 Laura Mercurio
Laura Mercurio Cristina Albanesi
Cristina Albanesi Stefania Madonna
Stefania Madonna
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 30 April 2021
Sec. Dermatology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.665647
This article is part of the Research Topic New Tools and Molecular Advances in Hyperproliferative Skin Disorders View all 6 articles
PhosphoInositide-3 Kinase (PI3K) represents a family of different classes of kinases which control multiple biological processes in mammalian cells, such as cell growth, proliferation, and survival. Class IA PI3Ks, the main regulators of proliferative signals, consists of a catalytic subunit (α, β, δ) that binds p85 regulatory subunit and mediates activation of AKT and mammalian Target Of Rapamycin (mTOR) pathways and regulation of downstream effectors. Dysregulation of PI3K/AKT/mTOR pathway in skin contributes to several pathological conditions characterized by uncontrolled proliferation, including skin cancers, psoriasis, and atopic dermatitis (AD). Among cutaneous cancers, basal cell carcinoma (BCC) and cutaneous squamous cell carcinoma (cSCC) display PI3K/AKT/mTOR signaling hyperactivation, implicated in hyperproliferation, and tumorigenesis, as well as in resistance to apoptosis. Upregulation of mTOR signaling proteins has also been reported in psoriasis, in association with enhanced proliferation, defective keratinocyte differentiation, senescence-like growth arrest, and resistance to apoptosis, accounting for major parts of the overall disease phenotypes. On the contrary, PI3K/AKT/mTOR role in AD is less characterized, even though recent evidence demonstrates the relevant function for mTOR pathway in the regulation of epidermal barrier formation and stratification. In this review, we provide the most recent updates on the role and function of PI3K/AKT/mTOR molecular axis in the pathogenesis of different hyperproliferative skin disorders, and highlights on the current status of preclinical and clinical studies on PI3K-targeted therapies.
Phosphatidylinositol 3-kinase (PI3K) represents a family of kinases which play vital roles in mammalian cells by regulating proliferation, growth, and survival initiated by many growth and survival factors (1, 2). Dysregulation of PI3K-dependent signaling and, in particular, of PI3K/AKT/mammalian target of rapamycin (mTOR) pathway has been observed in different pathological conditions characterized by uncontrolled proliferation, loss of cell growth control, and decreased apoptosis. Aberrant PI3K/AKT/mTOR signaling is also observed in pathological skin, in particular in cutaneous cancer, as well as in chronic inflammatory diseases, such as psoriasis and atopic dermatitis (AD).
Among skin tumors, non-melanoma skin cancers (NMSC) refer to keratinocyte carcinomas and are classified into two major groups, namely basal cell carcinoma (BCC) and cutaneous squamous cell carcinoma (cSCC). BCCs are the most common human skin cancers, comprising about 75–80% of all skin tumors, and originate mainly from basal cell layer of epidermis (3, 4). cSCCs, less frequent, arise from squamous cells of epidermis and hair follicle stem cells (5), but they can also originate from dysplastic epidermal areas known as actinic keratoses (AK) (6). cSCCs are more dangerous and aggressive than BCCs, being able to invade and metastasize the dermis and local lymph nodes (6). In these pathological contexts, PI3K/AKT/mTOR signaling is hyperactivated and implicated in hyperproliferation and tumorigenesis, as well as in resistance to apoptosis (7, 8). PI3K signaling is mostly activated in the epidermal compartments, specifically in keratinocytes, following their exposure to environmental agents determining DNA alterations, such as ultraviolet (UV) radiation, and/or to cytokines aberrantly produced by activated immune cells (9).
The immune-mediated skin diseases psoriasis and AD can be both considered as hyperproliferative disorders in which epidermal keratinocytes respond to T lymphocyte-derived cytokines by altering growth, proliferation, and differentiation responses, accounting for major parts of the overall disease phenotypes (10–12).
Psoriasis is a chronic inflammatory skin disorder, in which skin-infiltrating T-helper (Th1, Th17, and Th22) lymphocytes promote keratinocyte hyperproliferation and terminal differentiation by releasing the pro-inflammatory cytokines IL-17A, IL-22, TNF-α, and IFN-γ (12–15). IL-36 cytokines released by keratinocytes themselves also determine impaired keratinocyte maturation and cornification in psoriasis (16–19). In addition, these cytokines upregulate PI3K/AKT/mTOR pathway, which in turn controls secretion of pro-inflammatory mediators by keratinocytes (20), enhances proliferation and impairs keratinocyte differentiation in skin affected by psoriasis (21).
The role of PI3K/AKT/mTOR in AD is less characterized than in psoriasis. AD is an immune-mediated skin disease characterized by alterations of skin barrier primarily due to loss-of-function filaggrin (FLG) mutations (22). During the acute phase of AD, inflammatory infiltrate is mainly represented by Th2 lymphocytes releasing type-2 cytokines, such as IL-4 and IL-13 (23, 24), which impair keratinocyte terminal differentiation and proper epidermal stratification (25, 26). In chronic AD, lichenified lesions appear, typically exhibiting altered epidermal hyperplasia, parakeratosis, and hyperkeratosis with amplification of Th2 axis and concomitant presence of Th1 cells releasing IFN-γ and TNF-α (27, 28). Recent evidence demonstrates the relevant role for mTOR pathway in the regulation of epidermal barrier function in AD.
In this review, we provide an update on the latest research efforts on the roles and mechanisms of PI3K/AKT/mTOR molecular axis in regulating hyperproliferative processes in the epidermal compartment of diseased skin. We also highlight on the current status of preclinical and clinical studies for the development of PI3K-targeted therapies in NMSC and psoriasis.
The phosphatidylinositol 3-kinases (PI3Ks) are members of a unique and conserved family of intracellular lipid kinases that phosphorylate the 3′-hydroxyl group of phosphatidylinositol and phosphoinositides (29). This reaction leads to the activation of many intracellular signaling pathways that regulate cell metabolism, survival, and vesicle trafficking.
Among PI3Ks enzymes, class I PI3Ks are the most widely characterized. These kinases show similar structure and share a common specificity for phosphatidyl inositol phosphates (PIPs) as substrates (2, 30–32).
Class I PI3Ks are divided into two subfamilies, named IA and IB, depending on their receptors. Indeed, class I PI3Ks are cytosolic enzymes in resting cells, and in response to different stimuli they are recruited to membranes by interacting with specific receptors or adaptor proteins (33, 34).
Class I PI3Ks are heterodimers that comprise a catalytic p110 subunit and a regulatory/adaptor subunit (35, 36). Class IA consists of one of the three catalytic isoforms p110 α, β, and δ and p85 regulatory subunit (p85α, β or their splice variants p55α, p50α, or p55γ), whereas class IB PI3K consists of p110γ catalytic isoform and p101 regulatory subunit (35). p110α and p110β are ubiquitously expressed and display distinct roles in cellular signaling, cell growth, angiogenesis, and oncogenic transformation (37–39). In contrast, PI3K p110δ is mainly expressed by hematopoietic cells and is critical for full B- and T-cell antigen receptor signaling (2, 40). PI3Kδ expression has also been reported in non-leucocyte cell types, such as breast cancer cells (41), neurons (42), lung and synovial fibroblasts, and endothelial cells (43, 44). We have recently observed PI3Kδ expression also in human keratinocytes and in the epidermis of a mouse skin inflammation model.
Mechanistically, the p85 regulatory subunit is crucial in mediating the activation of class IA PI3K by RTKs, through its direct binding to receptors on cell membranes. Upon receptor stimulation, p85 subunit recruits p110 to the intracellular phosphorylated tyrosine residues of RTKs, leading to p110 activation (45, 46). Activated PI3K p110 phosphorylates PIP2 to generate PIP3 that regulates multiple downstream pathways and cellular processes, such as membrane trafficking, cell growth, proliferation, metabolism, and migration (32, 47, 48). Once generated, PIP3 binds to several proteins, including AKT, also known as protein kinase B (PKB) (33, 45, 46).
