 Xinyue Yao
Xinyue Yao Hong Shen2†
Hong Shen2† Haojun Zhang
Haojun Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med., 24 June 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.657918
This article is part of the Research TopicCell Cross-talk in Diabetic Kidney Diseases, Volume IIView all 8 articles
Diabetic nephropathy (DN) is the main cause of end stage renal disease (ESRD). Glomerulus damage is one of the primary pathological changes in DN. To reveal the gene expression alteration in the glomerulus involved in DN development, we screened the Gene Expression Omnibus (GEO) database up to December 2020. Eleven gene expression datasets about gene expression of the human DN glomerulus and its control were downloaded for further bioinformatics analysis. By using R language, all expression data were extracted and were further cross-platform normalized by Shambhala. Differentially expressed genes (DEGs) were identified by Student's t-test coupled with false discovery rate (FDR) (P < 0.05) and fold change (FC) ≥1.5. DEGs were further analyzed by the Database for Annotation, Visualization, and Integrated Discovery (DAVID) to enrich the Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. We further constructed a protein-protein interaction (PPI) network of DEGs to identify the core genes. We used digital cytometry software CIBERSORTx to analyze the infiltration of immune cells in DN. A total of 578 genes were identified as DEGs in this study. Thirteen were identified as core genes, in which LYZ, LUM, and THBS2 were seldom linked with DN. Based on the result of GO, KEGG enrichment, and CIBERSORTx immune cells infiltration analysis, we hypothesize that positive feedback may form among the glomerulus, platelets, and immune cells. This vicious cycle may damage the glomerulus persistently even after the initial high glucose damage was removed. Studying the genes and pathway reported in this study may shed light on new knowledge of DN pathogenesis.
Diabetic nephropathy (DN) is one of the most serious diabetic chronic microvascular complications and the major cause of end stage renal disease (ESRD) (1, 2). Glomerulus damage is one of the primary pathological changes in DN (3). The progression of DN is known to occur in a series of pathological changes in the glomerulus, such as expansion of glomerular mesangium, glomerular basement membrane (GBM) thickness, and podocytes loss. These changes damage glomerular filtration, causing proteinuria and glomerulosclerosis. Eventually, this may cause a decrease in the glomerular filtration rate (GFR) and the development of end stage renal disease (4). Now, DN is the main reason for dialysis or a kidney transplant and is a great global public health burden (5). Currently, drugs only target the renin-angiotensin-aldosterone system (RAAS) and sodium-glucose cotransporter 2 (SGLT2) inhibitors to treat DN (5–7). Therefore, it is urgent to explore the newly found molecular mechanism of DN and provide a new target for the diagnosis and treatment of DN.
Transcriptomic analysis is a powerful tool used to discover new targets and explore many diseases including DN (8). A lot of work has been done using the transcriptomic method, which has provided some novel targets and mechanisms for DN (9–17). However, as is known, the transcriptomic method has some limitations. This method can only use a single race sample and a small sample number, which is disproportionate to their high costs. And most transcriptomic methods have poor instability for their great measurement error (9). So, gene screening by different transcriptomic research methods vary and even conflict sometimes. Bioinformatics tools can integrate multiple transcriptomic methods to increase the statistical power. So, reduced population samples and many stable differentially expressed genes can be obtained (17). Bioinformatic tools were used in a lot of studies to analyze existing transcriptomics data and some important discoveries were found. Tang et al. analyzed glomerulus and renal tubule tissue transcription omics data, and found that NTNG1 and HGF were potential DN biomarkers of high specificity and sensitivity (18). Wang et al. found that the glomerulus in DN kidney tissue mainly caused changes in cell connectivity and tissue cell modification, while renal tubular tissue mainly caused abnormalities in energy metabolism, and changes in methylation status of core regulatory genes might be a potential factor for the pathogenesis of DN (19). As far as we know, research has seldom used bioinformatic tools to analyze all human existing glomerulus transcriptomics datasets to discover new potential biomarkers and the pathogenesis of DN. So it will be very attractive to do it.
Bioinformatic tools combining the information of multiple independent transcriptomic studies fundamentally include meta-analysis and cross-platform normalization (20). In the meta-analysis approach, each experiment is first analyzed separately and then combined by one of three types of statistics: p-value, effect size, and ranked gene lists. Cross-platform normalization considers all platform transcriptomic data as a single dataset. This approach normalizes transcriptomic data to remove the artifactual differences between transcriptomic studies and preserves biological differences between conditions. Cross-platform normalization is thought to have better performance than meta-analysis for “separate statistics” and “averaging is often less powerful than directly computing statistics from aggregated data” (20). We can always significantly find more differentially expressed genes in this process than meta-analysis (20). There is more than a dozen methods that can be used to undertake cross-platform normalization. But most of these cross-platform normalization methods can only process two different platforms, and transcriptomic data have comparable sample sizes. Recently a new method Shambhala (https://github.com/oncobox-admin/harmony) was found to solve this problem and may be the best choice to process gigantic transcriptomic datasets (21). Shambhala performs cross-platform normalization by using an auxiliary calibration dataset (P0) and a reference definitive dataset (Q). The initial data can be output into a generic form of a gene expression profile. This method can make experimental data independent of the experimental platform and the number of experiments (21). It can improve data comparability and reduce batch effect greatly (21). Above all, it is currently the only platform-independent data coordination technology that supports the processing of data obtained from multiple experimental platforms (21). Application of this technique may be the best choice to analysis all human existing glomerulus transcriptomics datasets.
Our objective is to comprehensively analyze transcriptomic profiles of all existing DN patient glomerular datasets in the GEO database for understanding of the pathogenesis of DN. In this study, we firstly downloaded all DN patients and their control glomerular original transcriptomic data. Then we used Bioconductor packages to extract the transcriptomic profiles and perform cross-platform normalization (Shambhala method), a static tests screening, and identification of differentially expressed genes (DEGs). We used the Database for Annotation, Visualization, and Integrated Discovery (DAVID) to enrich the Gene Ontology (GO) DEGs and the Kyoto Encyclopedia of Genes Genomes (KEGG) pathway. We constructed a protein-protein interaction (PPI) network and modules to screen core genes. We further used CIBERSORTx to explore the infiltration of immune cells in the DN glomerulus.
The Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) is an international public repository containing high-throughput microarray and next-generation sequence functional genomic datasets, which can provide researchers with a large number of gene expression profile data (22, 23). At present, a large number of microarray data of different diseases have been collected in the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/) database for sharing and learning by institutes around the world. In this report, in order to obtain transcripts related to human DN, we searched all GEO datasets in the NCBI database with details of “diabetic nephropathy [Mesh],” “glomerular [Mesh],” and “Homo sapiens [porgn:__txid9606]” before December 2020. A total of 21 datasets were retrieved. We defined the following exclusion criteria: (i) cell line sample; (ii) a sample of biological fluids, including blood, plasma, and urine; (iii) samples of tissues that have undergone special interventions, such as drug stimulation, hypoxia, and oxygen-enriched treatment, etc. After having filtered other tissues or diseases out, GSE96804 (24), GSE104948 (13), GSE99339 (25), GSE30528 (9), GSE21785 (26), and GSE47183 (15, 27) were selected for subsequent analysis. As there are fewer control samples in these datasets, we manually retrieved human glomerular datasets as control. Datasets GSE20602 (28), GSE121233 (29), GSE108109 (13), GSE104066 (30), and GSE32591 (31) were included. These dataset have similar characteristics as the control in the DN datasets, which were marked as “glomeruli from living human donor kidney biopsy” and “glomeruli from the unaffected portion of tumor nephrectomies.” Based on all the above datasets, a total of 90 DN and 95 healthy control glomerular samples were included in this study (see Table 1 for details). The samples were collected from multiple platforms, including Affymetrix GeneChip, Human Genome HG-U133A Custom CDF, and the Affymetrix Human Gene 2.1 ST Array.
