
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 19 March 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.661203
This article is part of the Research TopicImmunosenescence and Immunoexhaustion in Chronic Kidney Disease and Renal TransplantationView all 10 articles
 Elisavet Stavropoulou1,2*Konstantia Kantartzi3*Christina Tsigalou4Konstantina Aftzoglou5Chrysa Voidarou6Theocharis Konstantinidis7Mariana Carmen Chifiriuc8Elias Thodis3Eugenia Bezirtzoglou9
Elisavet Stavropoulou1,2*Konstantia Kantartzi3*Christina Tsigalou4Konstantina Aftzoglou5Chrysa Voidarou6Theocharis Konstantinidis7Mariana Carmen Chifiriuc8Elias Thodis3Eugenia Bezirtzoglou9The gut microbiome is known as an important predictive tool for perceiving characteristic shifts in disease states. Multiple renal diseases and pathologies seem to be associated with gut dysbiosis which directly affects host homeostasis. The gastrointestinal-kidney dialogue confers interesting information about the pathogenesis of multiple kidney diseases. Moreover, aging is followed by specific shifts in the human microbiome, and gradual elimination of physiological functions predisposes the microbiome to inflammaging, sarcopenia, and disease. Aging is characterized by a microbiota with an abundance of disease-associated pathobionts. Multiple factors such as the immune system, environment, medication, diet, and genetic endowment are involved in determining the age of the microbiome in health and disease. Our present review promotes recently acquired knowledge and is expected to inspire researchers to advance studies and investigations on the involved pathways of the gut microbiota and kidney axis.
The human newborn is devoid of bacteria at birth (1). Bacteria colonizing the sterile newborn either come from the hospital environment and staff as in the case of caesarian section or from normal maternal vaginal microflora (1). The establishment and progression of the human microflora is attributed to the influence of multiple epigenetic mechanisms (2). Personal habits and behavior, stress, hormones, antibiotics, vaccination, and infections (1) seem to be involved. However, nutrition remains the ultimate factor that can sway newborn development processes regulating epigenetic mechanisms during pregnancy and early life (1, 2). The importance of food intake variations is stated by a plethora of publications (1–4). Studies comparing children in rural Africa and Europe reported important variations in microbial populations due to eating habits (5). African children were colonized by more bacteria belonging to Actinobacteria phylum and Bacteroidetes than Firmicutes compared to European children group who carried more Firmicutes and Proteobacteria.
The importance of early life colonization is understood (1). The presence of beneficial bacteria such as Lactobacilli and Bifidobacteria protect against disease (6). The “Hygiene Hypothesis” was advanced to explain atopic disorders after immune dysregulation (7). Human microbiota evolve in parallel with the immune system supporting a bidirectional relationship resulting in normal immune development (8).
Nowadays, the term “microflora” is used less frequently in favor of the term “microbiota” as microbial genomes are also involved. The term was first used by the Nobel Laureate Joshua Lederberg (9).
Bacterial communities are involved in complex inter-communication and network models of unique microbiomes. In this vein, characterization of the different microbial communities in health and disease status was achieved due to new technological involvements and particularly 16S rRNA sequencing. This methodology permits the identification of complex microbial populations in the human body (10). Additionally, metagenomics Whole Genome Shotgun (WGS) sequencing has allowed for the identification of involved functions in relation to our microbiome (10, 11). It seems to be less crucial to confirm “who is there” than “what are they doing.” The Human Microbiome Project (USA) (12) as well as the metaHIT Consortium (Europe) (13) have shed light on the characterization of major healthy human sites in order to compare them with shifts occurring in disease states (14).
In developed countries, during the last century, improvements in healthcare have led to a population of higher age and life expectancy has risen (15). With the recognition of an aging population (16), geriatric research has gained the interest of multiple society sectors including topics such as social, work, and economic impact and nutrition and health issues.
It is known that frail and elderly people encounter more infections than younger people (17). Infections in elderly subjects are often complicated due to multi-morbidity (17), hormonal shifting, increased production of pro-inflammatory cytokines and chemokines, and abnormalities of the telomeres which finally could cause a dysfunction of the immune system called immunosenescence and malnutrition.
The impact of aging upon the intestinal microbiota is associated with a decrease in the anaerobic population (18, 19), specifically the Bifidobacterial population (20, 21), while an increase in Enterobacterial population has been reported (19, 21).
Age-related sequential changes were reported in the human microbiota (22) by 16SrRNA methodologies. Actinobacteria phylum (mainly Bifidobacteria) was decreased with age and after weaning (22), while Firmicutes (mainly Clostridium cluster XIVa and Faecalibacterium 57 prausnitzii) were more frequent in older children but at lower levels (23). Finally, Bacteroidetes and Proteobacteria were found in human recipients over 70 years old (22). Taking it one step further, in analysis focusing on bacterial co-abundance groups (CAGs) as defined by Kendall, correlations between genera showed that several transition types of microbiota were enriched in the adult population (22). Relative abundance of genera was registered in elderly-associated CAGs compared to infant- and adult-associated CAGs (22). Linkage clustering based on the abundance of genera indicated five age clusters with median ages 3, 33, 42, 77, and 94 years old (22). However, when clustering was based on the proportion of transporters evaluated by phylogenetic analysis of the bacterial communities by reconstruction of unobserved states (PICRUSt), the human recipients were classified into two age groups; the adult-enriched and the infant/elderly-enriched clusters (22).
It is known that the intestine possesses dual functions, firstly a role in nutrient absorption and also a function in the synthesis of substances such as amino acids, vitamins, and short chain fatty acids (SCFAs) (24). SCFAs exert beneficial effects, confer energy to epithelial cells, and engage in a potent role in the immunomodulation and barrier effect against pathogenic invaders (24). Particularly, they hold two basic signaling functions; the activation of G-protein-coupled receptors (GPCRs) and the inhibition of histone deacetylases (HDACs) (25). GPCRs are receptors of SCFAs which participate in metabolism, inflammation, and disease processes (25). Still, SCFAs are activated in the free fatty acid receptor-2 and−3 (FFAR2 and FFAR3) found in numerous human body sites (26). Additionally, SCFAs upset the physiology of the intestinal epithelial cells by inhibiting histone deacetylases (HDACs) resulting in chromatin remodeling and changes in transcription processes (27). Finally, HDACs seem to possess an anti-proliferative and anti-inflammatory action either in vitro or in vivo in developed models of inflammation (27).
