
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med., 10 June 2021
Sec. Dermatology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.658842
This article is part of the Research TopicThe Skin in Cancer Predisposition SyndromesView all 5 articles
 Agnes Lim1
Agnes Lim1 Joanne Ngeow1,2,3*
Joanne Ngeow1,2,3*Cowden syndrome (CS) is an autosomal dominant condition caused by mutations in the phosphatase and tensin homolog (PTEN) gene, and is characterized by multiple hamartomas and a predisposition to malignant tumors. Characteristic skin lesions include trichilemmomas, acral keratosis, mucocutaneous neuromas, oral papillomas, and penile macules, and are often the first clues to the underlying diagnosis. Here, we discuss the mucocutaneous manifestations of CS, differential diagnoses of genetic causes of each cutaneous finding, genetic analyses for patients with skin manifestations, management of patients with CS, and potential new targeted therapies for CS.
Cowden syndrome (CS) is a genodermatosis caused by mutations in the phosphatase and tensin homolog (PTEN) gene, with an autosomal dominant inheritance pattern. CS is part of a spectrum of disorders caused by germline mutations in the PTEN gene, collective named PTEN hamartoma tumor syndrome (PHTS), and includes Bannayan-Riley-Ruvalcaba syndrome (BRRS), Proteus and Proteus-like syndrome (1). The estimated prevalence of CS is ~1 in 200,000 live births, however this is likely an underestimation as the phenotypic variability makes diagnosis challenging (1–3). CS is characterized by multiple hamartomas, and an increased risk of breast, thyroid, endometrial, renal and colorectal carcinomas (2). Characteristic mucocutaneous manifestations of CS include trichilemmomas, acral keratoses, mucocutaneous neuromas, oral papillomas and macular pigmentation of the glans penis (4). As patients with CS can first present with cutaneous lesions with a wide range of differentials, it is important for dermatologists to be aware of the features of CS in order to make the diagnosis. Here, we discuss the mucocutaneous manifestations of CS, differential diagnoses of genetic causes of each cutaneous finding, genetic analyses for patients with skin manifestations, management of patients with CS, and potential new targeted therapies for CS.
Over 90% of individuals with CS develop some clinical manifestation of the disorder by the late 20s (3, 5, 6); 99% of affected individuals develop mucocutaneous stigmata by the third decade (5).
Trichilemmomas are benign hamartomatous lesions of the hair follicle outer root sheath (7). Early studies done prior to the establishment of International Cowden Consortium (ICC) criteria reported that all patients with multiple trichilemmomas had CS, and all patients with CS had multiple trichilemmomas (7, 8), however this is likely to be an overestimate due to the original focus on dermatologic manifestations in the diagnostic workup for CS. More recent studies have reported a prevalence of trichilemommas of 6–38% in patients with confirmed PTEN mutations (9–12). Clinically, trichilemmomas are well-defined, skin-colored papules 1–5 mm in diameter and located primarily on the face and neck (13). As solitary trichilemmomas can be found in patients without CS (14), the presence of multiple trichilemmomas and confirmation of diagnosis by skin biopsy is needed in order to fulfill criteria for CS (4, 5). Key histological findings are folliculocentric lobular proliferation of polygonal, clear, PAS-positive isthmic cells with nuclear palisading of the peripheral cells on a thickened hyaline eosinophilic basement membrane (14).
In addition to CS, multiple facial papules are also a feature of other inherited conditions. Differential diagnoses for genetic causes of multiple facial papules are summarized in Table 1. Some of these papules have characteristic dermoscopic findings, which can be a helpful aid for clinical diagnosis. For example, trichilemmomas have a radiated red iris-like structure on dermoscopy (15). Other genetic causes of multiple facial papules include Birt-Hogg-Dube syndrome (19–22), multiple familial trichoepitheloma (16–18), Brooke-Spiegler syndrome (16–18), tuberous sclerosis complex (TSC) (23, 24), and Muir-Torre syndrome (25, 26).