AKT is a serine/threonine kinase that consists of three isoforms involved in numerous cellular processes, such as cell cycle progression, protein synthesis, glucose metabolism, cell proliferation, and survival (49–53). For a full activation of AKT, Thr308 and Ser473 residues located in two different domains need to be phosphorylated (46). Following PIP3 binding to AKT, this last is recruited to plasma membrane, where it is phosphorylated in Thr308 by phosphoinositide-dependent kinase-1 (PDK1) (49). One of the key elements of PI3K/AKT network is the serine/threonine kinase mammalian target of rapamycin (mTOR). mTOR can form two distinct multi-protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (54). Following phosphorylation in Ser473 by mTORC2, AKT activates mTORC1 which in turn induces the phosphorylation of ribosomal protein S6 kinase beta-1 (S6K1), and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), a repressor of mRNA translation (55). As consequence, S6K1 phosphorylates S6 Ribosomal Protein (S6 Rb), whereas the inactivated 4E-BP1 repressor releases the eukaryotic translation initiation factor 4E (eIF4E) (55–57). Both phospho-S6Rb and eIF4E promote protein translation and cell proliferation (56). Of note, S6K1 can be phosphorylated also by PDK-1 without AKT involvement (58).
AKT not only regulates cell proliferation and protein synthesis, but also inhibits pro-apoptotic proteins, including BAD and caspase-9, two crucial pro-apoptotic components of cell death machinery, and forkhead box O (FOXO) transcription factor, a negative regulators of proliferation and cell survival (59–61). Moreover, AKT indirectly induces the transcription of anti-apoptotic genes via nuclear factor-κB (NF-κB) factors (62, 63).
Finally, PI3K-activated pathways play a key role in epidermal homeostasis by sustaining the proper epidermal formation, as well as keratinocyte differentiation and survival (64–67).
Alterations in PI3K/AKT/mTOR signaling has been implicated in the pathogenesis and progression of numerous cutaneous cancers, including NMSC (68–70). In particular, hyperactivation of the PI3K/AKT axis has been detected in both SCC and BCC skin tissues, where suggesting its potential involvement in the pathogenesis and malignancy of these tumors (7, 71, 72) (Figure 1A; Table 1). Chen et al. found that the percentage of phosphorylated AKT (Ser473) positive cells is significantly higher in SCC than AK, and further enhanced in SCCs with metastases. AKT hyperactivation correlated with an increased phosphorylation of mTOR and downstream effectors, such as 4E-BP1, 70S6K1, p70S6K1, and S6 (Ser6) (76). Consistently, increased AKT activity is associated to nuclear accumulation of molecules involved in cell cycle progression, such as Cyclin D1, phosphorylated-c-myc, and β-catenin in cutaneous head and neck SCC (91).
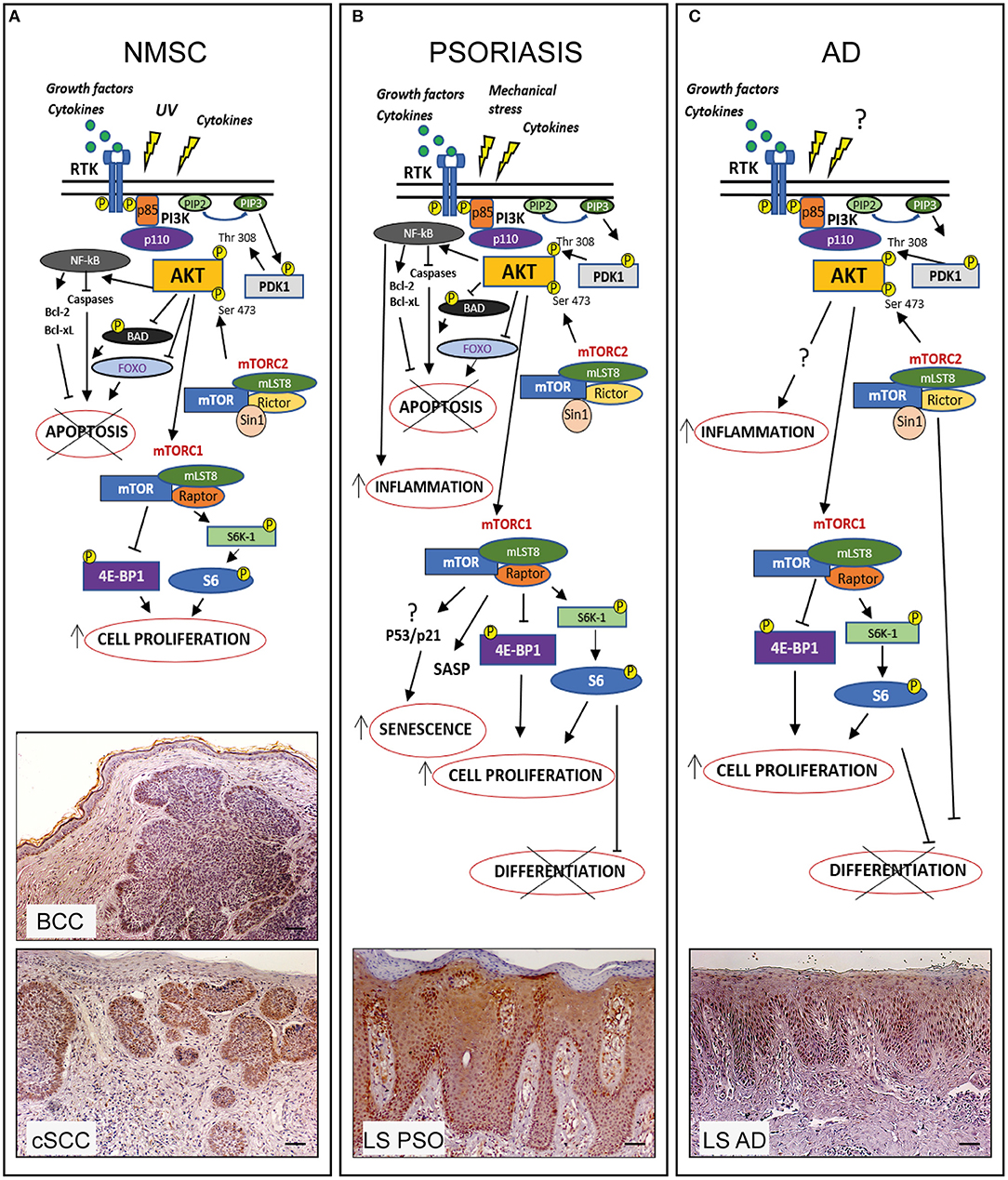
Figure 1. PI3K/AKT/mTOR pathway in hyperproliferative skin diseases. In healthy skin PI3K/AKT/mTOR pathway can be activated upon stimulation of receptor tyrosine kinases (RTK) leading to PI3K activation which in turn phosphorylates PIP2 to PIP3. Subsequently AKT is recruited to membrane and phosphorylated by PDK-1 and mTORC2. Phospho-AKT can induce mTORC1 activation by phosphorylating S6K-1 or 4E-BP1, thus controlling several cellular processes and maintaining the epidermal homeostasis. In hyperproliferative skin diseases as NMSC, psoriasis, and AD, several external stimuli are responsible for PI3K/AKT/mTOR over-expression and local increase of cytokines and growth factors lead to overexpression/upregulation of inflammatory molecular cascades contributing to progression of these skin disorders. In the three hyperproliferative skin conditions, PI3K/AKT/mTOR signaling is hyperactivated and involved in pathogenic processes (hyperproliferation, epidermal differentiation, inflammation, apoptosis, and senescence) depending on the disease context. The players of PI3K/AKT/mTOR pathway, as well as upstream/downstream mediators differentially activated in NMSC, psoriasis, and AD are schematically shown. Immunohistochemical analyses of p-AKT expression (red-brown color) of skin lesional areas of cSCC and BCC (A), psoriatic plaques (LS PSO) (B), and AD (LS AD) (C) show a wide expression of p-AKT in the epidermal layers of psoriasis and AD, as well as in cSCC and BCC, mostly expressed in tumor formations in both NMSCs types. Scale bars, 200 μm [Phospho-AKT stainings have been retrieved from Ref. (53, 73)].
