 Jingsheng Chang†
Jingsheng Chang† Xueling Li
Xueling Li- Department of Nephrology, Longhua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
Increasing evidence supports a role of proximal tubular (PT) injury in the progression of diabetic kidney disease (DKD), in patients with or without proteinuria. Research on the mechanisms of the PT injury in DKD could help us to identify potential new biomarkers and drug targets for DKD. A high glucose transport state and mismatched local hypoxia in the PT of diabetes patients may be the initiating factors causing PT injury. Other mechanism such as mitochondrial dysfunction, reactive oxygen species (ROS) overproduction, ER stress, and deficiency of autophagy interact with each other leading to more PT injury by forming a vicious circle. PT injury eventually leads to the development of tubulointerstitial inflammation and fibrosis in DKD. Many downstream signaling pathways have been demonstrated to mediate these diseased processes. This review focuses mostly on the novel mechanisms of proximal renal tubular injury in DKD and we believe such review could help us to better understand the pathogenesis of DKD and identify potential new therapies for this disease.
Introduction
Diabetic kidney disease (DKD) is a progressive microvascular complication of diabetes mellitus. Within the kidney, the glomeruli, tubules, vessels, and interstitium are disrupted, resulting in impaired renal functions and eventually end-stage renal disease (ESRD). Epidemiological studies have shown that the current global burden of diabetes affects more than 425 million people. Without intervention, the number of individuals with diabetes worldwide will rise to an estimated 629 million in 2045 (1). Given the high prevalence of diabetes, accordingly, the incidence of DKD is rising rapidly with ~30–40% of diabetic patients develop DKD and a third of these patients progress to ESRD, which brings tremendous impacts on the socio-economics (2, 3).
Based on the distinct pathological glomerular changes (4), DKD has previously been regarded as a glomerular disease, and the injury to the renal tubules considered as secondary to glomerular lesions. In the natural history of DKD, the development of persistent microalbuminuria (presence of albumin in the urine) progresses to overt proteinuria, followed by a gradual decline in the glomerular filtration rate (GFR) and eventually renal failure (5). Although albuminuria represents an independent risk factor for DKD, about 20% of patients with non-albuminuric DKD progress to advanced ESRD within 10 years (6). The in-depth understanding of the disease has enabled the identification of some patients with decreased renal function before the presence of microalbuminuria according to creatinine-based estimated glomerular filtration rate (eGFR) (7–10), and these are the patients that tend to progress more rapidly. When compared with patients with proteinuria, these patients tend to have more severe tubulointerstitial fibrosis and tubular atrophy, suggesting that renal tubular injury plays a key role in the progression of DKD in the absence of proteinuria. In general, renal tubular injury is closely correlated with the decline of eGFR in chronic kidney disease (CKD) patients. Recent evidence has suggested that the proximal tubular (PT) injury develops in the early stage of DKD and promotes DKD progression (11). Therefore, this review focuses on the mechanisms of PT injury in DKD.
Hypoxia
The kidney is an oxygen-intensive organ that receives 20% of the cardiac ejection fraction. The activity of renal tubular transport is accountable for major oxygen consumption in kidney metabolism. The process of renal oxygenation consists of a fine and balanced physiological process, which includes oxygen supply determined by renal blood flow as well as arterial oxygen content and oxygen consumption governed by renal tubular reabsorption. The oxygen in the renal cortex is mainly utilized for glomerular filtration and solute reabsorption. Majority of oxygen supply goes to the renal cortex with a low supply to the renal medulla. This ensures an effective countercurrent multiplication system while the oxygen supply of the medulla is extremely limited, albeit slightly higher than its oxygen utilization (12). Therefore, an imbalance between the oxygen supply and oxygen demand in the medulla will result in hypoxic damages to the medulla tubules (13), whereby the renal tubules in the medulla are the most vulnerable to renal ischemic injury (14).
The development of hypoxia depends on three factors: increased oxygen consumption, oxygen utilization disorder, and reduced oxygen supply, which often co-exists simultaneously and interacts with each other to form a vicious circle (15). Studies have shown that 60% of the overall energy consumption of kidneys is devoted to sodium reabsorption with the PT responsible for almost two-thirds through basal Na+/K+ ATPase activity primarily and quantified as ouabain-sensitive O2 consumption (16). The glucose in the tubule fluid is delivered into the cell by secondary active transport mostly via the sodium-dependent glucose transporters 2 (SGLT-2) in the apical membrane of the proximal tubular epithelial cell (PTEC). Although this is not an energy-dependent process, the sustainability of this activity demands a persistent electrochemical gradient of Na+ produced by Na+/K+ ATPase activity. Therefore, excessive glucose reabsorption in PT will invariably lead to increased oxygen consumption in type 2 diabetes (17). Moreover, the diabetic kidney is constantly in a state of high oxygen consumption due to hyperfiltration and increase tubular reabsorption, which increases further the severity of renal tubular hypoxia. This situation is then exacerbated by subsequent mitochondria dysfunction (15). Besides, the most classic complication of diabetes is systemic microangiopathy which is characterized by basement membrane thickening with hyaline deposition. This vascular injury will lead to decreased blood supply and oxygen supply in the kidney (18, 19). Furthermore, gluconeogenesis is a major source of oxygen and energy consumption in the kidney, accountable for 25% of the energy required for sodium reabsorption (20). For diabetic kidneys, the degree of gluconeogenesis in the kidney is increased significantly (14, 21).
In studies using diabetic animal models, outer medullary hypoxia has been demonstrated using blood oxygen level-dependent (BOLD) MRI. Also, both cortical and medullary hypoxia has been reported in the diabetic animal models as well as humans with DKD (22–25). With increased oxygen utilization, hypoxia inducible factor (HIF)-1 α has been implicated in the correlation of hypoxic and tubulointerstitial fibrosis (26–30). On the other hand, SGLT-2 inhibitors have been shown to poses a renal protective effect on diabetes patients by inhibiting glucose reabsorption and its associated high oxygen consumption (21, 31), in addition to targeting HIF-1 α protein to inhibit mitochondria oxygen consumption (32, 33). Furthermore, these factors inhibit and internalize megalin O-GlcNAcylation to reduce the reabsorption of plasma proteins (e.g., albumin and neutrophil gelatinase-associated lipoprotein) in PT, which is renal protective (34).
Mitochondrial Dysfunction
PTEC demands substantial energy to maintain a normal function (35), whereby 65% of the electrolytes and 100% of the glucose and amino acids filtered by the glomeruli are reabsorbed by the PT. PTEC is rich in mitochondria which is an important organelle performing oxidative metabolism in eukaryotic cells mainly through the β-oxidation of fatty acids to produce adenosine triphosphate (ATP) (35). Mitochondria is also a place for aerobic respiration and energy supply of cells, which produces 95% of the energy needed in cellular activities through oxidative phosphorylation and therefore is regarded as the power plant of a cell. Mitochondria is the center of ATP production and its dysfunction leads to apoptosis.
Mitochondrial homeostasis is strictly essential for an optimally functioning kidney, given that the kidney is an organ that demands high energy consumption (36). In diabetes, the epithelial cells of the S1 segment of the PT require a large amount of ATP as an energy source to reabsorb excess glucose. However, ATP production brings superoxide () production concurrently, which can be converted into excessive reactive oxygen species (ROS), leading to mitochondrial damage and disorders in ATP production (36). Indeed, a reduction in the ATP pool represents the initial event of PTEC damage, with the degree of ATP reduction correlates with the severity of the damage (37). Studies have demonstrated that the production level of ROS may exceed the capacity of the local antioxidants, which is the biomarker of renal mitochondrial dysfunction in diabetes (38–41). This is further supported by the changes of bioenergetics and kinetics of mitochondria that may precede the development of DKD (38).
In addition to the driving force of cells, mitochondria have also been regarded as the judge and executor of programmed cell death (42, 43). In mitochondrial homeostasis, a balance in the mitochondrial biogenesis, including fusion and mitophagy, is required (35). Both Fission and fusion complement each other to maintain the mitochondrial morphology under different metabolic conditions, while mitophagy removes damaged mitochondria from the network (35). Mitochondrial swelling is considered an indicator of mitochondria dysfunction (44), which can be confirmed by electron microscopy (45, 46). Uncontrolled mitochondria dysfunction eventually leads to the activation of the intrinsic cell death pathway and cell death (47, 48). Cell death may present in various forms, including apoptosis, autophagic cell death, pyroptosis (49).
In recent years, increasing research studies have been performed on the role of oxidative stress in cell death, given its integral role in tubule injury in DKD. Studies have shown that AOPPs (50, 51) induces oxidative stress and DKD mitochondria dysfunction through CD36/β-Catenin and PKC pathways, leading to tubulointerstitial fibrosis. On the contrary, in animal studies using DKD mice, PGC-1 α (52) ameliorates renal fibrosis via an antioxidant mechanism. Antioxidants (tempol and ramipril) inhibit NADPH upregulation by negatively regulating the endoplasmic reticulum stress (ERS) and inflammation to improve renal damage in DKD (53). Oxidative stress and endoplasmic reticulum stress positively regulate by each other, forming a vicious cycle (54). Sirt3-CD38 has also been shown to play a role in diabetic renal tubule damage by regulation of mitochondrial oxidative stress (55, 56).