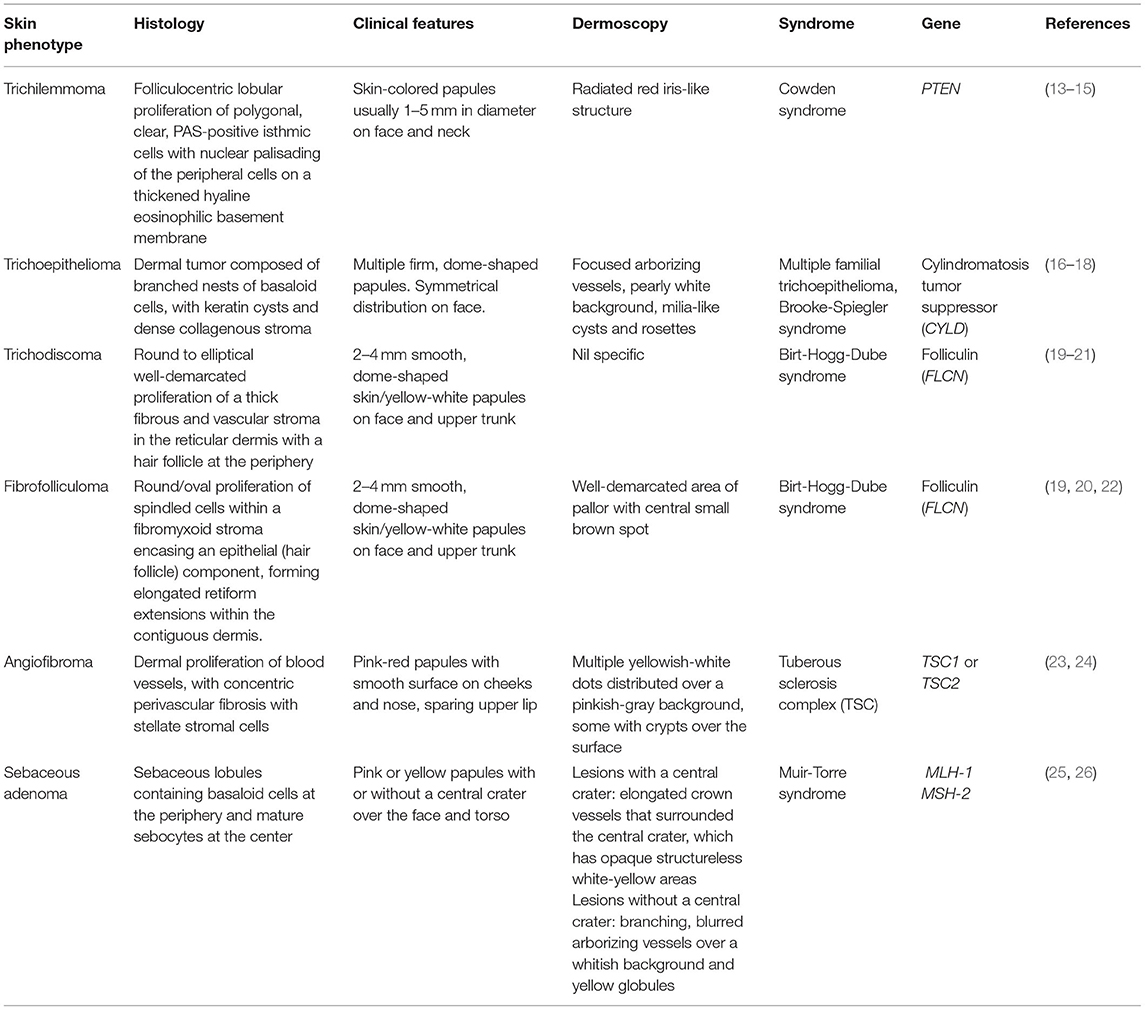
Table 1. Differential diagnoses for genetic causes of multiple facial papules.
Acral keratoses are located primarily on the dorsum of the hands and feet, and are wart-like, skin colored flat-topped papules 1–4 mm in diameter (27). Palmoplantar keratoses are translucent hard papules with or without a central pit (27). Earlier studies performed before establishment of ICC diagnostic criteria for CS reported a prevalence of acral keratoses of 63–73% in CS patients (27, 28). More recent studies report a prevalence of acral keratoses of 10.2–82% in CS patients with confirmed PTEN mutations (9, 11, 12). Few case reports observed keratosis appearing on non-acral sites such as the face and trunk, but no confirmatory skin biopsies were mentioned (27, 28). Further studies are needed to determine if non-acral keratoses are a common feature in CS.
Histologically, both acral keratoses and palmoplantar keratoses show non-specific orthokeratosis, hypergranulosis, and acanthosis (8). Differential diagnoses of genetic causes of acral keratoses and palmoplantar pits include Buschke-Fischer-Brauer keratoderma (29), Darier's disease (30–32), Cole disease (33), and nevoid basal cell carcinoma (Gorlin) syndrome (34–36) (Table 2).
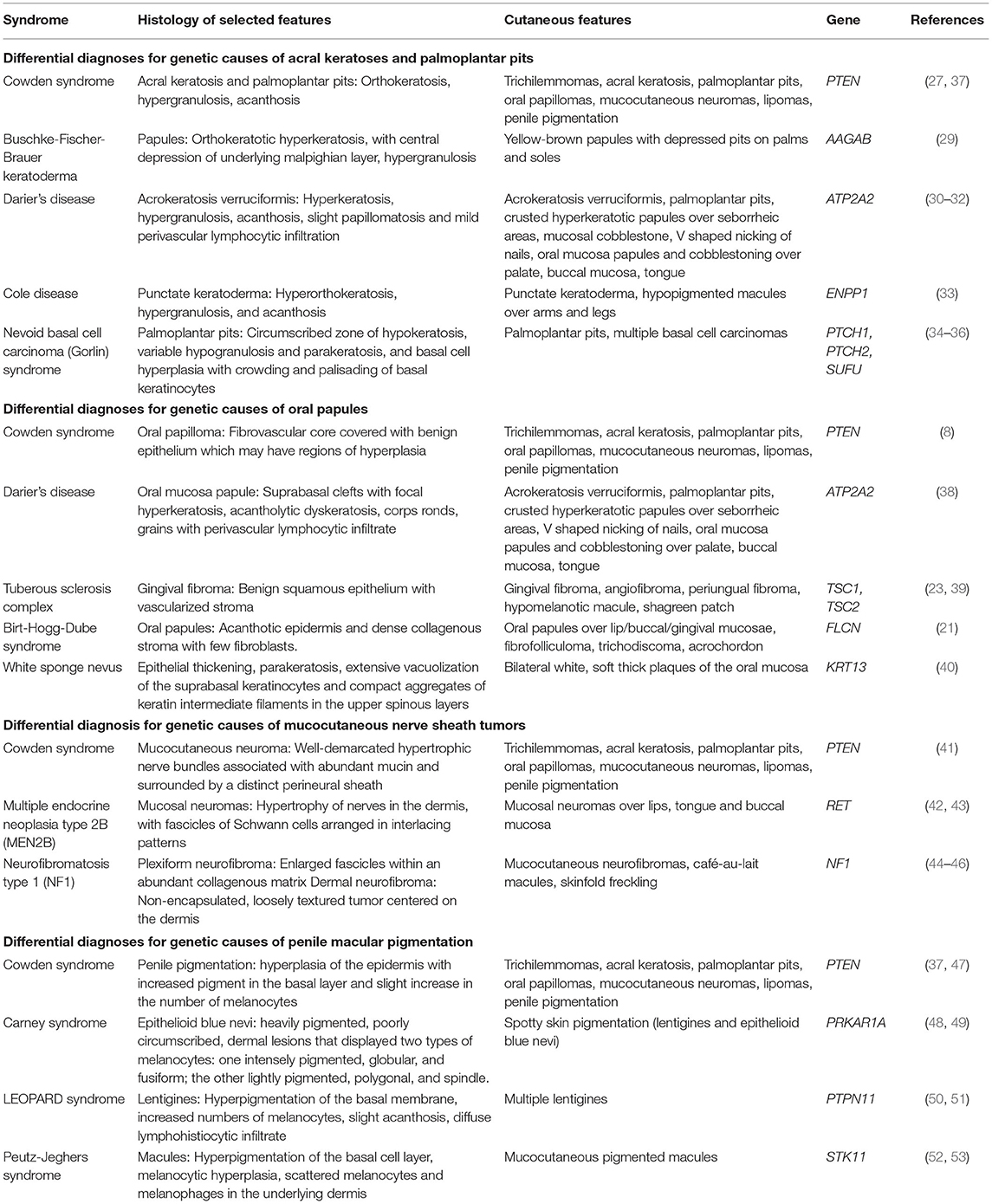
Table 2. Differential diagnoses for genetic causes of acral keratosis, palmoplantar pits, oral papules, mucocutaneous nerve sheath tumors, and penile macular pigmentation.
Oral papillomas are 1–3 mm diameter papules which may coalesce, giving the involved surface a cobble-stone pattern (27). Lesions are seen on the lips, buccal mucosae and gingivae; when the dorsum of the tongue is involved, it may take on a scrotal appearance (27). Papillomas have also been reported over the pharynx and larynx, which may require laryngoscopy for identification (54). Earlier studies performed before establishment of ICC diagnostic criteria for CS reported a prevalence of oral papillomas of 83–86% in CS patients (27, 28). More recent studies report a prevalence of oral papillomas of 15.2–85% in CS patients with confirmed PTEN mutations (9, 12). Histology of oral papillomas show a fibrovascular core covered with benign epithelium which may have regions of hyperplasia (8, 54). Differential diagnoses of genetic causes of oral papules include Darier's disease (38), tuberous sclerosis complex (23, 39), Birt-Hogg-Dube syndrome (21), and white sponge nevus (40) (Table 2).