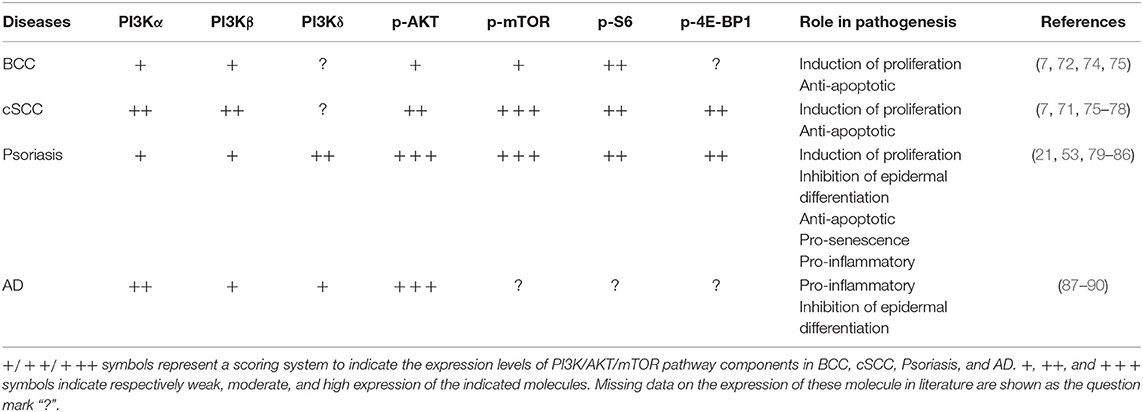
Table 1. Expression and role of PI3K class IA p110 isoforms and downstream molecules in hyperproliferative skin diseases.
Numerous reports have shown that PI3K/AKT/mTOR/S6K1 pathway can be activated in skin cancers by UV radiation exposure (54, 69, 70). In human and mouse epidermal keratinocytes, UV radiations induce the expression of p85 regulatory subunit and activates mTOR, with the consequent S6K1 phosphorylation (9, 92). UV radiation exposure can also determine the insurgence of mutations in PTEN gene, the major negative regulator of PI3Ks. PTEN commonly acts as a PI3K antagonist by dephosphorylating PIP3 to PIP2, and thus it inhibits AKT activation (93, 94). Recent evidence shows that chronic UVA radiation decreases PTEN expression, and this decrease is required for enhanced cell survival in transformed human keratinocytes, suggesting that PTEN might be critical for UVA-induced skin carcinogenesis (95). UVB was also reported to inhibit PTEN by increasing its stability and phosphorylation in human dermal fibroblasts (96). Thus, UV-mediated inhibition of PTEN further enhance AKT activation (92, 95, 96). Although a high frequency of PTEN mutations with consequent hyper-activation of AKT has been detected in malignant melanomas (97), the reduction of PTEN levels and the mechanism(s) by which its function and activity are regulated in NMSC remain to be established.
The mechanisms by which PI3K/AKT/mTOR pathway sustains NMSC development and progression involve both enhanced cell proliferation and resistance to apoptosis. In a recent study conducted on a transgenic mouse model developing multiple BCC, AKT1 isoform has been identified as obligatory for BCC tumorigenesis. Indeed, the pharmacological inhibition of AKT, as well as the genetic ablation of AKT1, diminished the growth of spontaneous and UV-induced tumors in this BCC murine model (74).
Previously, Zhao and colleagues investigated on the cutaneous expression of a series of pro-proliferative proteins, including AKT mediators, in transgenic mice expressing the tyrosine kinase Fyn, a model spontaneously forming keratotic lesions, scaly plaques, and large tumors, resembling AKs, carcinoma in situ (SCIS), and SCCs, respectively (98). They found increased levels of phosphorylated PDK1, together with STAT3 and ERK1/2, in both precancerous and SCCs lesions, compared with non-lesional epidermis. Of note, topical application of BEZ-235, a PI3K/mTOR inhibitor, induces regression of SCC in this disease model (77).
NMSC-infiltrating immune cells also indirectly contribute to tumor growth mediated by AKT pathway. Indeed, the release of pro-inflammatory cytokines, such as IL-22 by tumor-infiltrating lymphocytes (TILs) can promote in vitro keratinocyte hyperproliferation by sustaining AKT signaling the expression of cell cycle-related and anti-apoptotic molecules (99).
Hyperproliferation of epidermal keratinocytes contributes to the pathogenesis of several cutaneous disorders, including psoriasis and AD. Up-regulation of PI3K/AKT/mTOR pathway has been reported in skin of patients affected by psoriasis (Table 1), as well as in skin of imiquimod (IMQ)-induced psoriasiform mouse model (53, 79). In fact, Pike and colleagues measured a higher PI3K activity in epidermis of psoriatic patients than in healthy donors (80). Consistently, our group reported a strong expression of phosphorylated AKT in lesional psoriatic skin and in cytokine-activated keratinocytes derived from patients affected by psoriasis (53). Expression of the PI3K effectors phospho-AKT, -S6K1, -S6 Rb, and 4E-BP1, is more pronounced in suprabasal keratinocytes, whereas mTOR is hyperactivated in all epidermal layers of lesional psoriatic skin (53, 81). The wide expression of mTOR in psoriatic epidermis may be associated not only to a keratinocyte hyperproliferation (81, 82, 100), but also to aberrant differentiation, since AKT/mTOR pathway inactivation is requested during keratinocytes terminal differentiation (21, 101) (Figure 1B). In contrast, reduced levels of the FOXO1 and PTEN regulators have been observed in psoriatic epidermis (102–105).
AKT can also prevent cytokine-induced cellular apoptosis and promote senescence-like growth arrest in psoriasis (53) (Figure 1B). Indeed, psoriatic keratinocytes exhibit a senescent phenotype characterized by a peculiar resistance to apoptosis, secretion of inflammatory molecules, and expression of specific markers of senescence, which contributes to the epidermal thickening typically observed in psoriatic skin (106–108). Interestingly, the chemical inhibition of PI3K/AKT cascade by Ly294002 molecule renders psoriatic keratinocytes more susceptible to pro-apoptotic stimuli, such as pro-inflammatory Th1/17-released cytokines (53). However, the mechanism(s) by which PI3K/AKT axis sustains senescence phenotype in psoriatic keratinocytes remains to be established.
In support of our observation, Miyauchi et al. have reported that AKT promotes a senescence-like phenotype also in endothelial cells via tumor suppressor TP53 (P53) and cyclin-dependent kinase inhibitor p21WAF1/Cip1 (p21)-dependent pathway (109). In particular, constitutive activation of Akt inhibits the transcriptional activity of FOXO3a and thereby downregulates manganese superoxide dismutase, leading to an increase of ROS that promotes senescence-like growth arrest by inducing p53 activity and p21 expression (106). Additionally, chronic hyperactivation of AKT in human non-transformed fibroblasts results in a TORC1-dependent increase in p53 translation, and simultaneously stimulates MDM2 sequestration within the nucleous, thus inhibiting p53 ubiquitination and degradation. This event results in an accumulation of p53, leading to cellular senescence (110, 111). Finally, in a recent genome-wide RNAi screening study, three novel intracellular mediators of senescence induced by AKT have been identified in human fibroblasts, including the pro-apoptotic CCAR1 and FADD proteins, and NF1, the negative regulator of RAS/ERK signaling (112). Based on these data, we hypothesize that the hyperactivation PI3K/AKT pathway in psoriatic lesions could be implicated in the regulation of the senescent-like phenotype of epidermal keratinocytes.