Given that the mitochondria may be a target for therapeutic intervention, the mechanisms of some potential drugs have been explored. SS31, a novel antioxidative peptide that targets mitochondria, has been specially designed to concentrate in the inner mitochondrial membrane (57), which reduces renal tubulointerstitial damage in diabetic mice by decreasing mitochondrial fragments and restoring mitochondrial morphology through the inhibition of Drp1 expression and upregulation of Mfn1 expression in renal tubular epithelial cells. Also, the role of SS31 has been associated with CD36 (58). Besides, Na2S4, a polysulfide donor that directly sulfhydrates SIRT1, reduces high glucose-induced oxidative stress, cell apoptosis, inflammatory response in renal tubular epithelial cells, and the progression of epithelial-to-mesenchymal transition (EMT) (59). Also, Carnosine has been shown to significantly decrease the production of ROS, alleviate oxidative stress, and inhibit apoptosis through mitochondrial pathway in vitro (60) and in vivo (61). This may be a promising drug for the treatment of DKD. All these studies shed light on the new potential therapeutic agents in the prevention of renal tubulointerstitial damage through regulation of mitochondrial function and ROS production.
Innate Immunity
A persistently high glucose can cause abnormal activation of mitochondrial endoplasmic reticulum stress and intracellular signal transduction pathways, leading to cell stress and cellular dysfunction. The abnormal activation following each stress response promotes further activation of downstream inflammatory factors, the release of damps, and induction of innate immune response. The innate immune response induces a continuous process of chronic inflammatory reaction in the kidney, leading to substantial mesangial hyperplasia and renal interstitial fibrosis, which lays the foundation for the occurrence and development of DKD (62). Compared with adaptive immunity, the mechanism of an innate immune response plays an integral role in the occurrence of diabetic kidney injury (63, 64), which is composed of pattern recognition receptors that recognize pathogenic and endogenous ligands. The bindings of ligands trigger several complex inflammatory cascade reactions, including Toll-like receptor (TLR) signaling, nucleotide-binding domain and leucine-rich repeat containing receptors (NLRs), the kallikrein–kinin system (KKS), protease-activated receptor (PAR) signaling, and the complement cascade, resulting in further renal fibrosis and other renal damages (65). In particular, the complement cascade plays a key role in innate immunity that is responsible for the pathogenesis of several immune-mediated inflammatory diseases (66). A study has shown that the novel aptamer (NOX- D21) improves renal function and reduces tubulointerstitial fibrosis by inhibiting the expression of C5a in db/db mice (67). TAM receptors (Tyro3, Axl, and Mer) have been implicated in the innate immunity (68). Studies have demonstrated an obvious TAM shedding in DKD patients, though the mechanism of this observation remains unclear. Further research is warranted to establish the role of TAM in the development of renal injury and DKD.
Angiotensin II
Angiotensin II (AngII) is also recognized as a mediator of hyperglycemia-induced renal damage. The concentration of renal Ang II is ~1,000-folds higher than that of circulating AngII (69). An increased AngII level is implicated in the development of renal fibrosis by directly upregulating the pro-fibrosis genes (70). Early studies have revealed that AngII induces cellular hypertrophy of tubular cells that is mediated by the activation of endogenous TGF-β (71, 72). Also, the study of primary PT has demonstrated that glucose significantly increases the concentration of AngII in cell lysates, while angiotensin receptor blocker (ARB) significantly reduces this effect of AgnII (73, 74). Furthermore, AngII induces ROS (75) and EMT (76, 77), leading to tubular cell damage. Importantly, recent studies have revealed a high affinity of angiotensin II type 2 receptor (AT2R) in the mitochondria of renal tubules. In the early stage of diabetes, AT2R inhibits the production of mitochondrial reactive oxygen species and cell proliferation. Overexpression of AT2R in tubular epithelial cells contributes to the decreased mitochondrial bioenergy efficiency and increased mitochondrial superoxide production (78).
In the current clinical practice, angiotensin-converting enzyme inhibitors (ACEI), and ARB are the first-line drugs being used in the prevention of DKD. Several recent studies have shown that the combination therapy of renin-angiotensin system (RAS) inhibitor together with neprilysin inhibitor was more effective in preventing renal fibrosis than using RAS inhibitor alone in the development of DKD [LCZ696 and angiotensin receptor blocker (79); combination of sacubitril [NEPi] and valsartan (80); combination of thiorphan [NEPi]/telmisartan [ARB]; and thiorphan/Dize [ACE2 activator] therapies (81)]. Moreover, the combination of PGE1 with ACE inhibitor protects renal function more as compared with PGE1 or ACEI monotherapy (82). These studies provide evidence on the alternative options of effective clinical treatment with RAS blockers.
Fatty Acids
In healthy kidneys, ATP is primarily generated via oxidative phosphorylation (OXPHOS) of fatty acid (FA). However, in diabetics, the utilization of fatty acid is changed to glycolysis and lipid accumulation, which also represents an important pathway of DKD due to lipid accumulation in the renal tubular epithelia (83) via increased absorption and synthesis of fatty acids, in addition to decreased utilization. The toxic effect of FA on the renal tubular epithelial cells is associated with hypoxia and mitochondrial dysfunction (33, 84). A recent study has shown that FATP2, a member of the fatty acid transporter family, regulates DKD pathogenesis through a combined lipotoxicity and glucotoxicity (glucolipotoxicity) mechanism (85). Nevertheless, PBI-4050, which is a fatty acid receptor modulator, attenuates the development of DKD in type 2 diabetes (86). Saturated fatty acid (SFA)-related lipotoxicity is also the pathogenesis of diabetes-related PT cell damage. Therefore, increasing the enzymes that metabolize free fatty acid (FFA) can theoretically protect the PT cells from SFA-related lipotoxicity. The study by Iwai et al. has found a significantly lower expression of Stearoyl-CoA Desaturase-1 (SCD1) in the kidney of diabetic mice induced by a high-fat diet (HFD) than that of non-diabetic mice. Thus, enhancing SCD1-mediated desaturation of SFA and subsequent formation of neutral lipid droplets may provide a promising therapeutic target to reduce SFA-induced lipotoxicity (87). Besides, through restoring functional lymphatic vessels, SAR13175 was able to eliminate inflammatory cells and toxic lipid metabolites in the kidney that can also improve lipotoxicity-related fibrosis in diabetes (88).
Autophagy
Autophagy is a highly conserved pathway through which cells degrade and recycle macromolecules and organelles. Growing evidence shows dysregulated autophagy in DKD (89). The well-known autophagy regulation pathways include mammalian target of rapamycin (mTOR), Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and sirtuins (SIRT). In addition, a variety of stress conditions, including hypoxia, oxidative stress, ERS, and metabolism, have been shown to regulate autophagy (90, 91). In general, mild to moderate ERS and activation of autophagy play a protective role in kidney cells. When the harmful stimulus cannot be effectively alleviated, this leads to the sustained ERS creating an imbalance between ERS and autophagy. This will lead to kidney cell injury and progression of DKD (92).
mTOR can interact with several proteins to form two different complexes, namely mTORC1 and mTORC2, to regulate autophagy. There is ample evidence that mTORC1 is a key regulator of autophagy, which regulates different steps of autophagy such as nucleation, elongation, maturation, and termination (93). mTORC2 indirectly regulates autophagy by activating mTORC1. In general, mTORC1 is a negative regulator of autophagy by inhibiting the activity of Ulk1 complex through direct phosphorylation. On the contrary, AMPK and SIRT1 are effective positive regulator of autophagy (89). In recent years, some new findings have been made in this field. Huang et al. identified KCa3.1 (calcium-activated K+ channel) involved in renal tubular autophagy dysfunction through PI3K/Akt/mTOR signaling pathway in DKD (94). Theodomir et al. confirmed that P2Y2R deficiency increased the expression of sirtuin-1 and FOXO3a, which enhanced autophagy and improved renal insufficiency in DKD (95). In addition, Yang et al. found that Smad3, the downstream transcription factor activated by TGFβ (transforming growth factor β), suppressed lysosome biogenesis in a TFEB-dependent manner (96). Furthermore, ATF4 (activating transcription factor 4) (97), TRAIL (TNF related apoptosis inducing ligand) (98), Soluble epoxide hydrolase (sEH) and lys63 UB proteins were also confirmed to be involved in the regulation of autophagy in the kidney cells.
Targeting various components of autophagy pathway may become a new strategy for clinical treatment of DKD. As a potential target for regulating autophagy, Mikhail V blagosklony proposed rapamycin (sirolimus) for the treatment of diabetic kidney injury (99). However, clinical studies have found that rapamycin and its analogs can cause immunosuppression, glucose intolerance, increased risk of type 2 diabetes, and other side effects (100). In particular, it has been reported that long-term use of rapamycin can aggravate glomerular damage and increase albuminuria (101). Recently, Dudley W. Lamming group discovered the highly selective compound DL001, which inhibits mTORC1, could be developed for the treatment of DKD (100). In addition, several other drugs have been shown to improve DKD in vivo and in vitro models by regulating autophagy (102–104). SGLT2 inhibitors are also thought to increase autophagy in diabetic kidneys (105). The role of autophagy in the development of diabetes is still insufficient, and more experiments are needed to further elaborate in this field.