Mucocutaneous neuromas are hamartomas of the peripheral nerve sheath, and present clinically as painful, dome-shaped, translucent pink to skin-colored papules (41). In CS, mucocutaneous neuromas have been reported on the face, hands, trunk, and shin of patients (41, 55). Histology shows non-encapsulated neuromas, with well-demarcated hypertrophic nerve bundles associated with abundant mucin and surrounded by a distinct perineural sheath (41).
Earlier studies performed before establishment of ICC diagnostic criteria for CS reported a prevalence of mucocutaneous neuromas of 5–10.9% in CS patients (27, 28). More recent studies of CS patients with confirmed PTEN mutations did not report the incidence of mucocutaneous neuromas (9–12), possibly because the studies were done prior to the addition of mucocutaneous neuromas to CS diagnostic criteria in 2013 (4). In addition, neuromas may be in advertently unreported due to clinical misclassification in the absence of a biopsy (4, 41).
Differential diagnoses for genetic causes of multiple mucocutaneous nerve sheath tumors include multiple endocrine neoplasia type 2B (MEN2B) and neurofibromatosis type 1 (NF1) (Table 2). MEN2B is caused by mutations in the RET gene, and is associated with medullary thyroid carcinoma, pheochromocytoma, marfanoid habitus, and multiple mucosal neuromas involving the face (42, 43), as compared to CS, which presents with multiple neuromas affecting acral sites (41). NF1 is caused by mutations in the Neurofibromin gene, and is characterized by pigmentary lesions, peripheral nerve tumors (neurofibromas and malignant peripheral nerve sheath tumors), skeletal abnormalities and brain tumors (44). Neurofibromas can be classified into dermal or plexiform neurofibromas (45, 46). A study of 103 patients with NF1 found oral tumors (plexiform neurofibromas or discrete oral neurofibromas), overgrowth of gingival soft tissue and enlarged papillae of the tongue in 74% of NF1 patients (46), which can mimic the oral papillomas seen in CS.
Macular pigmentation of the glans penis is a major criterion for CS diagnosis in male patients. The prevalence of pigmented genital macules in the general population is about 15% in males and females (56).
In CS patients with confirmed PTEN mutations, the prevalence of macular pigmentation of the glans penis was reported at 19–54.3% (9–12, 57). Histology of penile macules from a patient with BRRS showed hyperplasia of the epidermis with increased pigment in the basal layer and slight increase in the number of melanocytes (47). In a case series of children with PTHS, genital lentiginosis was found in all patients with genital evaluation as early as age 2.5 years (58). A study of patients with PTEN mutations from 1 to 26 years found that penile macular pigmentation was present in 41% of males in the cohort, with earliest report of pigmentation occurring at 15 months of age (57).