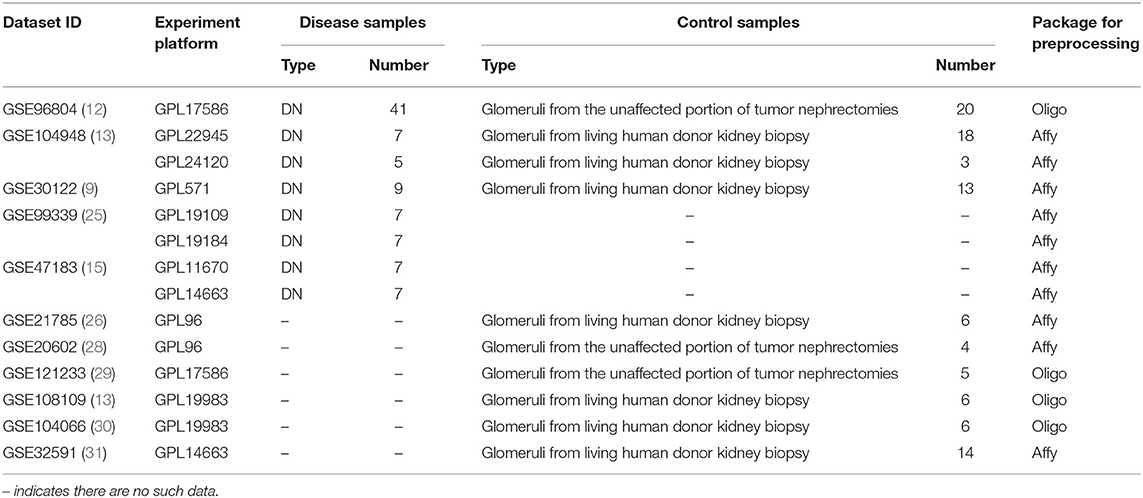
Table 1. The microarray datasets collected and used in this study.
For the preprocessing of a large number of multi-platform microarray datasets, we first used Affy1.64.0 (http://bioconductor.org/packages/release/bioc/html/affy.html) (32) and Oligo 1.50.0 (http://bioconductor.org/packages/release/bioc/html/oligo.html) (33) from Bioconductor in R (3.60) to extract gene expression value. Briefly, after downloading all raw data (Table 1) from the GEO repository, probe expression values were extracted by Affy according to the user guide. After reading the raw data, background correction (rma), normalization (quantiles), probe specific background correction (pmonly), and summary (liwong) were performed to obtain the probe expression value. If the raw data could not be extracted by Affy, the Oligo package was used following the user guide. After reading the raw data, further background subtraction, normalization, and summarization was performed by using rma. All probes were further annotated to genes by their own annotation data. The median of the probe expressions was calculated as the gene expression value. After merging all transcriptomics data into a large sample and removing none of the express genes, Shambhala (https://github.com/oncobox-admin/harmony) was used to perform cross-platform normalization according to the guidance of literature. In this research, a longer P0 was used in Shambhala which was kindly provided by developers and can be found in Supplementary Table 1. This auxiliary calibration dataset contains 13,645 genes which is much longer than what is provided on the website. The levels of each gene expression difference between control and DN were compared by Student's t-test coupled with a false discovery rate (FDR) correction. In this study, genes conforming to the fold change (FC) ≥1.5 and P < 0.05 (Student's t-test adjusted by FDR) were considered as DEGs.
GO and KEGG enrichment of the candidate genes were performed using the DAVID online tool (https://david.ncifcrf.gov) (34). GO analysis is a bioinformatics tool that presents information on the biological domain with respect to molecular function (MF), cellular components (CC), and biological processes (BP) (35). KEGG is a database that displays information of system integration gene functions (36). The enrichment significance threshold was set to P < 0.05. The visualization of GO enrichment results was conducted by using the GO plot package in the R software (37). To determine the changed tendency of pathways in DN, the Z-score was calculated in each term using the following formula:
The Nup and Ndown separately represent the number of upregulated and downregulated genes between DN and normal controls, and the count is the number of DEGs belonging to this term (37).
Cytoscape 3.8.0 was used for visualization and analysis of the complex network (38). In order to avoid the loss of the protein-protein interaction in a single database, we integrated PPI information collected from multiple databases. We imported network of DEGs by querying the Proteomics Standard Initiative Common QUery Interface (PSICQUIC) which is integrated in Cytoscape (39, 40). Four protein interaction databases were selected for analysis: (i) STRING (https://string-db.org/) (41) which integrates data from high-throughput experiments, text mining, bioinformatics prediction, and interaction databases, (ii) MINT (https://mint.bio.uniroma2.it/) (42) in which PPIs have been confirmed experimentally, (iii) IntAct (https://www.ebi.ac.uk/intact/) (43) which is directly submitted by users, and (iv) Reactome (http://www.reactome.org) (44) which is a pathway database that provides intuitive bioinformatics tools for the visualization, interpretation, and analysis of pathway knowledge. The former three databases focus on exploring the physical interactions between proteins and the last one focuses on biological pathways. After excluding non-human gene information, the analysis results were merged to obtain more comprehensive protein-protein interaction information. In a PPI network, degree and betweenness centrality (BC) are commonly used to evaluate the critical degree of nodes. Degree is the basic index of a node, which is used to indicate the number of links that interact with the node and the network (45). BC measures the importance of nodes based on the shortest paths, which represent the shortest distance between two nodes. A node with a greater BC value has a higher frequency of information exchange within the node (46, 47). In this study, nodes with a high degree and high BC were regarded as key nodes, and genes whose BC and degree were both in the top 10% in the total nodes of the network were regarded as important genes. In this study, we computed the properties of nodes and measured the default parameters with Cytoscape. Next, we used the Cytoscape plug-in Molecular Complex Detection tool (MCODE; version 1.5.1) (48) to identify the most important module in the network map. The criteria for MCODE analysis were a degree of cut-off = 2, MCODE scores >6, maximum depth = 100, node score cut-off = 0.2, and k-score = 2(48).
We selected DEGs that met the following three constraints as core genes: (i) DEGs that had a large fold change (top 100); (ii) the gene was located in key module; and (iii) nodes with top 10% BC values and degree determined by Cytoscape software.
CIBERSORTx (https://cibersortx.stanford.edu) (49) is a digital cytometry program that uses a machine learning method. It can provide an estimation of the abundances of member cell types in a mixed cell population by using gene expression data. We used a validated leukocyte gene signature matrix that contained 547 genes to distinguish 22 human hematopoietic cell phenotypes to identify glomerular immune cells infiltration. Seven T cell types, naive and memory B cells, plasma cells, natural killer (NK) cells, and myeloid subsets infiltration alteration were identified (50). After uploading the cross-platform normalized data to CIBERSORTx, permutations were set at 100 and absolute mode was selected. Absolute mode scales relative cellular fractions into a score that reflects the absolute proportion of each cell type in a mixture. When the p < 0.05, it indicates that the infiltration rate of the 22 immune cells types analyzed by CIBERSORTx is accurate. The accurately identified immune cell infiltration was further compared between normal control and DN by Wilcoxon signed-rank test. The steps of the whole process are shown in Figure 1.