Inflammation and EMT
In the development of tubulointerstitial fibrosis, the complicated process of inflammation not only is the initiating factor but also the result of the development of several other factors. Local inflammation in renal tubules is a marker of progressive renal disease (106). Additionally, systemic inflammation exists in patients with type 2 diabetes, which involves the production of a large variety of chemokines that promotes inflammation in the microenvironment, thus increasing renal damage. Inflammation promotes renal infiltration of monocytes and lymphocytes, which augments further the inflammatory response and the development of cell damage and fibrosis (71). Additionally, a large number of macrophages, lymphocytes, and mast cells infiltrate and secrete copious pro-inflammatory cytokines and oxygen-free radicals, which could provoke renal tissue damage and accelerate the process of renal fibrosis (107). Renal tubular inflammation is associated with several triggers, including local hyperglycemia, advanced glycation product, mitochondria oxidative stress, angiotensin II, PKC, and other factors (108). Recent evidence on the effects of histamines in renal function suggests that histamines may also contribute to glomerular hyperfiltration, inflammation, fibrosis, and tubule hypertrophy (109).
Other Pathways Discovered in Recent Years
Numerous cell signaling pathways have been confirmed to play a role in the progress of DKD. Here, we discussed some of the new pathways discovered in recent years.
Hippo Signal Pathway
The Hippo signal transduction pathway has been heavily researched in recent years. Experimental studies have shown the important roles of the Hippo signal transduction pathway in regulating organ size, carcinogenesis, tissue regeneration, and functions of stem cells. YAP (Yes-associated protein) and its homologous protein, TAZ (transcriptional coactivator with PDZ-binding motif), are the main effector molecules of the Hippo pathway. The study by Yang et al. has demonstrated that the activated YAP induced by the inhibition of MST1 up-regulates the activation of TEAD directly by binding to TEAD to form YAP-TEAD heterodimer, which promotes the expression of pro-fibrosis genes in the renal tubular epithelial cells (110). A high expression of YAP, TEAD, and CTGF was found in renal tissue of patients with type 2 DKD suggesting a key role of YAP in renal damage, while YAP expression is also correlated with Systolic BP, BUN, Cr, DKD stage, DKD pathological grade, serum albumin, and eGFR (111). The expression of YAP protein and its phosphorylation were also upregulated in the renal PTs of diabetic mice. Further studies have revealed that the activated EGFR-PI3K-Akt-CREB signaling pathway mediates the YAP gene expression, nuclear translocation, and interaction with the TEAD transcription factor complex (112). Besides, TAZ has been shown as a novel non-SMAD downstream effector of renal TGF-β1 signaling, which is activated in fibrotic kidney via TGF-β1-dependent mechanisms, while a sustained TAZ signaling promotes epithelial maladaptive repair (113).
Nod-Like Receptors (NLRs)
NLRs are a family of cytoplasmic pattern-recognition receptors, which play several key roles in both innate and adaptive immunity (114–116) by inducing inflammation and cell death while facilitating rapid removal of invasive pathogens. Different NLRs poses distinct roles in regulating immunity and inflammation (117). NLRC3 inflammasome aggravates tubular injury through promoting pro-inflammatory and pro-fibrotic response of renal tubular cells (118). A study demonstrated that the reduction of NLRP3 inflammasome suppressed by the TNF-α inhibition alleviated tubular injury in DKD rats (119). Moreover, NLRP3 exerts inflammasome-independent effects on TGFβ signaling, which contributes to renal fibrosis in DKD (120). The role of NLRC5 has been shown to be multifaceted in the progression of DKD. Under high-glucose conditions, NLRC5 enhances IκB phosphorylation and reprograms macrophages toward the M1 phenotype in addition to activating the TGFβ signaling (121). Macrophages are closely related to interstitial fibrosis (122). Among the variety of phenotypes of macrophages, macrophages of M1 phenotype infiltrated the diabetic kidneys at the early stage play mainly the pro-inflammatory role, while the activation of macrophages M2 occurs in the late stage to promote renal fibrosis in DKD (123–125).
PTEN
PTEN decreases in diabetic renal tubular epithelial cells when cultured with high glucose, contributing to impaired autophagy and renal fibrosis (126, 127). Animal studies have demonstrated that although the level of unmodified Pten decreases, the level of PtenK27−polyUb increases significantly with the damaged renal tubules. Sufficient serine/threonine phosphatase activity can be obtained after the modification of PtenK27−polyUb to remove the phosphate groups of TWIST, SNAI1, and YAP. Consequently, these pro-fibrosis transcriptional factors activate the pro-fibrosis genes (128). Li et al. have proposed that the unmodified PTEN (EMT prophylaxis) and PtenK27−polyUb (EMT promotion) are dynamically regulated in kidney disease, in which the identification of PtenK27−polyUb may help in the early diagnosis of DKD and represent a potential therapeutic target.
Zinc Transporter
Zinc transporters are categorized into Zrt/Irt-related protein (ZIP) and zinc transporters (ZnT), which function together to maintain intracellular zinc homeostasis. In the cytoplasm, both ZIP and ZnT are zinc transfer proteins (129). Studies have demonstrated the subcellular localization of ZnT8 on the insulin secretory vesicle membrane of the islet β cells, which promotes the synthesis, storage, and secretion of insulin and regulates the homeostasis of intracellular free zinc ions. The study by Zhang et al. has found that ZnT8 is highly expressed in the tubular epithelial cells but only weakly expressed in the glomeruli or podocytes, and confirmed the protective effect of ZnT8 against tubulointerstitial fibrosis by inhibiting the TGF-β1/Smads signal pathway. However, in normal circumstances, overexpression, or knock-down of ZnT7 does not alter the phosphorylation level of Smad2/3 (130). On the other hand, Zhang et al. (52) suggest the important anti-fibrotic role of Zn via the PI3K/Akt/GSK-3β signaling pathway. Nevertheless, the role of Zn in the pathogenesis of DKD requires further research and clarification.
Others
In addition to the above, several other signaling pathways have been studied in PT in DKD. These include FoxO1-STAT1 signaling (131), TSC1-mTORC1 signaling (132), HSP70-TLR4 axis (133), and PDGFRβ/Akt/mTORC1 nexus (134).
MicroRNA
MicroRNA (miRNA) is a small molecule that attracts great interest in the field of DKD research, given that it has been implicated in the occurrence and development of DKD. In particular, miRNAs participate in the progress of tubulointerstitial fibrosis, leading to structural changes and dysfunction of renal tubules. Also, miRNA and RNA-induced silencing complex (RISC) form a complex (135), which inhibits the expression of target genes by promoting mRNA degradation or inhibiting mRNA translation. Thus, whether miRNA promotes or inhibits fibrosis will depend on their specific target genes related to fibrosis as summarized in the table below (Table 1).

Table 1. miRNAs related to tubulointerstitial fibrosis in diabetic kidney disease.
Biomarkers of Tubular Cell Injury
Several biomarkers of tubular cell injury have been identified in patients with DKD. TNFR1 and TNFR2 have been shown as reliable biomarkers for predicting the progression of DKD (158) and their levels also correlate with tubular cell injury and inflammation (159). Kim1 is known as an early biomarker for DKD and its level increases even prior to the onset of microalbuminuria (160). Urinary N-acetyl-beta-d-glucosaminidase (NAG) is also considered as a potential early biomarker for DKD (161).A cross-sectional study shows that u-NGAL and RBP-4 are potential markers of tubular damage which can be used as complementary measurements to albuminuria and GFR in the early diagnosis of DKD (162).
Acute Kidney Injury (AKI) and DKD
Patients with DKD were susceptible to severe AKI and usually had a worse prognosis following AKI (163). Advani recently summarized clearly that diabetes may increase the risk of AKI while AKI may increase the risk of CKD in diabetes (164). PT suffers from more severe renal tubular hypoxia and mitochondria dysfunction in diabetic kidney. Inflammatory cytokines have been also reported to be upregulated in diabetic kidney leading to serial cascades of inflammation (165). Hyperglycemia, advanced glycation end products (AGEs) and albuminuria itself can induce the expression of adhesion molecules and chemokines in proximal tubular cells to aggravate injury. In a separate study (166), STZ-induced and Akita diabetic mouse models exhibited heightened susceptibility to increased tubule cell damage and programmed cell death caused by ischemia reperfusion injury (IRI). Proximal tubule cells exposed to high glucose exhibited higher apoptosis following depletion of ATP or exposure to severe hypoxia. The authors (166) identified activation of the intrinsic pathway of apoptosis characterized by mitochondrial Bax accumulation and cytochrome c release, and the activation of the intrinsic pathway of apoptosis which was induced by the upregulation of p53 in tubule cells exposed to high glucose and ischemic insult. The studies by Kelly et al. also showed that DKD patients are more susceptible to renal ischemia leading to more severe tubular cell apoptosis (167, 168).
In addition, miRNAs have been shown to be highly promising diagnostic markers for early DKD and it may have a potential role in the treatment of DKD. However, one miRNA often has multiple target genes, while one target gene may also be regulated by multiple miRNAs. Given this complexity, further studies are warranted to ascertain the specific roles of miRNA in renal fibrosis before considering their potentials application in clinical setting.
Conclusions
PT injury appears in the early stage of DKD and continues throughout the progression of DKD (169). Based on its structural and functional characteristics, PTs are vulnerable to injury in hyperglycemic states and difficult to recover. In diabetic patients, a high glucose transport state and local relative oxygen deficiency (primary and secondary) in PT may be the initial factors of tubular damage, while excessive mitochondria damages and ROS production are important contributors to the further damage of PTs in DKD. Abnormalities in hemodynamics, glucose and lipid metabolism, mitochondria, oxidative stress, inflammation, and many other factors interact with each other and form a vicious circle, leading to the renal tubular dysfunctions (Figure 1).