The role of PI3K/AKT/mTOR in AD is less characterized than in psoriasis, even though some evidences have accumulated so far (Figure 1C; Table 1). Topical application of the mTOR inhibitor, rapamycin, in experimental models of AD induced by different antigens in NC/Nga mice improves several clinical parameters, including epidermal thickness, dermal inflammatory infiltrate, serum IgE and Th2 and Th1 cytokine levels (87, 88).
Recent evidence has demonstrated the mTOR role in epidermal stratification and cornification. Indeed, Ding and colleagues, have shown that mTORC2 controls FLG processing and de novo epidermal lipid synthesis during cornification in mice lacking RICTOR in the epidermis (113). Furthermore, increased transcriptional levels of the regulatory-associated protein of mTORC1, RAPTOR, correlate with decreased FLG expression, barrier defects and presence of inflammatory markers in skin of patients with AD (114). Of interest, it has been recently reported that Th2-released IL-13 could activate the mTOR signaling pathway in human immortalized keratinocytes, and the pharmacological inhibition of mTORC1 by rapamycin blocks the IL-13-induced expression of p-mTOR, p-S6K1, and p-AKT. Concomitantly, in human keratinocytes rapamycin up-regulates the expression of terminal differentiation markers, including filaggrin, loricrin, and involucrin, typically impaired in AD skin lesions (89).
Finally, PI3K/AKT signaling is abnormally activated in peripheral T cells from pediatric AD patients. Of note, the PI3K inhibitor LY294002 significantly inhibits proliferation and release of the IL-10 and IL-6 cytokines in AD patient-derived T cells, thus suggesting a relevant role of PI3K pathways in AD inflammatory circuits (90). All these data suggest that PI3K/AKT blocking could be a potential effective therapeutic option in the management of AD.
In last years, targeting PI3K/AKT/mTOR axis proved to be a promising tool for treatment of NMSC, especially mTOR targeting by specific inhibitors. In BCC patients, treatment with the mTORC1 inhibitor Everolimus leads to a partial or complete tumor recession (70, 115–117). Furthermore, Itraconazole, previously discovered as an antifungal agent, has been found to have anticancer action by inhibiting mTOR signaling (118), and a recent clinical trial conducted in BCC patients have shown encouraging effectiveness (119). Other mTORC1 inhibitors, such as rapamycin and its analogs, showed a better clinical response in cSCC than in BCC, probably due to the higher mTOR expression in SCC epidermal tissue (70, 75, 78, 120).
However, the first generation mTOR inhibitors, selective for mTORC1 and with a poor action on mTORC2, led to a subsequent AKT activation. Thus, a second generation of mTOR inhibitors, targeting both mTORC1 and mTORC2, have been developed (121). Among these, GDC-0084 exhibited a potent anti-proliferative effect on cSCC in preclinical studies (122). LY3023414, a small PI3K-AKT dual inhibitor, showed a strong cytotoxic and anti-proliferative effect on SCC cell lines and in tumor xenografts models, and it is currently used in phase I and II clinical trials (123).
Of note, PI3K/AKT/mTOR signaling has been described to be involved in resistance to specific inhibitors classically employed in NMSCs, due to the intricate crosstalk between different pathways in these skin cancers (70, 117). Thus, the use of PI3K/AKT/mTOR inhibitors in combination with agents targeting other pathways is more effective in contrasting drug resistance (124).
In psoriasis, rapamycin (Sirolimus) has been employed via oral administration, alone and combination with cyclosporine. Despite enhanced ameliorative effects with the two drugs combined, rapamycin alone was ineffective (125). In contrast, a clinical trial with topical application of Sirolimus in psoriatic patients reported a decrease in clinical score, together with a significant reduction in CD4-positive T cells and proliferating Ki67+ cells in the epidermis. However, no effects on plaque thickness and erythema have been observed (126). These data suggest that mTOR inhibition does not exert significant improvement in psoriasis.
PI3K isoforms has been instead described as efficacious targets in treating psoriasis. PI3K inhibition results in reduction of epidermal thickness, number of infiltrating immune cells and levels of psoriasis-related cytokines in the IMQ-induced psoriasiform mouse model (83, 84). In addition, blocking of PI3Ks counteracts proliferation and activation processes in T cells derived from psoriatic patients (84). The selective PI3kδ inhibitor Seletalisib can reduce in vitro production of pro-inflammatory cytokines from IL-17-producing adaptive and innate-like lymphocytes (85, 86). Consistently, a recent first-in-human study of oral administration of the PI3Kδ inhibitor Seletalisib showed ameliorative effects on size and appearance of psoriatic lesions, together with a reduction in T cells and neutrophils, in skin from psoriasis patients undergone Seletalisib treatment (127). In line with these data, we observed that the topical administration of Seletalisib drastically reduced epidermal thickening and the number of infiltrating neutrophils in an IMQ-induced psoriasiform murine model. These findings support the clinical development of PI3K p110δ isoform inhibitors in psoriasis.
In AD, PI3K/AKT/mTOR inhibitors have not been yet tested. PI3K p110δ has been shown to be involved in type-2 inflammation associated to atopy/allergy (128), and, in particular, in the development of Th2 asthma, a common pathological symptom of many allergic diseases, including AD. In support of this, the selective inhibition of PI3K p110 δ and γ isoforms resulted in the attenuation of allergic airway inflammation in several preclinical models (129–133).
Therefore, blocking PI3K/AKT/mTOR could be an effective therapeutic strategy in AD treatment, being this pathway involved in the pathogenic mechanisms resulting in AD symptoms, as defective epidermal barrier, inflammation and allergic asthma. However, further investigations are needed to better understand the impact of PI3K/AKT/mTOR inhibition in AD clinical resolution.
PI3K/AKT pathway is implicated in NMSC development and progression, as well as in the pathogenic mechanisms associated to chronic inflammatory skin conditions, such as psoriasis and AD. However, our understanding of this complex network and its tight regulation is at its beginning and will need much more work to definitively assess the impact of its inhibition in the clinical outcomes of these hyperproliferative skin disorders. Targeting PI3K/AKT pathway in NMSCs, with synthetic small molecules alone or in various combinations, have been widely employed in clinical trials with effective clinical response, although several of these agents display limitations as undesired side effects. Thus, a careful selection and development of more potent and safer agents are needed. Moreover, a more in-depth characterization of the role of distinct PI3K isoforms in NMSCs are needed to determine whether targeting selective PI3Ks could represent a powerful strategy to counteract these diseases.
In the context of inflammatory skin condition, despite recent drug development has mainly centered on biological therapies for psoriasis and AD management, small molecule drugs targeting specific PI3K isoforms or combined drugs acting on multiple PI3K effectors, administrated orally or topically, could represent a valid alternative for treating psoriasis or AD patients undergone clinical failure with biologics or psoriasis patients affected by challenging-to-treat clinical subtypes.