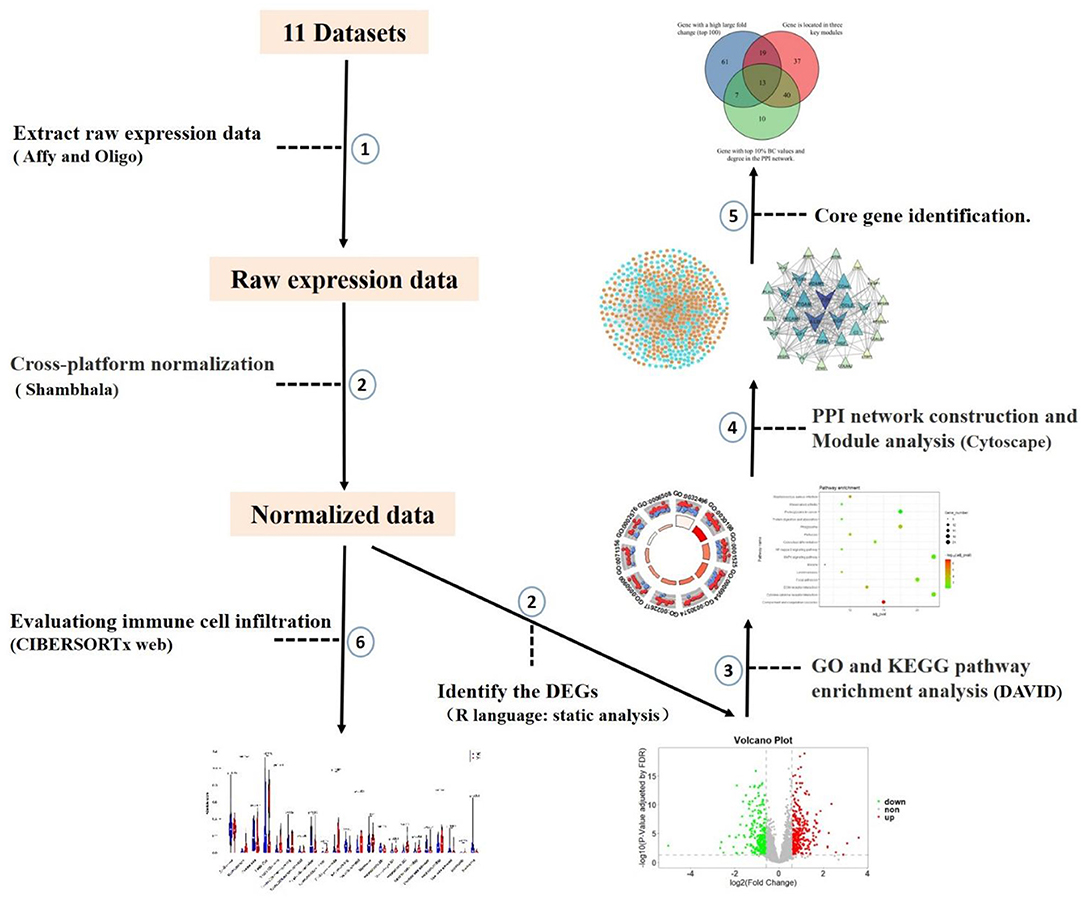
Figure 1. The workflow of microarray data preprocessing and subsequent analysis in this study. We selected 11 datasets based on the constraints. Firstly, raw expression data were extracted after background correction and quality control. Processing was carried out using the R package, and then the data were cross-platform normalized by coordination and transformation using Shambhala. Then DEGs of DN glomerular tissue and healthy control tissue were identified by static analysis. Enrichment results were obtained using DAVID, then PPI networks and modules were constructed, and core genes were identified. Finally, the dataset was brought into the CIBERSORTx web portal to evaluate immune cell infiltration.
All microarray datasets were standardized, and the results before and after standardization are shown in Figures 2A,B. According to values of p < 0.05 and FC ≥ 1.5, a total of 578 genes were identified to be differentially expressed in the DN group, including 334 upregulated and 244 downregulated genes (Figure 2C and Supplementary Table 2). DEGs with the top 100-fold change are shown in the heatmap (Figure 2D).
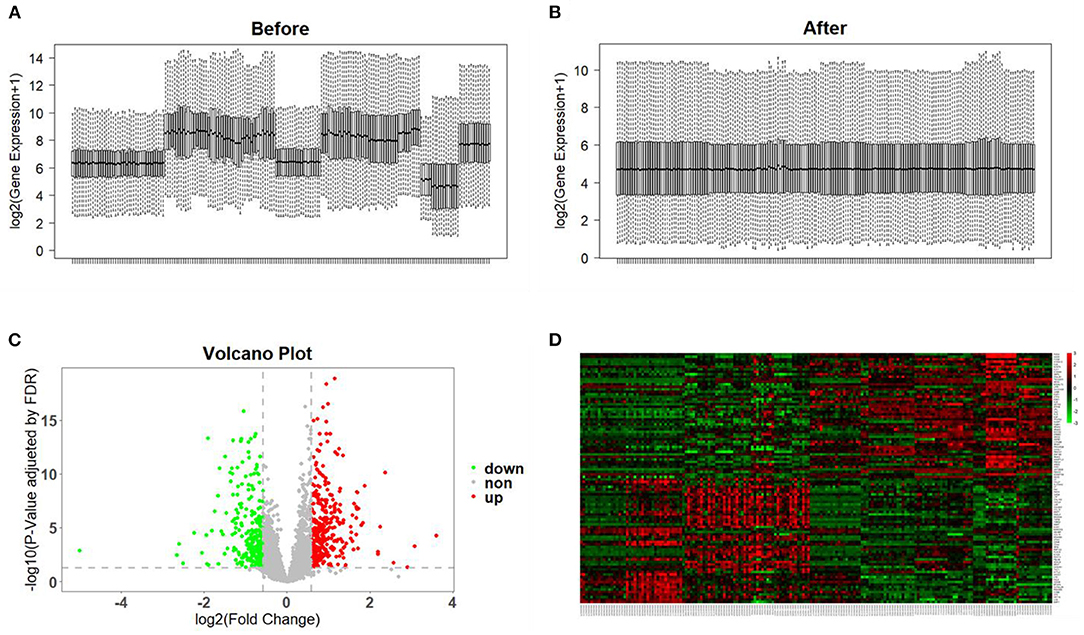
Figure 2. Data preprocessing and identification of DEGs. (A,B) Gene expression data before and after normalization. The horizontal axis represents the sample symbol and the vertical axis represents the gene expression values. The black line in the box plot represents the median value of gene expression. (C) Volcano plot analysis of DEGs. Red represents high expression, green represents low expression, and gray represents no difference. (D) The heatmap of top 100-fold-change DEGs. Red areas represent highly expressed genes and green areas represent lowly expressed genes in glomerular tissues from DN patients compared with normal controls.
GO analysis was performed based on the 578 DEGs, and circle graphs show the top 10 entries of each term. BP demonstrated that the DEGs were enriched in lipopolysaccharide, extracellular matrix organization, angiogenesis, inflammatory response, leukocyte migration, and platelet degranulation, etc. (Figure 3A). Variations in DEGs linked with CC were extracellular exosome, extracellular matrix, focal adhesion, and platelet alpha granule lumen, etc. (Figure 3B). Regarding MF, DEGs were significantly enriched in heparin binding, integrin binding, collagen binding, and extracellular matrix structural constituent, etc. (Figure 3C). Analysis of KEGG pathways indicated that canonical pathways associated with DEGs were complement and coagulation cascades, staphylococcus aureus infection, and ECM-receptor interaction, etc. The top 15 KEGG enrichment results are shown in Figure 3D. The complete enrichment analysis results are in Supplementary Table 3.