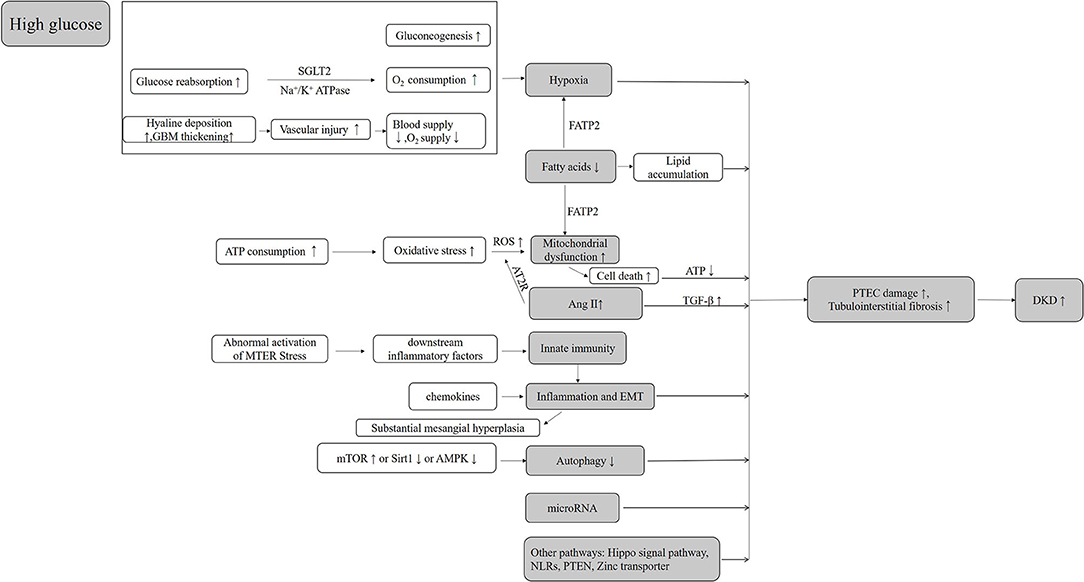
Figure 1. The mechanisms of tubular cell injury in DKD.
In this review, we discussed the potential mechanisms of renal tubular damage in DKD and potential therapeutic targets to prevent or treat the tubular cell injury. Renal tubular damage is a complex and dynamic process involving a “tubulocentric view” or “glomerulocentric view,” which represents a manifestation of different stages in the development of DKD. New studies are required to further understand the pathogenesis of tubular injury in DKD and to develop specific treatments to prevent and delay the tubular injury in DKD.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
YZ was supported by The National Natural Science Foundation of China [81973772]; The 2018–2020 Three-year Action Plan for Traditional Chinese Medicine Further Development in Shanghai [ZY(2018-2020)-CCCX-2002-02]; the Shanghai leadership program for Chinese Medicine [ZY(2018-2020)-RCPY-1007]; Shanghai Shuguang Scholar [16SG37]; Shanghai Municipal Key Clinical Specialty [shslczdzk04201]; RZ was supported by Senior Talents Program of Integrated Traditional Chinese and Western Medicine in Shanghai [ZY(2018-2020)-RCPY-2002].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. (2018) 138:271–81. doi: 10.1016/j.diabres.2018.02.023
2. Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. (2014) 103:137–49. doi: 10.1016/j.diabres.2013.11.002
3. Packham DK, Alves TP, Dwyer JP, Atkins R, de Zeeuw D, Cooper M, et al. Relative incidence of ESRD versus cardiovascular mortality in proteinuric type 2 diabetes and nephropathy: results from the DIAMETRIC (Diabetes Mellitus Treatment for Renal Insufficiency Consortium) database. Am J Kidney Dis. (2012) 59:75–83. doi: 10.1053/j.ajkd.2011.09.017
4. Kimmelstiel P, Wilson C. Intercapillary lesions in the glomeruli of the kidney. Am J Pathol. (1936) 12:83–98 7.
5. Krolewski AS. Progressive renal decline: the new paradigm of diabetic nephropathy in type 1 diabetes. Diabetes Care. (2015) 38:954–62. doi: 10.2337/dc15-0184
6. Yamanouchi M, Furuichi K, Hoshino J, Toyama T, Hara A, Shimizu M, et al. The Japan agency for medical, and development, nonproteinuric versus proteinuric phenotypes in diabetic kidney disease: a propensity score-matched analysis of a nationwide, biopsy-based cohort study. Diabetes Care. (2019) 42:891–902. doi: 10.2337/dc18-1320
7. Kramer HJ, Nguyen QD, Curhan G, Hsu CY. Renal insufficiency in the absence of albuminuria and retinopathy among adults with type 2 diabetes mellitus. JAMA. (2003) 289:3273–7. doi: 10.1001/jama.289.24.3273
8. MacIsaac RJ, Tsalamandris C, Panagiotopoulos S, Smith TJ, McNeil KJ, Jerums G. Nonalbuminuric renal insufficiency in type 2 diabetes. Diabetes Care. (2004) 27:195–200. doi: 10.2337/diacare.27.1.195
9. Yokoyama H, Sone H, Oishi M, Kawai K, Fukumoto Y, Kobayashi M, et al. Prevalence of albuminuria and renal insufficiency and associated clinical factors in type 2 diabetes: the Japan Diabetes Clinical Data Management study (JDDM15). Nephrol Dial Transplant. (2009) 24:1212–9. doi: 10.1093/ndt/gfn603
10. Thomas MC, Macisaac RJ, Jerums G, Weekes A, Moran J, Shaw JE, et al. Nonalbuminuric renal impairment in type 2 diabetic patients and in the general population (national evaluation of the frequency of renal impairment cO-existing with NIDDM [NEFRON] 11). Diabetes Care. (2009) 32:1497–502. doi: 10.2337/dc08-2186
11. Nowak N, Skupien J, Niewczas MA, Yamanouchi M, Major M, Croall S, et al. Increased plasma kidney injury molecule-1 suggests early progressive renal decline in non-proteinuric patients with type 1 diabetes. Kidney Int. (2016) 89:459–67. doi: 10.1038/ki.2015.314
12. Brezis M, Rosen S, Silva P, Epstein FH. Renal ischemia: a new perspective. Kidney Int. (1984) 26:375–83. doi: 10.1038/ki.1984.185
13. Korner A, Eklof AC, Celsi G, Aperia A. Increased renal metabolism in diabetes. mechanism and functional implications. Diabetes. (1994) 43:629–33. doi: 10.2337/diabetes.43.5.629
14. Scheen AJ. Sodium-glucose cotransporter type 2 inhibitors for the treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. (2020) 16:556–77. doi: 10.1038/s41574-020-0392-2
15. Gilbert RE. Proximal tubulopathy: prime mover and key therapeutic target in diabetic kidney disease. Diabetes. (2017) 66:791–800. doi: 10.2337/db16-0796
17. Meyer C, Stumvoll M, Nadkarni V, Dostou J, Mitrakou A, Gerich J. Abnormal renal and hepatic glucose metabolism in type 2 diabetes mellitus. J Clin Invest. (1998) 102:619–24. doi: 10.1172/JCI2415
18. Osterby R, Parving HH, Nyberg G, Hommel E, Jorgensen HE, Lokkegaard H, et al. A strong correlation between glomerular filtration rate and filtration surface in diabetic nephropathy. Diabetologia. (1988) 31:265–70.