In this vein, the intestinal microbiota via the intestinal barrier seem to adjust homeostasis and functions of both innate and adaptive immunity locally and systemically (28). However, when the intestinal barrier is breached, a situation called “leaky gut,” the gut bacteria and their toxins are able to infiltrate the intestinal mucosa and then through the blood stream circulate to different tissues and organs (1, 29). Moreover, activated immune cells penetrate the kidney and generate pro-and anti-inflammatory reactions and regulatory signals in order to induce a neutrophils response (30). Neutrophils together with macrophages are induced as part of the first line response in innate immunity against pathogens (31) and kidney disease (32).
Impairment of the macrophages' phagocytic ability has a negative effect on kidney function leading to chronic inflammation (31). Chronic systemic inflammation can be appraised using the neutrophil-to-lymphocyte ratio (NLR) which is associated with the risk of ESRD with stage 4 CKD. NLR could be a prognostic marker for cardiovascular risk and mortality in patients with CKD 3-5 and hemodialysis-peritoneal dialysis patients, respectively (33, 34).
To this end, the role of pattern recognition receptors (PRRs), and especially TLRs (toll like receptors) which are membrane glycoproteins, during inflammation processes is stated (31). TLRs are found in renal cells and activate mitogen-activated protein kinases, nuclear factor-κB, and activator protein-1 toward a pro-inflammatory status (35, 36). The importance of a dialogue between the acquired immune system and the innate system is understood (37, 38) through the production of cytokines.
Renal tubular epithelial cells participate in immunity processes by producing chemokines, cytokines, and antimicrobial substances (32). In their turn, cytokines participate in the immunological response by promoting the synthesis of acute phase proteins and tissue proteolysis and lipolysis. Moreover, they interact with T lymphocytes to generate the acquired immune response (39). A cell-mediated response to the antigen will take place and T lymphocytes will similarly produce cytokines in order to regulate the activity of immunocompetent cells and induce antibody production (40). Injured renal tubular epithelial cells dedifferentiate to achieve refit and thus they incite inflammation by recruiting myofibroblasts. In this way, tubular epithelial cell loss stimulates residual renal hypertrophy. Thus, the hypertrophied nephron is unable to cope with the increase in tubular transport as it overwhelms its energy-generating capacity, and anaerobic metabolism, acidosis, and hypoxia occurs (41, 42).
Renal tubular epithelial cells present a crucial role in inflammation, positively or negatively regulating T cell responses in an alternative way, as they express co-stimulators of T cells (ICOS-L) and B7-H1 molecules (43). Yet, macrophages, dendritic cells (DCs), and T regulatory cells (Tregs) induce an adaptive immune response and DC activation promotes the production of proinflammatory cytokines such as IL-12 and IL-6 (30). Clearly, DCs trigger the differentiation of naïve CD4+ T cells into regulatory T (Treg) cells and the maturation of B cells into IgA-secreting ones (30, 44). The role of Treg cells in renal disease seems to be crucial in protecting against inflammation processes and amplifying homeostasis by boosting microbiota (45). In support, T helper 17 (Th17) cells are activated inducing the production of pro-inflammatory interleukin-17 (IL-17) (46). To this end, nuclear factor-κB (NF-κB) is released by the renal tubular epithelial cells regulating pro-inflammatory response (47).
Furthermore, innate lymphoid cells (ILCs) tamper with pro-inflammatory cytokines IL-1β, IL-12, IL-23, IL-22, and IFNγ production (44, 48). It was also found that the aryl hydrocarbon receptor of IL-22 in innate lymphoid cell response (ILC3) suppresses inflammatory Th17 cell responses and regulates Treg-mediated gut homeostasis (49). So then, the suppression of Th17 cells in the intestine confers positively to the translocation and activation in the kidney (44). Gut expressed Th cells can be activated in the kidney through the CCL20/CCR6 axis (50).
The intestinal microbiota cooperate by means of microbial associated molecular patterns (MAMPs) or SCFAs as previously discussed to temper inflammation in the kidney (51). It is of note that by the aid of RT-PCR, four receptors (GPR41, GPR43, Olfr78, and GPR109a) expressed in the kidney are linked to particular pathologies (52) (Figure 1).