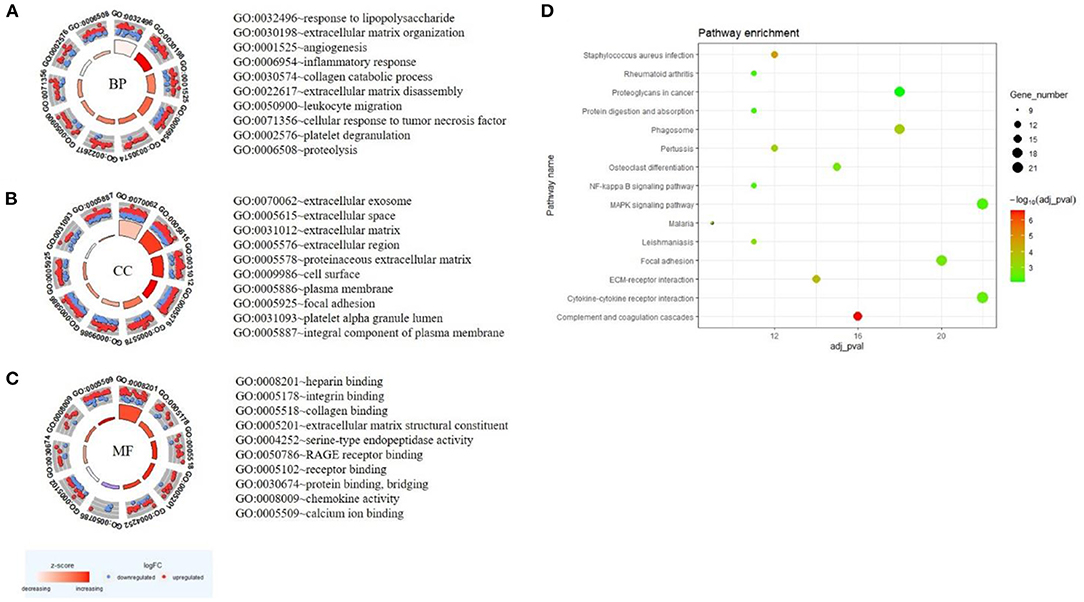
Figure 3. GO and KEGG enrichment result of DEGs. The result of the top 10 enrichment genes is shown in the GOCircle plot: BP (A), CC (B), MF (C). The inner ring is a bar plot where the bar height indicates the significance of the term (p-value) and the color indicates the z-score. The outer ring displays scatterplots of the expression levels (logFC) for the genes in each term. The blue node is the downregulated gene, and the red node is the upregulated gene. (D) The top 15 KEGG enrichment results. The x-axis represents gene number and the y-axis represents KEGG terms. The size of the circle represents gene count. Circles of different colors represent different adjusted p-values.
The DEG expression products in DN were constructed into PPI networks by merging the STRING, MINT, IntAct, and Reactome databases in Cytoscape software. By removing the separated and separately connected nodes, a complex network of DEGs was constructed and is presented in Figure 4A. Three modules were identified by MCODE (Figures 4B–D). Module 1 (score = 16.867) was composed of 31 nodes and 253 edges, module 2 (score = 10.051) was composed of 40 nodes and 196 edges, and module 3 (score = 6.216) was composed of 38 nodes and 115 edges.
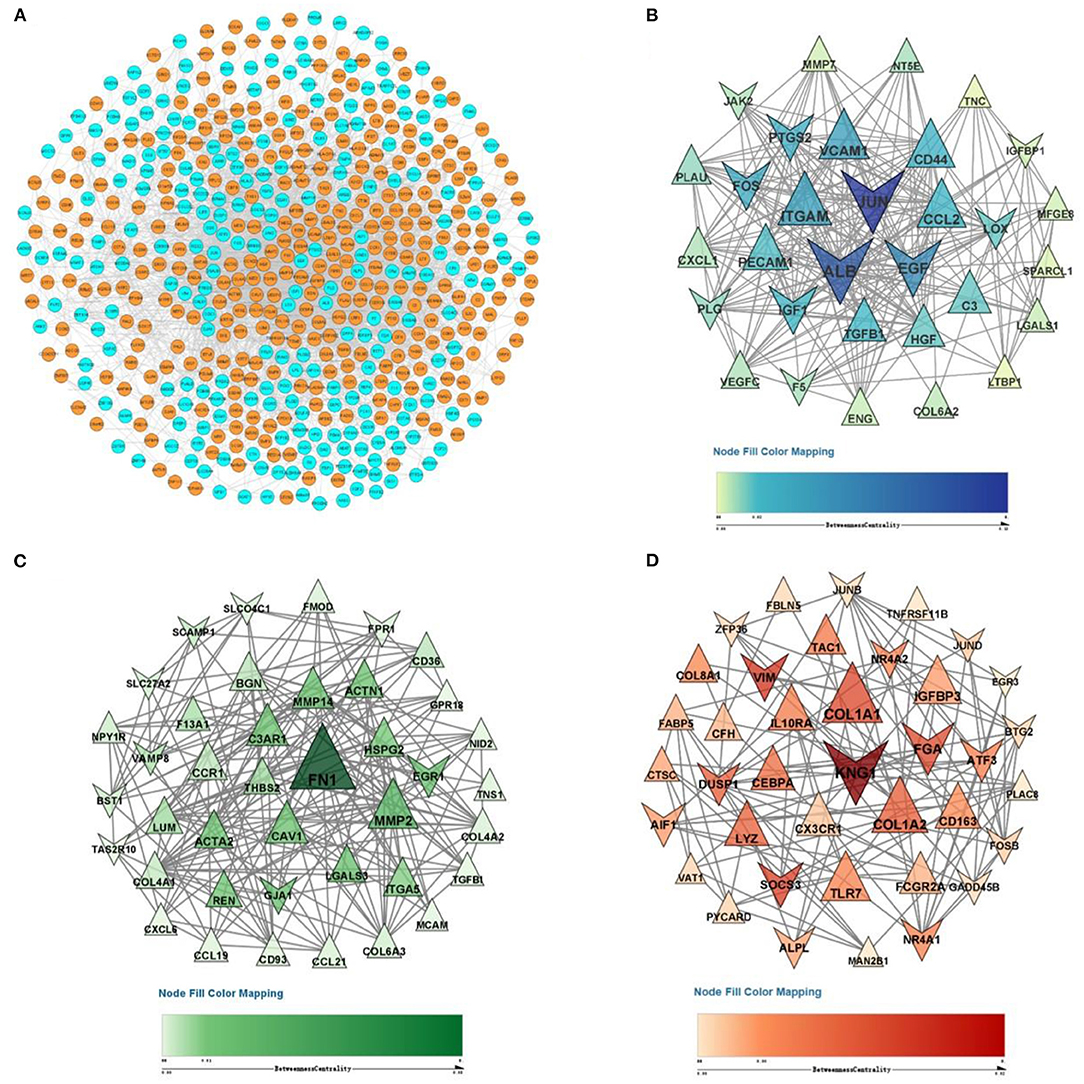
Figure 4. PPI network and three significant modules of DEGs. (A) PPI network of DEGs created by merging the STRING, MINT, IntAct, and Reactome databases. The orange purple nodes represent upregulated DEGs, and blue nodes represent downregulated DEGs. A total of 578 DEGs formed a PPI network consisting of 557 nodes and 3,882 edges. (B) The most significant module identified by MCODE (score = 16.867). (C) The second most significant module identified by MCODE (score = 10.051). (D) The third most significant module identified by MCODE (score = 6.216). The size of the nodes corresponds to their degree.
In this study, C3, CCL21, SLC34A2, C7, ALB, ESM1, ATF3, EGR1 etc. had large fold change, 109 genes (JUN, ALB, EGF, VCAM1, ITGAM, etc.) were located in the three key modules, 70 genes (JUN, ALB, FN1, EGF, VCAM1, ITGAM, FOS, etc.) were nodes in the PPI network with the top 10% of BC value and degree. The Venn diagram presented illustrates the overlaps between DEGs (Figure 5A). As shown in the Venn diagram, we selected 13 eligible DEGs as core genes, including complement C3 (C3), fibronectin 1 (FN1), collagen type I alpha 2 chain (COL1A2), lumican (LUM), thrombospondin 2 (THBS2), CD44 molecule (CD44), lysozyme (LYZ), Fos proto-oncogene (FOS), early growth response 1 (EGR1), albumin (ALB), plasminogen (PLG), epidermal growth factor (EGF), and dual-specificity protein phosphatase-1 (DUSP1). The details of core genes are shown in Figure 5B.
