particularly sensitive to epigenetic changes (e.g., DNA methylation, histone modification and microRNA expression) in response to the intrauterine environment (42). For example, maternal high-fat diet before, during and after pregnancy has been shown to alter miRNA expression and to induce a chronic dysregulation of insulin-like growth factor 2 (IGF-2) signaling in a mouse model (43). Such changes are not only detectable in adult mice (42), but also in human fetal umbilical cord blood (44). In addition, there are multiple reports of modified DNA methylation on sites of importance for metabolic processes after dysglycemia and/or high-fat diet during pregnancy (45–47).
In addition to metabolic consequences, the rising incidence of early childhood obesity is particularly concerning because of the association with respiratory symptoms and diseases in youth. One of the most common respiratory symptoms in childhood is wheezing, with ~30% of all children suffering from it (48). The risk of recurrent wheezing is especially high in children of obese mothers (49, 50). Obese children experience exertional dyspnea and often suffer from obstructive sleep apnea syndrome (OSAS) as well as obesity hypoventilation syndrome (OHS) (51, 52). In case of an emergency, obese children show dyspnea related to sedation and post-operative care (53). Furthermore, persistent asthma is strongly associated with high BMI throughout childhood (54–56), which manifests in a 92% higher prevalence of asthma in adult obese patients (57). In addition, the functional parameters of lung function such as respiratory muscle strength and endurance, airway resistance, lung volume and function or gas exchange are negatively influenced by obesity (58–60). Collectively, these studies indicate an adverse clinical course of respiratory diseases in children of obese mothers and after childhood obesity. In the following, we will discuss possible causes for the association between maternal obesity and child lung health.
Molecular Insights Into the Mechanical Effects of Obesity on Lung Function
Mechanical and physical influences on the lung play a significant role in the overall health of obese children and adults. Obese persons, including children, experience a lung restrictive syndrome, which goes along with increased overall body volume causing a narrowing of the upper airway and a reduced full inflation due to neck fat and an inadequate thoracic expansion, respectively (61). Accordingly, obese children suffer from obstructive sleep apnea hypoventilation syndrome (OSAHS) which is associated with hypoxemia, hypoventilation, sleep interruptions and chronic fatigue (62). Therefore, OSAHS has a drastic effect on oxygen supply and can induce hypoxia-associated changes in gene expression through the transcription factor hypoxia-inducible factor (HIF) (63). Under normoxia, prolyl hydroxylases (PHDs) hydroxylate proline residues on HIF-α subunits leading to subsequent proteasomal degradation through ubiquitination. In contrast, hypoxia reduces the O2-dependent hydroxylation of HIF-α subunits by PHDs, resulting in nuclear HIF-α accumulation (64). In a murine model, the effects of hypoxia-mediated HIF activity have been reported to be involved in the pathogenesis of pulmonary arterial hypertension (PAH), in part by upregulating the vasoconstrictor endothelin-1 (65, 66). Deficiency of HIF-2α, however, partly protected from the increase in endothelin and PAH (65, 66). In addition, stabilization of HIF-α induces alveolar epithelial type 2 cell (ATII) apoptosis and subsequent fibrotic lung diseases (67). In contrast, the use of PHD inhibitors in vivo to stabilize HIF-α improved lung growth and function in a model of prematurity (68, 69).
It has been shown that HIF-1α in part sustains the Warburg effect (70, 71). The Warburg effect describes a condition in which the cells obtain their energy mainly through glycolysis with subsequent excretion of lactate. This alternative metabolic state for energy production is used by cancer cells, but also by healthy cells under hypoxia (anaerobic glycolysis). As described above, obesity-associated mechanical forces can lead to an activation of HIF, a mediator of hypoxia. HIF can cause a shift toward glycolysis rather than oxidative phosphorylation, in order to meet the energy demands under hypoxic conditions (70). Interestingly, studies suggested a Warburg effect inversion, a condition in which cancer cells exposed to an adiposity environment increase energy production by aerobic respiration as well as gluconeogenesis (72). The authors suggest that the cells do not consume glucose in glycolysis, but produce glucose through gluconeogenesis. Moreover, it has been described that during hypoxia, mitochondria increase the production of reactive oxygen species (ROS) at complex III (73), leading to inhibition of PHD activity and subsequent stabilization of HIF-α (74, 75). The shift toward glycolysis by the Warburg effect and the increased production of ROS, both induced and maintained by hypoxia, resemble mitochondrial dysfunction (76, 77). Increasing evidence points toward a central role for mitochondrial dysfunction in the development of cancer as well as CLDs including asthma, COPD and PAH (71, 76, 77). Furthermore, recent studies have indicated that the hypoxia-induced increase of ROS in acute lung injury contributes to pulmonary fibrosis by triggering an epithelial-mesenchymal transition (EMT) (78, 79) via the stabilization of HIF-1α in several cell types, including alveolar epithelial cells (80).
These findings highlight the effect of obesity on oxygen sensing and energy metabolism as well as the subsequent consequences for the development of CLDs. HIF, as a central player in oxygen sensing, might serve as a potential therapeutic approach to target the rising incidences of obesity-related diseases. For example, preclinical data show that blocking HIF with digoxin in a mouse model prevented or slowed down the progression of PAH (81, 82). These promising findings demonstrate that not only preventing obesity itself, but also targeting specific metabolic processes might offer new preventive strategies for CLDs.
Cell Homeostasis and Inflammatory Response Under Obese Conditions
Obesity represents a state of low-grade chronic inflammation. The numbers of inflammatory cells such as CD8+, CD4+ and CD68+ cells are significantly elevated in adipose tissue (83, 84). These immune cells along with adipocytes release a wide range of inflammatory factors including leptin, tumor necrosis factor-alpha (TNF-α) (85), interleukin 6 (IL-6), and IL-8 (86), C-reactive protein, monocyte chemoattractant protein-1 (MCP-1), and Plasminogen activator inhibitor-1 (PAI-1) (87–89). Exposure of the lung to these pro-inflammatory cytokines can occur in three different ways at different time points during lung development: (1) through transplacental transport from the obese mother to the fetus; (2) through breast milk of the obese mother during lactation; and (3) through the child's own adipose tissue as a result of (early) postnatal obesity. For example, maternal high-fat diet in a murine model during lactation [postnatal day 1 (P1) to P21] induced an early-onset obesity in the offspring with elevated inflammatory cytokines, such as IL-4, IL-6, IL-13, IL-17A, and TNF-α. The early inflammatory response was related to increased airway hyperreactivity, similar to asthma (90). The adverse effect of IL-6 on the lung was further supported by a study that showed that elevated IL-6 could in part account for the development of emphysema through IL-6 trans-signaling-mediated apoptosis of ATII. Blocking IL-6/gp130 signaling, however, prevented features of lung emphysema (91, 92). Furthermore, elevated IL-6 levels contribute to PAH (93). For example, IL-6 induces a downstream activation of Stat3, which in turn causes a phosphorylation of the transcription factor forkhead box O (FoxO) 1. Phosphorylation of FoxO1 leads to its cytoplasmatic sequestration, subsequent inactivation and ultimately to a hyperproliferation of bronchial smooth muscle cells (SMC) (93, 94). In addition to IL-6, TNF-α is also a notable adipocytokine that is elevated under obese conditions (95). TNF-α modulates the effects of G-protein coupled receptor (GPCR)-induced hyperreactivity in cultured murine airway SMCs and increases contractility (96). By this mechanism, TNF-α may be contributing to SMC responsiveness and the development of asthma. Consequently, the anti-inflammatory adiponectin reduces TNF-α-induced nuclear factor κ B (NFκB) signaling. Thus, the obesity-related decrease in adiponectin further contributes to a dysregulated TNF-α signaling (97). Due to its potential impact on the development of asthma, TNF-α is under intense investigation as a therapeutic target (98–100). Another important functional aspect of TNF-α is the ability to contribute to insulin resistance by inhibiting tyrosine phosphorylation of insulin receptor substrate-1 (IRS-1) (95). Similarly, PAI-1 is produced and secreted by adipocytes and elevated in obesity serum levels (89). In a mouse model of airway hyperresponsiveness, PAI-1 was involved in airway remodeling after LPS-induced lung injury (101). Chronically elevated levels of PAI-1 affect the extracellular matrix turnover and contribute to collagen deposition in the airways (102). Moreover, dysfunction of the adipose tissue after perinatal obesity can further contribute to the maintenance of low-grade chronic inflammation. For example, a recent study indicated that maternal obesity induces metabolic programming of adipocytes in the offspring with lifelong dysfunctional adipose tissue and obesity (103). Collectively, obesity represents a state of low-grade chronic inflammation exposing the developing lung to pro-inflammatory cytokines which could adversely affect lung growth as a first “hit” and increase susceptibility for CLDs in later life.
Nutrient Sensing and Leptin Signaling as a Mechanism of Perinatal Obesity
Under physiological conditions, leptin is integrated in the complex mechanisms of airway and bronchial maturation. A recent study highlighted the importance of physiological non-obese levels of leptin in lung maturation through the upregulation of the expression and the secretion of surfactant protein A (Sftpa) in ATII (104, 105). Similarly, leptin promoted maturation of lung structure and contributed to postnatal lung remodeling and enlargement of the alveolar surface area via the induction of the genes Col1a1, Col3a1, Col6a3, Mmp2, Tieg1, and Stat1 (106). A lack of leptin signaling in ob/ob mice (induced by leptin deficiency) resulted in a significant reduction of alveolar surface, indicating a critical role of leptin in postnatal lung growth (106). These contradictory observations may be due to effects of high circulating concentrations of glucose and insulin during pregnancy in obese mothers, which might potentially overrule the beneficial effect of leptin on lung development (107). In addition, long-term exposure to leptin before birth could affect the expression of pulmonary leptin receptors, disturbing leptin-signaling, leading to defective lung maturation and respiratory function at birth (108, 109).
Leptin has a central role in the immune response as well. Leptin was linked to asthma in adults as well as in children; the severity of asthma was correlated to serum leptin levels in a meta-analysis of 13 studies (110–113). High leptin levels increased the T-helper cell type 2 (Th2)-type immune response in airways via a leptin-mediated and XBP1 (X-box binding protein 1) s-dependent activation of mTOR (mechanistic target of rapamycin) as well as MAPK (mitogen-activated protein kinase) signaling (114). A shift toward the Th2-type immune response in airways is characteristic for the pathogenesis of asthma, thus providing a relevant link between obesity-induced high circulating leptin levels and the development of asthma (114, 115). Adiponectin acts as an anti-inflammatory agent, counteracting leptin (88, 89). Circulating adiponectin levels are known to be reduced in obesity, possibly further contributing to the pathogenesis of obesity-associated asthma (116).
There have been several attempts to alter the high leptin and low adiponectin levels in order to restore the metabolic balance. For example, pharmacological elevation of adiponectin levels in obese mice protected from hyperglycemia, glucose intolerance, and insulin resistance (117) as well as increasing insulin sensitivity (118). However, to date, the effect of adiponectin supplementation on pulmonary development and function remains elusive. In diabetes, thiazolidinedione (TZD) is possibly the most extensively characterized regulator of adiponectin expression. TZDs, such as pioglitazone and rosiglitazone increase adiponectin expression through the activation of peroxisome proliferator-activated receptor gamma (PPARγ) (119, 120). Since it is already a well-established therapeutic intervention for diabetes, targeting adiponectin might be a new promising therapeutic approach for the prevention of long-term consequences of obesity such as pulmonary remodeling and reduced lung function.
Lipid Metabolism and Perinatal Obesity
Obesity is characterized by a dysregulation of the energy and lipid metabolism. Lipoproteins are responsible for the transport of fatty acids, cholesterol and phospholipids. Therefore, the lipoproteins in obese patients show a change in circulating protein levels (121). For example, apolipoprotein E (ApoE), which is part of the low-density lipoprotein (LDL), is elevated in the obese and contributes to fat mass accumulation (122). LDL/ApoE is internalized into cells by its receptor, the low-density lipoprotein receptors (LDLRs), and is the main source of cholesterol and phospholipids efflux out of cells. In the lung, ApoE is produced by lung macrophages and acts on ciliated airway epithelial cells, where it can modulate airway hyperreactivity, mucin gene expression, and goblet cell hyperplasia (121). Thereby, it is involved in reducing the susceptibility to airway hyperresponsiveness (121, 123). In line with this, genetic modified mice with an ApoE deletion show reduced alveologenesis and abnormal pulmonary function with increased airway resistance as well as high dynamic and static compliance (124). PPARγ is a nuclear receptor and considered one of the master regulators of adipogenesis, showing a high expression pattern in adipose tissue and in the lung (125–127). PPARγ is essential for normal lung development via the induction of alveolar epithelial-mesenchymal paracrine signaling (128, 129). Murine studies with genetically deactivated PPARγ demonstrated a spontaneous development of PAH. Here, PPARγ has an anti-proliferative effect on smooth muscle cell proliferation, which might give the opportunity to use PPARγ agonists in treating PAH (130, 131). Moreover, unsaturated fatty acids and several eicosanoids are regulators of PPARγ and induce expression of genes encoding lipoprotein lipase, CD36, phosphoenolpyruvate carboxykinase, aquaporin 7 and adiponectin (132). This is of particular interest since the western style diet has high concentrations of poly-unsaturated fatty acids (133). In this context of western style diet and a higher rate of obese individuals in industrial western countries, elevated fatty acid levels in obesity may be important regulators and modulators of normal and aberrant lung development.
Glucose Metabolism and Hyperinsulemia
Obesity is intimately linked to insulin resistance, accompanied by elevated circulating insulin concentrations. The transduction of insulin signaling is in part mediated through the downstream phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) and mTOR pathways (134–137). The mTOR cascade is integral in orchestrating the complex mechanism of lung development, balancing nutrient and energy supply in the early stages of embryogenesis and fine-tuning tissue growth during organogenesis. An elaborated and comprehensive article by Land et al. provides a broad overview of the role of mTOR in lung development (138). High levels of insulin from diabetic mothers have the potential to inhibit the Sftpa gene expression in lung epithelial cells and thereby delay the fetuses' lung development. This insulin-induced inhibition acts via the rapamycin-sensitive PI3K signaling pathway and not via mitogen-activated protein kinase (MAPK) (139). This notion is further supported by the fact that inhibition of PI3K can contribute to insulin resistance and diabetes (140). Moreover, Ikeda and colleagues demonstrated that insulin reduces vascular endothelial growth factor (VEGF) expression and the transcriptional activity of HIF-2 on the VEGF promoter in an AKT-mTOR-dependent manner in cultured lung epithelial cells. They further demonstrated that activation of the AKT-mTOR pathway in mice reduced alveolar capillarization, stressing the importance of this pathway in lung epithelium and in the development of infant respiratory distress syndrome (RDS) (141). Interestingly, moderate physical activity of obese mothers can rescue maternal and the offsprings' insulin sensitivity, overall improving the metabolic, as well as potential pulmonary outcome in the obese mother as well as her offspring (142).