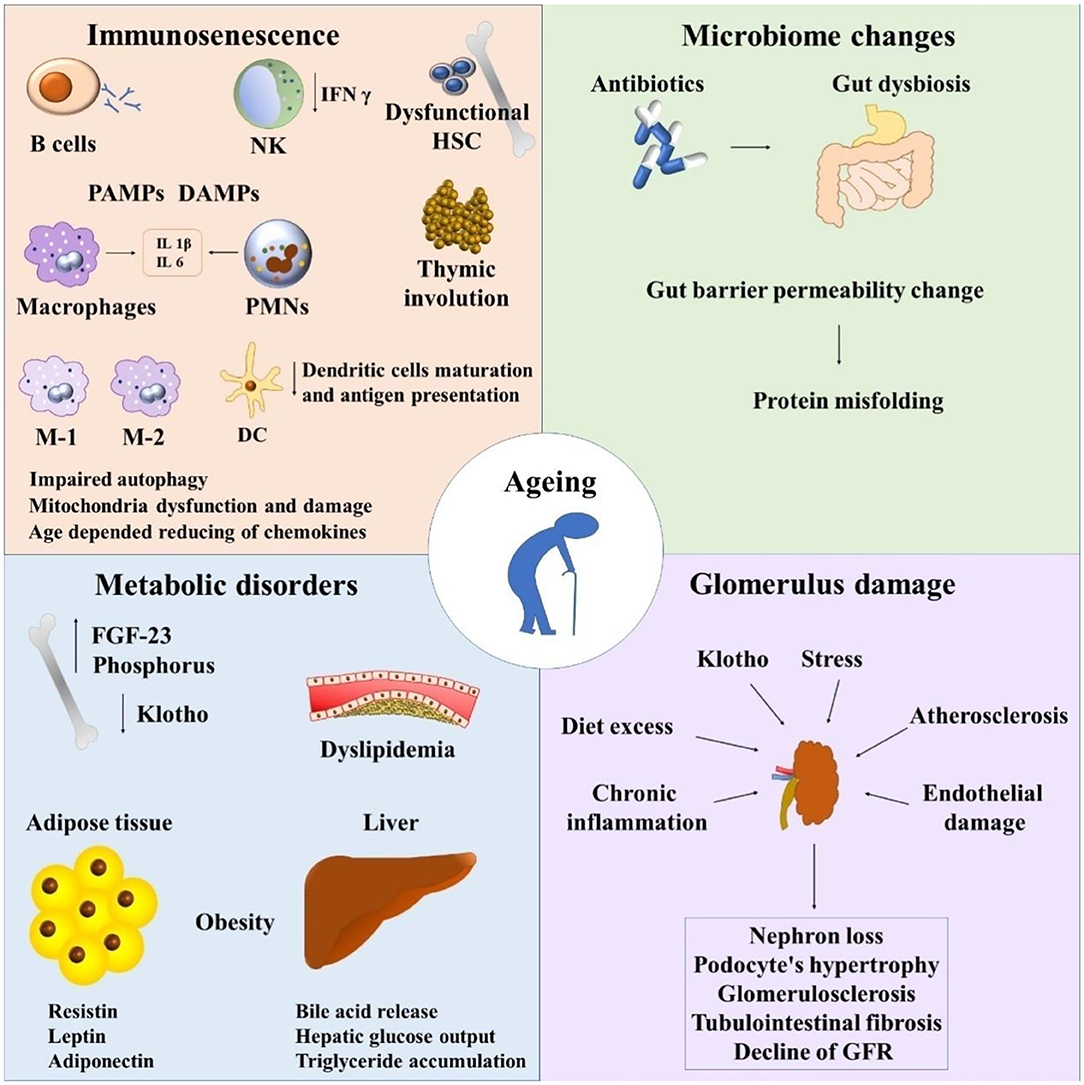
Figure 1. Immunosenescence and chronic kidney disease. NK, natural killers; HSC, hematopoietic stem cells; PAMPs, pathogen-associated molecular pattern molecules; DAMPs, damage-associated molecular pattern molecules; PMNs, polymorphonuclear leukocytes; FGF-23, fibroblast growth factor 23; GFR, glomerular filtration rate.
Without any doubt, important physiological changes occur in the kidney as a result of immunoactivation. Immune cells and inflammatory proteins contribute to the pathogenesis of kidney diseases (53). Finally, it is worth noting the importance of the dialogue between the kidney and gut, the so-called gut-kidney axis in health and disease (54).
Actually, in spite of the technological advancements in peritoneal dialysis (PD) and hemodialysis (HD) procedures, the mortality in ESRD remains high (55) as cardiovascular disease and infections occurred in these patients. It seems that both complications are associated with immunological shifts in ESRD such as uremia (55). Uremia is characterized by immune dysfunction and immunosuppression leading to multiple infections. The accumulation of pro-inflammatory cytokines takes place as a result of dropped renal elimination capacity, oxidative stress, and the accumulation of uremic toxins. Moreover, immunoactivation results in inflammation and cardiovascular disease. Immune dysfunction in uremia is linked to both innate and adaptive immunity (55). Yet, adaptive immunity is altered in ESRD patients. It seems to be caused by uremia per se and chronic renal failure. T cell proliferation is mitigated in an uremic environment. T helper lymphocytes (Th) have an impact on the immune response. Th1 cells activate macrophages, while Th2 cells promote humoral immunity (56). Interestingly, the maturation of Th cells in hemodialysis patients (HD) does occur, these subjects showed increased Th1 concentrations and an increased Th1/Th2 ratio (57). Studies state that ratio increase in HD is associated with the elevated production of IL-12 which effects T lymphocytes. This leads to an increase in IFN-γ and a decrease in IL-4, promoting their differentiation in Th1 cells (55). Yet, B cell lymphopenia is apparent due to apoptosis, despite the production of IgM and IgA in normal levels in dialysis patients (58). Following initiation of renal replacement therapy in HD or peritoneal dialysis (CAPD) subjects, the immunological status of patients was appraised (59). The percentage of CD4+CD28 null and CD8+CD28 null cells was found increased in ESRD patients. Therefore, CD4+CD28 null cells correlated with CRP and serum albumin levels while important differences in items of CD4+CD28 null and CD8+CD28 null cells were found in patients with cardiovascular disease. Shifts in the population of CD4+CD28 null cells was found following 6 months of dialysis. However, these changes showed significant differences between HD and CAPD patients (59), T cells subtypes are affected by CKD and a chronic inflammation disease is installed. This turmoil is enhanced in HD patients but alleviated in CAPD patients (59).
The intestinal microbiome of HD patients showed an increase in Proteobacteria, Actinobacteria, and Firmicutes with preponderance of the subphylum Clostridia, while a decrease in the taxa Firmicutes and Actinobacteria is found in CAPD patients (60). It is known that there is an interplay between the kidney and gut, called the gut-kidney axis (54, 61). Renal transplantation incites changes in the gut microbiota (62). Yet, hormones, environment, genetics, epigenetics, and pharmacogenetics seem to impact kidney allograft receivers (62).
Gut microbiota could incite antigen-presenting cells (APCs) and initiate immune response and alloimmune reactivity, as is the case in allogeneic bone marrow transplantation (HSCT) (63). However, when allograft recipients are submitted to gut decontamination, acute graft vs. host disease declines (64). A considerable shift in microbiota was found 1 month after transplantation. It is of note that patients hosting Faecalibacterium prausnitzii in their microbiota need higher tacrolimus therapeutic doses (65).