All authors have contributed in the ideation and writing of the manuscript and checked the final version of the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. (2017) 170:605–35. doi: 10.1016/j.cell.2017.07.029
2. Foster FM, Traer CJ, Abraham SM, Fry MJ. The phosphoinositide (PI) 3-kinase family. J Cell Sci. (2003) 116:3037–40. doi: 10.1242/jcs.00609
3. Kim DP, Kus KJB, Ruiz E. Basal cell carcinoma review. Hematol Oncol Clin North Am. (2019) 33:13–24. doi: 10.1016/j.hoc.2018.09.004
4. Feehan RP, Shantz LM. Molecular signaling cascades involved in nonmelanoma skin carcinogenesis. Biochem J. (2016) 473:2973–94. doi: 10.1042/BCJ20160471
5. Jian Z, Strait A, Jimeno A, Wang XJ. Cancer stem cells in squamous cell carcinoma. J Invest Dermatol. (2017) 137:31–7. doi: 10.1016/j.jid.2016.07.033
6. Madan V, Lear JT, Szeimies RM. Non-melanoma skin cancer. Lancet. (2010) 375:673–85. doi: 10.1016/S0140-6736(09)61196-X
7. Hafner C, Landthaler M, Vogt T. Activation of the PI3K/AKT signalling pathway in non-melanoma skin cancer is not mediated by oncogenic PIK3CA and AKT1 hotspot mutations. Exp Dermatol. (2010) 19:222–7. doi: 10.1111/j.1600-0625.2009.01056.x
8. Janus JM, O'Shaughnessy RFL, Harwood CA, Maffucci T. Phosphoinositide 3-kinase-dependent signalling pathways in cutaneous squamous cell carcinomas. Cancers (Basel). (2017) 9:86. doi: 10.3390/cancers9070086
9. Syed DN, Afaq F, Mukhtar H. Differential activation of signaling pathways by UVA and UVB radiation in normal human epidermal keratinocytes. Photochem Photobiol. (2012) 88:1184–90. doi: 10.1111/j.1751-1097.2012.01115.x
10. Guttman-Yassky E, Krueger JG. Atopic dermatitis and psoriasis: two different immune diseases or one spectrum? Curr Opin Immunol. (2017) 48:68–73. doi: 10.1016/j.coi.2017.08.008
11. Cibrian D, de la Fuente H, Sánchez-Madrid F. Metabolic pathways that control skin homeostasis and inflammation. Trends Mol Med. (2020) 26:975–86. doi: 10.1016/j.molmed.2020.04.004
12. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. (2009) 361:496–509. doi: 10.1056/NEJMra0804595
13. Albanesi C, Madonna S, Gisondi P, Girolomoni G. The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front Immunol. (2018) 9:1549. doi: 10.3389/fimmu.2018.01549
14. Chiricozzi A, Romanelli P, Volpe E, Borsellino G, Romanelli M. Scanning the immunopathogenesis of psoriasis. Int J Mol Sci. (2018) 19:179. doi: 10.3390/ijms19010179
15. Johnson-Huang LM, Lowes MA, Krueger JG. Putting together the psoriasis puzzle: an update on developing targeted therapies. DMM Dis Model Mech. (2012) 5:423–33. doi: 10.1242/dmm.009092
16. Madonna S, Girolomoni G, Dinarello CA, Albanesi C. The significance of il-36 hyperactivation and il-36r targeting in psoriasis. Int J Mol Sci. (2019) 20:3318. doi: 10.3390/ijms20133318
17. Mercurio L, Failla CM, Capriotti L, Scarponi C, Facchiano F, Morelli M, et al. Interleukin (IL)-17/IL-36 axis participates to the crosstalk between endothelial cells and keratinocytes during inflammatory skin responses. PLoS ONE. (2020) 15:e0222969. doi: 10.1371/journal.pone.0222969
18. Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G, et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. (2018) 9:1104. doi: 10.1038/s41419-018-1143-3
19. de Graaf DM, Jaeger M, van den Munckhof ICL, ter Horst R, Schraa K, Zwaag J, et al. Reduced concentrations of the B cell cytokine interleukin 38 are associated with cardiovascular disease risk in overweight subjects. Eur J Immunol. (2021) 51:662–71. doi: 10.1002/eji.201948390
20. Patel AB, Tsilioni I, Weng Z, Theoharides TC. TNF stimulates IL-6, CXCL8 and VEGF secretion from human keratinocytes via activation of mTOR, inhibited by tetramethoxyluteolin. Exp Dermatol. (2018) 27:135–43. doi: 10.1111/exd.13461
21. Buerger C, Shirsath N, Lang V, Berard A, Diehl S, Kaufmann R, et al. Inflammation dependent mTORC1 signaling interferes with the switch from keratinocyte proliferation to differentiation. PLoS ONE. (2017) 12:e0180853. doi: 10.1371/journal.pone.0180853
22. Jungersted JM, Scheer H, Mempel M, Baurecht H, Cifuentes L, Høgh JK, et al. Stratum corneum lipids, skin barrier function and filaggrin mutations in patients with atopic eczema. Allergy Eur J Allergy Clin Immunol. (2010) 65:911–8. doi: 10.1111/j.1398-9995.2010.02326.x
23. Wollenberg A, Klein E. Current aspects of innate and adaptive immunity in atopic dermatitis. Clin Rev Allergy Immunol. (2007) 33:35–44. doi: 10.1007/s12016-007-0032-9
24. Eyerich K, Pennino D, Scarponi C, Foerster S, Nasorri F, Behrendt H, et al. IL-17 in atopic eczema: linking allergen-specific adaptive and microbial-triggered innate immune response. J Allergy Clin Immunol. (2009) 123:59–66. doi: 10.1016/j.jaci.2008.10.031
25. Howell MD, Fairchild HR, Kim BE, Bin L, Boguniewicz M, Redzic JS, et al. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J Invest Dermatol. (2008) 128:2248–58. doi: 10.1038/jid.2008.74
26. Kim BE, Leung DYM, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. (2008) 126:332–7. doi: 10.1016/j.clim.2007.11.006
27. Renert-Yuval Y, Guttman-Yassky E. What's new in atopic dermatitis. Dermatol Clin. (2019) 37:205–13. doi: 10.1016/j.det.2018.12.007
28. Williamson S, Merritt J, De Benedetto A. Atopic dermatitis in the elderly: a review of clinical and pathophysiological hallmarks. Br J Dermatol. (2020) 182:47–54. doi: 10.1111/bjd.18652
29. Cantley LC. The phosphoinositide 3-kinase pathway. Science. (2002) 296:1655–7. doi: 10.1126/science.296.5573.1655
30. Rameh LE, Cantley LC. The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem. (1999) 274:8347–50. doi: 10.1074/jbc.274.13.8347
31. Fry MJ. Phosphoinositide 3-kinase signalling in breast cancer: how big a role might it play? Breast Cancer Res. (2001) 3:304–12. doi: 10.1186/bcr312
32. Katso R. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. (2001) 17:615–75. doi: 10.1146/annurev.cellbio.17.1.615
33. Hassan B, Akcakanat A, Holder AM, Meric-Bernstam F. Targeting the PI3-Kinase/Akt/mTOR signaling pathway. Surg Oncol Clin N Am. (2013) 22:641–64. doi: 10.1016/j.soc.2013.06.008
34. Chen PH, Yao H, Huang LJS. Cytokine receptor endocytosis: new kinase activity-dependent and -independent roles of PI3K. Front Endocrinol (Lausanne). (2017) 8:78. doi: 10.3389/fendo.2017.00078
35. Kriplani N, Hermida MA, Brown ER, Leslie NR. Class I PI 3-kinases: function and evolution. Adv Biol Regul. (2015) 59:53–64. doi: 10.1016/j.jbior.2015.05.002
36. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. (2010) 11:329–41. doi: 10.1038/nrm2882
37. Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, et al. Angiogenesis selectively requires the p110α isoform of PI3K to control endothelial cell migration. Nature. (2008) 453:662–6. doi: 10.1038/nature06892
38. Bénistant C, Chapuis H, Roche S. A specific function for phosphatidylinositol 3-kinase α (p85α-p110α) in cell survival and for phosphatidylinositol 3-kinase β (p85α-p110β) in de novo DNA synthesis of human colon carcinoma cells. Oncogene. (2000) 19:5083–90. doi: 10.1038/sj.onc.1203871
39. Utermark T, Rao T, Cheng H, Wang Q, Lee SH, Wang ZC, et al. The p110α and p110β isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev. (2012) 26:1573–86. doi: 10.1101/gad.191973.112
40. Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. (2002) 297:1031–4. doi: 10.1126/science.1073560
41. Sawyer C, Sturge J, Bennett DC, O'Hare MJ, Allen WE, Bain J, et al. Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110δ1. Cancer Res. (2003) 63:1667–75.