19. Bohle A, Mackensen-Haen S, Wehrmann M. Significance of postglomerular capillaries in the pathogenesis of chronic renal failure. Kidney Blood Press Res. (1996) 19:191–5. doi: 10.1159/000174072
20. Cohen JJ. Relationship between energy requirements for Na+ reabsorption and other renal functions. Kidney Int. (1986) 29:32–40. doi: 10.1038/ki.1986.5-
21. Meyer C, Woerle HJ, Dostou JM, Welle SL, Gerich JE. Abnormal renal, hepatic, and muscle glucose metabolism following glucose ingestion in type 2 diabetes. Am J Physiol Endocrinol Metab. (2004) 287:E1049–56. doi: 10.1152/ajpendo.00041.2004
22. Ries M, Basseau F, Tyndal B, Jones R, Deminiere C, Catargi B, et al. Renal diffusion and BOLD MRI in experimental diabetic nephropathy. blood oxygen level-dependent. J Magn Reson Imaging. (2003) 17:104–3. doi: 10.1002/jmri.10224
23. Edlund J, Hansell P, Fasching A, Liss P, Weis J, Glickson JD, et al. Reduced oxygenation in diabetic rat kidneys measured by T2* weighted magnetic resonance micro-imaging. Adv Exp Med Biol. (2009) 645:199–204. doi: 10.1007/978-0-387-85998-9_31
24. Inoue T, Kozawa E, Okada H, Inukai K, Watanabe S, Kikuta T, et al. Noninvasive evaluation of kidney hypoxia and fibrosis using magnetic resonance imaging. J Am Soc Nephrol. (2011) 22:1429–34. doi: 10.1681/ASN.2010111143
25. Yin WJ, Liu F, Li XM, Yang L, Zhao S, Huang ZX, et al. Noninvasive evaluation of renal oxygenation in diabetic nephropathy by BOLD-MRI. Eur J Radiol. (2012) 81:1426–31. doi: 10.1016/j.ejrad.2011.03.045
26. Singh DK, Winocour P, Farrington K. Mechanisms of disease: the hypoxic tubular hypothesis of diabetic nephropathy. Nat Clin Pract Nephrol. (2008) 4:216–26. doi: 10.1038/ncpneph0757
27. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. (2007) 117:3810–20. doi: 10.1172/JCI30487
28. Takiyama Y, Harumi T, Watanabe J, Fujita Y, Honjo J, Shimizu N, et al. Tubular injury in a rat model of type 2 diabetes is prevented by metformin: a possible role of HIF-1alpha expression and oxygen metabolism. Diabetes. (2011) 60:981–92. doi: 10.2337/db10-0655
29. Nayak BK, Shanmugasundaram K, Friedrichs WE, Cavaglierii RC, Patel M, Barnes J, et al. HIF-1 mediates renal fibrosis in OVE26 Type 1 diabetic mice. Diabetes. (2016) 65:1387–97. doi: 10.2337/db15-0519
30. Bhatt K, Wei Q, Pabla N, Dong G, Mi QS, Liang M, et al. MicroRNA-687 induced by hypoxia-inducible factor-1 targets phosphatase and tensin homolog in renal ischemia-reperfusion injury. J Am Soc Nephrol. (2015) 26:1588–96. doi: 10.1681/ASN.2014050463
31. Wanner C, Inzucchi SE, Zinman B. Empagliflozin and progression of kidney disease in Type 2 diabetes. N Engl J Med. (2016) 375:1801–2. doi: 10.1056/NEJMc1611290
32. Bessho R, Takiyama Y, Takiyama T, Kitsunai H, Takeda Y, Sakagami H, et al. Hypoxia-inducible factor-1alpha is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci Rep. (2019) 9:14754. doi: 10.1038/s41598-019-51343-1
33. Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1alpha-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. (2020) 11:390. doi: 10.1038/s41419-020-2544-7
34. Otomo H, Nara M, Kato S, Shimizu T, Suganuma Y, Sato T, et al. Sodium-glucose cotransporter 2 inhibition attenuates protein overload in renal proximal tubule via suppression of megalin O-GlcNacylation in progressive diabetic nephropathy. Metabolism. (2020) 113:154405. doi: 10.1016/j.metabol.2020.154405
35. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. (2017) 13:629–46. doi: 10.1038/nrneph.2017.107
36. Higgins G, Coughlan M. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol. (2014) 171:1917–42. doi: 10.1111/bph.12503
37. Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. (2003) 14:2199–210. doi: 10.1097/01.ASN.0000079785.13922.F6
38. Coughlan MT, Nguyen TV, Penfold SA, Higgins GC, Thallas-Bonke V, Tan SM, et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin Sci. (2016) 130:711–20. doi: 10.1042/CS20150838
39. Coughlan MT, Higgins GC, Nguyen TV, Penfold SA, Thallas-Bonke V, Tan SM, et al. Deficiency in apoptosis-inducing factor recapitulates chronic kidney disease via aberrant mitochondrial homeostasis. Diabetes. (2016) 65:1085–98. doi: 10.2337/db15-0864
40. Tan AL, Sourris KC, Harcourt BE, Thallas-Bonke V, Penfold S, Andrikopoulos S, et al. Disparate effects on renal and oxidative parameters following RAGE deletion, AGE accumulation inhibition, or dietary AGE control in experimental diabetic nephropathy. Am J Physiol Renal Physiol. (2010) 298:F763–70. doi: 10.1152/ajprenal.00591.2009
41. Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. (2013) 123:4888–99. doi: 10.1172/JCI66218-
42. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. (2010) 11:621–32. doi: 10.1038/nrm2952
43. Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. (2012) 111:1208–21. doi: 10.1161/CIRCRESAHA.112.265819
44. Romanello V, Sandri M. Mitochondrial quality control and muscle mass maintenance. Front Physiol. (2015) 6:422. doi: 10.3389/fphys.2015.00422
45. Kaneda K, Sakata N, Takebayashi S. Mitochondrial enlargement and basement membrane thickening of renal proximal tubules, possible initiators of microalbuminuria in non-insulin-dependent diabetics (NIDDM). Acta Pathol Jpn. (1992) 42:793–9. doi: 10.1111/j.1440-1827.1992.tb01880.x
46. Takebayashi S, Kaneda K. Mitochondrial derangement: possible initiator of microalbuminuria in NIDDM. J Diabet Complicat. (1991) 5:104–6. doi: 10.1016/0891-6632(91)90034-M
47. Davis RE, Williams M. Mitochondrial function and dysfunction: an update. J Pharmacol Exp Ther. (2012) 342:598–607. doi: 10.1124/jpet.112.192104
48. Smith RA, Hartley RC, Cocheme HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. (2012) 33:341–52. doi: 10.1016/j.tips.2012.03.010
49. Lin J, Cheng A, Cheng K, Deng Q, Zhang S, Lan Z, et al. New insights into the mechanisms of pyroptosis and implications for diabetic kidney disease. Int J Mol Sci. (2020) 21:7057. doi: 10.3390/ijms21197057
50. Li X, Xu L, Hou X, Geng J, Tian J, Liu X, et al. Advanced oxidation protein products aggravate tubulointerstitial fibrosis through protein Kinase C-dependent mitochondrial injury in early diabetic nephropathy. Antioxid Redox Signal. (2019) 30:1162–85. doi: 10.1089/ars.2017.7208
51. Li X, Zhang T, Geng J, Wu Z, Xu L, Liu J, et al. Advanced oxidation protein products promote lipotoxicity and tubulointerstitial fibrosis via CD36/beta-catenin pathway in diabetic nephropathy. Antioxid Redox Signal. (2019) 31:521–38. doi: 10.1089/ars.2018.7634
52. Zhang L, Liu J, Zhou F, Wang W, Chen N. PGC-1alpha ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Mol Med Rep. (2018) 17:4490–8. doi: 10.3892/mmr.2018.8433
53. De Blasio MJ, Ramalingam A, Cao AH, Prakoso D, Ye JM, Pickering R, et al. The superoxide dismutase mimetic tempol blunts diabetes-induced upregulation of NADPH oxidase and endoplasmic reticulum stress in a rat model of diabetic nephropathy. Eur J Pharmacol. (2017) 807:12–20. doi: 10.1016/j.ejphar.2017.04.026-
54. Sharma I, Deng F, Liao Y, Kanwar YS. Myo-inositol Oxygenase (MIOX) overexpression drives the progression of renal tubulointerstitial injury in diabetes. Diabetes. (2020) 69:1248–63. doi: 10.2337/db19-0935
55. Ogura Y, Kitada M, Monno I, Kanasaki K, Watanabe A, Koya D. Renal mitochondrial oxidative stress is enhanced by the reduction of Sirt3 activity, in Zucker diabetic fatty rats. Redox Rep. (2018) 23:153–9. doi: 10.1080/13510002.2018.1487174
56. Ogura Y, Kitada M, Xu J, Monno I, Koya D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD(+)/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging. (2020) 12:11325–36. doi: 10.18632/aging.103410-
57. Rocha M, Hernandez-Mijares A, Garcia-Malpartida K, Banuls C, Bellod L, Victor VM. Mitochondria-targeted antioxidant peptides. Curr Pharm Des. (2010) 16:3124–31. doi: 10.2174/138161210793292519
58. Hou Y, Shi Y, Han B, Liu X, Qiao X, Qi Y, et al. The antioxidant peptide SS31 prevents oxidative stress, downregulates CD36 and improves renal function in diabetic nephropathy. Nephrol Dial Transplant. (2018) 33:1908–18. doi: 10.1093/ndt/gfy021