Researchers found that gut-associated lymphoid tissue (GALT) plays a key role in the evolvement of immunoglobulin A (IgA) nephropathy (IgAN) (66).
Changes in gut microbiota and dysbiosis seem to be critical for immunoglobulin A nephropathy (IgAN) (54). In IgAN patients, an abundance of Fusobacteria is observed, while Synergistetes were decreased (67). Genome studies showed that IgAN and inflammatory bowel diseases are linked to the same loci (66). This observation involves a different clinical approach including a treatment option that focuses on subclinical intestinal inflammation or microbiota shifting (68).
Dysbiosis of the gut microbiota was also related to patients with idiopathic membranous nephrotic syndrome (INS) (69). Fusobacteria, Proteobacteria, and Parabacteroides are increased in INS patients, while Firmicutes dropped (69). At the genus level, Providencia and Myroides were found more frequently in INS patients (69). Yet, propionate acid and butyric acid are found in low concentrations in INS patients (69).
During aging, physiological and pathological changes emerge in contrast to senescence where mainly non-pathological changes occur. There is an impairment of multiple functions including the dermal, mucosal, and epithelial barrier and (50) the barrier effect (70). While most scientists have not found a quantitative variation in immunological cells with aging, B lymphocytes and T lymphocytes associated with adaptive immunity and natural killers cells, granulocytes, monocytes, and macrophages associated with the innate immunity were found in increased numbers (71, 72).
Yet, in elderly people, DCs showed a reduction in antigen presentation-function, impaired endocytosis, and reduced chemokine production (72). This reduced chemokine production leads to a decrease in cytotoxicity of the natural killer (NK) cells and a decreased killing capacity. While natural killer numbers do increase in healthy elderly people due to the enhanced activity of the markers cells CD56dim and CD57, function is impaired due to cytotoxicity. Therefore, an enhanced production of IL-4 and IL-10 and a decreased production of INF-γ in elderly subjects is observed.
The importance of natural killer (NK) cells in kidney infection and inflammation was previously discussed. Although, natural killer (NK) cells increase quantitatively in lymphatic organs, they showed a low proliferative capacity in the peripheral blood (73). Neutrophils make up 50–70% of human white blood cells and they play an essential role in the innate immune system. They remain stable in the peripheral blood and the bone marrow of the elderly, although they have low phagocytic and killing activity and are more vulnerable to apoptosis (74). Although monocytes also have stable quantitative levels in the peripheral blood of an aged subject, macrophage function is decreased (75, 76). Yet, a temperate phagocytosis, chemiotaxis, and oxidative activity is seen due to the release of ROS, as superoxide radical and hydrogen peroxide from different cells (76). Moreover, the antigen-presenting capacity is lower. Thus, infection occurring in the elderly will be long lasting and it is likely to develop into a chronic inflammation state more frequently. Similarly, the same profile was shown for dendritic cells (DCs) (77). Moreover, in the frail elderly, an extensive reactivity against auto-antigens and an enhanced release of the pro-inflammatory cytokines TNF-a and IL-6 was registered (78). It is of note that these pro-inflammatory cytokines are used as predictive biomarkers for comorbidities and mortality (17).
Shifts observed in the immunological structure during chronological aging induce a “prolific milieu” for the development of a chronic inflammation state, so-called “inflammaging.”
Age-related modifications are more pronounced in the adaptive immune system.
Chronological aging lends itself to the decrease of naïve T cells and the accumulation of oligoclonal memory and cytotoxic T cells (79). Upon the end of the thymus involution process at around 50 years of age, a drop in T cell levels is marked and globally observed age-related shifts are more noticeable (80). The decrease of CD8+ cells was more profound compared to CD4+ cell levels (81).
Although, B lymphocytes present a stable profile in the peripheral blood, the numbers of mnemonic B cells is enhanced in order to offset the drop in naïve B cells in the elderly. In support of that observation, insufficient production of specific antibodies following vaccination with advanced aged was shown (82).
Recapitalizing, important shifts are shown in immune system cells during aging which lead to thymic involution, clonal exhaustion, and rupture (83) (Figure 1).
The term immunosenescence was coined by Roy Walford (84) when he published his hallmark book entitled “The Immunologic Theory of Aging” (85). The term denotes the aging-related dysfunction of the immune system (72) associated with higher infection possibility.
However, there is some scientific disputation in defining the term “immunosenescence” (72). Scientists report immunosenescence as a dysfunction of the global immune system called the “damage theory of aging,” while others believe that only specific parameters are altered (72) entangling the telomere proliferation mechanisms (86). Telomeres seem to have a crucial role in aging via regulating cellular responses and DNA damage (87). Telomeres should “cap” chromosome ends to inhibit activation of DNA repair. As a result, apoptosis or cell senescence occurs when the number of “uncapped” telomeres accrues (87) due to shortening of each telomere length. This fact highlights the cessation of cellular proliferation which defines the aging status. Finally, a lack of telomeres is reported as an immunosenescence status (87). It is of note that amplified cancer cells have active telomerases and a stable telomere length and as a result they do not senescence and even when telomeres are linked to oncogenes, cells tend to immortalize (86–88).
Aging is linked to important shifts in gene expression. Overexpression of p16 and p21 gene inhibitors of the cellular cycle induce faster senescence (89). In this way, the induction of senescence induced by gene inhibitors may be a new therapeutic approach in the treatment of cancer (89).
The term “chronic kidney disease (CKD)” reflects lasting damage to the kidneys that can aggravate over time. Chronic kidney disease (CKD) and end-stage renal disease (ESRD) are a dominant medical challenge in the 21st century (90), as more than 1.2 million people died from CKD in 2017 showing a considerable increase in global-age prevalence and mortality in the last 20 years (91). In Oceania, sub-Saharan Africa, and Latin America, the burden of CKD was much higher compared to the disease burden in other countries (91). Patients may develop complications such as hypertension, anemia, heart and blood vessel diseases, and nerve damage (90). Diabetes and hypertension may cause CKD, susceptibility to infection, and other associated pathologies (90).
Early detection of the disease is important, as the disease develops and may lead to kidney impairment that necessities dialysis and finally kidney transplantation (90) to survive.