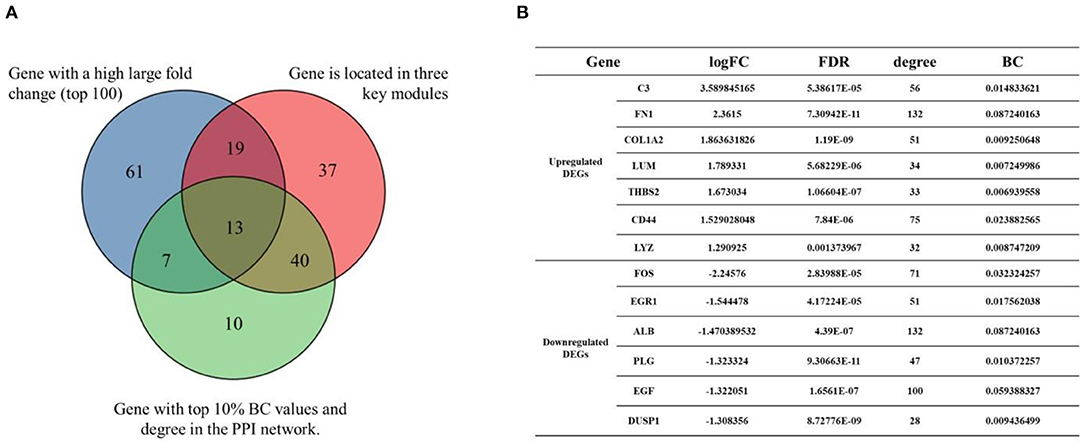
Figure 5. Venn diagram of core genes. (A) The blue circle represents DEGs that had a high large fold change (top 100). The red circle represents DEGs that were located in the three key modules. The green circle represents DEGs that had the top 10% BC values and degree in the PPI network. (B) Details of core genes (FC, FDR adjusted p-value, node degree, node BC value).
Since inflammation is enriched in GO and KEGG, it will be interesting to specify which immune cells infiltrated the glomerular under DN. CIBERSORTx is a bioinformatics tool that can specifically analyze the infiltration of immune cells in tissues. The results of CIBERSORTx analysis showed that there were 172 samples of glomerulus transcriptomic data at p < 0.05 (86 control and 86 DN). Indicating that most of the infiltration rates of the 22 immune cells types analyzed by CIBERSORTx were accurate. Compared with normal control glomerular tissues, the infiltration of plasma cells, follicular helper T cells, resting NK cells, macrophages M0, activated dendritic cells (DCs), and neutrophils were reduced in glomerular tissues affected by DN. Infantile CD4+ T cells, regulatory T cells, γδT cells, activated NK cells, macrophages M1, macrophages M2, resting DCs, and mast cells were increased in DN glomerular tissues (Figure 6).
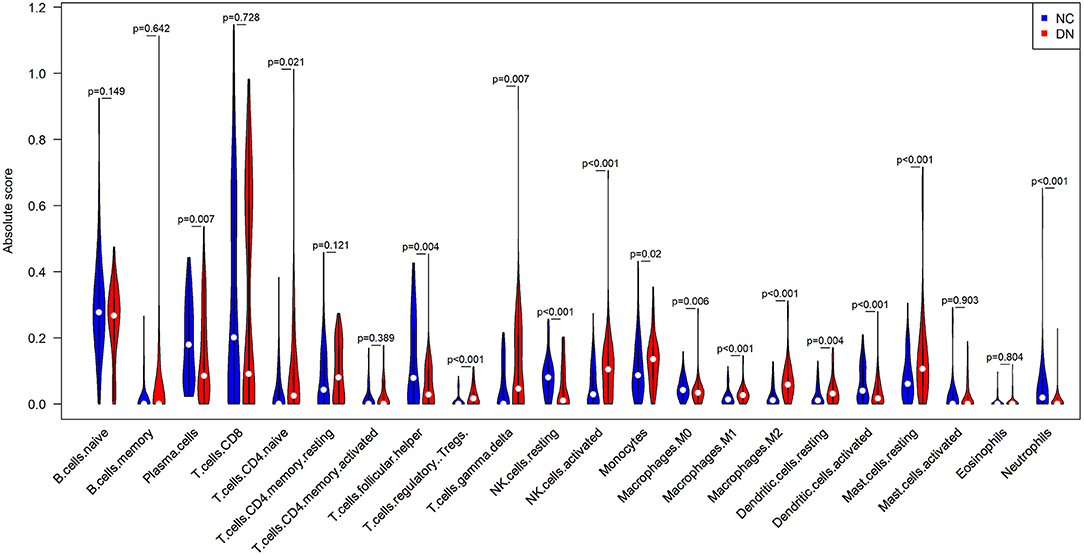
Figure 6. Comparison of infiltrated immune cell subpopulations in glomerulus tissues with or without DN. Violin plot of immune-infiltrating lymphocytes between DN glomerular tissues and healthy control glomerular samples, in which the red represents DN samples, and the blue represents control samples.
DN is a serious complication of long-term diabetes mellitus (DM) and a growing global economic burden (51). The glomerulus plays a key role in the development of DN. However, due to the complexity of etiology and ethnic differences, our understanding of the molecular mechanism in DN glomerular tissue is still incomplete. Therefore, it is urgent to explore the new molecular mechanism which may help DN treatment and diagnosis.
In this study, we used bioinformatics methods to analyze GEO transcriptomics datasets before December 2020, and attempted to explore the potential molecular mechanisms of the DN glomerulus. A total of 578 DEGs were identified in the glomerular samples between DN and normal samples, including 334 upregulated and 244 downregulated genes. Thirteen core genes were finally identified, including C3, FN1, COL1A2, LUM, THBS2, CD44, LYZ, FOS, EGR1, ALB, PLG, EGF, and DUSP1.
Among these core genes, some have been shown to play an important role in the pathogenesis of DN, C3 (52, 53), ALB (54–56), EGF (57), EGR1 (58–61), COL1A2 (62), FN1 (63, 64), CD44 (65, 66), FOS (67), PLG (68, 69), and DUSP1 (70). It is well-known that inflammation and fibrosis play an important role in the pathogenesis of the DN glomerulus (71).
Among these 13 core genes, there is little known about what roles LYZ, LUM, and THBS2 play in the development of DN. LYZ, which encodes lysozymes, is an antimicrobial agent found in human milk. It is also found in the spleen, lungs, kidneys, white blood cells, plasma, saliva, and tears. Gallo et al. found that LYZ downregulated the production and release of inflammatory mediators [such as interleukin (IL)-6] induced by late glycosylation end products in in vitro models of human proximal renal tubular epithelial cells, and prevented the recruitment of some macrophages at the inflammatory site (72). Indicating that locally expressed LYZ may take part in the pathogenesis of DN. LUM encodes members of the leucine-rich small proteoglycan (SLRP) family, which includes decorin, biglycan, and fibromodulin, etc. (73). The protein expressed by this gene partially binds collagen fibers, and highly charged hydrophilic glycosaminoglycans regulate the spacing between fibers (74). It has been reported that the LUM protein and its family member decorin accumulate strongly in the advanced glomerulosclerosis stage of DN (75). Decorin greatly affects the progression of DN by forming the ternary complex of decorin-type I collagen-transforming growth factor, beta (TGF-β) (75). It is speculated that LUM may also promote the development of DN by interacting with TGF-β. THBS2 proteins belong to the thrombospondin family. As a relatively special member of this family, it has an anti-angiogenic effect and interacts with various cell receptors and growth factors to regulate cell proliferation, apoptosis, and adhesion (76). It has been shown that THBS2 plays an important role in acute kidney injury (AKI) (77). It also has been found differently expressed in the plasma of type 2 diabetes patients (78). This indicates that THBS2 plays an important role in DN.
We enriched the GO terms and KEGG pathway of DEGs. Among the enriched pathways, inflammatory response, leukocyte migration, platelet degranulation, and platelet alpha particles attracted our attention. We further used CIBERSORTx to identify immune cell infiltration in DN glomerular tissues. Three types of T cells increased infiltration in the DN tissues, including naive CD4+ T cells, regulatory T cells, and γδT cells. The originally resting NK cells in the tissues were activated, and the macrophages were also differentiated from resting M0 into the classically activated and pro-inflammatory M1 and the alternatively activated M2. The active DCs were reduced to resting. The infiltration of mast cells was increased and the infiltration of plasma cells and neutrophils was decreased in DN glomerular tissue. These results imply that humoral immunity and cellular immunity is altered in DN patient glomerular tissue. These results imply that there may be crosstalk among the glomerulus, platelets, and immune cells.