42. Eickholt BJ, Ahmed AI, Davies M, Papakonstanti EA, Pearce W, Starkey ML, et al. Control of axonal growth and regeneration of sensory neurons by the p110δ PI 3-kinase. PLoS ONE. (2007) 2:1–9. doi: 10.1371/journal.pone.0000869
43. Conte E, Fruciano M, Fagone E, Gili E, Caraci F, Iemmolo M, et al. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: the role of class I P110 isoforms. PLoS ONE. (2011) 6:e24663. doi: 10.1371/journal.pone.0024663
44. Whitehead MA, Bombardieri M, Pitzalis C, Vanhaesebroeck B. Isoform-selective induction of human p110δ PI3K expression by TNFα: identification of a new and inducible PIK3CD promoter. Biochem J. (2012) 443:857–67. doi: 10.1042/BJ20112214
45. Vanhaesebroeck B, Alessi DR. The PI3K-PBK1 connection: more than just a road to PKB. Biochem J. (2000) 346:561–76. doi: 10.1042/bj3460561
46. Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. (1999) 253:239–54. doi: 10.1006/excr.1999.4701
47. Martin-Belmonte F, Mostov K. Regulation of cell polarity during epithelial morphogenesis. Curr Opin Cell Biol. (2008) 20:227–34. doi: 10.1016/j.ceb.2008.01.001
48. Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase-AKT pathway in humancancer. Nat Rev Cancer. (2002) 2:489–501. doi: 10.1038/nrc839
49. Nitulescu GM, Van De Venter M, Nitulescu G, Ungurianu A, Juzenas P, Peng Q, et al. The Akt pathway in oncology therapy and beyond (Review). Int J Oncol. (2018) 53:2319–31. doi: 10.3892/ijo.2018.4597
50. Bellacosa A, Kumar CC, Cristofano A Di, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. (2005) 94:29–86. doi: 10.1016/S0065-230X(05)94002-5
51. Fresno Vara JÁ, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. P13K/Akt signalling pathway and cancer. Cancer Treat Rev. (2004) 30:193–204. doi: 10.1016/j.ctrv.2003.07.007
52. Zhao GX, Pan H, Ouyang DY, He XH. The critical molecular interconnections in regulating apoptosis and autophagy. Ann Med. (2015) 47:305–15. doi: 10.3109/07853890.2015.1040831
53. Madonna S, Scarponi C, Pallotta S, Cavani A, Albanesi C. Anti-apoptotic effects of suppressor of cytokine signaling 3 and 1 in psoriasis. Cell Death Dis. (2012) 3:e334. doi: 10.1038/cddis.2012.69
54. Laplante M, Sabatini DM. MTOR signaling in growth control and disease. Cell. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
55. Dowling RJO, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, et al. mTORCI-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. (2010) 328:1172–6. doi: 10.1126/science.1187532
56. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. (2004) 18:1926–45. doi: 10.1101/gad.1212704
57. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. (2002) 4:648–57. doi: 10.1038/ncb839
58. Gagliardi PA, Puliafito A, Primo L. PDK1: at the crossroad of cancer signaling pathways. Semin Cancer Biol. (2018) 48:27–35. doi: 10.1016/j.semcancer.2017.04.014
59. Downward J. How bad phosphorylation is good for survival. Nat Cell Biol. (1999) 1:E33–5. doi: 10.1038/10026
60. Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. (1998) 282:1318–21. doi: 10.1126/science.282.5392.1318
61. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta - Mol Cell Res. (2011) 1813:1978–86. doi: 10.1016/j.bbamcr.2011.03.010
62. Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires tie Akt serine- threonine kinase. Nature. (1999) 401:82–5. doi: 10.1038/43466
63. Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-κB by the Akt/PKB kinase. Curr Biol. (1999) 9:601–4. doi: 10.1016/S0960-9822(99)80265-6
64. O'Shaughnessy RFL, Welti JC, Cooke JC, Avilion AA, Monks B, Birnbaum MJ, et al. AKT-dependent HspB1 (Hsp27) activity in epidermal differentiation. J Biol Chem. (2007) 282:17297–305. doi: 10.1074/jbc.M610386200
65. Sully K, Akinduro O, Philpott MP, Naeem AS, Harwood CA, Reeve VE, et al. The mTOR inhibitor rapamycin opposes carcinogenic changes to epidermal Akt1/PKBα isoform signaling. Oncogene. (2013) 32:3254–62. doi: 10.1038/onc.2012.338
66. Pankow S, Bamberger C, Klippel A, Werner S. Regulation of epidermal homeostasis and repair by phosphoinositide 3-kinase. J Cell Sci. (2006) 119:4033–46. doi: 10.1242/jcs.03175
67. Calautti E, Li J, Saoncella S, Brissette JL, Goetinck PF. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J Biol Chem. (2005) 280:32856–65. doi: 10.1074/jbc.M506119200
68. Pópulo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci. (2012) 13:1886–918. doi: 10.3390/ijms13021886
69. Leo MS, Sivamani RK. Phytochemical modulation of the Akt/mTOR pathway and its potential use in cutaneous disease. Arch Dermatol Res. (2014) 306:861–71. doi: 10.1007/s00403-014-1480-8
70. Chamcheu JC, Roy T, Uddin MB, Banang-Mbeumi S, Chamcheu RCN, Walker AL, et al. Role and therapeutic targeting of the PI3K/Akt/mTOR signaling pathway in skin cancer: a review of current status and future trends on natural and synthetic agents therapy. Cells. (2019) 8:803. doi: 10.3390/cells8080803
71. Einspahr JG, Calvert V, Alberts DS, Curiel-Lewandrowski C, Warneke J, Krouse R, et al. Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev Res. (2012) 5:403–13. doi: 10.1158/1940-6207.CAPR-11-0427
72. Brinkhuizen T, Weijzen CAH, Eben J, Thissen MR, Van Marion AM, Lohman BG, et al. Immunohistochemical analysis of the mechanistic target of rapamycin and hypoxia signalling pathways in basal cell carcinoma and trichoepithelioma. PLoS ONE. (2014) 9:e106427. doi: 10.1371/journal.pone.0106427
73. Madonna S, Scarponi C, Morelli M, Sestito R, Scognamiglio PL, Marasco D, et al. SOCS3 inhibits the pathological effects of IL-22 in nonmelanoma skin tumor-derived keratinocytes. Oncotarget. (2017) 8:24652–67. doi: 10.18632/oncotarget.15629
74. Kim AL, Back JH, Zhu Y, Tang X, Yardley NP, Kim KJ, et al. AKT1 activation is obligatory for spontaneous BCC tumor growth in a murine model that mimics some features of basal cell nevus syndrome. Cancer Prev Res. (2016) 9:794–802. doi: 10.1158/1940-6207.CAPR-16-0066
75. Karayannopoulou G, Euvrard S, Kanitakis J. Differential expression of p-mTOR in cutaneous basal and squamous cell carcinomas likely explains their different response to mTOR inhibitors in organ-transplant recipients. Anticancer Res. (2013) 33:3711–4.