CKD is defined according to the level of glomerular filtration rate (GFR) into five gradual stages from asymptomatic stage 1 to the end-stage renal disease (ESRD) stage 5. The stages 3–5 show a glomerular filtration rate (GFR) below 60 ml/min per 1.73 m2 for 3 months or more (90). Other pathological co-morbidities as well as additional markers of kidney damage, such as proteinuria or hematuria for 3 months or more are co-estimated (92). The disease seems to be more common in the elderly population considering chronological aging. CKD is characterized by senescence, and CKD and ESRD patients appear to be biologically older (93) due to their global malfunction status, when compared to a healthy population.
Kidney cellular shifts and immune cell dysfunction lead to immunosenescence and apoptosis as previously discussed. Moreover, important changes are registered in the kidney glomerular filtration barrier by loss of podocytes (94) which lead to renal impairment. Proteinuria and other lesions advance podocyte loss or induce death (75, 94).
The autophagy process has a central role in controlling homeostasis and adjacent involved mechanisms involved in glomerular disease and maintains podocytes homeostasis in aging (95, 96).
Tubulo-interstitial renal fibrosis is a condition of the aged kidney which is defined as a progressive detrimental connective tissue deposition on the kidney parenchyma leading to renal function damage (43, 97). The epithelial to mesenchymal transition (EMT) of tubular epithelial cells is converted to mesenchymal fibroblasts. Thus, fibroblasts proliferate to the adjacent interstitial parenchyma (97) (Figure 1).
As stated, advanced aging deteriorates the immune system, increases susceptibility to infection (98), and converges a low-grade activation of the inflammation system called inflammaging (79). Stimuli such as exposure to pathogens, cellular debris, nutrients, and the gut microbiota sustain inflammaging (53, 99).
The gut microbiota is the corner stone in inflammaging due to its capacity to produce inflammatory products and dialogue with other organs and systems (54). However, it is clear that the underlying aging mechanisms still need to be explained through this trajectory in order to gain a better understanding of this global dysregulation and provide more effective therapeutic approaches.
ES: conceptualization, writing, and editing. KK and CT: formal analysis and writing. CV and KA: resources and writing. TK: design and editing. MCC: writing and editing. EB and ET: supervision, original draft preparation, and editing. ET contribute to the reviewing and editing of the paper. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bezirtzoglou E, Stavropoulou E. Immunology and probiotic impact of the newborn and young children intestinal microflora. Anaerobe. (2011) 17:369–74. doi: 10.1016/j.anaerobe.2011.03.010
2. Li Y. Epigenetic mechanisms link maternal diets and gut microbiome to obesity in the offspring. Front Genet. (2018) 9:342. doi: 10.3389/fgene.2018.00342
3. Michaelsen KF, World Health Organization. Feeding and Nutrition of Infants and Young Children: Guidelines for the WHO European Region, With Emphasis on the Former Soviet Countries. Copenhagen: WHO Regional office for Europe (2000). p. 288.
4. Bezirtzoglou E, Romond C. Effect of the feeding practices on the establishment of bacterial interactions in the intestine of the newborn delivered by cesarean section. J Perinat Med. (1990) 18:139–43.
5. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. (2010) 107:14691–6. doi: 10.1073/pnas.1005963107
6. Wall R, Ross RP, Ryan CA, Hussey S, Murphy B, Fitzgerald GF, et al. Role of gut microbiota in early infant development. Clin Med Pediatr. (2009) 3:45–54. doi: 10.4137/CMPed.S2008
7. Rautava S, Ruuskanen O, Ouwehand A, Salminen S, Isolauri E. The hygiene hypothesis of atopic disease–an extended version. J Pediatr Gastroenterol Nutr. (2004) 38:378–88. doi: 10.1097/00005176-200404000-00004
8. Stiemsma LT, Reynolds LA, Turvey SE, Finlay BB. The hygiene hypothesis: current perspectives and future therapies. Immunotargets Ther. (2015) 4:143–57. doi: 10.2147/ITT.S61528
9. Lederberg J, McCray AT. “Ome Sweet” Omics-A genealogical treasury of words. Sci Phila. (2001) 15:8.
10. Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL. Analysis of the microbiome: advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun. (2016) 469:967–77. doi: 10.1016/j.bbrc.2015.12.083
11. Morgan XC, Huttenhower C. Human microbiome analysis. PLOS Comp Biol. (2012) 8:e1002808. doi: 10.1371/journal.pcbi.1002808
12. NIH Human Microbiome Project - Home. (2020). Available online at: https://hmpdacc.org/
13. MetaHIT Consortium (Metagenomics of the Human Intestinal Tract consortium) - Wellcome Sanger Institute. (2021). Available online at: https://www.sanger.ac.uk/resources/downloads/bacteria/metahit/
14. The Human Microbiome Project Consortium, Huttenhower C, Gevers, D. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 486:207–214. doi: 10.1038/nature11234
15. European Commission. Directorate-General for Economic and Financial Affairs. The 2012 Ageing Report : Underlying Assumptions and Projection Methodologies. LT: Publications Office (2011). Available online at: https://data.europa.eu/doi/10.2765/15373
16. WHO publications. global_health.pdf. Available online at: https://www.who.int/ageing/publications/global_health.pdf
17. Marengoni A, Angleman S, Melis R, Mangialasche F, Karp A, Garmen A, et al. Aging with multimorbidity: a systematic review of the literature. Ageing Res Rev. (2011) 10:430–9. doi: 10.1016/j.arr.2011.03.003
18. Samedi L, Linton Charles A. Evaluation of technological and probiotic abilities of local lactic acid bacteria. J Appl Environ Microbiol. (2019) 7:9–19. doi: 10.12691/jaem-7-1-3
19. Hébuterne X. Gut changes attributed to ageing: effects on intestinal microflora. Curr Opin Clin Nutr Metab Care. (2003) 6:49–54. doi: 10.1097/00075197-200301000-00008