Glomerular cells can recruit immune cells and activate platelets in DN. Glomerular cells suffer from oxidative stress (79), advanced glycation end products (AGEs) (80), abnormal lipid metabolism (81), and other damages in DN. These will lead the glomerular cells to lose their function and induce fibrosis (68). Injured glomerular mesangial cells, glomerular endothelial cells, and podocytes can produce inflammatory and adherence factors to recruit and activate immune cells (79, 82). These factors such as C-C motif chemokine ligand 2 (CCL2) (83–86), C-X3-C motif chemokine receptor 1 (CX3CR1) (83), inter-cellular adhesion molecule-1 (ICAM-1) (87, 88), vascular cell adhesion molecule-1 (VCAM-1) (89), and tumor necrosis factor-alpha (TNF-α) (90, 91) will recruit and activate lymphocytes, monocytes, and other immune cells (83). Injured glomerular mesangial cells, glomerular endothelial cells, and podocytes can also activate platelets (92, 93). In the development of DN, collagen is accumulated in the glomerulus (94, 95). Collagen has long been considered as an important activator of platelet activation. Collagen can directly bind to the glycoprotein VI (GPVI) receptor or integrate the von Willebrand factor (vWF) to activate the glycoprotein Ib-IX-V complex (GPIb-IX) receptor to activate platelets (96, 97). In DN, the increase of AGEs (98), chemokines (such as CCL2, C-X-C motif chemokine ligand 1 (CXCL1)) (99–101), very low density lipoprotein (VLDL) (102), and abnormal metabolism of nitric oxide (NO) can also active platelets (92, 103–105).
Recruited immune cells can release a variety of chemokines, these will damage glomerular cells, cause fibrosis in DN, and activate platelets. Recruited macrophages can be induced by locally secreted TNF-α, and differentiated from resting M0 to activated pro-inflammatory M1 and M2 (90, 91). Once activated, macrophages will release reactive oxygen species (ROS), IL-1, TNF-α, complement factors, and metalloproteinases (106). Recruited mast cells and macrophages can release CXCL1 (107–109). Recruited CD4+ T cells (110), γδ T cells, and NK cells (111) secrete inflammatory factors (interferon gamma (IFN-γ) and IL-17A) (112, 113) and chemokines to promote the proliferation and differentiation of B cells and the formation of immune complexes (114). We all know those factors will damage the glomerulus, cause fibrosis, and promote the progression of DN (82, 83). Immune cells release chemokines (such as CCL2, CXCL1), and ROS can also activate platelets (99–101, 115).
Activated platelets may further activate other platelets (94), recruit immune cells, damage glomerular cell, and cause fibrosis in DN. Once platelets are activated, platelets will express the CD36 molecule (CD36), protein kinase C eta (PRKCH), and coagulation factor II thrombin receptor like 2 (F2RL2) on the surface of platelets, which will cause more platelets to be activated (116–123). Platelet hyper function is observed in DM (92, 93) and DN (93) patients, indicating that platelets may play an important role in the development of DN. Activated platelets release a variety of pro-inflammatory cytokines (TNF-α, P-selectin, TGF-β, FN1, ILs, VCAM-1) and chemokines (CCL-2, CXCL1, and CX3CR1) (96). As described before, some of those factors can recruit and active immune cells (124), and those active immune cells will damage glomerular cells. Some of these factors even can damage glomerular cells directly, such as TGF-β (125), TNF-α (126), ILs (127), and FN1 (63).
Therefore, there may be positive feedback among the glomerulus, platelets, and immune cells. This vicious cycle may damage the glomerulus for a long time even after the initial high glucose damages have been removed. This may be a reason why renal damage in DN patients still progresses even after blood glucose was strictly controlled. The hypothesized crosstalk among platelets, immune cells, and glomerular cells are shown in the schema (Figure 7).
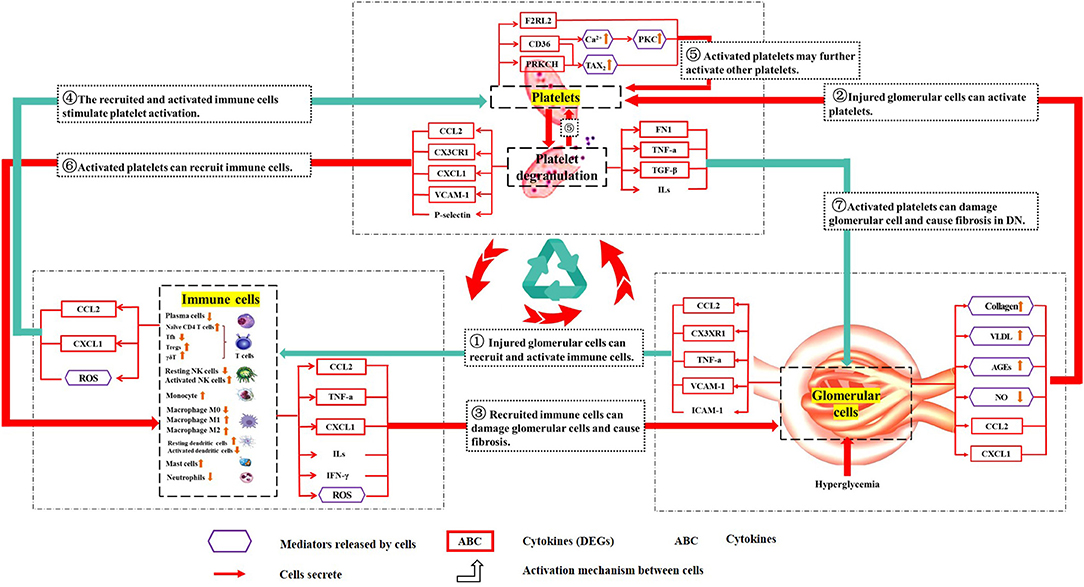
Figure 7. Schematic diagram describing the crosstalk among platelets, immune cells, and the glomerulus. The purple hexagonal box lines represent cellular mediators. The red box indicates the protein expressed by DEGs. The frameless protein was not expressed by DEGs. “ → ” cells that secrete; “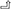 ” the activation mechanism between cells (the red line is glomerular cells-immune cells-platelets, and the blue line is glomerular cells-platelets-immune cells). ① Injured glomerular cells can recruit and activate immune cells. ② Injured glomerular cells can activate platelets. ③ Recruited immune cells can damage glomerular cells and cause fibrosis. ④ The recruited and activated immune cells stimulate platelet activation. ⑤ Activated platelets may further activate other platelets. ⑥ Activated platelets can recruit immune cells. ⑦ Activated platelets can damage glomerular cells and cause fibrosis in DN. The above is the hypothetical crosstalk among the glomerulus, immune cells, and platelets.
” the activation mechanism between cells (the red line is glomerular cells-immune cells-platelets, and the blue line is glomerular cells-platelets-immune cells). ① Injured glomerular cells can recruit and activate immune cells. ② Injured glomerular cells can activate platelets. ③ Recruited immune cells can damage glomerular cells and cause fibrosis. ④ The recruited and activated immune cells stimulate platelet activation. ⑤ Activated platelets may further activate other platelets. ⑥ Activated platelets can recruit immune cells. ⑦ Activated platelets can damage glomerular cells and cause fibrosis in DN. The above is the hypothetical crosstalk among the glomerulus, immune cells, and platelets.
In summary, we found three core genes that may be associated with the pathogenesis of DN (LYZ, LUM, and THBS2). Furthermore, our further bioinformatics analysis suggested that there might be positive feedback among platelets, immune cells, and the glomerulus. And this feedback may damage the glomerulus for a long time even after the initial high glucose damages have been removed. These findings may provide new ideas for the pathogenesis and treatment of DN. However, due to the lack of experimental verification in this study, further studies are needed.
The related microarray datasets GSE96804, GSE104948, GSE99339, GSE30528, GSE21785, GSE47183, GSE20602, GSE121233, GSE108109, GSE104066 and GSE32591 were downloaded from the GEO (https://www.ncbi.nlm.nih.gov/gds/).