Insulin does not only affect the alveolar epithelial cells, but also increases the expression of genes related to the contractile phenotype of airway SMC through a Rho kinase- and PI3K-dependent mechanism (143). Apart from these direct effects on pulmonary cells, insulin is involved in the modulation of the immune response and thereby in the pathogenesis of asthma. For example, in mast cells, insulin induces PI3K-dependent signaling, which could contribute to allergic bronchoconstriction (144). On the other hand, Viardot and colleagues demonstrated that insulin influences T cell differentiation promoting a shift toward a Th2-type response. They state, that this effect may contribute to insulin's anti-inflammatory role in chronic inflammation associated with obesity and type 2 diabetes (145). Insulin further exhibits anti-inflammatory properties in acute Th1-type inflammation, where insulin diminishes acute lung injury and reduces levels of inflammatory cytokines (146). Taken together, insulin plays an important role in physiological lung development, supporting alveolarization. In obese patients, however, elevated insulin levels interfere with lung development and maturation, while facilitating a pro-asthmatic immune environment, which could affect the outcome of CLDs in later life.
Collectively, these studies show that perinatal obesity resulting from maternal and early childhood obesity may determine individual susceptibility for CLDs later in life. In addition to mechanical factors due to increased body mass, adipose tissue dysfunction and its consequences play a particularly important role. Low-grade chronic inflammation with increased levels of adipocytokines, impaired insulin signaling, and altered lipid metabolism can be important in metabolic programming of CLDs. In the future, further elucidation of the fat-lung axis is imperative for a better understanding of metabolic mechanisms in the development of CLDs and to develop new preventive and therapeutic approaches.
The Impact of Perinatal Nutritive Deficiency on the Origins of Chronic Lung Disease
Nutrient Deprivation and Lung Development: the Role of Intrauterine Growth Restriction
Intrauterine growth restriction (IUGR) was first described as “dysmaturity” and indicates an abnormally low birth weight for the gestational age. Classically, IUGR was defined as a birthweight below 2,500 g (147). More recently, it has been characterized as “not reaching the biologically based potential,” often due to reduced perfusion or malnutrition in utero (148–150). IUGR and “small for gestational age” (SGA, birthweight of-2 SD/mean) are often used interchangeably; however, SGA neither excludes nor proves IUGR but serves as an easily quantifiable proxy for IUGR. Pathological intrauterine circumstances induce IUGR, resulting in an infant with low birth weight, often followed by a period of rapid postnatal weight gain, also called “catch-up growth.” Catch-up growth is associated with altered nutrient supply, and overlaps with the final stages of pulmonary alveolarization and vascular maturation (151, 152). Moreover, infants with catch-up growth after IUGR have a higher risk to become overweight or obese and to develop metabolic disorders later in life (4, 153). These clinical findings have been supported by experimental models of IUGR (154–157).
The etiology of IUGR can be divided in (1) fetal origins, such as genetic abnormalities (e.g., chromosomal abnormalities), (2) maternal factors (e.g., vascular diseases, persistent hypoxia or undernutrition, and toxins), and (3) placental etiologies (e.g., placental insufficiency, inflammation) (158). It is thought that 40% of birth weight is ascribable to genetic factors and that the remaining 60% is due to fetal environmental exposures (159). Several historical events have caused a surge of IUGR cases in a defined birth cohort, which has provided deeper insight into the clinical sequelae of IUGR. The latest temporary surge of IUGR caused by maternal malnutrition in Europe was caused by the Second World War. Investigations of the Dutch Famine Birth Cohort (Amsterdam, 1944-1946) have shown that low birth weight infants often have a lower FEV1 and FVC, but not FEV1/FVC ratios, indicative of restrictive lung alterations (160, 161). Other cohorts, however, including an Indian study demonstrate an association of small head circumference (indicative of early gestational growth restriction) with reduced FEV1/FVC ratios (162, 163). These data show the diverse impact of intrauterine nutrient deprivation on lung health that could be in part accounted to the window of injury or the type of nutrient restriction (e.g., protein, vitamins). Overall, these observational and experimental studies highlight that being born IUGR represents a pathologic condition with far-reaching consequences for the child's health and disease, especially regarding metabolism and the lung.
The Interplay Between IUGR and Obesity
Maternal obesity and GDM are often associated with macrosomic offspring (164). However, in uncontrolled or badly controlled GDM, diabetic vasculopathy and nephropathy may lead to placental insufficiency-induced IUGR (165, 166). In addition, experimental data have shown that overnutrition of pregnant sheep causes IUGR in the fetus, likely due to major restriction in placental growth and relative hypoglycemia and fetal hypoinsulinemia during late pregnancy (167). This might be partly related to fetal hypoxia, in turn inducing fetal catecholamine expression and reducing circulating insulin concentrations (168). On the contrary, IUGR induces metabolic changes to the growing fetus that cause a risk for developing obesity, diabetes and metabolic syndrome later in life (4, 153). These changes are passed onto the next generation; female IUGR rat offspring exhibit symptoms of gestational diabetes, and their offspring has increased fasting glucose and insulin levels despite having a normal birth weight when compared to controls (169). These transgenerational changes might be attributed to epigenetic changes, not only affecting the IUGR offspring, but also the second generation by direct exposition of the offspring germ-line to the IUGR environment (170, 171). More specifically, the increased risk for childhood and adult obesity in IUGR offspring could be in part due to programming of the adipocytes toward lipogenesis and proliferation (172, 173). Moreover, the combination of IUGR (induced by surgical bilateral artery ligation) with maternal obesity increased hepatic cholesterol accumulation and LDLR expression when compared to non-IUGR controls (156). These data further support the notion that maternal obesity along with IUGR provides an additional risk for metabolic complications.
The Adverse Effects of IUGR on Pulmonary Structure and Function
IUGR causes structural changes to the lung. Multiple animal studies have shown that IUGR impairs alveolar formation and lung growth, leading to reduced lung function (155, 174–179). In addition, a recent study from our group has demonstrated that IUGR also negatively influences angiogenesis and extracellular matrix formation (157). The intimate link between angiogenesis and alveologenesis has been shown in various animal studies, where alveolar formation was reduced after blocking angiogenesis (180–182). Conversely, the positive influence of angiogenesis on alveolar growth and regeneration is of great therapeutic importance (181, 183). Structural alveolar and vascular changes during lung development could account for the functional alterations that were reported after IUGR in epidemiological studies: several cohort studies have shown that school-children born IUGR have a significantly lower FEV1 and airway resistance as well as a higher susceptibility to airway infections, independent of catch-up growth (184–189). Moreover, in long-term follow-up studies it was shown that a low birthweight decreases lung function in adulthood, with a reduction of lung capacity and elasticity, resembling a COPD phenotype (9, 190). In summary, there is compelling epidemiological and experimental evidence that IUGR determines lung structure and function and could thereby predispose for CLDs.
Endocrine Effects of IUGR and Catch-Up Growth Resemble those of Obesity
Children born SGA have an increased risk of reduced embryonic β-cell growth, glucose intolerance, insulin resistance, type II diabetes and obesity in childhood as well as later in life (191–197). The effect of IUGR on the regulation of insulin levels has been extensively studied, as insulin is not only important for euglycemia in the fetus but also serves as a major fetal (pulmonary-) growth factor (159). The stable glucose flow over the placenta during healthy pregnancy causes fetal insulin secretion that regulates normal adipose tissue development and deposition (198). As stated before, IUGR fetus can exhibit hypoglycemia and hypoinsulinemia due to inhibition of endocrine signaling by catecholamines (168). In addition, the pancreatic function can be decreased after IUGR, resulting in lower levels of intrauterine insulin secretion as well (199). In contrast, reports on postnatal insulin levels in IUGR newborns are contradictive, they might be slightly lower or equal to healthy controls (200, 201). Thus, IUGR causes a deregulation of intrauterine insulin levels, an important mediator in adipose tissue development and fat deposition.
The phase of catch-up growth after IUGR appears to be a strong determinant of future (lung) health. A key fetal adaptation to nutrient deprivation is the intrauterine upregulation of the insulin receptor under hypoinsulinemic circumstances in fetal skeletal muscle (202). After birth and under nutrient surplus, this upregulated receptor is activated by an abundance of glucose and insulin, inducing accelerated body growth (202, 203). The closely related insulin-like growth factor 1 (IGF-1) is induced by growth hormone (GH)/somatropin and is an essential regulator of body growth. The inhibition of the GH/IGF-1 axis has been shown to dysregulate alveologenesis, mainly through disruption of the physiological deposition of the extracellular matrix (204). Work by our group has shown that inhibition of the GH/IGF-1 axis by IUGR was associated with an arrest of lung development; in contrast, catch-up growth caused a significant increase of GH/IGF-1 expression (174). Interestingly, recent work demonstrated that postnatal treatment with recombinant human IGF-1 improves lung growth and structure in a model for bronchopulmonary dysplasia (BPD) (205). In conclusion, there is a postnatal reactive upregulation of both the insulin receptor and the insulin-signaling (including IGF-1) pathway after IUGR, resulting in an initially increased insulin sensitivity during postnatal catch-up growth (4). However, school-aged and adolescent children with accelerated weight gain and catch-up growth after IUGR show increased levels of insulin and reduced insulin sensitivity, indicating the long-lasting effects of prenatal metabolic programming (206, 207).
In addition to the dysregulation of prenatal and postnatal insulin signaling, leptin has been identified to be dysregulated after IUGR as well. Animal studies have shown that IUGR rat pups rapidly develop leptin resistance during their catch-up growth, thereby stimulating weight gain through hyperphagia (208–210). An important molecular link between nutrient status, insulin/leptin signaling and metabolic outcome is the mTOR pathway, controlling cell growth in response to its environment (e.g., stress, oxygen, nutrient status) through protein synthesis as well as lipid, nucleotide, and glucose metabolism (211, 212). A study from our group has shown that nutrient sensing via the mTOR signaling pathway is dysregulated in lungs from a rat model of nutrient deprivation-induced IUGR (157). Recent reports demonstrated that the mTOR signaling pathway is also altered in the placenta of humans and in experimental IUGR studies, enforcing adaptive mechanisms from both the maternal nutrient supply and the fetus's energy demands (213, 214). These studies suggest that both the placenta and the fetus react to nutrient availability by regulating this key nutrient sensor. The mTOR pathway is postnatally essential for pancreatic β-cell and islet maturation (215). Furthermore, mTOR is a potent mediator of endocrine responses, translating signals from leptin and insulin to a negative feedback for insulin (216). Interestingly, studies demonstrate that mTOR is involved in lung development as well, by regulating cell growth for proper organ development (211, 212) and by interfering with essential developmental signaling pathways, such as pulmonary angiogenesis (VEGF) and extracellular matrix deposition (bone morphogenetic protein, BMP) (217, 218). Collectively, these data highlight the eminent impact of intrauterine nutrient deprivation on endocrine function. Of note are the converging similarities between IUGR and obesity with regard to the endocrine system and the long-term metabolic and pulmonary sequelae.
IUGR Causes Transgenerational Metabolic Programing
Epidemiological studies as well as animal studies have shown transgenerational effects of IUGR on metabolic function (219–221). In part, these effects can be attributed to epigenetic programming (222, 223). For example, Fu et al. as well as Tosh et al. described histone modification along the IGF-1 gene and subsequently altered mRNA expression of IGF-1 in a rat model for IUGR induced by placental insufficiency or maternal malnutrition, respectively (224, 225). In addition, Tosh et al. showed that the restriction of early postnatal nutrient intake partly prevents these epigenetic changes (224). Park et al. observed consistent epigenetic adaptations related to differential binding of dinucleotide methyl transferase 1 and 3a together with changes in histone acetylation and methylation in the promoter region of the Pdx1 homeobox gene in a rat model of IUGR (226). Recent studies by Gonzalez-Rodriguez et al., demonstrated the genetic imprinting of H19/IGF2 in second-generation IUGR offspring. This genetic imprinting was associated with altered H19 and IGF2 expression, which is in turn related to an increased risk for obesity and associated metabolic diseases (220, 227). Interestingly, this effect is reversible with postnatal essential nutrient supplementation (220, 228). These studies highlight the influence of perinatal nutrition in the development but also the primary prevention of metabolic diseases, including their secondary pulmonary complications as described in the previous chapter. In summary, these data indicate the great potential of perinatal nutrition and metabolism as a preventive and therapeutic target for metabolic health and CLDs.
Chronic Inflammation in IUGR-Associated CLD
One of the vital connections between the metabolic consequences of intrauterine nutrient deprivation and altered lung development is chronic inflammation. Chronic inflammation has been associated with (1) IUGR (229–232), (2) obesity, type 2 diabetes and metabolic syndrome (233–235) as well as (3) CLDs (236–239). IUGR, followed by catch-up growth, shows similar endocrine dysregulation and activation of inflammatory mechanisms as obesity. For example, both obesity and IUGR exhibit similar levels of leptin and insulin resistance in response to their prenatal nutritional status and postnatal accelerated weight gain (191–197). A possible shift of the Th2 immune response might be another link between CLDs and metabolic changes, e.g., elevated leptin (240) and insulin (206, 207) levels after IUGR. Interestingly, a study in IUGR mice has shown that the Th2 shift and consequent recruitment of macrophages cause inflammation in the pancreatic β-cell islets, causing type 2 diabetes (230). To date, there is no conclusive evidence whether IUGR-associated chronic inflammation is causative for or a consequence of metabolic distress, but the reports support an intimate link between both conditions.
A clinical study on the cord blood of 20 SGA neonates showed that IUGR causes a low-grade inflammatory response: infants born IUGR had significantly increased levels of inflammatory markers IL-6, TNF-α, CRP and thrombopoetin (232). Moreover, animal studies have demonstrated IUGR-associated systemic inflammation in various organs: adult (uteroplacental) IUGR rats exhibited increased pancreatic β-cell inflammation, increasing the risk of diabetes (230, 241); a sheep model for hypothermia-induced IUGR showed a decrease of NF-κB as key regulator of immune-responses (231); and finally, a recent study in IUGR lambs demonstrated increased inflammatory markers and expression of inflammatory as well as pro-apoptotic genes in liver tissue (229). Along with these reports, prior work from our group has shown that IUGR causes dysregulation of key developmental signaling pathways such as NPY(neuropeptide Y)/PKC(protein kinase C), IL-6/AMPKα and TGFβ (transforming growth factor β) signaling as well as the associated inflammatory response (178, 179, 242).
A highly relevant comorbidity for IUGR infants is prematurity. About 30–50% of all extremely premature infants display symptoms of IUGR (243, 244). The causes of prematurity are multifactorial, but there is a strong correlation with maternal obesity. A meta-analysis of 84 clinical studies has shown a significantly increased risk of (induced) preterm labor in overweight and obese pregnancies (245). In addition, the risk of neonatal respiratory complications after premature birth is higher in obese vs. non-obese pregnancies (246, 247).
Premature birth and perinatal inflammatory responses have been intimately linked to pathological processes (248). The lungs of preterm infants are often in the late-saccular to early-alveolar phase at birth and require respiratory support (249). Mechanical ventilation, continuous positive airway pressure (CPAP) or oxygen supplementation are necessary treatments, but cause inflammation, acute lung injury and lead to a neonatal CLD, also known as BPD (250–254). Lungs of infants with BPD are characterized by vascular and alveolar hypoplasia (255). As stated previously, IUGR alone adversely affects lung microvascular and alveolar formation. Interestingly, the combination of IUGR with the immature lung in premature infants increases the risk for the clinical manifestation of BPD (i.e., prolonged need of oxygen supplementation >36 weeks of gestation) (251, 256). These reports indicate that IUGR might be an initial “hit” to the organism, raising susceptibility to CLDs such as BPD.