20. Ljungh A, Wadstrom T. Lactic acid bacteria as probiotics. Curr Issues Intest Microbiol. (2006) 7:73–89.
21. Stavropoulou E, Bezirtzoglou E. Human microbiota in aging and infection: a review. Crit Rev Food Sci Nutr. (2019) 59:537–45. doi: 10.1080/10408398.2017.1379469
22. Odamaki T, Kato K, Sugahara H, Hashikura N, Takahashi S, Xiao J, et al. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. (2016) 16:90. doi: 10.1186/s12866-016-0708-5
23. Salazar N, Valdés-Varela L, González S, Gueimonde M, Reyes-Gavilán CG de Los. Nutrition and the gut microbiome in the elderly. Gut Microbes. (2017) 8:82–97. doi: 10.1080/19490976.2016.1256525
24. Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. (2017) 19:29–41. doi: 10.1111/1462-2920.13589
25. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. (2014) 121:91–119. doi: 10.1016/B978-0-12-800100-4.00003-9
26. Layden BT, Angueira AR, Brodsky M, Durai V, Lowe WL. Short chain fatty acids and their receptors: new metabolic targets. Transl Res J Lab Clin Med. (2013) 161:131–40. doi: 10.1016/j.trsl.2012.10.007
27. Schilderink R, Verseijden C, de Jonge WJ. Dietary Inhibitors of Histone Deacetylases in Intestinal Immunity and Homeostasis. Front Immunol. (2013) 4:226. doi: 10.3389/fimmu.2013.00226
28. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. (2012) 489:231–41. doi: 10.1038/nature11551
29. Romond M-B, Colavizza M, Mullié C, Kalach N, Kremp O, Mielcarek C, et al. Does the intestinal bifidobacterial colonisation affect bacterial translocation? Anaerobe. (2008) 14:43–8. doi: 10.1016/j.anaerobe.2007.09.003
30. Dhana E, Ludwig-Portugall I, Kurts C. Role of immune cells in crystal-induced kidney fibrosis. Matrix Biol J Int Soc Matrix Biol. (2018) 68–69:280–92. doi: 10.1016/j.matbio.2017.11.013
31. Guiteras R, Flaquer M, Cruzado JM. Macrophage in chronic kidney disease. Clin Kidney J. (2016) 9:765–71. doi: 10.1093/ckj/sfw096
32. Tang PC-T, Zhang Y-Y, Chan MK-K, Lam WW-Y, Chung JY-F, Kang W, et al. The emerging role of innate immunity in chronic kidney diseases. Int J Mol Sci. (2020) 21:4018. doi: 10.3390/ijms21114018
33. C-STRIDE study group, Yuan Q, Wang J, Peng Z, Zhou Q, Xiao X, et al. Neutrophil-to-lymphocyte ratio and incident end-stage renal disease in Chinese patients with chronic kidney disease: results from the Chinese Cohort Study of Chronic Kidney Disease (C-STRIDE). J Transl Med. (2019) 17:86. doi: 10.1186/s12967-019-1808-4
34. An X, Mao H-P, Wei X, Chen J-H, Yang X, Li Z-B, et al. Elevated neutrophil to lymphocyte ratio predicts overall and cardiovascular mortality in maintenance peritoneal dialysis patients. Int Urol Nephrol. (2012) 44:1521–8. doi: 10.1007/s11255-012-0130-3
35. Cowan KJ, Storey KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol. (2003) 206:1107–15. doi: 10.1242/jeb.00220
36. de Heer E, Peters DJM. Innate immunity as a driving force in renal disease. Kidney Int. (2008) 73:7–8. doi: 10.1038/sj.ki.5002658
37. Rivera A, Siracusa MC, Yap GS, Gause WC. Innate cell communication kick-starts pathogen-specific immunity. Nat Immunol. (2016) 17:356–63. doi: 10.1038/ni.3375
38. IQWiG (Institute for Quality and Efficiency in Health Care). The Innate and Adaptive Immune Systems. InformedHealth.org (2020). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK279396/
39. Novais FO, Scott P. Immunity to pathogens and tumors -proinflammatory cytokine - an overview. In: Encyclopedia of Immunobiology 2016. Science Direct Topics. Toronto, ON (2021).
40. Cano RLE, Lopera HDE. Introduction to T and B lymphocytes. In: Autoimmunity: From Bench to Bedside. Bogota: El Rosario University Press (2013).