76. Chen SJ, Nakahara T, Takahara M, Kido M, Dugu L, Uchi H, et al. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin-dependent kinase 2. Br J Dermatol. (2009) 160:442–5. doi: 10.1111/j.1365-2133.2008.08903.x
77. Yang X, Daifallah AEM, Shankar S, Beer J, Marshall C, Dentchev T, Seykora F, et al. Topical kinase inhibitors induce regression of cutaneous squamous cell carcinoma. Exp Dermatol. (2019) 28:609–13. doi: 10.1111/exd.13902
78. Wu N, Du Z, Zhu Y, Song Y, Pang L, Chen Z. The expression and prognostic impact of the PI3K/AKT/mTOR signaling pathway in advanced esophageal squamous cell carcinoma. Technol Cancer Res Treat. (2018) 17:1533033818758772. doi: 10.1177/1533033818758772
79. Chamcheu JC, Adhami VM, Siddiqui A, Wood GS, Longley BJ, Mukhtar H, et al. Upregulation of PI3K/AKT/mTOR, FABP5 and PPARβ/δ in human psoriasis and imiquimod-induced murine psoriasiform dermatitis model. Acta Derm Venereol. (2016) 96:854–6. doi: 10.2340/00015555-2359
80. Pike MC, Lee CS, Elder JT, Voorhees JJ, Fisher GJ. Increased phosphatidylinositol kinase activity in psoriatic epidermis. J Invest Dermatol. (1989) 92:791–7. doi: 10.1111/1523-1747.ep12696808
81. Buerger C, Malisiewicz B, Eiser A, Hardt K, Boehncke WH. Mammalian target of rapamycin and its downstream signalling components are activated in psoriatic skin. Br J Dermatol. (2013) 169:156–9. doi: 10.1111/bjd.12271
82. Ochaion A, Bar-Yehuda S, Cohen S, Barer F, Patoka R, Amital H, et al. The anti-inflammatory target A(3) adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn's disease. Cell Immunol. (2009) 258:115–22. doi: 10.1016/j.cellimm.2009.03.020
83. Chamcheu JC, Adhami VM, Esnault S, Sechi M, Siddiqui IA, Satyshur KA, et al. Dual inhibition of PI3K/Akt and mTOR by the dietary antioxidant, delphinidin, ameliorates psoriatic features in vitro and in an imiquimod-induced psoriasis-like disease in mice. Antioxidants Redox Signal. (2017) 26:49–69. doi: 10.1089/ars.2016.6769
84. Roller A, Perino A, Dapavo P, Soro E, Okkenhaug K, Hirsch E, et al. Blockade of phosphatidylinositol 3-kinase (PI3K)δ or PI3Kγ reduces IL-17 and ameliorates imiquimod-induced psoriasis-like dermatitis. J Immunol. (2012) 189:4612–20. doi: 10.4049/jimmunol.1103173
85. Yager N, Haddadeen C, Powell M, Payne A, Allen R, Healy E. Expression of PI3K signaling associated with T cells in psoriasis is inhibited by seletalisib, a PI3Kδ Inhibitor, and is required for functional activity. J Invest Dermatol. (2018) 138:1435–9. doi: 10.1016/j.jid.2017.12.028
86. Chen S, Paveley R, Kraal L, Sritharan L, Stevens E, Dedi N, et al. Selective targeting of PI3Kδ suppresses human IL-17-producing T cells and innate-like lymphocytes and may be therapeutic for IL-17-mediated diseases. J Autoimmun. (2020) 111:102435. doi: 10.1016/j.jaut.2020.102435
87. Jung KE, Lee YJ, Ryu YH, Kim JE, Kim HS, Kim BJ, et al. Effects of topically applied rapamycin and mycophenolic acid on TNCB-induced atopic dermatitis-like skin lesions in NC/Nga mice. Int Immunopharmacol. (2015) 26:432–8. doi: 10.1016/j.intimp.2015.03.007
88. Yang F, Tanaka M, Wataya-Kaneda M, Yang L, Nakamura A, Matsumoto S, et al. Topical application of rapamycin ointment ameliorates Dermatophagoides farina body extract-induced atopic dermatitis in NC/Nga mice. Exp Dermatol. (2014) 23:568–72. doi: 10.1111/exd.12463
89. Jia QN, Zeng YP. Rapamycin blocks the Il-13-induced deficiency of epidermal barrier related proteins via upregulation of MiR-143 in hacat keratinocytes. Int J Med Sci. (2020) 17:2087–94. doi: 10.7150/ijms.45765
90. Xiao DQ, Xu PH, Deng H, Chen LS, Ding XA, Bai M, et al. The characterization and clinical significance of PI3K/Akt signaling pathway activation in the peripheral T cells of pediatric patients with atopic dermatitis. Int J Clin Exp Med. (2017) 10:2904–10.
91. Segrelles C, Moral M, Fernanda Lara M, Ruiz S, Santos M, Leis H, et al. Molecular determinants of Akt-induced keratinocyte transformation. Oncogene. (2006) 25:1174–85. doi: 10.1038/sj.onc.1209155
92. Huang C, Li J, Ke Q, Leonard SS, Jiang BH, Zhong XS, et al. Ultraviolet-induced phosphorylation of p70S6K at Thr389 and Thr421/Ser424 involves hydrogen peroxide and mammalian target of rapamycin but not Akt and atypical protein kinase C. Cancer Res. (2002) 62:5689–97.
93. Newton AC, Trotman LC. Turning off AKT: PHLPP as a drug target. Annu Rev Pharmacol Toxicol. (2014) 54:537–58. doi: 10.1146/annurev-pharmtox-011112-140338
94. Haddadi N, Lin Y, Travis G, Simpson AM, McGowan EM, Nassif NT. PTEN/PTENP1: “Regulating the regulator of RTK-dependent PI3K/Akt signalling”, new targets for cancer therapy. Mol Cancer. (2018) 17:37. doi: 10.1186/s12943-018-0803-3
95. He Y-Y, Pi J, Huang J-L, Diwan BA, Waalkes MP, Chignell CF. Chronic UVA irradiation of human HaCaT keratinocytes induces malignant transformation associated with acquired apoptotic resistance. Oncogene. (2006) 25:3680–8. doi: 10.1038/sj.onc.1209384
96. Oh JH, Kim A, Park JM, Kim SH, Chung AS. Ultraviolet B-induced matrix metalloproteinase-1 and−3 secretions are mediated via PTEN/Akt pathway in human dermal fibroblasts. J Cell Physiol. (2006) 209:775–85. doi: 10.1002/jcp.20754
97. Bonneau D, Longy M. Mutations of the human PTEN gene. Hum Mutat. (2000) 16:109–22. doi: 10.1002/1098-1004(200008)16:2<109::AID-HUMU3>3.0.CO;2-0
98. Zhao L, Li W, Marshall C, Griffin T, Hanson M, Hick R, et al. Srcasm inhibits fyn-induced cutaneous carcinogenesis with modulation of Notch1 and p53. Cancer Res. (2009) 69:9439–47. doi: 10.1158/0008-5472.CAN-09-2976
99. Nardinocchi L, Sonego G, Passarelli F, Avitabile S, Scarponi C, Failla CM, et al. Interleukin-17 and interleukin-22 promote tumor progression in human nonmelanoma skin cancer. Eur J Immunol. (2015) 45:922–31. doi: 10.1002/eji.201445052
100. Huang T, Lin X, Meng X, Lin M. Phosphoinositide-3 kinase/protein kinase-B/mammalian target of rapamycin pathway in psoriasis pathogenesis. A potential therapeutic target? Acta Derm Venereol. (2014) 94:371–9. doi: 10.2340/00015555-1737
101. Buerger C. Epidermal mTORC1 signaling contributes to the pathogenesis of psoriasis and could serve as a therapeutic target. Front Immunol. (2018) 9:2786. doi: 10.3389/fimmu.2018.02786
102. Liu Y, Luo W, Chen S. Comparison of gene expression profiles reveals aberrant expression of FOXO1, Aurora A/B and EZH2 in lesional psoriatic skins. Mol Biol Rep. (2011) 38:4219–24. doi: 10.1007/s11033-010-0544-x
103. Zhang M, Zhang X. The role of PI3K/AKT/FOXO signaling in psoriasis. Arch Dermatol Res. (2019) 311:83–91. doi: 10.1007/s00403-018-1879-8
104. Hong KK, Gwak MJ, Song J, Kim NI. Nuclear factor-κB pathway activation and phosphatase and tensin homolog downregulation in psoriasis. Br J Dermatol. (2016) 174:433–5. doi: 10.1111/bjd.14106
105. Li Y, Man X, You L, Xiang Q, Li H, Xu B, et al. Downregulation of PTEN expression in psoriatic lesions. Int J Dermatol. (2014) 53:855–60. doi: 10.1111/ijd.12061
106. Wrone-Smith T, Mitra RS, Thompson CB, Jasty R, Castle VP, Nickoloff BJ. Keratinocytes derived from psoriatic plaques are resistant to apoptosis compared with normal skin. Am J Pathol. (1997) 151:1321–9.