In conclusion, similar to perinatal obesity and GDM, IUGR leads to acute as well as long-term functional and structural changes in the lung. A distinction must be made between an intrauterine and a postnatal phase in the process of perinatal programming caused by IUGR. While the intrauterine phase is characterized by nutritional deprivation, the postnatal phase is usually characterized by a catch-up growth. With regard to the pathomechanisms, metabolic signaling pathways, inflammation, and nutrient-sensing processes play an essential role, ultimately controlling alveolar and vascular formation and lung growth. However, the different phases of injury in IUGR also provide windows of opportunity for preventive strategies, therapeutic interventions and reprogramming in the future (Figure 2).
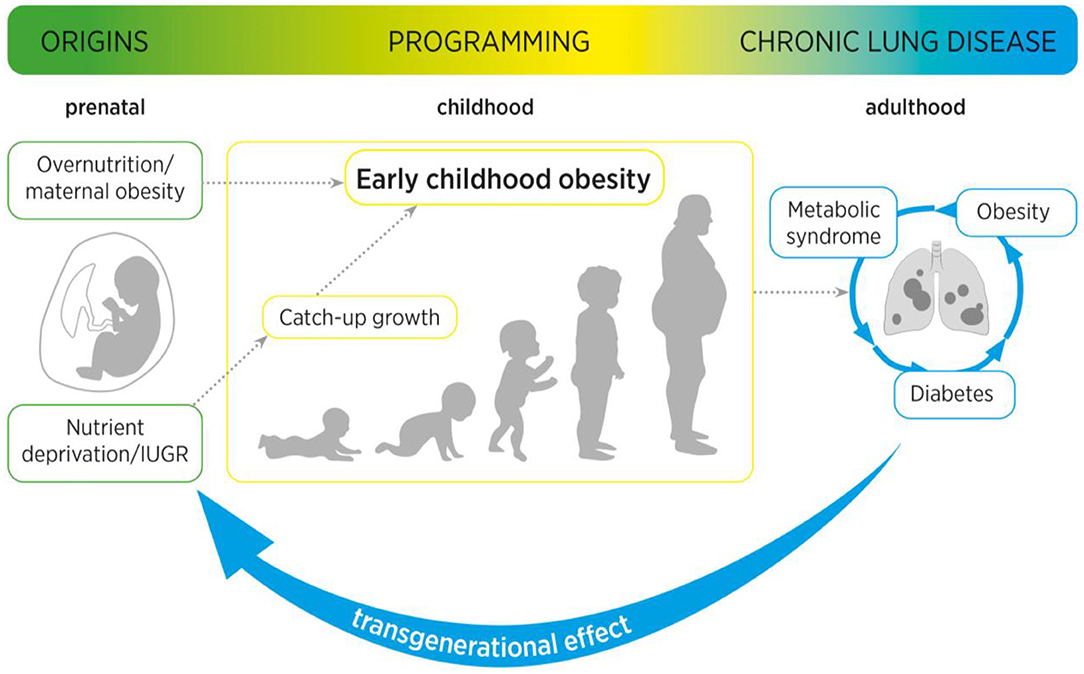
Figure 2. Maternal obesity as well as intrauterine growth restriction (IUGR) increase the risk of (catch-up growth mediated) early childhood obesity. Maternal and early childhood obesity are associated with long-term adverse metabolic effects, including type 2 diabetes mellitus and metabolic syndrome. These pathological metabolic processes are not only intimately linked to an increased risk for pulmonary diseases, but can cause a transgenerational effect from mother to child, to second-generation offspring.
The Microbiome as a Link Between Nutrition and Lung Health: Opportunity for Intervention
As stated before, obesity represents a state of low-grade chronic systemic inflammation, as illustrated by the increased amount of circulating inflammatory cells (83, 84) as well as elevated expression levels of inflammatory factors (85–88). External influences on the lung health such as airway pollution and cigarette smoking have been extensively studied in the last decade. Recently however, another field of interest has gained momentum: the gut-lung microbiome. This represents an extremely important link between nutrition, chronic inflammation and pulmonary health. In the following, we will detail the role of the microbiome in the early origins of CLDs.
The infant's microbiome is predominantly established during and shortly after birth, where it is exposed to the maternal and environmental microbiome (257). The introduction of solid foods into the children's diet is the next essential step in the microbiome development. Western diet (rich in meat and fat) has been linked to decreased bacterial gut richness, whereas a diet-based on fruits and vegetables is associated with increased bacterial richness (258, 259). The human microbiome is closely related to the nutritional status and chronic inflammatory processes of the individual (260, 261). Studies have shown that it is possible to predict if an individual is lean or obese based on a classification of the gut microbiome with an accuracy of over 90% (262). In humans for example, the abundance of bacteria of the taxa Christensenella is negatively correlated with BMI; in contrast, in in vivo experiments feeding mice Christensenella bacteria induces weight loss (263). Another human study revealed that the gut microbiome can influence leptin concentrations, indicating that the microbiome might regulate appetite (264). Interestingly, it has been shown that the same dietary ingredients have different effects on the blood glucose levels in humans, which is thought to be mediated by the microbiome as well (265). In addition, new studies have shown that the fecal transplantation of lean to obese patients improves insulin sensitivity (266).
The microbiome alters the immune system and future immune response. For example, it has been shown that the yeast Candida Albicans in particular has a prominent effect on the TNF-α response of the host; and the palmitoleic acid metabolism of bacteria has been associated with lower systemic responses (267). An overall decrease of bacterial richness is linked to a variety of diseases including obesity, coronary vascular disease, metabolic syndrome insulin resistance, dyslipidemia, and inflammatory disorders (268, 269). When the development of the infants' microbiome is perturbed by the use of antibiotics it can lead to the development of obesity or asthma in later life (270). These examples highlight the mutual relationship between the immune system and the microbiome, creating a finely tuned balance (271). As a result, an imbalance between both creates a lifelong signature of the infants' microbiome (272).
The gut microbiome has been extensively studied, but the lung microbiome has only recently gained interest with the first reports of altered microbiome in asthma (273). The lung microbiome has a strong influence on the susceptibility to a wide array of chronic lung diseases, including COPD, asthma, Idiopathic Pulmonary Fibrosis (IPF) as well as altering the prognosis of cystic fibrosis (CF) (274). In healthy individuals, the lung microbiome is well-regulated by the environment (high clearance, low immigration and low nutrient availability). However, processes that favor alterations of the microbiome and inflammation include the increased production of mucus, creating a moist and warm bacterial niche, increased vascular permeability which increases the nutrient availability and selective growth promotion as well as selective clearance due to the altered immune response to airway colonization (275, 276). These factors promote the bacterial colonization of the airways as well as the selective overgrowth of certain well-adapted species, thereby creating a shift of the microbiome from healthy to diseased and inducing the “dysbiosis-inflammation cycle” as introduced by Dickson et al. (275–278). Thus, a perpetual cycle of microbial changes, possibly due to initial nutritional changes before and early after birth, along with inflammation has a significant impact on the development and prognosis of CLDs.
To date, the gut-lung axis remains elusive, especially with regard to clinical interventions. Nonetheless, several initial successes have been reported in the recent years. For example, stimulation of the gut microbiome with a high-fiber diet in COPD patients has been shown to increase the production of anti-inflammatory short chain fatty acids (SCFAs). These anti-inflammatory factors might reduce chronic inflammation of the lungs, prevent or decrease lung remodeling and therefore improve the lung health of COPD patients (279). Meanwhile, the Canadian Healthy Infant Longitudinal Development (CHILD) Study revealed that bacterial genera Lachnospira, Veillonella, Faecalibacterium, and Rothia are significantly reduced in infants at risk for asthma. Inoculation with these four bacteria reduced airway inflammation in a mouse model, possibly lowering the risk for asthma (280). These studies highlight the promising benefits of dietary changes or adjustment of the gut microbiome for the improvement of lung health.
The virome, including the genes of pathogenic viruses, resident viruses and bacteriophages, is of interest for CLDs as well (281, 282). Viral infection is the predominant reason for acute respiratory infections and the exacerbation of CLDs such as asthma, COPD and CF (283, 284). Next generation sequencing has made it possible to assess the viral DNA or RNA load in respiratory samples (285, 286). Obesity influences the virome of the host; it has been reported that increased viral RNA abundance is closely correlated to an increase of fat mass and hyperglycemia in mice (287). In line with this finding, obese patients show a higher susceptibility to dengue fever (288). Moreover, the adenovirus Ad-36 interferes with adipocyte differentiation, leptin production and glucose metabolism (289). Special interest is drawn to the fact that viral presence in the gut influences the host's immune response (290) by interfering with the host's microbiome and immune-modulatory actions (e.g., through the TNF-α pathway) (267, 291). This crosstalk between the bacterial microbiome and virome and their immune-modulating properties have also been reported in the lung (277, 292).
In conclusion, nutritional changes can directly and indirectly influence the intestinal and pulmonary microbiome (including the virome), modulating the immune response and increasing inflammation, and ultimately the risk of severe CLD. Targeting the microbiome might offer new preventive and therapeutic avenues for CLDs early in life.
Systemic Consequences of Fetal Programming
Maternal obesity, maternal malnutrition or fetal nutrient deficiency through placental insufficiency naturally not only affect the lung, but all other organs as well. The organ-specific susceptibility to metabolic influences varies, and the “window of exposure” plays a crucial role. In other words, the timeframe during which organ development and/or -function is especially vulnerable differs from organ to organ. While a lot of research has been performed with traditional technologies, the ever-growing possibilities of bioinformatics will improve the longitudinal integration of transcriptomic, proteomic and lipidomic data with clinical parameters. These analyses will help to increase the understanding of the complex interaction of internal and external variables during development, resulting in a healthy individual or one with programmed disease. While the strength of animal models lies in the possibility to elucidate molecular mechanisms, it will be a challenge for clinicians to identify individuals at risk. Harmonization and standardization of cohorts like in the LifeCycle-Project (293) improves the epidemiological basis to translate hypotheses on the early origins of disease from animal models in to the human context and to confirm their clinical relevance. In this context, research on biomarkers is of high relevance to improve the diagnostic options in early detection of aberrant organ development. Candidates have been studied in clinical situations of known organ damage, e.g., BDP (294, 295) and neonatal kidney injury (296–298) and will have to be tested in the context of metabolic programming. Importantly, the goal is not to label an individual organ function as pathological beyond classical criteria but to identify individuals who are at risk to develop disease later in life in order to provide targeted prevention strategies.
Looking at molecular mechanisms, it is important to note that early metabolic origins of disease are based on a complex encounter of small-scale dysregulations rather than one single dysregulated pathway. Interestingly, apparently distinct causes of nutritional programming can cause similar molecular alterations. As discussed in detail for the lung, both IUGR (299) and maternal obesity-associated (300) models seem to induce inflammation in other organs as well. Briefly summarized, it is demonstrated that neuroinflammation is an important mechanism contributing to neurocognitive impairment after IUGR and maternal obesity (301, 302) and the window of vulnerability extends well-beyond birth (303, 304). Circulating inflammatory proteins were even tested as biomarkers for later cognitive impairment in preterm infants (305). Experimental studies have also linked perinatal inflammation to adverse kidney development (306, 307) and cardiac dysfunction (308). Taken together, these studies highlight the need to consider inter-organ communication as an important contributor to health and disease.
Conclusion
The mission of this article was to provide a comprehensive review of the impact of perinatal nutrition and metabolism on lung development and early origins of CLD. The current literature provides compelling evidence that maternal obesity, early childhood obesity and IUGR are intimately linked to increased risk for lung disease. These perinatal nutritional alterations of the fetus and infant converge in similar metabolic, endocrine, nutrient sensing and inflammatory signaling pathways. Key regulators are insulin and leptin, and their respective downstream signaling cascades. Both hormones are essential for physiological growth and development during pregnancy; in contrast, interruption of the concerted interaction and balance of hormones, cytokines and growth factors during a critical window of development can disrupt developmental processes and adversely affect child (lung) health throughout life. For example, maternal obesity and early childhood obesity cause hyperinsulinemia and hyperleptinemia combined with insulin- and leptin-insensitivity. On the other hand, IUGR is characterized by a transient prenatal downregulation of insulin- and leptin signaling, followed by a postnatal upregulation during catch-up growth resulting in the same pathology as obesity. These two endocrine factors subsequently cause a cascade of pro-inflammatory programing with the release of (adipo-) cytokines and can contribute to metabolic and pulmonary disease (Figure 3, Table 1). These pulmonary sequelae span from aberrant alveolarization and angiogenesis to remodeling of the extracellular matrix and ultimately reducing lung function. While the present review primarily focused on the lung, other organs are affected as well, highlighting the importance of inter-organ communication. Beyond the metabolo-inflammatory stress response after perinatal nutritional alterations, smoking, air pollution as well as a consecutive dysbiosis of the intestinal and pulmonary microbiome contribute to the susceptibility and early origins of CLDs such as COPD, PAH and Asthma.
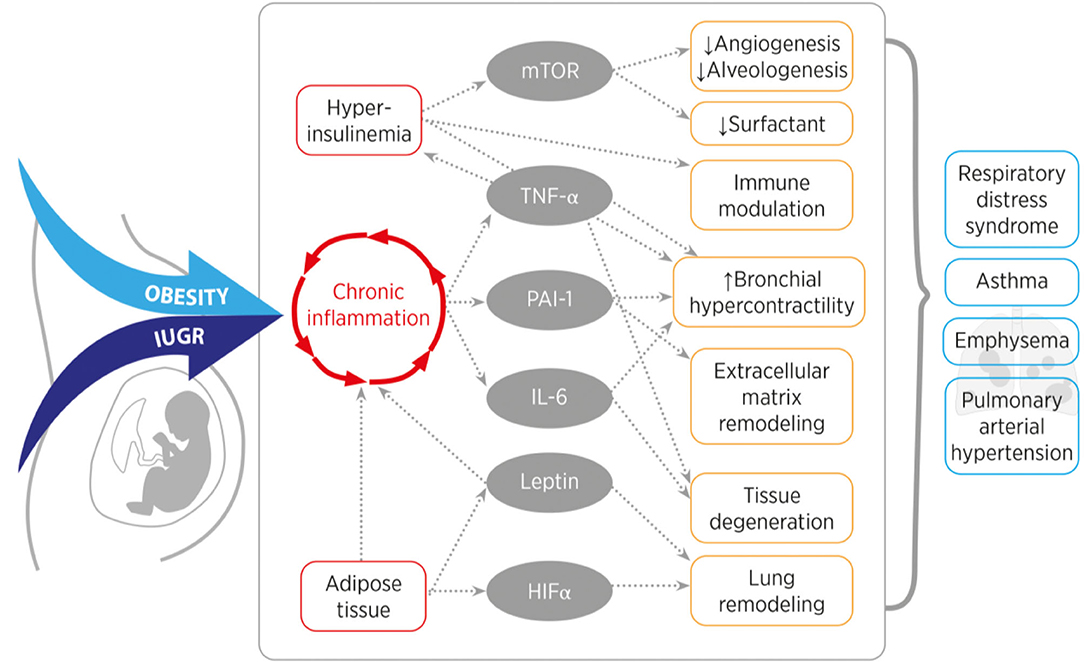
Figure 3. Overview of the converging inflammatory signaling and nutrient sensing pathways of obesity and intrauterine growth restriction (IUGR). Obesity or IUGR lead to chronic inflammation and hyperinsulinemia, which induces mTOR, TNF-α, PAI-1, IL-6, Leptin, and HIF-α signaling. In the lung, these signaling molecules cause for example tissue remodeling, reduce alveolarization and induce smooth muscle cell hyperreactivity. These features are characteristics for a higher susceptibility to develop a chronic lung disease in later life. [mTOR (mechanistic target of rapamycin), TNF-α (tumor necrosis factor alpha), PAI-1 (plasminogen activator inhibitor-1), IL-6 (interleukin-6), HIF-α (hypoxia inducible hypoxia-inducible factor alpha)].