41. Schnaper HW. The tubulointerstitial pathophysiology of progressive kidney disease. Adv Chronic Kidney Dis. (2017) 24:107–16. doi: 10.1053/j.ackd.2016.11.011
43. De Haij S, Woltman AM, Trouw LA, Bakker AC, Kamerling SW, Van Der Kooij SW, et al. Renal tubular epithelial cells modulate T-cell responses via ICOS-L and B7-H1. Kidney Int. (2005) 68:2091–102. doi: 10.1111/j.1523-1755.2005.00665.x
44. Gong J, Noel S, Pluznick JL, Hamad ARA, Rabb H. Gut microbiota-kidney cross-talk in acute kidney injury. Semin Nephrol. (2019) 39:107–16. doi: 10.1016/j.semnephrol.2018.10.009
45. Alikhan MA, Huynh M, Kitching AR, Ooi JD. Regulatory T cells in renal disease. Clin Transl Immunol. (2018) 7:e1004. doi: 10.1002/cti2.1004
46. Mitsdoerffer M, Lee Y, Jäger A, Kim H-J, Korn T, Kolls JK, et al. Proinflammatory thelper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci USA. (2010) 107:14292–7. doi: 10.1073/pnas.1009234107
47. Zheng L, Sinniah R, Hsu SI-H. Pathogenic role of NF-kappaB activation in tubulointerstitial inflammatory lesions in human lupus nephritis. J Histochem Cytochem Off J Histochem Soc. (2008) 56:517–29. doi: 10.1369/jhc.7A7368.2008
48. Hazenberg MD, Spits H. Human innate lymphoid cells. Blood. (2014) 124:700–9. doi: 10.1182/blood-2013-11-427781
49. Li S, Bostick JW, Zhou L. Regulation of innate lymphoid cells by aryl hydrocarbon receptor. Front Immunol. (2018) 8:1909. doi: 10.3389/fimmu.2017.01909
50. Krebs CF, Paust H-J, Krohn S, Koyro T, Brix SR, Riedel J-H, et al. Autoimmune renal disease is exacerbated by S1P-receptor-1-dependent intestinal Th17 cell migration to the kidney. Immunity. (2016) 45:1078–92. doi: 10.1016/j.immuni.2016.10.020
51. Luu M, Monning H, Visekruna A. Exploring the molecular mechanisms underlying the protective effects of microbial SCFAs on intestinal tolerance and food allergy. Front Immunol. (2020) 11:1225. doi: 10.3389/fimmu.2020.01225
52. Mielenz M, Kuhla B, Hammon HM. Abundance of adiponectin system and G-protein coupled receptor GPR109A mRNA in adipose tissue and liver of F2 offspring cows of Charolais × German Holstein crosses that differ in body fat accumulation. J Dairy Sci. (2013) 96:278–89. doi: 10.3168/jds.2012-5816
53. Imig JD, Ryan MJ. Immune and inflammatory role in renal disease. Compr Physiol. (2013) 3:957–76. doi: 10.1002/cphy.c120028
54. Stavropoulou E, Kantartzi K, Tsigalou C, Konstantinidis T, Romanidou G, Voidarou C, et al. Focus on the gut–kidney axis in health and disease. Front Med. (2021) 7:e620102. doi: 10.3389/fmed.2020.620102
55. Kato S, Chmielewski M, Honda H, Pecoits-Filho R, Matsuo S, Yuzawa Y, et al. Aspects of immune dysfunction in end-stage renal disease. Clin J Am Soc Nephrol. (2008) 3:1526–33. doi: 10.2215/CJN.00950208
56. Ando M, Shibuya A, Yasuda M, Azuma N, Tsuchiya K, Akiba T, et al. Impairment of innate cellular response to in vitro stimuli in patients on continuous ambulatory peritoneal dialysis. Nephrol Dial Transplant. (2005) 20:2497–503. doi: 10.1093/ndt/gfi048
57. Sester U, Sester M, Hauk M, Kaul H, Köhler H, Girndt M. T-cell activation follows Th1 rather than Th2 pattern in haemodialysis patients. Nephrol Dial Transplant. (2000) 15:1217–23. doi: 10.1093/ndt/15.8.1217
58. Eleftheriadis T, Antoniadi G, Liakopoulos V, Kartsios C, Stefanidis I. Basic science and dialysis: disturbances of acquired immunity in hemodialysis patients. Semin Dial. (2007) 20:440–51. doi: 10.1111/j.1525-139X.2007.00283.x
59. Sampani E, Stangou M, Daikidou DV, Nikolaidou V, Asouchidou D, Dimitriadis C, et al. P0297 changes of CD4CD28null and CD8CD28null T cells in chronic kidney disease and the impact of different dialysis modalities. Nephrol Dial Transplant. (2020) 35:gfaa142.P0297. doi: 10.1093/ndt/gfaa142.P0297
60. Nallu A, Sharma S, Ramezani A, Muralidharan J, Raj D. Gut microbiome in chronic kidney disease: challenges and opportunities. Transl Res. (2017) 179:24–37. doi: 10.1016/j.trsl.2016.04.007
61. Ramezani A, Raj DS. The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol. (2014) 25:657–70. doi: 10.1681/ASN.2013080905
62. Ardalan M, Vahed SZ. Gut microbiota and renal transplant outcome. Biomed Pharmacother. (2017) 90:229–36. doi: 10.1016/j.biopha.2017.02.114
63. Vossen JM, Guiot HFL, Lankester AC, Vossen ACTM, Bredius RGM, Wolterbeek R, et al. Complete suppression of the gut microbiome prevents acute graft-versus-host disease following allogeneic bone marrow transplantation. PLoS ONE. (2014) 9:e105706. doi: 10.1371/journal.pone.0105706
64. Lee JR, Muthukumar T, Dadhania D, Toussaint NC, Ling L, Pamer E, et al. Gut microbial community structure and complications after kidney transplantation: a pilot study. Transplantation. (2014) 98:697–705. doi: 10.1097/TP.0000000000000370
65. Lee JR, Muthukumar T, Dadhania D, Taur Y, Jenq RR, Toussaint NC, et al. Gut microbiota and tacrolimus dosing in kidney transplantation. PLOS ONE. (2015) 10:e0122399. doi: 10.1371/journal.pone.0122399
66. Coppo R. The gut–kidney axis in IgA nephropathy: role of microbiota and diet on genetic predisposition. Pediatr Nephrol. (2018) 33:53–61. doi: 10.1007/s00467-017-3652-1
67. Hu X, Du J, Xie Y, Huang Q, Xiao Y, Chen J, et al. Fecal microbiota characteristics of Chinese patients with primary IgA nephropathy: a cross-sectional study. BMC Nephrol. (2021) 21:97. doi: 10.1186/s12882-020-01741-9
68. Coppo R. The intestine-renal connection in IgA nephropathy. Nephrol Dial Transplant. (2015) 30:360–6. doi: 10.1016/j.semnephrol.2018.05.020