59. Sun HJ, Xiong SP, Cao X, Cao L, Zhu MY, Wu ZY, et al. Polysulfide-mediated sulfhydration of SIRT1 prevents diabetic nephropathy by suppressing phosphorylation and acetylation of p65 NF-kappaB and STAT3. Redox Biol. (2021) 38:101813. doi: 10.1016/j.redox.2020.101813
60. Cao Y, Xu J, Cui D, Liu L, Zhang S, Shen B, et al. Protective effect of carnosine on hydrogen peroxide-induced oxidative stress in human kidney tubular epithelial cells. Biochem Biophys Res Commun. (2021) 534:576–582. doi: 10.1016/j.bbrc.2020.11.037
61. Liu XQ, Jiang L, Lei L, Nie ZY, Zhu W, Wang S, et al. Carnosine alleviates diabetic nephropathy by targeting GNMT, a key enzyme mediating renal inflammation and fibrosis. Clin Sci. (2020) 134:3175–93. doi: 10.1042/CS20201207
62. Tesch GH. Diabetic nephropathy - is this an immune disorder? Clin Sci. (2017) 131:2183–99. doi: 10.1042/CS20160636
63. Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol. (2006) 17:368–77. doi: 10.1681/ASN.2005080859
64. Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. (2011) 7:327–40. doi: 10.1038/nrneph.2011.51-
65. Panchapakesan U, Pollock C. The role of toll-like receptors in diabetic kidney disease. Curr Opin Nephrol Hypertens. (2018) 27:30–4. doi: 10.1097/MNH.0000000000000377
66. Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. (2017) 18:1288–98. doi: 10.1038/ni.3858
67. Yiu WH, Li RX, Wong DWL, Wu HJ, Chan KW, Chan LYY, et al. Complement C5a inhibition moderates lipid metabolism and reduces tubulointerstitial fibrosis in diabetic nephropathy. Nephrol Dial Transplant. (2018) 33:1323–32. doi: 10.1093/ndt/gfx336
68. Ochodnicky P, Lattenist L, Ahdi M, Kers J, Uil M, Claessen N, et al. Increased circulating and urinary levels of soluble TAM receptors in diabetic nephropathy. Am J Pathol. (2017) 187:1971–83. doi: 10.1016/j.ajpath.2017.05.004-
69. Klahr S, Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int Suppl. (2000) 75:S7–14. doi: 10.1046/j.1523-1755.57.s75.9.x-
70. Nogueira A, Pires MJ, Oliveira PA. Pathophysiological mechanisms of renal fibrosis: a review of animal models and therapeutic strategies. In Vivo. (2017) 31:1–22. doi: 10.21873/invivo.11019-
71. Wolf G, Neilson EG, Goldfarb S, Ziyadeh FN. The influence of glucose concentration on angiotensin II-induced hypertrophy of proximal tubular cells in culture. Biochem Biophys Res Commun. (1991) 176:902–9. doi: 10.1016/S0006-291X(05)80271-2-
72. Wolf G, Mueller E, Stahl RA, Ziyadeh FN. Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-beta. J Clin Invest. (1993) 92:1366–72. doi: 10.1172/JCI116710-
73. Li XC, Zhuo JL. Nuclear factor-kappaB as a hormonal intracellular signaling molecule: focus on angiotensin II-induced cardiovascular and renal injury. Curr Opin Nephrol Hypertens. (2008) 17:37–43. doi: 10.1097/MNH.0b013e3282f2903c-
74. Takao T, Horino T, Kagawa T, Matsumoto R, Shimamura Y, Ogata K, et al. Possible involvement of intracellular angiotensin II receptor in high-glucose-induced damage in renal proximal tubular cells. J Nephrol. (2011) 24:218–24. doi: 10.5301/JN.2010.5785
75. Feliers D, Gorin Y, Ghosh-Choudhury G, Abboud HE, Kasinath BS. Angiotensin II stimulation of VEGF mRNA translation requires production of reactive oxygen species. Am J Physiol Renal Physiol. (2006) 290:F927–36. doi: 10.1152/ajprenal.00331.2005
76. Yang F, Huang XR, Chung AC, Hou CC, Lai KN, Lan HY. Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J Pathol. (2010) 221:390–401. doi: 10.1002/path.2721
77. Zhou L, Xue H, Yuan P, Ni J, Yu C, Huang Y, et al. Angiotensin AT1 receptor activation mediates high glucose-induced epithelial-mesenchymal transition in renal proximal tubular cells. Clin Exp Pharmacol Physiol. (2010) 37:e152–7. doi: 10.1111/j.1440-1681.2010.05421.x
78. Micakovic T, Papagiannarou S, Clark E, Kuzay Y, Abramovic K, Peters J, et al. The angiotensin II type 2 receptors protect renal tubule mitochondria in early stages of diabetes mellitus. Kidney Int. (2018) 94:937–50. doi: 10.1016/j.kint.2018.06.006-
79. Rahman A, Sherajee SJ, Rafiq K, Kobara H, Masaki T, Nakano D, et al. The angiotensin II receptor-neprilysin inhibitor LCZ696 attenuates the progression of proteinuria in type 2 diabetic rats. J Pharmacol Sci. (2020) 142:124–6. doi: 10.1016/j.jphs.2019.09.014
80. Habibi J, Aroor AR, Das NA, Manrique-Acevedo CM, Johnson MS, Hayden MR, et al. The combination of a neprilysin inhibitor (sacubitril) and angiotensin-II receptor blocker (valsartan) attenuates glomerular and tubular injury in the Zucker Obese rat. Cardiovasc Diabetol. (2019) 18:40. doi: 10.1186/s12933-019-0847-8
81. Malek V, Sharma N, Sankrityayan H, Gaikwad AB. Concurrent neprilysin inhibition and renin-angiotensin system modulations prevented diabetic nephropathy. Life Sci. (2019) 221:159–167. doi: 10.1016/j.lfs.2019.02.027
82. Mou Y, Zhang Y, Guo C, Zhao J, Zhang Z, Zhou X, et al. Integrated treatment of prostaglandin E1 and angiotensin-converting enzyme inhibitor in diabetic kidney disease rats: possible role of antiapoptosis in renal tubular epithelial cells. DNA Cell Biol. (2018) 37:133–41. doi: 10.1089/dna.2017.3690
83. Stadler K, Goldberg IJ, Susztak K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr Diab Rep. (2015) 15:40. doi: 10.1007/s11892-015-0611-8
84. Murea M, Freedman BI, Parks JS, Antinozzi PA, Elbein SC, Ma L. Lipotoxicity in diabetic nephropathy: the potential role of fatty acid oxidation. Clin J Am Soc Nephrol. (2010) 5:2373–9. doi: 10.2215/CJN.08160910
85. Khan S, Gaivin R, Abramovich C, Boylan M, Calles J, Schelling JR. Fatty acid transport protein-2 regulates glycemic control and diabetic kidney disease progression. JCI Insight. (2020) 5:e136845. doi: 10.1172/jci.insight.136845
86. Li Y, Chung S, Li Z, Overstreet JM, Gagnon L, Grouix B, et al. Fatty acid receptor modulator PBI-4050 inhibits kidney fibrosis and improves glycemic control. JCI Insight. (2018) 3:e120365. doi: 10.1172/jci.insight.120365
87. Jia Y, Zheng Z, Yang Y, Zou M, Li J, Wang L, et al. MiR-4756 promotes albumin-induced renal tubular epithelial cell epithelial-to-mesenchymal transition and endoplasmic reticulum stress via targeting Sestrin2. J Cell Physiol. (2019) 234:2905–15. doi: 10.1002/jcp.27107
88. Hwang SD, Song JH, Kim Y, Lim JH, Kim MY, Kim EN, et al. Inhibition of lymphatic proliferation by the selective VEGFR-3 inhibitor SAR131675 ameliorates diabetic nephropathy in db/db mice. Cell Death Dis. (2019) 10:219. doi: 10.1038/s41419-019-1436-1
89. Ding Y, Choi ME. Autophagy in diabetic nephropathy. J Endocrinol. (2015) 224:R15–30. doi: 10.1530/JOE-14-0437
90. De Rechter S, Decuypere JP, Ivanova E, van den Heuvel LP, De Smedt H, Levtchenko E, et al. Autophagy in renal diseases. Pediatr Nephrol. (2016) 31:737–52. doi: 10.1007/s00467-015-3134-2
91. Kitada M, Ogura Y, Monno I, Koya D. Regulating autophagy as a therapeutic target for diabetic nephropathy. Curr Diabet Rep. (2017) 17:53. doi: 10.1007/s11892-017-0879-y
92. Yang D, Livingston MJ, Liu Z, Dong G, Zhang M, Chen JK, et al. Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cell Mol Life Sci. (2018) 75:669–88. doi: 10.1007/s00018-017-2639-1
93. Melia TJ, Lystad AH, Simonsen A. Autophagosome biogenesis: from membrane growth to closure. J Cell Biol. (2020) 219:e202002085. doi: 10.1083/jcb.202002085
94. Huang C, Lin MZ, Cheng D, Braet F, Pollock CA, Chen XM. KCa3.1 mediates dysfunction of tubular autophagy in diabetic kidneys via PI3k/Akt/mTOR signaling pathways. Sci Rep. (2016) 6:23884. doi: 10.1038/srep23884
95. Dusabimana T, Kim SR, Park EJ, Je J, Jeong K, Yun SP, et al. P2Y2R contributes to the development of diabetic nephropathy by inhibiting autophagy response. Mol Metab. (2020) 42:101089. doi: 10.1016/j.molmet.2020.101089
96. Yang C, Chen XC, Li ZH, Wu HL, Jing KP, Huang XR, et al. SMAD3 promotes autophagy dysregulation by triggering lysosome depletion in tubular epithelial cells in diabetic nephropathy. Autophagy. (2020). doi: 10.1080/15548627.2020.1824694. [Epub ahead of print].