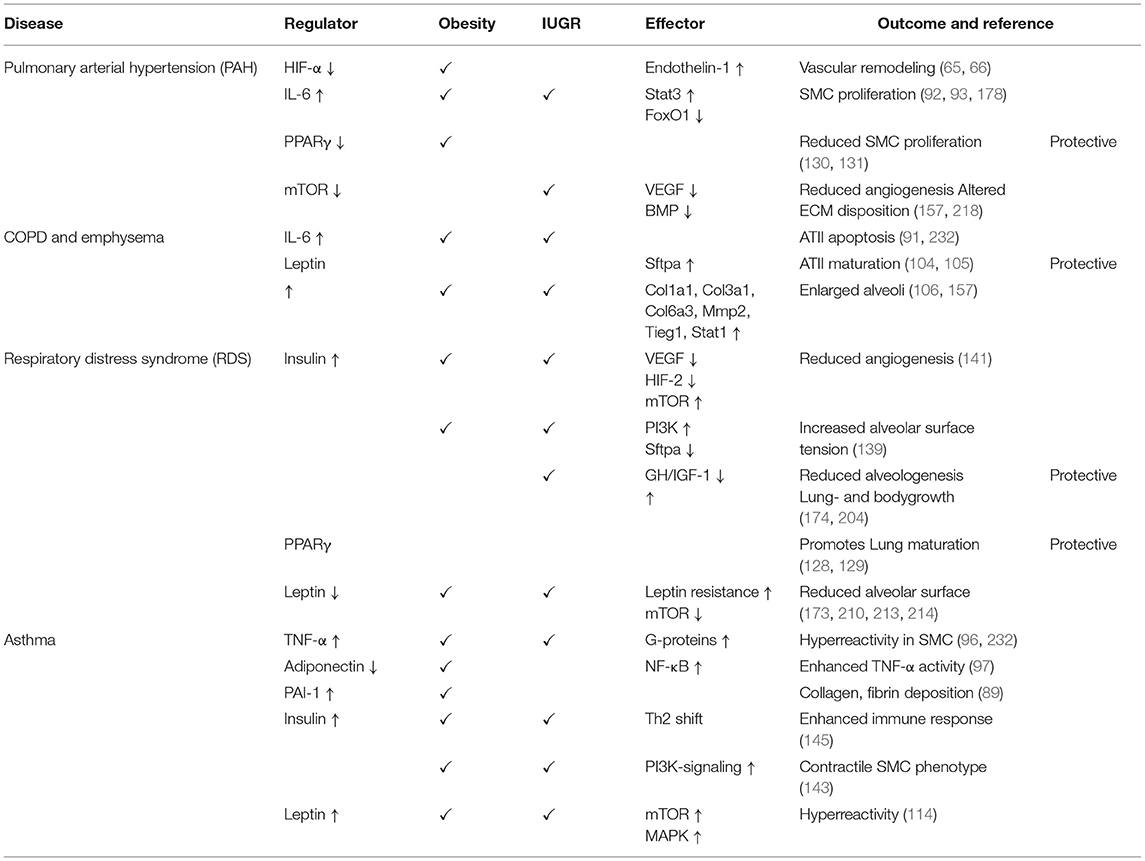
Table 1. Overview of the signaling molecules and pathways involved in the perinatal nutritional and metabolic origins of chronic lung diseases.
Both obesity-related comorbidities and CLDs are a relevant socioeconomic and individual burden. The alarmingly increasing rates of overweight and obese adults, pregnant women, and children emphasize the need to investigate and decipher the crosstalk between nutrition metabolism and the early origins of CLDs. Elucidation of the metabolo-pulmonary axis with subsequent identification of novel targets will provide new avenues to prevent metabolic programming and the early origins of CLDs.
Author Contributions
CK-M, JS, EN, and MA conceived, designed, and drafted the manuscript. CK-M, JS, EN, JD, and MA edited and revised the manuscript and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Deutsche Forschungsgemeinschaft AL1632/2-1 (MA), the Marga and Walter Boll Stiftung, the Stiftung Oskar-Helene Heim, and by the Center for Molecular Medicine Cologne, faculty of medicine and university hospital Cologne, Germany (CMMC; MA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We greatly thank Mrs. Petra Kleinwächter (MedizinFoto Köln, University Hospital Cologne, University of Cologne) for her extraordinary support in graphical design and preparing the illustrations.
References
1. May SM, Li JT. Burden of chronic obstructive pulmonary disease: healthcare costs and beyond. Allergy Asthma Proc. (2015) 36:4–10. doi: 10.2500/aap.2015.36.3812
2. Maleki-Yazdi MR, Kelly SM, Lam SY, Marin M, Barbeau M, Walker V. The burden of illness in patients with moderate to severe chronic obstructive pulmonary disease in Canada. Can Respir J. (2012) 19:319–24. doi: 10.1155/2012/328460
3. Poston L, Caleyachetty R, Cnattingius S, Corvalan C, Uauy R, Herring S, et al. Preconceptional and maternal obesity: epidemiology and health consequences. Lancet Diabetes Endocrinol. (2016) 4:1025–36. doi: 10.1016/S2213-8587(16)30217-0
4. Longo S, Bollani L, Decembrino L, Di Comite A, Angelini M, Stronati M. Short-term and long-term sequelae in intrauterine growth retardation (IUGR). J Matern Fetal Neonatal Med. (2013) 26:222–5. doi: 10.3109/14767058.2012.715006
5. Nardozza LM, Caetano AC, Zamarian AC, Mazzola JB, Silva CP, Marcal VM, et al. Fetal growth restriction: current knowledge. Arch Gynecol Obstet. (2017) 295:1061–77. doi: 10.1007/s00404-017-4341-9
7. Plagemann A. Perinatal programming and functional teratogenesis: impact on body weight regulation and obesity. Physiol Behav. (2005) 86:661–8. doi: 10.1016/j.physbeh.2005.08.065
8. Barker DJ. Intrauterine programming of adult disease. Mol Med Today. (1995) 1:418–23. doi: 10.1016/S1357-4310(95)90793-9
9. Edwards CA, Osman LM, Godden DJ, Campbell DM, Douglas JG. Relationship between birth weight and adult lung function: controlling for maternal factors. Thorax. (2003) 58:1061–5. doi: 10.1136/thorax.58.12.1061
10. Harpsoe MC, Basit S, Bager P, Wohlfahrt J, Benn CS, Nohr EA, et al. Maternal obesity, gestational weight gain, and risk of asthma and atopic disease in offspring: a study within the Danish national birth cohort. J Allergy Clin Immunol. (2013) 131:1033–40. doi: 10.1016/j.jaci.2012.09.008
11. Haataja P, Korhonen P, Ojala R, Hirvonen M, Paassilta M, Gissler M, et al. Asthma and atopic dermatitis in children born moderately and late preterm. Eur J Pediatr. (2016) 175:799–808. doi: 10.1007/s00431-016-2708-8
12. Aspberg S, Dahlquist G, Kahan T, Kallen B. Confirmed association between neonatal phototherapy or neonatal icterus and risk of childhood asthma. Pediatr Allergy Immunol. (2010) 21:e733–9. doi: 10.1111/j.1399-3038.2010.01038.x
13. Dyer JS, Rosenfeld CR. Metabolic imprinting by prenatal, perinatal, and postnatal overnutrition: a review. Semin Reprod Med. (2011) 29:266–76. doi: 10.1055/s-0031-1275521
14. Schittny JC. Development of the lung. Cell Tissue Res. (2017) 367:427–44. doi: 10.1007/s00441-016-2545-0
15. Burri PH. Structural aspects of postnatal lung development - alveolar formation and growth. Biol Neonate. (2006) 89:313–22. doi: 10.1159/000092868
16. Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metabolism. (2019) 92:6–10. doi: 10.1016/j.metabol.2018.09.005
17. Albuquerque D, Stice E, Rodriguez-Lopez R, Manco L, Nobrega C. Current review of genetics of human obesity: from molecular mechanisms to an evolutionary perspective. Mol Genet Genomics. (2015) 290:1191–221. doi: 10.1007/s00438-015-1015-9
18. Hopkins M, Blundell JE. Energy balance, body composition, sedentariness and appetite regulation: pathways to obesity. Clin Sci. (2016) 130:1615–28. doi: 10.1042/CS20160006
19. Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. (2000) 106:473–81. doi: 10.1172/JCI10842
20. Reaven GM. Pathophysiology of insulin resistance in human disease. Physiol Rev. (1995) 75:473–86. doi: 10.1152/physrev.1995.75.3.473
21. Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. (2000) 404:661–71. doi: 10.1038/35007534
22. El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. (2000) 105:1827–32. doi: 10.1172/JCI9842
23. Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. (1997) 389:374–7. doi: 10.1038/38717
24. King NA, Caudwell P, Hopkins M, Byrne NM, Colley R, Hills AP, et al. Metabolic and behavioral compensatory responses to exercise interventions: barriers to weight loss. Obesity. (2007) 15:1373–83. doi: 10.1038/oby.2007.164
25. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global burden of disease study 2013. Lancet. (2014) 384:766–81. doi: 10.1016/S0140-6736(14)60460-8
26. Ogden CL, Carroll MD, Fryar CD, Flegal KM. Prevalence of obesity among adults and youth: United States, 2011-2014. NCHS Data Brief. (2015) 219:1–8.
27. Venn AJ, Thomson RJ, Schmidt MD, Cleland VJ, Curry BA, Gennat HC, et al. Overweight and obesity from childhood to adulthood: a follow-up of participants in the 1985 Australian schools health and fitness survey. Med J Aust. (2007) 186:458–60. doi: 10.5694/j.1326-5377.2007.tb01436.x
28. Santangeli L, Sattar N, Huda SS. Impact of maternal obesity on perinatal and childhood outcomes. Best Pract Res Clin Obstet Gynaecol. (2015) 29:438–48. doi: 10.1016/j.bpobgyn.2014.10.009
29. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA. (2012) 307:491–7. doi: 10.1001/jama.2012.39
30. Knight B, Shields BM, Hill A, Powell RJ, Wright D, Hattersley AT. The impact of maternal glycemia and obesity on early postnatal growth in a nondiabetic Caucasian population. Diabetes Care. (2007) 30:777–83. doi: 10.2337/dc06-1849
31. Cheng CW, Rifai A, Ka SM, Shui HA, Lin YF, Lee WH, et al. Calcium-binding proteins annexin A2 and S100A6 are sensors of tubular injury and recovery in acute renal failure. Kidney Int. (2005) 68:2694–703. doi: 10.1111/j.1523-1755.2005.00740.x
32. Parsons TJ, Power C, Manor O. Fetal and early life growth and body mass index from birth to early adulthood in 1958 British cohort: longitudinal study. BMJ. (2001) 323:1331–5. doi: 10.1136/bmj.323.7325.1331
33. Reilly MP, Rader DJ. The metabolic syndrome: more than the sum of its parts? Circulation. (2003) 108:1546–51. doi: 10.1161/01.CIR.0000088846.10655.E0
34. Han JC, Lawlor DA, Kimm SY. Childhood obesity. Lancet. (2010) 375:1737–48. doi: 10.1016/S0140-6736(10)60171-7
35. Godfrey KM, Reynolds RM, Prescott SL, Nyirenda M, Jaddoe VW, Eriksson JG, et al. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. (2017) 5:53–64. doi: 10.1016/S2213-8587(16)30107-3
36. Ehrenberg HM, Mercer BM, Catalano PM. The influence of obesity and diabetes on the prevalence of macrosomia. Am J Obstet Gynecol. (2004) 191:964–8. doi: 10.1016/j.ajog.2004.05.052
37. Patel MS, Srinivasan M, Laychock SG. Metabolic programming: role of nutrition in the immediate postnatal life. J Inherit Metab Dis. (2009) 32:218–28. doi: 10.1007/s10545-008-1033-4
38. Eising JB, Uiterwaal CS, van der Ent CK. Maternal body mass index, neonatal lung function and respiratory symptoms in childhood. Eur Respir J. (2015) 46:1342–9. doi: 10.1183/13993003.00784-2014
39. Aalinkeel R, Srinivasan M, Song F, Patel MS. Programming into adulthood of islet adaptations induced by early nutritional intervention in the rat. Am J Physiol Endocrinol Metab. (2001) 281:E640–8. doi: 10.1152/ajpendo.2001.281.3.E640
40. Vadlamudi S, Hiremagalur BK, Tao L, Kalhan SC, Kalaria RN, Kaung HL, et al. Long-term effects on pancreatic function of feeding a HC formula to rats during the preweaning period. Am J Physiol. (1993) 265:E565–71. doi: 10.1152/ajpendo.1993.265.4.E565
41. Srinivasan M, Dodds C, Ghanim H, Gao T, Ross PJ, Browne RW, et al. Maternal obesity and fetal programming: effects of a high-carbohydrate nutritional modification in the immediate postnatal life of female rats. Am J Physiol Endocrinol Metab. (2008) 295:E895–903. doi: 10.1152/ajpendo.90460.2008
42. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. (2002) 16:6–21. doi: 10.1101/gad.947102
43. Zhang J, Zhang F, Didelot X, Bruce KD, Cagampang FR, Vatish M, et al. Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring. BMC Genomics. (2009) 10:478. doi: 10.1186/1471-2164-10-478
44. Jing J, Wang Y, Quan Y, Wang Z, Liu Y, Ding Z. Maternal obesity alters C19MC microRNAs expression profile in fetal umbilical cord blood. Nutr Metab. (2020) 17:52. doi: 10.1186/s12986-020-00475-7
45. Antoun E, Kitaba NT, Titcombe P, Dalrymple KV, Garratt ES, Barton SJ, et al. Maternal dysglycaemia, changes in the infant's epigenome modified with a diet and physical activity intervention in pregnancy: Secondary analysis of a randomised control trial. PLoS Med. (2020) 17:e1003229. doi: 10.1371/journal.pmed.1003229
46. Finer S, Mathews C, Lowe R, Smart M, Hillman S, Foo L, et al. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum Mol Genet. (2015) 24:3021–9. doi: 10.1093/hmg/ddv013
47. Moody L, Wang H, Jung PM, Chen H, Pan YX. Maternal and post-weaning high-fat diets produce distinct DNA methylation patterns in hepatic metabolic pathways within specific genomic contexts. Int J Mol Sci. (2019) 20:3229. doi: 10.3390/ijms20133229
48. Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, Morgan WJ. Asthma and wheezing in the first six years of life. The group health medical associates. N Engl J Med. (1995) 332:133–8. doi: 10.1056/NEJM199501193320301
49. Zugna D, Galassi C, Annesi-Maesano I, Baiz N, Barros H, Basterrechea M, et al. Maternal complications in pregnancy and wheezing in early childhood: a pooled analysis of 14 birth cohorts. Int J Epidemiol. (2015) 44:199–208. doi: 10.1093/ije/dyu260
50. Kumar R, Story RE, Pongracic JA, Hong X, Arguelles L, Wang G, et al. Maternal pre-pregnancy obesity and recurrent wheezing in early childhood. Pediatr Allergy Immunol Pulmonol. (2010) 23:183–90. doi: 10.1089/ped.2010.0032
51. Arens R, Muzumdar H. Childhood obesity and obstructive sleep apnea syndrome. J Appl Physiol. (2010) 108:436–44. doi: 10.1152/japplphysiol.00689.2009
52. Rosen CL. Clinical features of obstructive sleep apnea hypoventilation syndrome in otherwise healthy children. Pediatr Pulmonol. (1999) 27:403–9. doi: 10.1002/(SICI)1099-0496(199906)27:6<403::AID-PPUL7>3.0.CO;2-811
53. Adams JP, Murphy PG. Obesity in anaesthesia and intensive care. Br J Anaesth. (2000) 85:91–108. doi: 10.1093/bja/85.1.91
54. Ekstrom S, Magnusson J, Kull I, Andersson N, Bottai M, Besharat Pour M, et al. Body mass index development and asthma throughout childhood. Am J Epidemiol. (2017) 186:255–63. doi: 10.1093/aje/kwx081
55. Black MH, Smith N, Porter AH, Jacobsen SJ, Koebnick C. Higher prevalence of obesity among children with asthma. Obesity. (2012) 20:1041–7. doi: 10.1038/oby.2012.5
56. Porter M, Wegienka G, Havstad S, Nageotte CG, Johnson CC, Ownby DR, et al. Relationship between childhood body mass index and young adult asthma. Ann Allergy Asthma Immunol. (2012) 109:408–11. e401. doi: 10.1016/j.anai.2012.09.009
57. Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med. (2007) 175:661–6. doi: 10.1164/rccm.200611-1717OC