69. Zhang J, Luo D, Lin Z, Zhou W, Rao J, Li Y, et al. Dysbiosis of gut microbiota in adult idiopathic membranous nephropathy with nephrotic syndrome. Microb Pathog. (2020) 147:104359. doi: 10.1016/j.micpath.2020.104359
70. Gavazzi G, Krause K-H. Ageing and infection. Lancet Infect Dis. (2002) 2:659–66. doi: 10.1016/S1473-3099(02)00437-1
71. Fülöp T, Larbi A, Pawelec G. Human T Cell aging and the impact of persistent viral infections. Front Immunol. (2013) 4:271. doi: 10.3389/fimmu.2013.00271
72. Vasto S, Caruso C. Immunity & ageing: a new journal looking at ageing from an immunological point of view. Immun Ageing. (2004) 1:1. doi: 10.1186/1742-4933-1-1
73. Culley FJ. Natural killer cells in infection and inflammation of the lung. Immunology. (2009) 128:151–63. doi: 10.1111/j.1365-2567.2009.03167.x
74. Butcher S, Chahel H, Lord JM. Ageing and the neutrophil: no appetite for killing? Immunology. (2000) 100:411–6. doi: 10.1046/j.1365-2567.2000.00079.x
75. Yang J, Zhang L, Yu C, Yang X-F, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res. (2014) 2:1. doi: 10.1186/2050-7771-2-1
76. Dunston CR, Griffiths HR. The effect of ageing on macrophage Toll-like receptor-mediated responses in the fight against pathogens. Clin Exp Immunol. (2010) 161:407–16. doi: 10.1111/j.1365-2249.2010.04213.x
77. Agrawal A, Sridharan A, Prakash S, Agrawal H. Dendritic cells and aging: consequences for autoimmunity. Expert Rev Clin Immunol. (2012) 8:73–80. doi: 10.1586/eci.11.77
78. Dunn AJ, Swiergiel AH. The role of cytokines in infection-related behavior. Ann N Y Acad Sci. (1998) 840:577–85. doi: 10.1111/j.1749-6632.1998.tb09596.x
79. Nikolich-Zugich J, Messaoudi I. Mice and flies and monkeys too: caloric restriction rejuvenates the aging immune system of non-human primates. Exp Gerontol. (2005) 40:884–93. doi: 10.1016/j.exger.2005.06.007
80. Lynch LA, O'Connell JM, Kwasnik AK, Cawood TJ, O'Farrelly C, O'Shea DB. Are natural killer cells protecting the metabolically healthy obese patient? Obes Silver Spring Md. (2009) 17:601–5. doi: 10.1038/oby.2008.565
81. Hersperger AR, Martin JN, Shin LY, Sheth PM, Kovacs CM, Cosma GL, et al. Increased HIV-specific CD8+ T-cell cytotoxic potential in HIV elite controllers is associated with T-bet expression. Blood. (2011) 117:3799–808. doi: 10.1182/blood-2010-12-322727
82. Garner ID. The impact of aging on susceptobility to infection. Rev Inf Dis. (1980) 2:801–10. doi: 10.1093/clinids/2.5.801
83. Lynch HE, Goldberg GL, Chidgey A, Van den Brink MRM, Boyd R, Sempowski GD. Thymic involution and immune reconstitution. Trends Immunol. (2009) 30:366–73. doi: 10.1016/j.it.2009.04.003
84. Walford RL. The immunologic theory of aging. Immunol Rev. (1969) 2:171–7. doi: 10.1111/j.1600-065X.1969.tb00210.x
85. Effros RB. Roy Walford and the immunologic theory of aging. Immun Ageing A. (2005) 2:7. doi: 10.1186/1742-4933-2-7
86. Freitas AA, de Magalhães JP. A review and appraisal of the DNA damage theory of ageing. Mutat Res. (2011) 728:12–22. doi: 10.1016/j.mrrev.2011.05.001
87. Aubert G, Lansdorp PM. Telomeres and aging. Physiol Rev. (2008) 88:557–79. doi: 10.1152/physrev.00026.2007
88. Shammas MA. Telomeres, lifestyle, cancer, and aging. Curr Opin Clin Nutr Metab Care. (2011) 14:28–34. doi: 10.1097/MCO.0b013e32834121b1
89. Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. (2012) 11:3599–610. doi: 10.4161/cc.21884
90. Bello AK, Nwankwo E, El Nahas AM. Prevention on chronic kidney disease: a global challenge. Kidney Int. (2005) 68:S11–7. doi: 10.1111/j.1523-1755.2005.09802.x
91. Bikbov B, Purcell CA, Levey AS, Smith M, Abdoli A, Abebe M, et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2020) 395:709–33. doi: 10.1016/S0140-6736(20)30045-3
92. National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis Off J Natl Kidney Found. (2002) 39(2 Suppl. 1):S1–266.
93. Lin Lang C. A review of chronic kidney disease and the immune system: a special form of immunosenescence. J Gerontol Geriat Res. (2014) 3:2. doi: 10.4172/2167-7182.1000144
94. Liapis H, Romagnani P, Anders H-J. New Insights into the pathology of podocyte loss. Am J Pathol. (2013) 183:1364–74. doi: 10.1016/j.ajpath.2013.06.033
95. Weide T, Huber TB. Implications of autophagy for glomerular aging and disease. Cell Tissue Res. (2011) 343:467–73. doi: 10.1007/s00441-010-1115-0
96. Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. (2010) 120:1084–96. doi: 10.1172/JCI39492
97. Efstratiadis G, Divani M, Katsioulis E, Vergoulas G. Renal fibrosis. Hippokratia. (2009) 13:224–9.
98. Linehan E, Fitzgerald DC. Ageing and the immune system: focus on macrophages. Eur J Microbiol Immunol. (2015) 5:14–24. doi: 10.1556/EuJMI-D-14-00035
Keywords: gut, kidney, gut-kidney axis, microbiome, ageing, immunosenescence, chronic kidney disease, microbiota
Citation: Stavropoulou E, Kantartzi K, Tsigalou C, Aftzoglou K, Voidarou C, Konstantinidis T, Chifiriuc MC, Thodis E and Bezirtzoglou E (2021) Microbiome, Immunosenescence, and Chronic Kidney Disease. Front. Med. 8:661203. doi: 10.3389/fmed.2021.661203
Received: 30 January 2021; Accepted: 23 February 2021;
Published: 19 March 2021.
Edited by:
Maria J. Stangou, Aristotle University of Thessaloniki, GreeceReviewed by:
Evangelos Papachristou, University of Patras, GreeceCopyright © 2021 Stavropoulou, Kantartzi, Tsigalou, Aftzoglou, Voidarou, Konstantinidis, Chifiriuc, Thodis and Bezirtzoglou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisavet Stavropoulou, elisabeth.stavropoulou@gmail.com; Konstantia Kantartzi, kkantart@med.duth.gr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.