107. Tchkonia T, Zhu Y, Van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. (2013) 123:966–72. doi: 10.1172/JCI64098
108. Mercurio L, Lulli D, Mascia F, Dellambra E, Scarponi C, Morelli M, et al. Intracellular Insulin-like growth factor binding protein 2 (IGFBP2) contributes to the senescence of keratinocytes in psoriasis by stabilizing cytoplasmic p21. Aging (Albany NY). (2020) 12:6823–51. doi: 10.18632/aging.103045
109. Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. (2004) 23:212–20. doi: 10.1038/sj.emboj.7600045
110. Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, et al. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene. (2012) 31:1949–62. doi: 10.1038/onc.2011.394
111. Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. (2008) 14:458–70. doi: 10.1016/j.ccr.2008.11.003
112. Chan KT, Blake S, Zhu H, Kang J, Trigos AS, Madhamshettiwar PB, et al. A functional genetic screen defines the AKT-induced senescence signaling network. Cell Death Differ. (2020) 27:725–41. doi: 10.1038/s41418-019-0384-8
113. Ding X, Willenborg S, Bloch W, Wickström SA, Wagle P, Brodesser S, et al. Epidermal mammalian target of rapamycin complex 2 controls lipid synthesis and filaggrin processing in epidermal barrier formation. J Allergy Clin Immunol. (2020) 145:283–300. doi: 10.1016/j.jaci.2019.07.033
114. Naeem AS, Tommasi C, Cole C, Brown SJ, Zhu Y, Way B, et al. A mechanistic target of rapamycin complex 1/2 (mTORC1)/V-Akt murine thymoma viral oncogene homolog 1 (AKT1)/cathepsin H axis controls filaggrin expression and processing in skin, a novel mechanism for skin barrier disruption in patients with atopic dermat. J Allergy Clin Immunol. (2017) 139:1228–41. doi: 10.1016/j.jaci.2016.09.052
115. Eibenschutz L, Colombo D, Catricalà C. Everolimus for compassionate use in multiple basal cell carcinomas. Case Rep Dermatol Med. (2013) 2013:1–4. doi: 10.1155/2013/604301
116. Atwood SX, Whitson RJ, Oro AE. Advanced treatment for basal cell carcinomas. Cold Spring Harb Perspect Med. (2014) 4:a013581. doi: 10.1101/cshperspect.a013581
117. Piérard-Franchimont C, Hermanns-Lê T, Paquet P, Herfs M, Delvenne P, Piérard GE. Hedgehog- and mTOR-targeted therapies for advanced basal cell carcinomas. Futur Oncol. (2015) 11:2997–3002. doi: 10.2217/fon.15.181
118. Tsubamoto H, Ueda T, Inoue K, Sakata K, Shibahara H, Sonoda T. Repurposing itraconazole as an anticancer agent (Review). Oncol Lett. (2017) 14:1240–6. doi: 10.3892/ol.2017.6325
119. Kim DJ, Kim J, Spaunhurst K, Montoya J, Khodosh R, Chandra K, et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J Clin Oncol. (2014) 32:745–51. doi: 10.1200/JCO.2013.49.9525
120. Euvrard S, Morelon E, Rostaing L, Goffin E, Brocard A, Tromme I, et al. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med. (2012) 367:329–68. doi: 10.1056/NEJMoa1204166
121. Zhang YJ, Duan Y, Zheng XF. Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today. (2011) 16:325–31. doi: 10.1016/j.drudis.2011.02.008
122. Ding LT, Zhao P, Yang ML, Lv GZ, Zhao TL. GDC-0084 inhibits cutaneous squamous cell carcinoma cell growth. Biochem Biophys Res Commun. (2018) 503:1941–8. doi: 10.1016/j.bbrc.2018.07.139
123. Zou Y, Ge M, Wang X. Targeting PI3K-AKT-mTOR by LY3023414 inhibits human skin squamous cell carcinoma cell growth in vitro and in vivo. Biochem Biophys Res Commun. (2017) 490:385–92. doi: 10.1016/j.bbrc.2017.06.052
124. Bakshi A, Chaudhary SC, Rana M, Elmets CA, Athar M. Basal cell carcinoma pathogenesis and therapy involving hedgehog signaling and beyond. Mol Carcinog. (2017) 56:2543–57. doi: 10.1002/mc.22690
125. Reitamo S, Spuls P, Sassolas B, Lahfa M, Claudy A, Griffiths CEM. Efficacy of sirolimus (rapamycin) administered concomitantly with a subtherapeutic dose of cyclosporin in the treatment of severe psoriasis: a randomized controlled trial. Br J Dermatol. (2001) 145:438–45. doi: 10.1046/j.1365-2133.2001.04376.x
126. Ormerod AD, Shah SAA, Copeland P, Omar G, Winfield A. Treatment of psoriasis with topical sirolimus: preclinical development and a randomized, double-blind trial. Br J Dermatol. (2005) 152:758–64. doi: 10.1111/j.1365-2133.2005.06438.x
127. Helmer E, Watling M, Jones E, Tytgat D, Jones M, Allen R, et al. First-in-human studies of seletalisib, an orally bioavailable small-molecule PI3Kδ inhibitor for the treatment of immune and inflammatory diseases. Eur J Clin Pharmacol. (2017) 73:581–91. doi: 10.1007/s00228-017-2205-7
128. Jeong JS, Kim JS, Kim SR, Lee YC. Defining bronchial asthma with phosphoinositide 3-kinase delta activation: towards endotype-driven management. Int J Mol Sci. (2019) 20:3525. doi: 10.3390/ijms20143525
129. Stark AK, Sriskantharajah S, Hessel EM, Okkenhaug K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr Opin Pharmacol. (2015) 23:82–91. doi: 10.1016/j.coph.2015.05.017
130. Yoo EJ, Ojiaku CA, Sunder K, Panettieri RA. Phosphoinositide 3-kinase in asthma: novel roles and therapeutic approaches. Am J Respir Cell Mol Biol. (2017) 56:700–7. doi: 10.1165/rcmb.2016-0308TR
131. Tsang F, Wong WSF. Inhibitors of tyrosine kinase signaling cascade attenuated antigen challenge of guinea-pig airways in vitro. Am J Respir Crit Care Med. (2000) 162:126–33. doi: 10.1164/ajrccm.162.1.9908105
132. Lee KS, Jeong JS, Kim SR, Cho SH, Kolliputi N, Ko YH, et al. Phosphoinositide 3-kinase-δ regulates fungus-induced allergic lung inflammation through endoplasmic reticulum stress. Thorax. (2016) 71:52–63. doi: 10.1136/thoraxjnl-2015-207096
Keywords: PI3K, AKT, non-melanoma skin cancer, psoriasis, atopic dermatitis, hyperproliferation, apoptosis
Citation: Mercurio L, Albanesi C and Madonna S (2021) Recent Updates on the Involvement of PI3K/AKT/mTOR Molecular Cascade in the Pathogenesis of Hyperproliferative Skin Disorders. Front. Med. 8:665647. doi: 10.3389/fmed.2021.665647
Received: 08 February 2021; Accepted: 29 March 2021;
Published: 30 April 2021.
Edited by:
Cristina Pellegrini, University of L'Aquila, ItalyReviewed by:
Elisabetta Palazzo, University of Modena and Reggio Emilia, ItalyCopyright © 2021 Mercurio, Albanesi and Madonna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Albanesi, Yy5hbGJhbmVzaUBpZGkuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.