58. Koenig SM. Pulmonary complications of obesity. Am J Med Sci. (2001) 321:249–79. doi: 10.1097/00000441-200104000-00006
59. Lazarus R, Colditz G, Berkey CS, Speizer FE. Effects of body fat on ventilatory function in children and adolescents: cross-sectional findings from a random population sample of school children. Pediatr Pulmonol. (1997) 24:187–94. doi: 10.1002/(SICI)1099-0496(199709)24:3<187::AID-PPUL4>3.0.CO;2-K
60. Li AM, Chan D, Wong E, Yin J, Nelson EA, Fok TF. The effects of obesity on pulmonary function. Arch Dis Child. (2003) 88:361–3. doi: 10.1136/adc.88.4.361
61. Watson RA, Pride NB, Thomas EL, Fitzpatrick J, Durighel G, McCarthy J, et al. Reduction of total lung capacity in obese men: comparison of total intrathoracic and gas volumes. J Appl Physiol. (2010) 108:1605–12. doi: 10.1152/japplphysiol.01267.2009
62. Gislason T, Benediktsdottir B. Snoring, apneic episodes, and nocturnal hypoxemia among children 6 months to 6 years old. An epidemiologic study of lower limit of prevalence. Chest. (1995) 107:963–6. doi: 10.1378/chest.107.4.963
63. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. (1992) 12:5447–54. doi: 10.1128/MCB.12.12.5447
64. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. (2001) 292:464–8. doi: 10.1126/science.1059817
65. Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, et al. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. (2003) 111:1519–27. doi: 10.1172/JCI15496
66. Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. (1999) 103:691–6. doi: 10.1172/JCI5912
67. Krick S, Eul BG, Hanze J, Savai R, Grimminger F, Seeger W, et al. Role of hypoxia-inducible factor-1alpha in hypoxia-induced apoptosis of primary alveolar epithelial type II cells. Am J Respir Cell Mol Biol. (2005) 32:395–403. doi: 10.1165/rcmb.2004-0314OC
68. Asikainen TM, Waleh NS, Schneider BK, Clyman RI, White CW. Enhancement of angiogenic effectors through hypoxia-inducible factor in preterm primate lung in vivo. Am J Physiol Lung Cell Mol Physiol. (2006) 291:L588–95. doi: 10.1152/ajplung.00098.2006
69. Asikainen TM, Chang LY, Coalson JJ, Schneider BK, Waleh NS, Ikegami M, et al. Improved lung growth and function through hypoxia-inducible factor in primate chronic lung disease of prematurity. FASEB J. (2006) 20:1698–700. doi: 10.1096/fj.06-5887fje
70. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. (2010) 330:1340–4. doi: 10.1126/science.1193494
71. Burns JS, Manda G. Metabolic pathways of the warburg effect in health and disease: perspectives of choice, chain or chance. Int J Mol Sci. (2017) 18:2755. doi: 10.3390/ijms18122755
72. Luis C, Duarte F, Faria I, Jarak I, Oliveira PF, Alves MG, et al. Warburg effect inversion: adiposity shifts central primary metabolism in MCF-7 breast cancer cells. Life Sci. (2019) 223:38–46. doi: 10.1016/j.lfs.2019.03.016
73. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. (2000) 275:25130–8. doi: 10.1074/jbc.M001914200
74. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. (2003) 3:721–32. doi: 10.1038/nrc1187
75. Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci USA. (2002) 99:13459–64. doi: 10.1073/pnas.192342099
76. Prakash YS, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest. (2017) 152:618–26. doi: 10.1016/j.chest.2017.03.020
77. Rowlands DJ. Mitochondria dysfunction: A novel therapeutic target in pathological lung remodeling or bystander? Pharmacol Ther. (2016) 166:96–105. doi: 10.1016/j.pharmthera.2016.06.019
78. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. (2006) 103:13180–5. doi: 10.1073/pnas.0605669103
79. Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, et al. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L1120–30. doi: 10.1152/ajplung.00007.2009
80. Schroedl C, McClintock DS, Budinger GR, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. (2002) 283:L922–31. doi: 10.1152/ajplung.00014.2002
81. Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci USA. (2008) 105:19579–86. doi: 10.1073/pnas.0809763105
82. Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, et al. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA. (2012) 109:1239–44. doi: 10.1073/pnas.1120385109
83. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/JCI200319246
84. Travers RL, Motta AC, Betts JA, Bouloumie A, Thompson D. The impact of adiposity on adipose tissue-resident lymphocyte activation in humans. Int J Obes. (2015) 39:762–9. doi: 10.1038/ijo.2014.195
85. Bullo M, Garcia-Lorda P, Salas-Salvado J. Plasma soluble tumor necrosis factor alpha receptors and leptin levels in normal-weight and obese women: effect of adiposity and diabetes. Eur J Endocrinol. (2002) 146:325–31. doi: 10.1530/eje.0.1460325
86. Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, et al. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab. (2000) 85:3338–42. doi: 10.1210/jcem.85.9.6839
87. Roth CL, Kratz M, Ralston MM, Reinehr T. Changes in adipose-derived inflammatory cytokines and chemokines after successful lifestyle intervention in obese children. Metabolism. (2011) 60:445–52. doi: 10.1016/j.metabol.2010.03.023
88. Rajala MW, Scherer PE. Minireview: the adipocyte–at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. (2003) 144:3765–73. doi: 10.1210/en.2003-0580
89. Nawrocki AR, Scherer PE. The delicate balance between fat and muscle: adipokines in metabolic disease and musculoskeletal inflammation. Curr Opin Pharmacol. (2004) 4:281–9. doi: 10.1016/j.coph.2004.03.003
90. Dinger K, Kasper P, Hucklenbruch-Rother E, Vohlen C, Jobst E, Janoschek R, et al. Early-onset obesity dysregulates pulmonary adipocytokine/insulin signaling and induces asthma-like disease in mice. Sci Rep. (2016) 6:24168. doi: 10.1038/srep24168
91. Ruwanpura SM, McLeod L, Dousha LF, Seow HJ, Alhayyani S, Tate MD, et al. Therapeutic targeting of the IL-6 trans-signaling/mechanistic target of rapamycin complex 1 axis in pulmonary emphysema. Am J Respir Crit Care Med. (2016) 194:1494–505. doi: 10.1164/rccm.201512-2368OC
92. Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. (2011) 121:3375–83. doi: 10.1172/JCI57158
93. Savai R, Al-Tamari HM, Sedding D, Kojonazarov B, Muecke C, Teske R, et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med. (2014) 20:1289–300. doi: 10.1038/nm.3695
94. Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM, et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest. (2018) 128:1956–70. doi: 10.1172/JCI96462
95. Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord. (2003) 27(Suppl. 3):S53–5. doi: 10.1038/sj.ijo.0802502
96. Chen H, Tliba O, Van Besien CR, Panettieri RA, Jr Amrani Y. TNF-[alpha] modulates murine tracheal rings responsiveness to G-protein-coupled receptor agonists and KCl. J Appl Physiol. (2003) 95:864–72. doi: 10.1152/japplphysiol.00140.2003
97. Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Walsh K. Obesity, adiponectin and vascular inflammatory disease. Curr Opin Lipidol. (2003) 14:561–6. doi: 10.1097/00041433-200312000-00003
98. Kim J, Remick DG. Tumor necrosis factor inhibitors for the treatment of asthma. Curr Allergy Asthma Rep. (2007) 7:151–6. doi: 10.1007/s11882-007-0013-3
99. Desai D, Brightling C. TNF-alpha antagonism in severe asthma? Recent Pat Inflamm Allergy Drug Discov. (2010) 4:193–200. doi: 10.2174/187221310793564218
100. Antoniu SA, Mihaltan F, Ulmeanu R. Anti-TNF-alpha therapies in chronic obstructive pulmonary diseases. Expert Opin Investig Drugs. (2008) 17:1203–11. doi: 10.1517/13543784.17.8.1203
101. Savov JD, Brass DM, Berman KG, McElvania E, Schwartz DA. Fibrinolysis in LPS-induced chronic airway disease. Am J Physiol Lung Cell Mol Physiol. (2003) 285:L940–8. doi: 10.1152/ajplung.00102.2003
102. Oh CK, Ariue B, Alban RF, Shaw B, Cho SH. PAI-1 promotes extracellular matrix deposition in the airways of a murine asthma model. Biochem Biophys Res Commun. (2002) 294:1155–60. doi: 10.1016/S0006-291X(02)00577-6
103. Litzenburger T, Huber EK, Dinger K, Wilke R, Vohlen C, Selle J, et al. Maternal high-fat diet induces long-term obesity with sex-dependent metabolic programming of adipocyte differentiation, hypertrophy and dysfunction in the offspring. Clin Sci. (2020) 134:921–39. doi: 10.1042/CS20191229
104. Chen H, Zhang JP, Huang H, Wang ZH, Cheng R, Cai WB. Leptin promotes fetal lung maturity and upregulates SP-A expression in pulmonary alveoli type-II epithelial cells involving TTF-1 activation. PLoS ONE. (2013) 8:e69297. doi: 10.1371/journal.pone.0069297
105. Kirwin SM, Bhandari V, Dimatteo D, Barone C, Johnson L, Paul S, et al. Leptin enhances lung maturity in the fetal rat. Pediatr Res. (2006) 60:200–4. doi: 10.1203/01.pdr.0000227478.29271.52
106. Huang K, Rabold R, Abston E, Schofield B, Misra V, Galdzicka E, et al. Effects of leptin deficiency on postnatal lung development in mice. J Appl Physiol. (2008) 105:249–59. doi: 10.1152/japplphysiol.00052.2007
107. Lock M, McGillick EV, Orgeig S, McMillen IC, Morrison JL. Regulation of fetal lung development in response to maternal overnutrition. Clin Exp Pharmacol Physiol. (2013) 40:803–16. doi: 10.1111/1440-1681.12166
108. Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. (2008) 70:537–56. doi: 10.1146/annurev.physiol.70.113006.100707
109. Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci USA. (1997) 94:8878–83. doi: 10.1073/pnas.94.16.8878
110. Zhang L, Yin Y, Zhang H, Zhong W, Zhang J. Association of asthma diagnosis with leptin and adiponectin: a systematic review and meta-analysis. J Investig Med. (2017) 65:57–64. doi: 10.1136/jim-2016-000127
111. Guler N, Kirerleri E, Ones U, Tamay Z, Salmayenli N, Darendeliler F. Leptin: does it have any role in childhood asthma? J Allergy Clin Immunol. (2004) 114:254–9. doi: 10.1016/j.jaci.2004.03.053
112. Gurkan F, Atamer Y, Ece A, Kocyigit Y, Tuzun H, Mete N. Serum leptin levels in asthmatic children treated with an inhaled corticosteroid. Ann Allergy Asthma Immunol. (2004) 93:277–80. doi: 10.1016/S1081-1206(10)61501-3
113. Tanju A, Cekmez F, Aydinoz S, Karademir F, Suleymanoglu S, Gocmen I. Association between clinical severity of childhood asthma and serum leptin levels. Indian J Pediatr. (2011) 78:291–5. doi: 10.1007/s12098-010-0281-0
114. Zheng H, Wu D, Wu X, Zhang X, Zhou Q, Luo Y, et al. Leptin promotes allergic airway inflammation through targeting the unfolded protein response pathway. Sci Rep. (2018) 8:8905. doi: 10.1038/s41598-018-27278-4
115. Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. (1995) 1:1155–61. doi: 10.1038/nm1195-1155
116. Shore SA, Fredberg JJ. Obesity, smooth muscle, and airway hyperresponsiveness. J Allergy Clin Immunol. (2005) 115:925–7. doi: 10.1016/j.jaci.2005.01.064
117. Xu A, Wang H, Hoo RL, Sweeney G, Vanhoutte PM, Wang Y, et al. Selective elevation of adiponectin production by the natural compounds derived from a medicinal herb alleviates insulin resistance and glucose intolerance in obese mice. Endocrinology. (2009) 150:625–33. doi: 10.1210/en.2008-0999
118. Combs TP, Pajvani UB, Berg AH, Lin Y, Jelicks LA, Laplante M, et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology. (2004) 145:367–83. doi: 10.1210/en.2003-1068
119. Riera-Guardia N, Rothenbacher D. The effect of thiazolidinediones on adiponectin serum level: a meta-analysis. Diabetes Obes Metab. (2008) 10:367–75. doi: 10.1111/j.1463-1326.2007.00755.x
120. Amin RH, Mathews ST, Camp HS, Ding L, Leff T. Selective activation of PPARgamma in skeletal muscle induces endogenous production of adiponectin and protects mice from diet-induced insulin resistance. Am J Physiol Endocrinol Metab. (2010) 298:E28–37. doi: 10.1152/ajpendo.00446.2009
121. Yao X, Remaley AT, Levine SJ. New kids on the block: the emerging role of apolipoproteins in the pathogenesis and treatment of asthma. Chest. (2011) 140:1048–54. doi: 10.1378/chest.11-0158
122. Gao J, Katagiri H, Ishigaki Y, Yamada T, Ogihara T, Imai J, et al. Involvement of apolipoprotein E in excess fat accumulation and insulin resistance. Diabetes. (2007) 56:24–33. doi: 10.2337/db06-0144