Differential diagnoses for genetic causes of penile macular pigmentation include Carney syndrome, LEOPARD syndrome, and Peutz-Jeghers syndrome (Table 2). Carney syndrome is caused by mutations in PRKAR1A, presents with spotty skin pigmentation (lentigines and epithelioid blue nevi), endocrine tumors and schwannomas, and can be differentiated from CS by the presence of cardiac myxomas (48, 49). LEOPARD syndrome is named based on the acronym of its manifestations- lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormal genitalia, retardation of growth, sensorineural deafness, and is caused by mutations in PTPN11 (50, 51). Peutz-Jeghers syndrome is characterized by gastrointestinal hamartomatous polyps and mucocutaneous pigmentated macules, which are most often found on the lips, buccal mucosa and digits, but can also be found on genitalia (52, 53).
Cutaneous features that are associated with CS but not part of major criteria include lipomas and vascular anomalies.
The incidence of single lipomas in the general population is 1% (59). In contrast, the incidence of lipomas in CS is 31–39.1% based on early studies without PTEN analysis (27, 28), and 34.6–56.7% in patients with CS and PTEN mutations (9–11). In order to fulfill minor criteria for CS, a patient has to have at least 3 lipomas (4).
Vascular anomalies have been reported in soft tissue, skin, bone, viscera, and the central nervous system (60) of patients with PTEN mutations. Vascular anomalies in the skin present as hemangiomas, and have a prevalence of 21.7% in early studies (27). Prevalence of arteriovenous malformations in CS patients with PTEN mutations in a single study was 6.4–11.4% (61), but prevalence of hemangiomas specifically is not known.
The ICC operational diagnostic criteria for CS was developed in 1996 (62), and has since undergone revisions (1, 6) and incorporation into the National Comprehensive Cancer Network (NCCN) guidelines. Under NCCN 2020 guidelines, patients meeting one of six testing criteria should undergo PTEN genetic testing (63). These testing criteria are: (1) Family history of known PTEN pathogenic/likely pathogenic variant; (2) Personal history of Bannayan-Riley-Ruvalcaba syndrome; (3) Individual meeting clinical diagnostic criteria for CS [described by Pilarski et al. (4)]; (4) Individual not meeting clinical diagnostic criteria for CS but has a personal history of adult Lhermitte-Duclos disease or autism spectrum disorder and macrocephaly or two or more biopsy-proven trichilemmomas or one of several possible combinations of major and minor criteria; (5) At-risk individual with a relative with a clinical diagnosis of CS/PHTS or BRRS for whom testing has not been performed; (6) PTEN pathogenic/likely pathogenic variant detected by tumor profiling on any tumor type in the absence of germline analysis.
Given the multisystem involvement of CS and evolving clinical spectrum, use of the current NCCN criteria may be challenging outside a specialist community. A clinical scoring system based on a prospective study of over 3,000 patients was developed to aid selection of patients for PTEN mutation testing (9). The Cleveland Clinic (CC) score is a weighted semi-quantitative score that predicts the individual-level pre-test probability of harboring a germline PTEN mutation, based on age-adjusted phenotypic characteristics. Patients with a threshold CC score of 10 (or pre-test probability of 3%) and above are recommended for referral to medical genetics for specialist evaluation. Compared to the NCCN 2010 guidelines, the CC scoring system demonstrated increased sensitivity and specificity for the detection of PTEN mutations (9).
Following referral to a medical geneticist, diagnosis of CS is established in a patient by identification of a heterozygous germline pathogenic variant in PTEN on molecular genetic testing (5). However, prospective studies estimate that only ~25% of CS patients who meet clinical criteria harbor pathogenic PTEN mutations (9, 64, 65). For patients who meet CS clinical diagnostic criteria and have known familial pathogenic variants, genetic testing for familial pathogenic variants is conducted. If these patients are positive for familial pathogenic variants, management and cancer surveillance should be performed as per NCCN guidelines for CS. If these patients are negative for familial pathogenic variants, cancer screening should occur as per NCCN screening guidelines (63). For patients who meet CS clinical diagnostic criteria and do not have known familial pathogenic variants, genetic testing using a multi-gene panel should be conducted. If patients are positive, management and cancer surveillance should be performed as per NCCN guidelines for the appropriate gene. If patients are negative, they should be managed as likely pathogenic variant carriers (63).
Research efforts to elucidate non-PTEN etiologies for CS have identified SDHx, KLLN, AKT1, PIK3CA, SEC23B, and WWP1 as candidate predisposition genes for CS (66–70), but these genes have yet to be validated for use in a clinical setting. Identification of susceptibility genes is important because it allows patients to have gene-specific genetic counseling, predictive testing of family members and precise risk assessment.
As CS is associated with increased risk of breast, endometrial, thyroid, gastrointestinal and renal cell carcinoma, cancer screening is a crucial component of management, and has been described in NCCN guidelines (63). Briefly, women should begin regular breast self-examination at age 25 years, and commence annual mammography starting at age 30–35 or 5–10 years before onset of known breast cancer in the family. Endometrial cancer screening should be decided on an individual basis starting at age 35 years. Annual thyroid ultrasound should start at age 7 years. Colonoscopy should start at age 35 or 5–10 years before earliest colon cancer in the family, and repeated at least every 5 years. Renal ultrasound should be considered every 1–2 years from age 40 years. Although data on melanoma risk in CS is limited, there is reported increased melanoma standardized incidence risk (SIR) of 8.5 in PTEN mutation positive CS patients (61), with median age of diagnosis of 40 years, compared with 63 years in the general population (71). Hence, annual dermatology examination of CS patients is recommended.