97. Liang Q, Liu T, Guo T, Tao W, Chen X, Chen W, et al. ATF4 promotes renal tubulointerstitial fibrosis by suppressing autophagy in diabetic nephropathy. Life Sci. (2021) 264:118686. doi: 10.1016/j.lfs.2020.118686
98. Toffoli B, Tonon F, Tisato V, Michelli A, Zauli G, Secchiero P, et al. TRAIL treatment prevents renal morphological changes and TGF-beta-induced mesenchymal transition associated with diabetic nephropathy. Clin Sci. (2020) 134:2337–52. doi: 10.1042/CS20201004
99. Blagosklonny MV. Fasting and rapamycin: diabetes versus benevolent glucose intolerance. Cell Death Dis. (2019) 10:607. doi: 10.1038/s41419-019-1822-8
100. Schreiber KH, Arriola Apelo SI, Yu D, Brinkman JA, Velarde MC, Syed FA, et al. A novel rapamycin analog is highly selective for mTORC1 in vivo. Nat Commun. (2019) 10:3194. doi: 10.1038/s41467-019-11174-0
101. Yao Y, Inoki K. The role of mechanistic target of rapamycin in maintenance of glomerular epithelial cells. Curr Opin Nephrol Hypertens. (2016) 25:28–34. doi: 10.1097/MNH.0000000000000181
102. Chen F, Sun Z, Zhu X, Ma Y. Astilbin inhibits high glucose-induced autophagy and apoptosis through the PI3K/Akt pathway in human proximal tubular epithelial cells. Biomed Pharmacother. (2018) 106:1175–81. doi: 10.1016/j.biopha.2018.07.072
103. Kim H, Dusabimana T, Kim SR, Je J, Jeong K, Kang MC, et al. Supplementation of abelmoschus manihot ameliorates diabetic nephropathy and hepatic steatosis by activating autophagy in mice. Nutrients. (2018) 10:1703. doi: 10.3390/nu10111703
104. Wei Y, Gao J, Qin L, Xu Y, Shi H, Qu L, et al. Curcumin suppresses AGEs induced apoptosis in tubular epithelial cells via protective autophagy. Exp Ther Med. (2017) 14:6052–8. doi: 10.3892/etm.2017.5314
105. Packer M. Role of impaired nutrient and oxygen deprivation signaling and deficient autophagic flux in diabetic CKD development: implications for understanding the effects of sodium-glucose cotransporter 2-Inhibitors. J Am Soc Nephrol. (2020) 31:907–19. doi: 10.1681/ASN.2020010010
106. Tang SC, Lai KN. The pathogenic role of the renal proximal tubular cell in diabetic nephropathy. Nephrol Dial Transplant. (2012) 27:3049–56. doi: 10.1093/ndt/gfs260
107. Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. (2011) 7:327–40. doi: 10.1038/nrneph.2011.51
108. Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. (2011) 6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150
109. Pini A, Verta R, Grange C, Gurrieri M, Rosa AC. Histamine and diabetic nephropathy: an up-to-date overview. Clin Sci. (2019) 133:41–54. doi: 10.1042/CS20180839
110. Yang T, Heng C, Zhou Y, Hu Y, Chen S, Wang H, et al. Targeting mammalian serine/threonine-protein kinase 4 through Yes-associated protein/TEA domain transcription factor-mediated epithelial-mesenchymal transition ameliorates diabetic nephropathy orchestrated renal fibrosis. Metabolism. (2020) 108:154258. doi: 10.1016/j.metabol.2020.154258
111. Ma R, Ren JM, Li P, Zhou YJ, Zhou MK, Hu Z, et al. Activated YAP causes renal damage of type 2 diabetic nephropathy. Eur Rev Med Pharmacol Sci. (2019) 23:755–63. doi: 10.26355/eurrev_201901_16890
112. Chen J, Harris RC. Interaction of the EGF receptor and the hippo pathway in the diabetic kidney. J Am Soc Nephrol. (2016) 27:1689–700. doi: 10.1681/ASN.2015040415
113. Anorga S, Overstreet JM, Falke LL, Tang J, Goldschmeding RG, Higgins PJ, et al. Deregulation of Hippo-TAZ pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. (2018) 32:2644–57. doi: 10.1096/fj.201700722R
114. Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol. (2012) 12:813–20. doi: 10.1038/nri3339
115. Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. (2010) 141:483–96. doi: 10.1016/j.cell.2010.03.040
116. Kuenzel S, Till A, Winkler M, Hasler R, Lipinski S, Jung S, et al. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J Immunol. (2010) 184:1990–2000. doi: 10.4049/jimmunol.0900557
117. Yao Y, Qian Y. Expression regulation and function of NLRC5. Protein Cell. (2013) 4:168–75. doi: 10.1007/s13238-012-2109-3
118. Song S, Qiu D, Luo F, Wei J, Wu M, Wu H, et al. Knockdown of NLRP3 alleviates high glucose or TGFB1-induced EMT in human renal tubular cells. J Mol Endocrinol. (2018) 61:101–13. doi: 10.1530/JME-18-0069
119. Cheng D, Liang R, Huang B, Hou J, Yin J, Zhao T, et al. Tumor necrosis factor-α blockade ameliorates diabetic nephropathy in rats. Clin Kidney J. (2019) 14:301–8. doi: 10.1093/ckj/sfz137
120. Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol. (2013) 190:1239–49. doi: 10.4049/jimmunol.1201959
121. Luan P, Zhuang J, Zou J, Li H, Shuai P, Xu X, et al. NLRC5 deficiency ameliorates diabetic nephropathy through alleviating inflammation. FASEB J. (2018) 32:1070–84. doi: 10.1096/fj.201700511RR
122. Nguyen D, Ping F, Mu W, Hill P, Atkins R, Chadban SJN. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology. (2006) 11:226–31. doi: 10.1111/j.1440-1797.2006.00576.x
123. Tesch GH. Role of macrophages in complications of type 2 diabetes. Clin Exp Pharmacol Physiol. (2007) 34:1016–9. doi: 10.1111/j.1440-1681.2007.04729.x-
124. Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. (2008) 118:3522–30. doi: 10.1172/JCI36150
125. Anders HJ, Ryu M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. (2011) 80:915–25. doi: 10.1038/ki.2011.217
126. Liu X, Zhang Y, Shi M, Wang Y, Zhang F, Yan R, et al. Notch1 regulates PTEN expression to exacerbate renal tubulointerstitial fibrosis in diabetic nephropathy by inhibiting autophagy via interactions with Hes1. Biochem Biophys Res Commun. (2018) 497:1110–6. doi: 10.1016/j.bbrc.2018.02.187
127. Zhang Y, Zhao S, Wu D, Liu X, Shi M, Wang Y, et al. MicroRNA-22 promotes renal tubulointerstitial fibrosis by targeting PTEN and suppressing autophagy in diabetic nephropathy. J Diabetes Res. (2018) 2018:4728645. doi: 10.1155/2018/4728645
128. Li Y, Hu Q, Li C, Liang K, Xiang Y, Hsiao H, et al. PTEN-induced partial epithelial-mesenchymal transition drives diabetic kidney disease. J Clin Invest. (2019) 129:1129–51. doi: 10.1172/JCI121987
129. Kambe T, Tsuji T, Hashimoto A, Itsumura N. The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol Rev. (2015) 95:749–84. doi: 10.1152/physrev.00035.2014-
130. Zhang X, Lian X, Liang D, Zhang L, Liu S, Yang L, et al. Protective effect of Znt7 on High glucose-induced epithelial-to-mesenchymal transition in renal tubular epithelial cells. Kidney Blood Press Res. (2018) 43:500–12. doi: 10.1159/000488697
131. Huang F, Wang Q, Guo F, Zhao Y, Ji L, An T, et al. FoxO1-mediated inhibition of STAT1 alleviates tubulointerstitial fibrosis and tubule apoptosis in diabetic kidney disease. EBioMedicine. (2019) 48:491–504. doi: 10.1016/j.ebiom.2019.09.002
132. Lu Q, Chen YB, Yang H, Wang WW, Li CC, Wang L, et al. Inactivation of TSC1 promotes epithelial-mesenchymal transition of renal tubular epithelial cells in mouse diabetic nephropathy. Acta Pharmacol Sin. (2019) 40:1555–67. doi: 10.1038/s41401-019-0244-6
133. Jheng HF, Tsai PJ, Chuang YL, Shen YT, Tai TA, Chen WC, et al. Albumin stimulates renal tubular inflammation through an HSP70-TLR4 axis in mice with early diabetic nephropathy. Dis Model Mech. (2015) 8:1311–21. doi: 10.1242/dmm.019398
134. Das F, Ghosh-Choudhury N, Venkatesan B, Kasinath BS G. Ghosh Choudhury, PDGF receptor-beta uses Akt/mTORC1 signaling node to promote high glucose-induced renal proximal tubular cell collagen I (alpha2) expression. Am J Physiol Renal Physiol. (2017) 313:F291–F307. doi: 10.1152/ajprenal.00666.2016
135. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. (2009) 136:642–55. doi: 10.1016/j.cell.2009.01.035
136. Fu Y, Zhang Y, Wang Z, Wang L, Wei X, Zhang B, et al. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am J Nephrol. (2010) 32:581–9. doi: 10.1159/000322105
137. Wan RJ, Li YH. MicroRNA146a/NAPDH oxidase4 decreases reactive oxygen species generation and inflammation in a diabetic nephropathy model. Mol Med Rep. (2018) 17:4759–66. doi: 10.3892/mmr.2018.8407