123. Yao X, Gordon EM, Figueroa DM, Barochia AV, Levine SJ. Emerging roles of Apolipoprotein E and Apolipoprotein A-I in the pathogenesis and treatment of lung disease. Am J Respir Cell Mol Biol. (2016) 55:159–69. doi: 10.1165/rcmb.2016-0060TR
124. Massaro D, Massaro GD. Apoetm1Unc mice have impaired alveologenesis, low lung function, and rapid loss of lung function. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L991–7. doi: 10.1152/ajplung.00013.2008
125. Chen H, Jackson S, Doro M, McGowan S. Perinatal expression of genes that may participate in lipid metabolism by lipid-laden lung fibroblasts. J Lipid Res. (1998) 39:2483–92. doi: 10.1016/S0022-2275(20)33329-0
126. Tontonoz P, Hu E, Spiegelman BM. Regulation of adipocyte gene expression and differentiation by peroxisome proliferator activated receptor gamma. Curr Opin Genet Dev. (1995) 5:571–6. doi: 10.1016/0959-437X(95)80025-5
127. Lazar MA. PPAR gamma, 10 years later. Biochimie. (2005) 87:9–13. doi: 10.1016/j.biochi.2004.10.021
128. Simon DM, Arikan MC, Srisuma S, Bhattacharya S, Tsai LW, Ingenito EP, et al. Epithelial cell PPAR[gamma] contributes to normal lung maturation. FASEB J. (2006) 20:1507–9. doi: 10.1096/fj.05-5410fje
129. Torday JS, Torres E, Rehan VK. The role of fibroblast transdifferentiation in lung epithelial cell proliferation, differentiation, and repair in vitro. Pediatr Pathol Mol Med. (2003) 22:189–207. doi: 10.1080/pdp.22.3.189.207
130. Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. (2008) 118:1846–57. doi: 10.1172/JCI32503
131. Rabinovitch M. PPARgamma and the pathobiology of pulmonary arterial hypertension. Adv Exp Med Biol. (2010) 661:447–58. doi: 10.1007/978-1-60761-500-2_29
132. Koutnikova H, Cock TA, Watanabe M, Houten SM, Champy MF, Dierich A, et al. Compensation by the muscle limits the metabolic consequences of lipodystrophy in PPAR gamma hypomorphic mice. Proc Natl Acad Sci USA. (2003) 100:14457–62. doi: 10.1073/pnas.2336090100
133. Ailhaud G, Massiera F, Weill P, Legrand P, Alessandri JM, Guesnet P. Temporal changes in dietary fats: role of n-6 polyunsaturated fatty acids in excessive adipose tissue development and relationship to obesity. Prog Lipid Res. (2006) 45:203–36. doi: 10.1016/j.plipres.2006.01.003
134. Kanai F, Ito K, Todaka M, Hayashi H, Kamohara S, Ishii K, et al. Insulin-stimulated GLUT4 translocation is relevant to the phosphorylation of IRS-1 and the activity of PI3-kinase. Biochem Biophys Res Commun. (1993) 195:762–8. doi: 10.1006/bbrc.1993.2111
135. Czech MP, Corvera S. Signaling mechanisms that regulate glucose transport. J Biol Chem. (1999) 274:1865–8. doi: 10.1074/jbc.274.4.1865
136. Courtneidge SA, Heber A. An 81 kd protein complexed with middle T antigen and pp60c-src: a possible phosphatidylinositol kinase. Cell. (1987) 50:1031–7. doi: 10.1016/0092-8674(87)90169-3
137. Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. (2013) 15:555–64. doi: 10.1038/ncb2763
138. Land SC, Scott CL, Walker D. mTOR signalling, embryogenesis and the control of lung development. Semin Cell Dev Biol. (2014) 36:68–78. doi: 10.1016/j.semcdb.2014.09.023
139. Miakotina OL, Goss KL, Snyder JM. Insulin utilizes the PI 3-kinase pathway to inhibit SP-A gene expression in lung epithelial cells. Respir Res. (2002) 3:27. doi: 10.1186/rr191
140. Maffei A, Lembo G, Carnevale D. PI3Kinases in diabetes mellitus and its related complications. Int J Mol Sci. (2018) 19:4098. doi: 10.3390/ijms19124098
141. Ikeda H, Shiojima I, Oka T, Yoshida M, Maemura K, Walsh K, et al. Increased Akt-mTOR signaling in lung epithelium is associated with respiratory distress syndrome in mice. Mol Cell Biol. (2011) 31:1054–65. doi: 10.1128/MCB.00732-10
142. Fernandez-Twinn DS, Gascoin G, Musial B, Carr S, Duque-Guimaraes D, Blackmore HL, et al. Exercise rescues obese mothers' insulin sensitivity, placental hypoxia and male offspring insulin sensitivity. Sci Rep. (2017) 7:44650. doi: 10.1038/srep44650
143. Schaafsma D, McNeill KD, Stelmack GL, Gosens R, Baarsma HA, Dekkers BG, et al. Insulin increases the expression of contractile phenotypic markers in airway smooth muscle. Am J Physiol Cell Physiol. (2007) 293:C429–39. doi: 10.1152/ajpcell.00502.2006
144. Lessmann E, Grochowy G, Weingarten L, Giesemann T, Aktories K, Leitges M, et al. Insulin and insulin-like growth factor-1 promote mast cell survival via activation of the phosphatidylinositol-3-kinase pathway. Exp Hematol. (2006) 34:1532–41. doi: 10.1016/j.exphem.2006.05.022
145. Viardot A, Grey ST, Mackay F, Chisholm D. Potential antiinflammatory role of insulin via the preferential polarization of effector T cells toward a T helper 2 phenotype. Endocrinology. (2007) 148:346–53. doi: 10.1210/en.2006-0686
146. Shapiro H, Kagan I, Shalita-Chesner M, Singer J, Singer P. Inhaled aerosolized insulin: a “topical” anti-inflammatory treatment for acute lung injury and respiratory distress syndrome? Inflammation. (2010) 33:315–9. doi: 10.1007/s10753-010-9187-2
147. Wigglesworth JS. Foetal growth retardation. Br Med Bull. (1966) 22:13–5. doi: 10.1093/oxfordjournals.bmb.a070429
148. Kesavan K, Devaskar SU. Intrauterine growth restriction: postnatal monitoring and outcomes. Pediatr Clin North Am. (2019) 66:403–23. doi: 10.1016/j.pcl.2018.12.009
149. Gordijn SJ, Beune IM, Ganzevoort W. Building consensus and standards in fetal growth restriction studies. Best Pract Res Clin Obstet Gynaecol. (2018) 49:117–26. doi: 10.1016/j.bpobgyn.2018.02.002
150. Beune IM, Bloomfield FH, Ganzevoort W, Embleton ND, Rozance PJ, van Wassenaer-Leemhuis AG, et al. Consensus based definition of growth restriction in the newborn. J Pediatr. (2018) 196:71–6. e71. doi: 10.1016/j.jpeds.2017.12.059
151. Ong KK, Ahmed ML, Emmett PM, Preece MA, Dunger DB. Association between postnatal catch-up growth and obesity in childhood: prospective cohort study. BMJ. (2000) 320:967–71. doi: 10.1136/bmj.320.7240.967
152. Healy MJ, Lockhart RD, Mackenzie JD, Tanner JM, Whitehouse RH. Aberdeen growth study. I. The prediction of adult body measurements from measurements taken each year from birth to 5 years. Arch Dis Child. (1956) 31:372–81. doi: 10.1136/adc.31.159.372
153. Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. (1976) 295:349–53. doi: 10.1056/NEJM197608122950701
154. Rozance PJ, Seedorf GJ, Brown A, Roe G, O'Meara MC, Gien J, et al. Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am J Physiol Lung Cell Mol Physiol. (2011) 301:L860–71. doi: 10.1152/ajplung.00197.2011
155. Joss-Moore L, Carroll T, Yang Y, Fitzhugh M, Metcalfe D, Oman J, et al. Intrauterine growth restriction transiently delays alveolar formation and disrupts retinoic acid receptor expression in the lung of female rat pups. Pediatr Res. (2013) 73:612–20. doi: 10.1038/pr.2013.38
156. Zinkhan EK, Zalla JM, Carpenter JR, Yu B, Yu X, Chan G, et al. Intrauterine growth restriction combined with a maternal high-fat diet increases hepatic cholesterol and low-density lipoprotein receptor activity in rats. Physiol Rep. (2016) 4:e12862. doi: 10.14814/phy2.12862
157. Kuiper-Makris C, Zanetti D, Vohlen C, Fahle L, Muller M, Odenthal M, et al. Mendelian randomization and experimental IUGR reveal the adverse effect of low birth weight on lung structure and function. Sci Rep. (2020) 10:22395. doi: 10.1038/s41598-020-79245-7
158. Brodsky D, Christou H. Current concepts in intrauterine growth restriction. J Intensive Care Med. (2004) 19:307–19. doi: 10.1177/0885066604269663
159. Devaskar SU, Chu A. Intrauterine growth restriction: hungry for an answer. Physiology. (2016) 31:131–46. doi: 10.1152/physiol.00033.2015
160. Lumey LH, Stein AD. Offspring birth weights after maternal intrauterine undernutrition: a comparison within sibships. Am J Epidemiol. (1997) 146:810–9. doi: 10.1093/oxfordjournals.aje.a009198
161. Lopuhaa CE, Roseboom TJ, Osmond C, Barker DJ, Ravelli AC, Bleker OP, et al. Atopy, lung function, and obstructive airways disease after prenatal exposure to famine. Thorax. (2000) 55:555–61. doi: 10.1136/thorax.55.7.555
162. Stein CE, Kumaran K, Fall CH, Shaheen SO, Osmond C, Barker DJ. Relation of fetal growth to adult lung function in south India. Thorax. (1997) 52:895–9. doi: 10.1136/thx.52.10.895
163. Lawlor DA, Ebrahim S, Davey Smith G. Association of birth weight with adult lung function: findings from the British Women's Heart and Health Study and a meta-analysis. Thorax. (2005) 60:851–8. doi: 10.1136/thx.2005.042408
164. Baeten JM, Bukusi EA, Lambe M. Pregnancy complications and outcomes among overweight and obese nulliparous women. Am J Public Health. (2001) 91:436–40. doi: 10.2105/AJPH.91.3.436
165. Hayes EK, Lechowicz A, Petrik JJ, Storozhuk Y, Paez-Parent S, Dai Q, et al. Adverse fetal and neonatal outcomes associated with a life-long high fat diet: role of altered development of the placental vasculature. PLoS ONE. (2012) 7:e33370. doi: 10.1371/journal.pone.0033370
166. Gutaj P, Wender-Ozegowska E, Iciek R, Zawiejska A, Pietryga M, Brazert J. Maternal serum placental growth factor and fetal SGA in pregnancy complicated by type 1 diabetes mellitus. J Perinat Med. (2014) 42:629–33. doi: 10.1515/jpm-2013-0227
167. Wallace JM, Bourke DA, Aitken RP, Palmer RM, Da Silva P, Cruickshank MA. Relationship between nutritionally-mediated placental growth restriction and fetal growth, body composition and endocrine status during late gestation in adolescent sheep. Placenta. (2000) 21:100–8. doi: 10.1053/plac.1999.0440
168. Limesand SW, Rozance PJ. Fetal adaptations in insulin secretion result from high catecholamines during placental insufficiency. J Physiol. (2017) 595:5103–13. doi: 10.1113/JP273324
169. Thamotharan M, Garg M, Oak S, Rogers LM, Pan G, Sangiorgi F, et al. Transgenerational inheritance of the insulin-resistant phenotype in embryo-transferred intrauterine growth-restricted adult female rat offspring. Am J Physiol Endocrinol Metab. (2007) 292:E1270–9. doi: 10.1152/ajpendo.00462.2006
170. Skinner MK. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod Toxicol. (2008) 25:2–6. doi: 10.1016/j.reprotox.2007.09.001
171. Horsthemke B. A critical view on transgenerational epigenetic inheritance in humans. Nat Commun. (2018) 9:2973. doi: 10.1038/s41467-018-05445-5
172. Meng R, Lv J, Yu C, Guo Y, Bian Z, Yang L, et al. Prenatal famine exposure, adulthood obesity patterns and risk of type 2 diabetes. Int J Epidemiol. (2018) 47:399–408. doi: 10.1093/ije/dyx228
173. Desai M, Ross MG. Fetal programming of adipose tissue: effects of intrauterine growth restriction and maternal obesity/high-fat diet. Semin Reprod Med. (2011) 29:237–45. doi: 10.1055/s-0031-1275517
174. Nawabi J, Vohlen C, Dinger K, Thangaratnarajah C, Klaudt C, Lopez Garcia E, et al. Novel functional role of GH/IGF-I in neonatal lung myofibroblasts and in rat lung growth after intrauterine growth restriction. Am J Physiol Lung Cell Mol Physiol. (2018) 315:L623–37. doi: 10.1152/ajplung.00413.2017
175. Khazaee R, McCaig LA, Yamashita C, Hardy DB, Veldhuizen RAW. Maternal protein restriction during perinatal life affects lung mechanics and the surfactant system during early postnatal life in female rats. PLoS ONE. (2019) 14:e0215611. doi: 10.1371/journal.pone.0215611
176. Joss-Moore LA, Wang Y, Baack ML, Yao J, Norris AW, Yu X, et al. IUGR decreases PPARgamma and SETD8 Expression in neonatal rat lung and these effects are ameliorated by maternal DHA supplementation. Early Hum Dev. (2010) 86:785–91. doi: 10.1016/j.earlhumdev.2010.08.026
177. Dravet-Gounot P, Morin C, Jacques S, Dumont F, Ely-Marius F, Vaiman D, et al. Lung microRNA deregulation associated with impaired alveolarization in rats after intrauterine growth restriction. PLoS ONE. (2017) 12:e0190445. doi: 10.1371/journal.pone.0190445
178. Alejandre Alcazar MA, Ostreicher I, Appel S, Rother E, Vohlen C, Plank C, et al. Developmental regulation of inflammatory cytokine-mediated Stat3 signaling: the missing link between intrauterine growth restriction and pulmonary dysfunction? J Mol Med. (2012) 90:945–57. doi: 10.1007/s00109-012-0860-9