Treatment of cutaneous manifestation of CS is the same as for their sporadic counterparts (5). Trichilemmomas are benign lesions and do not require treatment, although carbon dioxide laser ablation or simple surgical excision have been utilized (72). There are case reports of the use of oral retinoids for treatment of oral papillomas or acral keratosis, but the lesions recurred following cessation of treatment (73, 74). Treatment of severe oral papillomatosis include surgical excision, carbon dioxide laser debulking, or localized radiotherapy (75).
PTEN functions both in the cytoplasm to regulate signaling pathways, and in the nucleus to maintain chromosomal stability and DNA repair (76). PTEN is a dual-specificity phosphatase that dephosphorylates both protein and lipid substrates. For example, PTEN dephosphorylates focal adhesion kinase (FAK), resulting in inhibition of cell migration (77). PTEN also negatively regulates the PI3K/AKT/mTOR pathway. In the absence of PTEN inhibition, phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3) causes AKT activation, resulting in phosphorylation of proteins such as mTOR that affect cell growth, cell cycle entry and cell survival (78). PTEN dephosphorylates PIP3 to PIP2 (79), hence loss of PTEN lipid phosphatase activity results in constitutive activation of the PI3K/AKT/mTOR pathway.
Inhibition of mTOR is thus a reasonable therapeutic strategy. A pilot study involving 18 CS patients showed that administration of oral mTOR-inhibitor sirolimus 2 mg daily for 56 days was tolerable in terms of side effect profile, and was sufficient to down-regulate mTOR signaling in skin and gastrointestinal tissue (80). Of note, 14 patients demonstrated improvement in skin lesions based on dermatologic examination, although the specific lesion type (trichilemmoma, oral papilloma, etc.) or method of assessing improvement were not mentioned and will require larger patient populations for confirmatory studies (80). One limitation of targeting the PI3K/AKT/mTOR pathway is feedback activation of other signaling pathways, causing drug resistance (81). In theory, this could be overcome by the use of combination drug therapies. For example, rebound increase in AKT activation has been observed in rapamycin-resistance breast cancer cell lines, and this can be overcome with concomitant administration of resveratrol (82).
Given the myriad PTEN mutations and resultant diverse disruption in cellular function, a possible approach is to target therapeutics to the specific mutation each patient has. For example, mutation p.Lys289Glu identified in a CS patient resulted in a nuclear import defect of PTEN (83). Nuclear PTEN is involved in DNA double-stranded break repair (76), and loss of PTEN causes increased sensitivity of cells to the inhibition of DNA repair enzyme PARP1 both in vitro and in mouse models, resulting in reduced cell growth (84). However, given that PTEN germline mutations in CS affect all cells, targeting the PAPR1 inhibitor to tumor cells would be key.
Mucocutaneous manifestations of CS may appear similar to that of other inherited conditions, hence it is imperative for dermatologists to be aware of the constellation of signs that typify CS. Expedient referrals to cancer geneticists for genetic testing and initiation of cancer screening is paramount. Further studies to identify organ-specific therapeutic strategies for CS are still needed.
All authors contributed to the conceptualization, writing, and editing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. (2004) 41:323–6. doi: 10.1136/jmg.2004.018036
2. Ngeow J, Sesock K, Eng C. Breast cancer risk and clinical implications for germline PTEN mutation carriers. Breast Cancer Res Treat. (2017) 165:1–8. doi: 10.1007/s10549-015-3665-z
3. Nelen MR, Padberg GW, Peeters EAJ, Lin AY, van den Helm B, Frants RR, et al. Localization of the gene for Cowden disease to chromosome 10q22–23. Nat Genet. (1996) 13:114–6. doi: 10.1038/ng0596-114
4. Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Ins. (2013) 105:1607–16. doi: 10.1093/jnci/djt277
5. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al. PTENHamartoma Tumor Syndrome. GeneReviews® [Internet]. Seattle (WA): University of Washington (1993).
6. Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. (2000) 37:828–30. doi: 10.1136/jmg.37.11.828
7. Brownstein MH, Mehregan AH, Bikowski JB. Trichilemmomas in Cowden's disease. Jama. (1977) 238:26. doi: 10.1001/jama.1977.03280010026004
8. Brownstein MH, Mehregan AH, Bikowski JB, Lupulescu A, Patterson JC. The dermatopathology of Cowden's syndrome. Br J Dermatol. (1979) 100:667–73. doi: 10.1111/j.1365-2133.1979.tb08070.x
9. Tan M-H, Mester J, Peterson C, Yang Y, Chen J-L, Rybicki LA, et al. A clinical scoring system for selection of patients for pten mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Human Genet. (2011) 88:42–56. doi: 10.1016/j.ajhg.2010.11.013
10. Pilarski R, Stephens JA, Noss R, Fisher JL, Prior TW. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan–Riley–Ruvalcaba syndrome clinical features. J Med Genet. (2011) 48:505–12. doi: 10.1136/jmg.2011.088807
11. Heald B, Mester J, Rybicki L, Orloff MS, Burke CA, Eng C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology. (2010) 139:1927–33. doi: 10.1053/j.gastro.2010.06.061
12. Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. (2013) 50:255–63. doi: 10.1136/jmedgenet-2012-101339
13. Farooq A, Walker LJ, Bowling J, Audisio RA. Cowden syndrome. Cancer Treat Rev. (2010) 36:577–83. doi: 10.1016/j.ctrv.2010.04.002
14. Tellechea O, Cardoso JC, Reis JP, Ramos L, Gameiro AR, Coutinho I, et al. Benign follicular tumors. An Bras Dermatol. (2015) 90:780–96; quiz 797–8. doi: 10.1590/abd1806-4841.20154114
15. Horcajada-Reales C, Avilés-Izquierdo JA, Ciudad-Blanco C, Conde-Montero E, Parra-Blanco V, Lázaro-Ochaita P, et al. Dermoscopic pattern in facial trichilemmomas: red iris-like structure. J Am Acad Dermatol. (2015) 72:S30–2. doi: 10.1016/j.jaad.2014.04.064
16. Dissanayaka DWVN, Dassanayaka DKB, Jayasooriya PR. Clinical, histopathological, and management challenges of multiple familial trichoepithelioma: a case report of a patient presenting with multiple facial papules. Case Rep Dentistry. (2020) 2020:1–6. doi: 10.1155/2020/5648647
17. Dubois A, Wilson V, Bourn D, Rajan N. CYLD Genetic testing for brooke-spiegler syndrome, familial cylindromatosis and multiple familial trichoepitheliomas. PLoS Curr. (2015) 7: doi: 10.1371/currents.eogt.45c4e63dd43d62e12228cc5264d6a0db
18. Navarrete-Dechent C, Bajaj S, Marghoob AA, González S, Muñoz D. Multiple familial trichoepithelioma: confirmation via dermoscopy. Dermatol Pract Concept. (2016) 6:51–4. doi: 10.5826/dpc.0603a10
19. Collins GL, Somach S, Morgan MB. Histomorphologic and immunophenotypic analysis of fibrofolliculomas and trichodiscomas in Birt-Hogg-Dube syndrome and sporadic disease. J Cutan Pathol. (2002) 29:529–33. doi: 10.1034/j.1600-0560.2002.290904.x
20. Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. (2002) 2:157–64. doi: 10.1016/S1535-6108(02)00104-6
21. Toro JR, Glenn G, Duray P, Darling T, Weirich G, Zbar B, et al. Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol. (1999) 135:1195–202. doi: 10.1001/archderm.135.10.1195
22. Iwabuchi C, Ebana H, Ishiko A, Negishi A, Mizobuchi T, Kumasaka T, et al. Skin lesions of Birt-Hogg-Dubé syndrome: clinical and histopathological findings in 31 Japanese patients who presented with pneumothorax and/or multiple lung cysts. J Dermatol Sci. (2018) 89:77–84. doi: 10.1016/j.jdermsci.2017.10.014
23. Schwartz RA, Fernández G, Kotulska K, Józwiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. (2007) 57:189–202. doi: 10.1016/j.jaad.2007.05.004
24. Behera B, Kumari R, Gochhait D, Sathya AB, Thappa DM. Dermoscopy of adenoma sebaceum. J Am Acad Dermatol. (2017) 76:S86–8. doi: 10.1016/j.jaad.2016.05.023
25. Moscarella E, Argenziano G, Longo C, Cota C, Ardigò M, Stigliano V, et al. Clinical, dermoscopic and reflectance confocal microscopy features of sebaceous neoplasms in Muir–Torre syndrome. J Eur Acad Dermatol Venereol. (2013) 27:699–705. doi: 10.1111/j.1468-3083.2012.04539.x
26. Ponti G, de Leon MP. Muir-Torre syndrome. Lancet Oncol. (2005) 6:980–7. doi: 10.1016/S1470-2045(05)70465-4
27. Salem OS, Steck WD. Cowden's disease (multiple hamartoma and neoplasia syndrome). A case report and review of the English literature. J Am Acad Dermatol. (1983) 8:686–96. doi: 10.1016/S0190-9622(83)70081-2
28. Starink TM, van der Veen JPW, Arwert F, de Waal LP, de Lange GG, Gille JJP, et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet. (1986) 29:222–33. doi: 10.1111/j.1399-0004.1986.tb00816.x
29. Maione V, Stinco G, Orsaria M, Errichetti E. Keratotic papules of palms and soles. Dermatol Pract Concept. (2015) 5:67–8. doi: 10.5826/dpc.0502a10
30. Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. (1992) 27:40–50. doi: 10.1016/0190-9622(92)70154-8
31. Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, Monk S, et al. Mutations in ATP2A2, encoding a Ca 2+ pump, cause Darier disease. Nat Genet. (1999) 21:271–7. doi: 10.1038/6784
32. Behera B, Prabhakaran N, Naveed S, Kumari R, Thappa DM, Gochhait D. Dermoscopy of acrokeratosis verruciformis of Hopf. J Am Acad Dermatol. (2017) 77:e33–5. doi: 10.1016/j.jaad.2017.01.003