138. Wang Y, Zheng ZJ, Jia YJ, Yang YL, Xue YM. Role of p53/miR-155-5p/sirt1 loop in renal tubular injury of diabetic kidney disease. J Transl Med. (2018) 16:146. doi: 10.1186/s12967-018-1486-7
139. Xu H, Sun F, Li X, Sun L. Down-regulation of miR-23a inhibits high glucose-induced EMT and renal fibrogenesis by up-regulation of SnoN. Hum Cell. (2018) 31:22–32. doi: 10.1007/s13577-017-0180-z
140. Hou X, Tian J, Geng J, Li X, Tang X, Zhang J, et al. MicroRNA-27a promotes renal tubulointerstitial fibrosis via suppressing PPARgamma pathway in diabetic nephropathy. Oncotarget. (2016) 7:47760–76. doi: 10.18632/oncotarget.10283
141. Wang Y, Liu Y, Zhang L, Bai L, Chen S, Wu H, et al. miR-30b-5p modulate renal epithelial-mesenchymal transition in diabetic nephropathy by directly targeting SNAI1. Biochem Biophys Res Commun. (2020) 535:12–8. doi: 10.1016/j.bbrc.2020.10.096
142. Zhao Y, Yin Z, Li H, Fan J, Yang S, Chen C, et al. MiR-30c protects diabetic nephropathy by suppressing epithelial-to-mesenchymal transition in db/db mice. Aging Cell. (2017) 16:387–400. doi: 10.1111/acel.12563
143. Gao BH, Wu H, Wang X, Ji LL, Chen C. MiR-30c-5p inhibits high glucose-induced EMT and renal fibrogenesis by down-regulation of JAK1 in diabetic nephropathy. Eur Rev Med Pharmacol Sci. (2020) 24:1338–49. doi: 10.26355/eurrev_202002_20191
144. Fu JX, Sun GQ, Wang HL, Jiang HX. LncRNA OIP5-AS1 induces epithelial-to-mesenchymal transition and renal fibrosis in diabetic nephropathy via binding to miR-30c-5p. J Biol Regul Homeost Agents. (2020) 34:961–8. doi: 10.23812/20-199-A-68
145. Xue M, Li Y, Hu F, Jia YJ, Zheng ZJ, Wang L, et al. High glucose up-regulates microRNA-34a-5p to aggravate fibrosis by targeting SIRT1 in HK-2cells. Biochem Biophys Res Commun. (2018) 498:38–44. doi: 10.1016/j.bbrc.2017.12.048-
146. Zeng Y, Feng Z, Liao Y, Yang M, Bai Y, He Z. Diminution of microRNA-98 alleviates renal fibrosis in diabetic nephropathy by elevating Nedd4L and inactivating TGF-beta/Smad2/3 pathway. Cell Cycle. (2020) 19:3406–18. doi: 10.1080/15384101.2020.1838780
147. Bai X, Geng J, Zhou Z, Tian J, Li X. MicroRNA-130b improves renal tubulointerstitial fibrosis via repression of snail-induced epithelial-mesenchymal transition in diabetic nephropathy. Sci Rep. (2016) 6:20475. doi: 10.1038/srep20475
148. Sun Z, Ma Y, Chen F, Wang S, Chen B, Shi J. miR-133b and miR-199b knockdown attenuate TGF-beta1-induced epithelial to mesenchymal transition and renal fibrosis by targeting SIRT1 in diabetic nephropathy. Eur J Pharmacol. (2018) 837:96–104. doi: 10.1016/j.ejphar.2018.08.022
149. Liu B, Qiang L, Wang GD, Duan Q, Liu J. LncRNA MALAT1 facilities high glucose induced endothelial to mesenchymal transition and fibrosis via targeting miR-145/ZEB2 axis. Eur Rev Med Pharmacol Sci. (2019) 23:3478–86. doi: 10.26355/eurrev_201904_17713
150. Xu P, Guan MP, Bi JG, Wang D, Zheng ZJ, Xue YM. High glucose down-regulates microRNA-181a-5p to increase pro-fibrotic gene expression by targeting early growth response factor 1 in HK-2 cells. Cell Signal. (2017) 31:96–104. doi: 10.1016/j.cellsig.2017.01.012-
151. Zanchi C, Macconi D, Trionfini P, Tomasoni S, Rottoli D, Locatelli M, et al. MicroRNA-184 is a downstream effector of albuminuria driving renal fibrosis in rats with diabetic nephropathy. Diabetologia. (2017) 60:1114–25. doi: 10.1007/s00125-017-4248-9
152. Liu F, Zhang ZP, Xin GD, Guo LH, Jiang Q, Wang ZX. miR-192 prevents renal tubulointerstitial fibrosis in diabetic nephropathy by targeting Egr1. Eur Rev Med Pharmacol Sci. (2018) 22:4252–60. doi: 10.26355/eurrev_201807_15420
153. Krupa A, Jenkins R, Luo DD, Lewis A, Phillips A, Fraser D. Loss of MicroRNA-192 promotes fibrogenesis in diabetic nephropathy. J Am Soc Nephrol. (2010) 21:438–47. doi: 10.1681/ASN.2009050530
154. Zhang R, Qin L, Shi J. MicroRNA199a3p suppresses high glucoseinduced apoptosis and inflammation by regulating the IKKbeta/NFkappaB signaling pathway in renal tubular epithelial cells. Int J Mol Med. (2020) 46:2161–71. doi: 10.3892/ijmm.2020.4751
155. Tang WB, Zheng L, Yan R, Yang J, Ning J, Peng L, et al. miR302a-3p may modulate renal epithelial-mesenchymal transition in diabetic kidney disease by targeting ZEB1. Nephron. (2018) 138:231–42. doi: 10.1159/000481465
156. Wang B, Jha JC, Hagiwara S, McClelland AD, Jandeleit-Dahm K, Thomas MC, et al. Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. (2014) 85:352–61. doi: 10.1038/ki.2013.372
157. Docherty NG, Murphy M, Martin F, Brennan EP, Godson C. Targeting cellular drivers and counter-regulators of hyperglycaemia- and transforming growth factor-beta1-associated profibrotic responses in diabetic kidney disease. Exp Physiol. (2014) 99:1154–62. doi: 10.1113/expphysiol.2014.078774
158. Coca SG, Nadkarni GN, Huang Y, Moledina DG, Rao V, Zhang J, et al. Plasma biomarkers and kidney function decline in early and established diabetic kidney disease. J Am Soc Nephrol. (2017) 28:2786–93. doi: 10.1681/ASN.2016101101
159. Greenberg JH, Abraham AG, Xu Y, Schelling JR, Feldman HI, Sabbisetti VS, et al. Plasma biomarkers of tubular injury and inflammation are associated with ckd progression in children. J Am Soc Nephrol. (2020) 31:1067–77. doi: 10.1681/ASN.2019070723
160. Gohda T, Kamei N, Koshida T, Kubota M, Tanaka K, Yamashita Y, et al. Circulating kidney injury molecule-1 as a biomarker of renal parameters in diabetic kidney disease. J Diabet Investigat. (2020) 11:435–40. doi: 10.1111/jdi.13139
161. Siddiqui K, Al-Malki B, George TP, Nawaz SS, Rubeaan KA. Urinary N-acetyl-beta-d-glucosaminidase (NAG) with neutrophil gelatinase-associated lipocalin (NGAL) improves the diagnostic value for proximal tubule damage in diabetic kidney disease. Biotech. (2019) 9:66. doi: 10.1007/s13205-019-1593-z
162. Abbasi F, Moosaie F, Khaloo P, Dehghani Firouzabadi F, Fatemi Abhari SM, Atainia B, et al. Neutrophil gelatinase-associated lipocalin and retinol-binding Protein-4 as biomarkers for diabetic kidney disease. Kidney Blood Pressure Res. (2020) 45:222–32. doi: 10.1159/000505155-
163. Prabhu RA, Shenoy SV, Nagaraju SP, Rangaswamy D, Rao IR, Bhojaraja MV, et al. Acute kidney injury and progressive diabetic kidney disease: an epidemiological perspective. Int J Nephrol Renovasc Dis. (2021) 14:23–31. doi: 10.2147/IJNRD.S291319
164. Advani A. Acute kidney injury: a bona fide complication of diabetes. Diabetes. (2020) 69:2229–37. doi: 10.2337/db20-0604
165. Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti V, Ichimura T, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. (2012) 82:172–83. doi: 10.1038/ki.2012.20
166. Peng J, Li X, Zhang D, Chen JK, Su Y, Smith SB, et al. Hyperglycemia, p53, and mitochondrial pathway of apoptosis are involved in the susceptibility of diabetic models to ischemic acute kidney injury. Kidney Int. (2015) 87:137–50. doi: 10.1038/ki.2014.226
167. Kelly KJ, Burford JL, Dominguez JH. Postischemic inflammatory syndrome: a critical mechanism of progression in diabetic nephropathy. Am J Physiol Renal Physiol. (2009) 297:F923–31. doi: 10.1152/ajprenal.00205.2009
168. Kelly KJ, Dominguez JH. Treatment of the post-ischaemic inflammatory syndrome of diabetic nephropathy. Nephrol Dialysis Trans. (2010) 25:3204–12. doi: 10.1093/ndt/gfq217
Keywords: tubulointerstitial fibrosis, diabetic kidney disease, proximal tubular cell, proximal tubular, pathogenesis
Citation: Chang J, Yan J, Li X, Liu N, Zheng R and Zhong Y (2021) Update on the Mechanisms of Tubular Cell Injury in Diabetic Kidney Disease. Front. Med. 8:661076. doi: 10.3389/fmed.2021.661076
Received: 30 January 2021; Accepted: 08 March 2021;
Published: 30 March 2021.
Edited by:
Tingting Zhao, China-Japan Friendship Hospital, ChinaReviewed by:
Niansong Wang, Shanghai Jiao Tong University, ChinaKun Gao, Affiliated Hospital of Nanjing University of Chinese Medicine, China
Copyright © 2021 Chang, Yan, Li, Liu, Zheng and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rong Zheng, enJvbmdfbWRAMTI2LmNvbQ==; Yifei Zhong, eWlmZWlsaWx5QDEyNi5jb20=
†These authors have contributed equally to this work