179. Alejandre Alcazar MA, Morty RE, Lendzian L, Vohlen C, Oestreicher I, Plank C, et al. Inhibition of TGF-beta signaling and decreased apoptosis in IUGR-associated lung disease in rats. PLoS ONE. (2011) 6:e26371. doi: 10.1371/journal.pone.0026371
180. Thebaud B, Abman SH. Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med. (2007) 175:978–85. doi: 10.1164/rccm.200611-1660PP
181. Tang JR, Markham NE, Lin YJ, McMurtry IF, Maxey A, Kinsella JP, et al. Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor. Am J Physiol Lung Cell Mol Physiol. (2004) 287:L344–51. doi: 10.1152/ajplung.00291.2003
182. Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, et al. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol. (2000) 279:L600–7. doi: 10.1152/ajplung.2000.279.3.L600
183. Yun EJ, Lorizio W, Seedorf G, Abman SH, Vu TH. VEGF and endothelium-derived retinoic acid regulate lung vascular and alveolar development. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L287–98. doi: 10.1152/ajplung.00229.2015
184. Svanes C, Omenaas E, Heuch JM, Irgens LM, Gulsvik A. Birth characteristics and asthma symptoms in young adults: results from a population-based cohort study in Norway. Eur Respir J. (1998) 12:1366–70. doi: 10.1183/09031936.98.12061366
185. Rona RJ, Gulliford MC, Chinn S. Effects of prematurity and intrauterine growth on respiratory health and lung function in childhood. BMJ. (1993) 306:817–20. doi: 10.1136/bmj.306.6881.817
186. Greenough A, Yuksel B, Cheeseman P. Effect of in utero growth retardation on lung function at follow-up of prematurely born infants. Eur Respir J. (2004) 24:731–3. doi: 10.1183/09031936.04.00060304
187. Ronkainen E, Dunder T, Kaukola T, Marttila R, Hallman M. Intrauterine growth restriction predicts lower lung function at school age in children born very preterm. Arch Dis Child Fetal Neonatal Ed. (2016) 101:F412–7. doi: 10.1136/archdischild-2015-308922
188. Kotecha SJ, Watkins WJ, Heron J, Henderson J, Dunstan FD, Kotecha S. Spirometric lung function in school-age children: effect of intrauterine growth retardation and catch-up growth. Am J Respir Crit Care Med. (2010) 181:969–74. doi: 10.1164/rccm.200906-0897OC
189. He B, Kwok MK, Au Yeung SL, Lin SL, Leung JYY, Hui LL, et al. Birth weight and prematurity with lung function at ~17.5 years: “Children of 1997” birth cohort. Sci Rep. (2020) 10:341. doi: 10.1038/s41598-019-56086-7
190. Cai Y, Shaheen SO, Hardy R, Kuh D, Hansell AL. Birth weight, early childhood growth and lung function in middle to early old age: 1946 British birth cohort. Thorax. (2016) 71:916–22. doi: 10.1136/thoraxjnl-2014-206457
191. Simmons RA, Templeton LJ, Gertz SJ. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes. (2001) 50:2279–86. doi: 10.2337/diabetes.50.10.2279
192. Ravelli AC, van Der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr. (1999) 70:811–6. doi: 10.1093/ajcn/70.5.811
193. Newsome CA, Shiell AW, Fall CH, Phillips DI, Shier R, Law CM. Is birth weight related to later glucose and insulin metabolism?–A systematic review. Diabet Med. (2003) 20:339–48. doi: 10.1046/j.1464-5491.2003.00871.x
194. Mohan R, Baumann D, Alejandro EU. Fetal undernutrition, placental insufficiency, and pancreatic beta-cell development programming in utero. Am J Physiol Regul Integr Comp Physiol. (2018) 315:R867–78. doi: 10.1152/ajpregu.00072.2018
195. Laitinen J, Pietilainen K, Wadsworth M, Sovio U, Jarvelin MR. Predictors of abdominal obesity among 31-y-old men and women born in Northern Finland in 1966. Eur J Clin Nutr. (2004) 58:180–90. doi: 10.1038/sj.ejcn.1601765
196. Crume TL, Scherzinger A, Stamm E, McDuffie R, Bischoff KJ, Hamman RF, et al. The long-term impact of intrauterine growth restriction in a diverse US. cohort of children: the EPOCH study. Obesity. (2014) 22:608–15. doi: 10.1002/oby.20565
197. Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. (1993) 36:62–7. doi: 10.1007/BF00399095
198. Illsley NP, Baumann MU. Human placental glucose transport in fetoplacental growth and metabolism. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165359. doi: 10.1016/j.bbadis.2018.12.010
199. Boehmer BH, Limesand SW, Rozance PJ. The impact of IUGR on pancreatic islet development and beta-cell function. J Endocrinol. (2017) 235:R63–76. doi: 10.1530/JOE-17-0076
200. Yada KK, Gupta R, Gupta A, Gupta M. Insulin levels in low birth weight neonates. Indian J Med Res. (2003) 118:197–203.
201. Wolf HJ, Ebenbichler CF, Huter O, Bodner J, Lechleitner M, Foger B, et al. Fetal leptin and insulin levels only correlate inlarge-for-gestational age infants. Eur J Endocrinol. (2000) 142:623–9. doi: 10.1530/eje.0.1420623
202. Muhlhausler BS, Duffield JA, Ozanne SE, Pilgrim C, Turner N, Morrison JL, et al. The transition from fetal growth restriction to accelerated postnatal growth: a potential role for insulin signalling in skeletal muscle. J Physiol. (2009) 587:4199–211. doi: 10.1113/jphysiol.2009.173161
203. Dunlop K, Cedrone M, Staples JF, Regnault TR. Altered fetal skeletal muscle nutrient metabolism following an adverse in utero environment and the modulation of later life insulin sensitivity. Nutrients. (2015) 7:1202–16. doi: 10.3390/nu7021202
204. Beyea JA, Sawicki G, Olson DM, List E, Kopchick JJ, Harvey S. Growth hormone (GH) receptor knockout mice reveal actions of GH in lung development. Proteomics. (2006) 6:341–8. doi: 10.1002/pmic.200500168
205. Seedorf G, Kim C, Wallace B, Mandell EW, Nowlin T, Shepherd D, et al. rhIGF-1/BP3 preserves lung growth and prevents pulmonary hypertension in experimental bronchopulmonary dysplasia. Am J Respir Crit Care Med. (2020) 201:1120–34. doi: 10.1164/rccm.201910-1975OC
206. Invitti C, Gilardini L, Mazzilli G, Sartorio A, Viberti GC, Ong KK, et al. Insulin sensitivity and secretion in normal children related to size at birth, postnatal growth, and plasma insulin-like growth factor-I levels. Diabetologia. (2004) 47:1064–70. doi: 10.1007/s00125-004-1565-6
207. Ibanez L, Potau N, Marcos MV, de Zegher F. Exaggerated adrenarche and hyperinsulinism in adolescent girls born small for gestational age. J Clin Endocrinol Metab. (1999) 84:4739–41. doi: 10.1210/jc.84.12.4739
208. Gurugubelli Krishna R, Vishnu Bhat B. Molecular mechanisms of intrauterine growth restriction. J Matern Fetal Neonatal Med. (2018) 31:2634–40. doi: 10.1080/14767058.2017.1347922
209. Desai M, Gayle D, Han G, Ross MG. Programmed hyperphagia due to reduced anorexigenic mechanisms in intrauterine growth-restricted offspring. Reprod Sci. (2007) 14:329–37. doi: 10.1177/1933719107303983
210. Coupe B, Grit I, Hulin P, Randuineau G, Parnet P. Postnatal growth after intrauterine growth restriction alters central leptin signal and energy homeostasis. PLoS ONE. (2012) 7:e30616. doi: 10.1371/journal.pone.0030616
211. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 169:361–71. doi: 10.1016/j.cell.2017.03.035
212. Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. (2019) 21:63–71. doi: 10.1038/s41556-018-0205-1
213. Ganguly A, Collis L, Devaskar SU. Placental glucose and amino acid transport in calorie-restricted wild-type and Glut3 null heterozygous mice. Endocrinology. (2012) 153:3995–4007. doi: 10.1210/en.2011-1973
214. Brett KE, Ferraro ZM, Yockell-Lelievre J, Gruslin A, Adamo KB. Maternal-fetal nutrient transport in pregnancy pathologies: the role of the placenta. Int J Mol Sci. (2014) 15:16153–85. doi: 10.3390/ijms150916153
215. Sinagoga KL, Stone WJ, Schiesser JV, Schweitzer JI, Sampson L, Zheng Y, et al. Distinct roles for the mTOR pathway in postnatal morphogenesis, maturation and function of pancreatic islets. Development. (2017) 144:2402–14. doi: 10.1242/dev.146316
216. Hu F, Xu Y, Liu F. Hypothalamic roles of mTOR complex I: integration of nutrient and hormone signals to regulate energy homeostasis. Am J Physiol Endocrinol Metab. (2016) 310:E994–1002. doi: 10.1152/ajpendo.00121.2016
217. Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. (2000) 60:1541–5. doi: 10.1002/cyto.990020515
218. Wahdan-Alaswad RS, Song K, Krebs TL, Shola DT, Gomez JA, Matsuyama S, et al. Insulin-like growth factor I suppresses bone morphogenetic protein signaling in prostate cancer cells by activating mTOR signaling. Cancer Res. (2010) 70:9106–17. doi: 10.1158/0008-5472.CAN-10-1119
219. Pinheiro AR, Salvucci ID, Aguila MB, Mandarim-de-Lacerda CA. Protein restriction during gestation and/or lactation causes adverse transgenerational effects on biometry and glucose metabolism in F1 and F2 progenies of rats. Clin Sci. (2008) 114:381–92. doi: 10.1042/CS20070302
220. Gonzalez-Rodriguez P, Cantu J, O'Neil D, Seferovic MD, Goodspeed DM, Suter MA, et al. Alterations in expression of imprinted genes from the H19/IGF2 loci in a multigenerational model of intrauterine growth restriction (IUGR). Am J Obstet Gynecol. (2016) 214:625. doi: 10.1016/j.ajog.2016.01.194
221. Berends LM, Ozanne SE. Early determinants of type-2 diabetes. Best Pract Res Clin Endocrinol Metab. (2012) 26:569–80. doi: 10.1016/j.beem.2012.03.002
222. Aiken CE, Ozanne SE. Transgenerational developmental programming. Hum Reprod Update. (2014) 20:63–75. doi: 10.1093/humupd/dmt043
223. Joss-Moore LA, Lane RH, Albertine KH. Epigenetic contributions to the developmental origins of adult lung disease. Biochem Cell Biol. (2015) 93:119–27. doi: 10.1139/bcb-2014-0093
224. Tosh DN, Fu Q, Callaway CW, McKnight RA, McMillen IC, Ross MG, et al. Epigenetics of programmed obesity: alteration in IUGR rat hepatic IGF1 mRNA expression and histone structure in rapid vs. delayed postnatal catch-up growth. Am J Physiol Gastrointest Liver Physiol. (2010) 299:G1023–9. doi: 10.1152/ajpgi.00052.2010
225. Fu Q, Yu X, Callaway CW, Lane RH, McKnight RA. Epigenetics: intrauterine growth retardation (IUGR) modifies the histone code along the rat hepatic IGF-1 gene. FASEB J. (2009) 23:2438–49. doi: 10.1096/fj.08-124768
226. Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. (2008) 118:2316–24. doi: 10.1172/JCI33655
227. Deodati A, Inzaghi E, Liguori A, Puglianiello A, Germani D, Brufani C, et al. IGF2 methylation is associated with lipid profile in obese children. Horm Res Paediatr. (2013) 79:361–7. doi: 10.1159/000351707
228. Goodspeed D, Seferovic MD, Holland W, McKnight RA, Summers SA, Branch DW, et al. Essential nutrient supplementation prevents heritable metabolic disease in multigenerational intrauterine growth-restricted rats. FASEB J. (2015) 29:807–19. doi: 10.1096/fj.14-259614
229. Zhang H, Fan Y, Elsabagh M, Guo S, Wang M, Jiang H. Dietary supplementation of L-Arginine and N-carbamylglutamate attenuated the hepatic inflammatory response and apoptosis in suckling lambs with intrauterine growth retardation. Mediators Inflamm. (2020) 2020:2453537. doi: 10.1155/2020/2453537
230. Jaeckle Santos LJ, Li C, Doulias PT, Ischiropoulos H, Worthen GS, Simmons RA. Neutralizing Th2 inflammation in neonatal islets prevents beta-cell failure in adult IUGR rats. Diabetes. (2014) 63:1672–84. doi: 10.2337/db13-1226
231. Dodson RB, Powers KN, Gien J, Rozance PJ, Seedorf G, Astling D, et al. Intrauterine growth restriction decreases NF-kappaB signaling in fetal pulmonary artery endothelial cells of fetal sheep. Am J Physiol Lung Cell Mol Physiol. (2018) 315:L348–59. doi: 10.1152/ajplung.00052.2018
232. Amarilyo G, Oren A, Mimouni FB, Ochshorn Y, Deutsch V, Mandel D. Increased cord serum inflammatory markers in small-for-gestational-age neonates. J Perinatol. (2011) 31:30–2. doi: 10.1038/jp.2010.53
233. Fresno M, Alvarez R, Cuesta N. Toll-like receptors, inflammation, metabolism and obesity. Arch Physiol Biochem. (2011) 117:151–64. doi: 10.3109/13813455.2011.562514
234. Bhargava P, Lee CH. Role and function of macrophages in the metabolic syndrome. Biochem J. (2012) 442:253–262. doi: 10.1042/BJ20111708
235. Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. (2006) 17:4–12.
236. Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. (2019) 25:1153–63. doi: 10.1038/s41591-019-0468-5
237. Speer CP. Chorioamnionitis, postnatal factors and proinflammatory response in the pathogenetic sequence of bronchopulmonary dysplasia. Neonatology. (2009) 95:353–61. doi: 10.1159/000209301
238. Ramakrishna L, de Vries VC, Curotto de Lafaille MA. Cross-roads in the lung: immune cells and tissue interactions as determinants of allergic asthma. Immunol Res. (2012) 53:213–28. doi: 10.1007/s12026-012-8296-4
239. Bhat TA, Panzica L, Kalathil SG, Thanavala Y. Immune dysfunction in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. (2015) 12(Suppl. 2):S169–75. doi: 10.1513/AnnalsATS.201503-126AW
240. Kyriakakou M, Malamitsi-Puchner A, Militsi H, Boutsikou T, Margeli A, Hassiakos D, et al. Leptin and adiponectin concentrations in intrauterine growth restricted and appropriate for gestational age fetuses, neonates, and their mothers. Eur J Endocrinol. (2008) 158:343–8. doi: 10.1530/EJE-07-0692
241. Rashid CS, Lien YC, Bansal A, Jaeckle-Santos LJ, Li C, Won KJ, et al. Transcriptomic analysis reveals novel mechanisms mediating islet dysfunction in the intrauterine growth-restricted rat. Endocrinology. (2018) 159:1035–49. doi: 10.1210/en.2017-00888
242. Thangaratnarajah C, Dinger K, Vohlen C, Klaudt C, Nawabi J, Lopez Garcia E, et al. Novel role of NPY in neuroimmune interaction and lung growth after intrauterine growth restriction. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L491–506. doi: 10.1152/ajplung.00432.2016
243. Rosenberg A. The IUGR newborn. Semin Perinatol. (2008) 32:219–24. doi: 10.1053/j.semperi.2007.11.003
244. Regev RH, Reichman B. Prematurity and intrauterine growth retardation–double jeopardy? Clin Perinatol. (2004) 31:453–73. doi: 10.1016/j.clp.2004.04.017
245. McDonald SD, Han Z, Mulla S, Beyene J, Knowledge Synthesis G. Overweight and obesity in mothers and risk of preterm birth and low birth weight infants: systematic review and meta-analyses. BMJ. (2010) 341:c3428. doi: 10.1136/bmj.c3428
246. Lynch TA, Malshe A, Colihan S, Meyers J, Li D, Holloman C, et al. Impact of maternal obesity on perinatal outcomes in preterm prelabor rupture of membranes >/=34 weeks. Am J Perinatol. (2020) 37:467–74. doi: 10.1055/s-0039-1698833
247. Vincent S, Czuzoj-Shulman N, Spence AR, Abenhaim HA. Effect of pre-pregnancy body mass index on respiratory-related neonatal outcomes in women undergoing elective cesarean prior to 39 weeks. J Perinat Med. (2018) 46:905–12. doi: 10.1515/jpm-2017-0384
248. Goldenberg RL, Culhane JF. Prepregnancy health status and the risk of preterm delivery. Arch Pediatr Adolesc Med. (2005) 159:89–90. doi: 10.1001/archpedi.159.1.89
249. Smith LJ, McKay KO, van Asperen PP, Selvadurai H, Fitzgerald DA. Normal development of the lung and premature birth. Paediatr Respir Rev. (2010) 11:135–42. doi: 10.1016/j.prrv.2009.12.006
250. Kumar VH, Lakshminrusimha S, Kishkurno S, Paturi BS, Gugino SF, Nielsen L, et al. Neonatal hyperoxia increases airway reactivity and inflammation in adult mice. Pediatr Pulmonol. (2016) 51:1131–41. doi: 10.1002/ppul.23430
251. Jobe AH. The new bronchopulmonary dysplasia. Curr Opin Pediatr. (2011) 23:167–72. doi: 10.1097/MOP.0b013e3283423e6b
252. Dumpa V, Bhandari V. Surfactant, steroids and non-invasive ventilation in the prevention of BPD. Semin Perinatol. (2018) 42:444–52. doi: 10.1053/j.semperi.2018.09.006
253. Collins JJP, Tibboel D, de Kleer IM, Reiss IKM, Rottier RJ. The future of bronchopulmonary dysplasia: emerging pathophysiological concepts and potential new avenues of treatment. Front Med (Lausanne). (2017) 4:61. doi: 10.3389/fmed.2017.00061
254. Bustani P, Kotecha S. Role of cytokines in hyperoxia mediated inflammation in the developing lung. Front Biosci. (2003) 8:s694–704. doi: 10.2741/1113
255. Hwang JS, Rehan VK. Recent advances in bronchopulmonary dysplasia: pathophysiology, prevention, and treatment. Lung. (2018) 196:129–38. doi: 10.1007/s00408-018-0084-z
256. Garite TJ, Clark R, Thorp JA. Intrauterine growth restriction increases morbidity and mortality among premature neonates. Am J Obstet Gynecol. (2004) 191:481–7. doi: 10.1016/j.ajog.2004.01.036
257. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA. (2011) 108(Suppl. 1):4578–85. doi: 10.1073/pnas.1000081107
258. Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. (2010) 464:908–912. doi: 10.1038/nature08937
259. Craig WJ. Health effects of vegan diets. Am J Clin Nutr. (2009) 89:1627S−33S. doi: 10.3945/ajcn.2009.26736N
260. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. (2011) 334:105–8. doi: 10.1126/science.1208344
261. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. (2017) 17:219–32. doi: 10.1038/nri.2017.7
262. Knights D, Parfrey LW, Zaneveld J, Lozupone C, Knight R. Human-associated microbial signatures: examining their predictive value. Cell Host Microbe. (2011) 10:292–6. doi: 10.1016/j.chom.2011.09.003
263. Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. (2014) 159:789–99. doi: 10.1016/j.cell.2014.09.053
264. Zhang C, Yin A, Li H, Wang R, Wu G, Shen J, et al. Dietary modulation of gut microbiota contributes to alleviation of both genetic and simple obesity in children. EBioMedicine. (2015) 2:968–84. doi: 10.1016/j.ebiom.2015.07.007
265. Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, et al. Personalized nutrition by prediction of glycemic responses. Cell. (2015) 163:1079–94. doi: 10.1016/j.cell.2015.11.001
266. Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. (2012) 143:913–6.e917. doi: 10.1053/j.gastro.2012.06.031
267. Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. (2016) 167:1897. doi: 10.1016/j.cell.2016.10.020
268. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. (2013) 500:541–6. doi: 10.1038/nature12506
269. Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, et al. Dietary intervention impact on gut microbial gene richness. Nature. (2013) 500:585–8. doi: 10.1038/nature12480
270. Trasande L, Blustein J, Liu M, Corwin E, Cox LM, Blaser MJ. Infant antibiotic exposures and early-life body mass. Int J Obes. (2013) 37:16–23. doi: 10.1038/ijo.2012.132
271. Karczewski J, Poniedzialek B, Adamski Z, Rzymski P. The effects of the microbiota on the host immune system. Autoimmunity. (2014) 47:494–504. doi: 10.3109/08916934.2014.938322
272. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. (2012) 489:231–41. doi: 10.1038/nature11551
273. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS ONE. (2010) 5:e8578. doi: 10.1371/journal.pone.0008578
274. O'Dwyer DN, Dickson RP, Moore BB. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J Immunol. (2016) 196:4839–47. doi: 10.4049/jimmunol.1600279
275. Siwicka-Gieroba D, Czarko-Wicha K. Lung microbiome - a modern knowledge. Cent Eur J Immunol. (2020) 45:342–5. doi: 10.5114/ceji.2020.101266
276. Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet. (2014) 384:691–702. doi: 10.1016/S0140-6736(14)61136-3
277. Sencio V, Machado MG, Trottein F. The lung-gut axis during viral respiratory infections: the impact of gut dysbiosis on secondary disease outcomes. Mucosal Immunol. (2021) 14:296–304. doi: 10.1038/s41385-020-00361-8
278. Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. (2013) 7:245–57. doi: 10.1586/ers.13.24
279. Vaughan A, Frazer ZA, Hansbro PM, Yang IA. COPD and the gut-lung axis: the therapeutic potential of fibre. J Thorac Dis. (2019) 11:S2173–80. doi: 10.21037/jtd.2019.10.40
280. Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med. (2015) 7:307ra152. doi: 10.1126/scitranslmed.aab2271
281. Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. (2009) 138:30–50. doi: 10.1016/j.cell.2009.06.036
282. Duerkop BA, Hooper LV. Resident viruses and their interactions with the immune system. Nat Immunol. (2013) 14:654–9. doi: 10.1038/ni.2614
283. Hewitt R, Farne H, Ritchie A, Luke E, Johnston SL, Mallia P. The role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthma. Ther Adv Respir Dis. (2016) 10:158–74. doi: 10.1177/1753465815618113
284. Billard L, Le Berre R, Pilorge L, Payan C, Hery-Arnaud G, Vallet S. Viruses in cystic fibrosis patients' airways. Crit Rev Microbiol. (2017) 43:690–708. doi: 10.1080/1040841X.2017.1297763
285. van Boheemen S, van Rijn AL, Pappas N, Carbo EC, Vorderman RHP, Sidorov I, et al. retrospective validation of a metagenomic sequencing protocol for combined detection of RNA and DNA viruses using respiratory samples from pediatric patients. J Mol Diagn. (2020) 22:196–207. doi: 10.1016/j.jmoldx.2019.10.007
286. Prachayangprecha S, Schapendonk CM, Koopmans MP, Osterhaus AD, Schurch AC, Pas SD, et al. Exploring the potential of next-generation sequencing in detection of respiratory viruses. J Clin Microbiol. (2014) 52:3722–30. doi: 10.1128/JCM.01641-14
287. Yadav H, Jain S, Nagpal R, Marotta F. Increased fecal viral content associated with obesity in mice. World J Diabetes. (2016) 7:316–20. doi: 10.4239/wjd.v7.i15.316
288. Tan VPK, Ngim CF, Lee EZ, Ramadas A, Pong LY, Ng JI, et al. The association between obesity and dengue virus (DENV) infection in hospitalised patients. PLoS ONE. (2018) 13:e0200698. doi: 10.1371/journal.pone.0200698
289. Vangipuram SD, Yu M, Tian J, Stanhope KL, Pasarica M, Havel PJ, et al. Adipogenic human adenovirus-36 reduces leptin expression and secretion and increases glucose uptake by fat cells. Int J Obes. (2007) 31:87–96. doi: 10.1038/sj.ijo.0803366
290. Mukhopadhya I, Segal JP, Carding SR, Hart AL, Hold GL. The gut virome: the 'missing link' between gut bacteria and host immunity? Therap Adv Gastroenterol. (2019) 12:1756284819836620. doi: 10.1177/1756284819836620
291. Tian Y, Jennings J, Gong Y, Sang Y. Viral infections and interferons in the development of obesity. Biomolecules. (2019) 9:726. doi: 10.3390/biom9110726
292. Russell CD, Unger SA, Walton M, Schwarze J. The human immune response to respiratory syncytial virus infection. Clin Microbiol Rev. (2017) 30:481–502. doi: 10.1128/CMR.00090-16
293. Jaddoe VWV, Felix JF, Andersen AN, Charles MA, Chatzi L, Corpeleijn E, et al. The LifeCycle project-EU child cohort network: a federated analysis infrastructure and harmonized data of more than 250,000 children and parents. Eur J Epidemiol. (2020) 35:709–24. doi: 10.1007/s10654-020-00662-z
294. Piersigilli F, Lam TT, Vernocchi P, Quagliariello A, Putignani L, Aghai ZH, et al. Identification of new biomarkers of bronchopulmonary dysplasia using metabolomics. Metabolomics. (2019) 15:20. doi: 10.1007/s11306-019-1482-9
295. Sahni M, Yeboah B, Das P, Shah D, Ponnalagu D, Singh H, et al. Novel biomarkers of bronchopulmonary dysplasia and bronchopulmonary dysplasia-associated pulmonary hypertension. J Perinatol. (2020) 40:1634–43. doi: 10.1038/s41372-020-00788-8
296. Askenazi DJ, Koralkar R, Patil N, Halloran B, Ambalavanan N, Griffin R. Acute kidney injury urine biomarkers in very low-birth-weight infants. Clin J Am Soc Nephrol. (2016) 11:1527–35. doi: 10.2215/CJN.13381215
297. Baumert M, Surmiak P, Wiecek A, Walencka Z. Serum NGAL and copeptin levels as predictors of acute kidney injury in asphyxiated neonates. Clin Exp Nephrol. (2017) 21:658–64. doi: 10.1007/s10157-016-1320-6
298. Jung YH, Han D, Shin SH, Kim EK, Kim HS. Proteomic identification of early urinary-biomarkers of acute kidney injury in preterm infants. Sci Rep. (2020) 10:4057. doi: 10.1038/s41598-020-60890-x
299. Watanabe IKM, Jara ZP, Volpini RA, Franco MDC, Jung FF, Casarini DE. Up-regulation of renal renin-angiotensin system and inflammatory mechanisms in the prenatal programming by low-protein diet: beneficial effect of the post-weaning losartan treatment. J Dev Origins Health Dis. (2018) 9:530–5. doi: 10.1017/S2040174418000296
300. Flynn ER, Alexander BT, Lee J, Hutchens ZM, Jr Maric-Bilkan C. High-fat/fructose feeding during prenatal and postnatal development in female rats increases susceptibility to renal and metabolic injury later in life. Am J Physiol Regul Integr Comp Physiol. (2013) 304:R278–85. doi: 10.1152/ajpregu.00433.2012
301. Wixey JA, Chand KK, Colditz PB, Bjorkman ST. Review: neuroinflammation in intrauterine growth restriction. Placenta. (2017) 54:117–24. doi: 10.1016/j.placenta.2016.11.012
302. Bangma JT, Hartwell H, Santos HP, O'Shea TM, Fry RC. Placental programming, perinatal inflammation, and neurodevelopment impairment among those born extremely preterm. Pediatr Res. (2021) 89:326–35. doi: 10.1038/s41390-020-01236-1
303. Leviton A, Fichorova RN, O'Shea TM, Kuban K, Paneth N, Dammann O, et al. Two-hit model of brain damage in the very preterm newborn: small for gestational age and postnatal systemic inflammation. Pediatr Res. (2013) 73:362–70. doi: 10.1038/pr.2012.188
304. Leviton A, Allred EN, Fichorova RN, Kuban KC, Michael O'Shea T, Dammann O. Systemic inflammation on postnatal days 21 and 28 and indicators of brain dysfunction 2years later among children born before the 28th week of gestation. Early Hum Dev. (2016) 93:25–32. doi: 10.1016/j.earlhumdev.2015.11.004
305. Kuban KC, Joseph RM, O'Shea TM, Heeren T, Fichorova RN, Douglass L, et al. Circulating inflammatory-associated proteins in the first month of life and cognitive impairment at age 10 years in children born extremely preterm. J Pediatr. (2017) 180:116–23.e111. doi: 10.1016/j.jpeds.2016.09.054
306. Guo W, Guan X, Pan X, Sun X, Wang F, Ji Y, et al. Post-natal inhibition of NF-kappaB activation prevents renal damage caused by prenatal LPS exposure. PLoS ONE. (2016) 11:e0153434. doi: 10.1371/journal.pone.0153434
307. Nüsken E, Fink G, Lechner F, Voggel J, Wohlfarth M, Sprenger L, et al. Altered molecular signatures during kidney development after intrauterine growth restriction of different origins. J Mol Med. (2020) 98:395–407. doi: 10.1007/s00109-020-01875-1
Keywords: lung development and pulmonary diseases, perinatal nutrition, maternal obesity, intrauterine growth restriction, chronic lung disease, bronchopulmonary dysplasia (BPD)
Citation: Kuiper-Makris C, Selle J, Nüsken E, Dötsch J and Alejandre Alcazar MA (2021) Perinatal Nutritional and Metabolic Pathways: Early Origins of Chronic Lung Diseases. Front. Med. 8:667315. doi: 10.3389/fmed.2021.667315
Received: 12 February 2021; Accepted: 12 May 2021;
Published: 15 June 2021.
Edited by:
Hsiao-Chi Chuang, Taipei Medical University, TaiwanReviewed by:
Robbert J. Rottier, Erasmus Medical Center, NetherlandsDaan Ophelders, Maastricht University, Netherlands
Copyright © 2021 Kuiper-Makris, Selle, Nüsken, Dötsch and Alejandre Alcazar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel A. Alejandre Alcazar, bWlndWVsLmFsZWphbmRyZS1hbGNhemFyQHVrLWtvZWxuLmRl
†These authors share first authorship