33. Eytan O, Morice-Picard F, Sarig O, Ezzedine K, Isakov O, Li Q, et al. Cole disease results from mutations in ENPP1. Am J Hum Genet. (2013) 93:752–7. doi: 10.1016/j.ajhg.2013.08.007
34. Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. (1996) 85:841–51. doi: 10.1016/S0092-8674(00)81268-4
35. Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C, et al. Identification of a SUFUgermline mutation in a family with Gorlin syndrome. Am J Med Genet A. (2009) 149A:1539–43. doi: 10.1002/ajmg.a.32944
36. North JP, McCalmont TH, LeBoit P. Palmar pits associated with the nevoid basal cell carcinoma syndrome. J Cutan Pathol. (2012) 39:736–8. doi: 10.1111/j.1600-0560.2012.01984.x
37. Pilarski R. Cowden syndrome: a critical review of the clinical literature. J Genet Counsel. (2009) 18:13–27. doi: 10.1007/s10897-008-9187-7
38. Dharman S, Arvind M. Darier's disease–oral, general and histopathological features in a 7 year old child. J Indian Soc Pedod Prev Dent. (2016) 34:177–9. doi: 10.4103/0970-4388.180449
39. Sparling JD, Hong C-H, Brahim JS, Moss J, Darling TN. Oral findings in 58 adults with tuberous sclerosis complex. J Am Acad Dermatol. (2007) 56:786–90. doi: 10.1016/j.jaad.2006.11.019
40. Richard G, De Laurenzi V, Didona B, Bale SJ, Compton JG. Keratin 13 point mutation underlies the hereditary mucosal epithelia disorder white sponge nevus. Nat Genet. (1995) 11:453–5. doi: 10.1038/ng1295-453
41. Schaffer JV, Kamino H, Witkiewicz A, McNiff JM, Orlow SJ. Mucocutaneous neuromas: an underrecognized manifestation of PTEN hamartoma-tumor syndrome. Arch Dermatol. (2006) 142:625–32. doi: 10.1001/archderm.142.5.625
42. Lee MJ, Chung KH, Park JS, Chung H, Jang HC, Kim JW. Multiple endocrine neoplasia type 2B: early diagnosis by multiple mucosal neuroma and its DNA analysis. Ann Dermatol. (2010) 22:452–5. doi: 10.5021/ad.2010.22.4.452
43. Hofstra RMW, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. (1994) 367:375–6. doi: 10.1038/367375a0
44. Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. (2017) 3:17004. doi: 10.1038/nrdp.2017.4
45. Megahed M. Histopathological variants of neurofibroma. A study of 114 lesions. Am J Dermatopathol. (1994) 16:486–95. doi: 10.1097/00000372-199410000-00003
46. Jouhilahti E-M, Visnapuu V, Soukka T, Aho H, Peltonen S, Happonen R-P, et al. Oral soft tissue alterations in patients with neurofibromatosis. Clin Oral Invest. (2012) 16:551–8. doi: 10.1007/s00784-011-0519-x
47. Fargnoli MC, Orlow SJ, Semel-Concepcion J, Bolognia JL. Clinicopathologic findings in the Bannayan-Riley-Ruvalcaba syndrome. Arch Dermatol. (1996) 132:1214–8. doi: 10.1001/archderm.132.10.1214
48. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. (2000) 26:89–92. doi: 10.1038/79238
49. Carney JA, Ferreiro JA. The epithelioid blue nevus. A multicentric familial tumor with important associations, including cardiac myxoma and psammomatous melanotic schwannoma. Am J Surg Pathol. (1996) 20:259–72. doi: 10.1097/00000478-199603000-00001
50. Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Human Genet. (2002) 71:389–94. doi: 10.1086/341528
51. Martínez-Quintana E, Rodríguez-González F. LEOPARD syndrome: clinical features and gene mutations. Mol Syndromol. (2012) 3:145–57. doi: 10.1159/000342251
52. Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. (1998) 18:38–43. doi: 10.1038/ng0198-38
53. Gass JD, Glatzer RJ. Acquired pigmentation simulating Peutz-Jeghers syndrome: initial manifestation of diffuse uveal melanocytic proliferation. Br J Ophthalmol. (1991) 75:693–5. doi: 10.1136/bjo.75.11.693
54. Smid L, Zargi M. Cowden's disease–its importance for otolaryngologists. J Laryngol Otol. (1993) 107:1063–5. doi: 10.1017/S0022215100125307
55. Ferran M, Bussaglia E, Lazaro C, Guiu XM, Pujol RM. Acral papular neuromatosis: an early manifestation of Cowden syndrome. Br J Dermatol. (2008) 158:174–6. doi: 10.1111/j.1365-2133.2007.08237.x
56. Barnhill RL, Albert LS, Shama SK, Goldenhersh MA, Rhodes AR, Sober AJ. Genital lentiginosis: a clinical and histopathologic study. J Am Acad Dermatol. (1990) 22:453–60. doi: 10.1016/0190-9622(90)70064-O
57. Hansen-Kiss E, Beinkampen S, Adler B, Frazier T, Prior T, Erdman S, et al. A retrospective chart review of the features of PTEN hamartoma tumour syndrome in children. J Med Genet. (2017) 54:471–8. doi: 10.1136/jmedgenet-2016-104484
58. Martin H, Bessis D, Bourrat E, Mazereeuw Hautier J, Morice-Picard F, Balguerie X, et al. Cutaneous lipomas and macrocephaly as early signs of PTEN hamartoma tumor syndrome. Pediatr Dermatol. (2020) 37:839–43. doi: 10.1111/pde.14265
59. Elsherif MA, Babovic-Vuksanovic D, Spinner RJ. An association of peripheral nerve sheath tumors and lipomas. Acta Neurochir (Wien). (2017) 159:185–90. doi: 10.1007/s00701-016-3038-3
60. Kurek KC, Howard E, Tennant LB, Upton J, Alomari AI, Burrows PE, et al. PTEN hamartoma of soft tissue: a distinctive lesion in PTEN syndromes. Am J Surg Pathol. (2012) 36:671–87. doi: 10.1097/PAS.0b013e31824dd86c
61. Tan M-H, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. (2012) 18:400–7. doi: 10.1158/1078-0432.CCR-11-2283
63. Genetic/Familial High-Risk Assessment: Breast Ovarian Pancreatic (Version 1.2021) (2020). Available online at: www.nccn.org (accessed September 26, 2020).
64. Yehia L, Ni Y, Sesock K, Niazi F, Fletcher B, Chen HJL, et al. Unexpected cancer-predisposition gene variants in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome patients without underlying germline PTEN mutations. PLoS Genet. (2018) 14:e1007352. doi: 10.1371/journal.pgen.1007352
65. Yehia L, Eng C. PTEN hamartoma tumour syndrome: what happens when there is no PTEN germline mutation? Hum Mol Genet. (2020) 29:R150–7. doi: 10.1093/hmg/ddaa127
66. Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, Edelman E, et al. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. (2008) 83:261–8. doi: 10.1016/j.ajhg.2008.07.011
67. Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. Jama. (2010) 304:2724–31. doi: 10.1001/jama.2010.1877
68. Orloff MS, He X, Peterson C, Chen F, Chen J-L, Mester JL, et al. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Human Genet. (2013) 92:76–80. doi: 10.1016/j.ajhg.2012.10.021
69. Yehia L, Niazi F, Ni Y, Ngeow J, Sankunny M, Liu Z, et al. Germline heterozygous variants in SEC23B are associated with Cowden syndrome and enriched in apparently sporadic thyroid cancer. Am J Human Genet. (2015) 97:661–76. doi: 10.1016/j.ajhg.2015.10.001
70. Lee Y-R, Yehia L, Kishikawa T, Ni Y, Leach B, Zhang J, et al. WWP1 gain-of-function inactivation of PTEN in cancer predisposition. N Eng J Med. (2020) 382:2103–16. doi: 10.1056/NEJMoa1914919
71. Hendricks LAJ, Hoogerbrugge N, Hoeijmakers JHMS, Vos JR. A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin Genet. (2020) 99:219–25. doi: 10.1111/cge.13875
72. Zaid Al T, Ditelberg JS, Prieto VG, Lev D, Luthra R, Davies MA, et al. Trichilemmomas show loss of PTEN in Cowden syndrome but only rarely in sporadic tumors. J Cutan Pathol. (2012) 39:493–9. doi: 10.1111/j.1600-0560.2012.01888.x
73. Masmoudi A, Chermi ZM, Marrekchi S, Raida BS, Boudaya S, Mseddi M, et al. Cowden syndrome. J Dermatol Case Rep. (2011) 5:8–13. doi: 10.3315/jdcr.2011.1063
74. Cnudde F, Boulard F, Muller P, Chevallier J, Teron-Abou B. Cowden disease: treatment with acitretine. Ann Dermatol Venereol. (1996) 123:739–41.
75. Botma M, Russell DI, Kell RA. Cowden's disease: a rare cause of oral papillomatosis. J Laryngol Otol. (2006) 116:221–3. doi: 10.1258/0022215021910393
76. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. (2007) 128:157–70. doi: 10.1016/j.cell.2006.11.042
77. Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. (1998) 280:1614–7. doi: 10.1126/science.280.5369.1614
78. Cantley LC. The phosphoinositide 3-kinase pathway. Science. (2002) 296:1655–7. doi: 10.1126/science.296.5573.1655
79. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. (1998) 273:13375–8. doi: 10.1074/jbc.273.22.13375
80. Komiya T, Blumenthal GM, DeChowdhury R, Fioravanti S, Ballas MS, Morris J, et al. A pilot study of sirolimus in subjects with Cowden syndrome or other syndromes characterized by germline mutations in PTEN. Oncologist. (2019) 24:1510–65. doi: 10.1634/theoncologist.2019-0514
81. Ngeow J, Eng C. PTEN in hereditary and sporadic cancer. Cold Spring Harb Perspect Med. (2020) 10:a036087. doi: 10.1101/cshperspect.a036087
82. He X, Wang Y, Zhu J, Orloff M, Eng C. Resveratrol enhances the anti-tumor activity of the mTOR inhibitor rapamycin in multiple breast cancer cell lines mainly by suppressing rapamycin-induced AKT signaling. Cancer Lett. (2011) 301:168–76. doi: 10.1016/j.canlet.2010.11.012
83. Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. (2007) 128:141–56. doi: 10.1016/j.cell.2006.11.040
Keywords: genodermatoses, cancer genetics, Cowden syndrome, PTEN, cancer predisposition
Citation: Lim A and Ngeow J (2021) The Skin in Cowden Syndrome. Front. Med. 8:658842. doi: 10.3389/fmed.2021.658842
Received: 26 January 2021; Accepted: 17 May 2021;
Published: 10 June 2021.
Edited by:
Isabella Mammi, Ospedale Dolo, ItalyReviewed by:
Artem Vorobyev, University Medical Center Schleswig-Holstein, GermanyCopyright © 2021 Lim and Ngeow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanne Ngeow, am9hbm5lLm5nZW93QG50dS5lZHUuc2c=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.