ZL, HS, and HZ conceived and designed the study. XY, HS, FC, HH, BL, XZ, HZ, and ZL performed bioinformatics and correlation analyses. XY wrote the first draft. ZL and HS revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Hebei Province (H2020209243) and the National Natural Science Foundation of China (81773958 and 81873140).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Nicolas Borisov for his gift of the long P0 datasets.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.657918/full#supplementary-material
Supplementary Table 1. Long P0 datasets.
Supplementary Table 2. Identification of DEGs.
Supplementary Table 3. The complete enrichment analysis results.
1. Zhang A, Fang H, Chen J, He L, Chen Y. Role of VEGF-A and LRG1 in abnormal angiogenesis associated with diabetic nephropathy. Front Physiol. (2020) 11:1064. doi: 10.3389/fphys.2020.01064
2. Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2·7 million participants. Lancet. (2011) 378:31–40. doi: 10.1016/S0140-6736(11)60679-X
3. Letelier CEM, San Martin Ojeda CA, Ruiz Provoste JJ, Frugone Zaror CJ. [Pathophysiology of diabetic nephropathy: a literature review]. Medwave. (2017) 17:e6839. doi: 10.5867/medwave.2017.01.6839
4. Tung CW, Hsu YC, Shih YH, Chang PJ, Lin CL. Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrology. (2018) 23(Suppl. 4):32–7. doi: 10.1111/nep.13451
5. Chen Y, Lee K, Ni Z, He JC. Diabetic kidney disease: challenges, advances, and opportunities. Kidney Dis. (2020) 6:215–25. doi: 10.1159/000506634
6. Brenneman J, Hill J, Pullen S. Emerging therapeutics for the treatment of diabetic nephropathy. Bioorg Med Chem Lett. (2016) 26:4394–402. doi: 10.1016/j.bmcl.2016.07.079
7. Johnson SA, Spurney RF. Twenty years after ACEIs and ARBs: emerging treatment strategies for diabetic nephropathy. Am J Physiol Renal Physiol. (2015) 309 F807–20. doi: 10.1152/ajprenal.00266.2015
8. Fan Y, Yi Z, D'Agati VD, Sun Z, Zhong F, Zhang W, et al. Comparison of kidney transcriptomic profiles of early and advanced diabetic nephropathy reveals potential new mechanisms for disease progression. Diabetes. (2019) 68:2301–14. doi: 10.2337/db19-0204
9. Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. (2011) 60:2354–69. doi: 10.2337/db10-1181
10. Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, et al. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci USA. (2019) 116:19619–25. doi: 10.1073/pnas.1908706116
11. Levin Reznichenko A, Witasp A, Liu P, Greasley PJ, Sorrentino A, Blondal T, et al. Novel insights into the disease transcriptome of human diabetic glomeruli and tubulointerstitium. Nephrol Dial Transpl. (2020) 35:2059–72. doi: 10.1093/ndt/gfaa121
12. Pan Y, Jiang S, Hou Q, Qiu D, Shi J, Wang L, et al. Dissection of glomerular transcriptional profile in patients with diabetic nephropathy: SRGAP2a protects podocyte structure and function. Diabetes. (2018) 67:717–30. doi: 10.2337/db17-0755
13. Grayson PC, Eddy S, Taroni JN, Lightfoot YL, Mariani L, Parikh H, et al. Metabolic pathways and immunometabolism in rare kidney diseases. Ann Rheum Dis. (2018) 77:1226–33. doi: 10.1136/annrheumdis-2017-212935
14. Sircar M, Rosales IA, Selig MK, Xu D, Zsengeller ZK, Stillman IE, et al. Complement 7 is up-regulated in human early diabetic kidney disease. Am J Pathol. (2018) 188:2147–54. doi: 10.1016/j.ajpath.2018.06.018
15. Ju W, Greene CS, Eichinger F, Nair V, Hodgin JB, Bitzer M, et al. Defining cell-type specificity at the transcriptional level in human disease. Genome Res. (2013) 23:1862–73. doi: 10.1101/gr.155697.113
16. Hodgin JB, Nair V, Zhang H, Randolph A, Harris RC, Nelson RG, et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes. (2013) 62:299–308. doi: 10.2337/db11-1667
17. Ouzounis CA, Valencia A. Early bioinformatics: the birth of a discipline–a personal view. Bioinformatics. (2003) 19:2176–90. doi: 10.1093/bioinformatics/btg309
18. Tang YL, Dong XY, Zeng ZG, Feng Z. Gene expression-based analysis identified NTNG1 and HGF as biomarkers for diabetic kidney disease. Medicine. (2020) 99:e18596. doi: 10.1097/MD.0000000000018596
19. Wang YZ, Xu WW, Zhu DY, Zhang N, Wang YL, Ding M, et al. Specific expression network analysis of diabetic nephropathy kidney tissue revealed key methylated sites. J Cell Physiol. (2018) 233:7139–47. doi: 10.1002/jcp.26638
20. Walsh CJ, Hu P, Batt J, Santos CC. Microarray meta-analysis and cross-platform normalization: integrative genomics for robust biomarker discovery. Microarrays. (2015) 4:389–406. doi: 10.3390/microarrays4030389
21. Borisov N, Shabalina I, Tkachev V, Sorokin M, Garazha A, Pulin A, et al. Shambhala: a platform-agnostic data harmonizer for gene expression data. BMC Bioinformatics. (2019) 20:66. doi: 10.1186/s12859-019-2641-8
22. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. (2013) 41:D991–5. doi: 10.1093/nar/gks1193
23. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. (2002) 30:207–10. doi: 10.1093/nar/30.1.207
24. Shi JS, Qiu DD, Le WB, Wang H, Li S, Lu YH, et al. Identification of transcription regulatory relationships in diabetic nephropathy. Chin Med J. (2018) 131:2886–90. doi: 10.4103/0366-6999.246063
25. Shved N, Warsow G, Eichinger F, Hoogewijs D, Brandt S, Wild P, et al. Transcriptome-based network analysis reveals renal cell type-specific dysregulation of hypoxia-associated transcripts. Sci Rep. (2017) 7:8576. doi: 10.1038/s41598-017-08492-y
26. Lindenmeyer MT, Eichinger F, Sen K, Anders HJ, Edenhofer I, Mattinzoli D, et al. Systematic analysis of a novel human renal glomerulus-enriched gene expression dataset. PloS ONE. (2010) 5:e11545. doi: 10.1371/journal.pone.0011545
27. Martini S, Nair V, Keller BJ, Eichinger F, Hawkins JJ, Randolph A, et al. Integrative biology identifies shared transcriptional networks in CKD. J Am Soc Nephrol. (2014) 25:2559–72. doi: 10.1681/ASN.2013080906
28. Neusser MA, Lindenmeyer MT, Moll AG, Segerer S, Edenhofer I, Sen K, et al. Human nephrosclerosis triggers a hypoxia-related glomerulopathy. Am J Pathol. (2010) 176:594–607. doi: 10.2353/ajpath.2010.090268
29. Hu S, Han R, Shi J, Zhu X, Qin W, Zeng C, et al. The long noncoding RNA LOC105374325 causes podocyte injury in individuals with focal segmental glomerulosclerosis. J Biol Chem. (2018) 293:20227–39. doi: 10.1074/jbc.RA118.005579
30. Mitrofanova Molina J, Varona Santos J, Guzman J, Morales XA, Ducasa GM, Bryn J, et al. Hydroxypropyl-β-cyclodextrin protects from kidney disease in experimental Alport syndrome and focal segmental glomerulosclerosis. Kidney Int. (2018) 94:1151–9. doi: 10.1016/j.kint.2018.06.031
31. Berthier CC, Bethunaickan R, Gonzalez-Rivera T, Nair V, Ramanujam M, Zhang W, et al. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol. (2012) 1950) 189:988–1001. doi: 10.4049/jimmunol.1103031
32. Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. (2004) 20:307–15. doi: 10.1093/bioinformatics/btg405
33. Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. (2010) 26:2363–7. doi: 10.1093/bioinformatics/btq431
34. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. (2009) 37:1–13. doi: 10.1093/nar/gkn923
35. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. (2000) 25:25–9. doi: 10.1038/75556
36. Altermann E, Klaenhammer TR. PathwayVoyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics. (2005) 6:60. doi: 10.1186/1471-2164-6-60
37. Walter W, Sánchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. (2015) 31:2912–4. doi: 10.1093/bioinformatics/btv300
38. Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, Lotia S, et al. A travel guide to Cytoscape plugins. Nature methods. (2012) 9:1069–76. doi: 10.1038/nmeth.2212
39. Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol. (2011) 696:291–303. doi: 10.1007/978-1-60761-987-1_18
40. Sardiu ME, Washburn MP. Building protein-protein interaction networks with proteomics and informatics tools. J Biol Chem. (2011) 286:23645–51. doi: 10.1074/jbc.R110.174052
41. Snel Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. (2000) 28:3442–4. doi: 10.1093/nar/28.18.3442
42. Chatr-aryamontri, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, et al. MINT: the Molecular INTeraction database. Nucleic Acids Res. (2007) 35:D572–4. doi: 10.1093/nar/gkl950
43. Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, Derow C, et al. IntAct–open source resource for molecular interaction data. Nucleic Acids Res. (2007) 35:D561–5. doi: 10.1093/nar/gkl958
44. Jassal Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, et al. The reactome pathway knowledgebase. Nucleic Acids Res. (2020) 48:D498–503. doi: 10.1093/nar/gkz1031
45. Jeanquartier F, Jean-Quartier C, Holzinger A. Integrated web visualizations for protein-protein interaction databases. BMC Bioinformatics. (2015) 16:195. doi: 10.1186/s12859-015-0615-z
46. Chen SJ, Liao DL, Chen CH, Wang TY, Chen KC. Construction and analysis of protein-protein interaction network of heroin use disorder. Sci Rep. (2019) 9:4980. doi: 10.1038/s41598-019-41552-z
47. Raman K. Construction and analysis of protein-protein interaction networks. Automat Exp. (2010) 2:2. doi: 10.1186/1759-4499-2-2
48. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. (2003) 4:2. doi: 10.1186/1471-2105-4-2
50. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. (2015) 12:453–7. doi: 10.1038/nmeth.3337
51. Thomas B. The global burden of diabetic kidney disease: time trends and gender gaps. Curr Diabetes Rep. (2019) 19:18. doi: 10.1007/s11892-019-1133-6
52. Li L, Chen L, Zang J, Tang X, Liu Y, Zhang J, et al. C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism. (2015) 64:597–610. doi: 10.1016/j.metabol.2015.01.014
53. Gao S, Cui Z, Zhao MH. The complement C3a and C3a receptor pathway in kidney diseases. Front Immunol. (2020) 11:1875. doi: 10.3389/fimmu.2020.01875
54. Tessari P, Kiwanuka E, Barazzoni R, Vettore M, Zanetti M. Diabetic nephropathy is associated with increased albumin and fibrinogen production in patients with type 2 diabetes. Diabetologia. (2006) 49:1955–61. doi: 10.1007/s00125-006-0288-2
55. Sueud T, Hadi NR, Abdulameer R, Jamil DA, Al-Aubaidy HA. Assessing urinary levels of IL-18, NGAL and albumin creatinine ratio in patients with diabetic nephropathy. Diabetes Metab Syndr. (2019) 13:564–8. doi: 10.1016/j.dsx.2018.11.022
56. Jamal M, Shahwan Hassan NAG, Shaheen RA. Assessment of kidney function and associated risk factors among type 2 diabetic patients. Diabetes Metab Syndr. (2019) 13:2661–5. doi: 10.1016/j.dsx.2019.07.025
57. Perlman AS, Chevalier JM, Wilkinson P, Liu H, Parker T, Levine DM, et al. Serum inflammatory and immune mediators are elevated in early stage diabetic nephropathy. Ann Clin Lab Sci. (2015) 45:256–63.
58. Wang Guan MP, Zheng ZJ, Li WQ, Lyv FP, Pang RY, Xue YM. Transcription factor Egr1 is involved in high glucose-induced proliferation and fibrosis in rat glomerular mesangial cells. Cell Physiol Biochem. (2015) 36:2093–107. doi: 10.1159/000430177
59. Wu C, Ma X, Zhou Y, Liu Y, Shao Y, Wang Q. Klotho REstraining Egr1/TLR4/mTOR axis to reducing the expression of fibrosis and inflammatory cytokines in high glucose cultured rat mesangial cells. Exp Clin Endocrinol Diabetes. (2019) 127:630–40. doi: 10.1055/s-0044-101601
60. Liu Zhang ZP, Xin GD, Guo LH, Jiang Q, Wang ZX. miR-192 prevents renal tubulointerstitial fibrosis in diabetic nephropathy by targeting Egr1. Eur Rev Med Pharmacol Sci. (2018) 22:4252–60. doi: 10.1155/2018/4728645
61. Hu Xue M, Li Y, Jia YJ, Zheng ZJ, Yang YL, Guan MP, et al. Early growth response 1 (Egr1) is a transcriptional activator of NOX4 in oxidative stress of diabetic kidney disease. J Diabetes Res. (2018) 2018:3405695. doi: 10.1155/2018/3405695
62. Ninichuk V, Kulkarni O, Clauss S, Anders H. Tubular atrophy, interstitial fibrosis, and inflammation in type 2 diabetic db/db mice. An accelerated model of advanced diabetic nephropathy. Eur J Med Res. (2007) 12:351–5. doi: 10.1016/j.vaccine.2007.05.046
63. Mou X, Zhou DY, Liu YH, Liu K, Zhou D. Identification of potential therapeutic target genes in mouse mesangial cells associated with diabetic nephropathy using bioinformatics analysis. Exp Ther Med. (2019) 17:4617–27. doi: 10.3892/etm.2019.7524
64. Ma X, Lu C, Lv C, Wu C, Wang Q. The expression of miR-192 and its significance in diabetic nephropathy patients with different urine albumin creatinine ratio. J Diabetes Res. (2016) 2016:6789402. doi: 10.1155/2016/6789402
65. Zhao X, Chen X, Chima A, Zhang Y, George J, Cobbs A, et al. Albumin induces CD44 expression in glomerular parietal epithelial cells by activating extracellular signal-regulated kinase 1/2 pathway. J Cell Physiol. (2019) 234:7224–35. doi: 10.1002/jcp.27477
66. Marhaba R, Zöller M. CD44 in cancer progression: adhesion, migration and growth regulation. J Mol Histol. (2004) 35:211–31. doi: 10.1023/B:HIJO.0000032354.94213.69
67. Mao CP, Gu ZL. Puerarin reduces increased c-fos, c-jun, and type IV collagen expression caused by high glucose in glomerular mesangial cells. Acta Pharmacol Sin. (2005) 26:982–6. doi: 10.1111/j.1745-7254.2005.00133.x
68. Zeng LF, Xiao Y, Sun L. A glimpse of the mechanisms related to renal fibrosis in diabetic nephropathy. Adv Exp Med Biol. (2019) 1165:49–79. doi: 10.1007/978-981-13-8871-2_4
69. Dai Liu Q, Liu B. Research progress on mechanism of podocyte depletion in diabetic nephropathy. J Diabetes Res. (2017) 2017:2615286. doi: 10.1155/2017/2615286
70. Sheng J, Li H, Dai Q, Lu C, Xu M, Zhang J, et al. DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways. J Cell Physiol. (2019) 234:3043–57. doi: 10.1002/jcp.27124
71. Tuttle KR. Linking metabolism and immunology: diabetic nephropathy is an inflammatory disease. J Am Soc Nephrol. (2005) 16:1537–8. doi: 10.1681/ASN.2005040393
72. Gallo D, Cocchietto M, Masat E, Agostinis C, Harei E, Veronesi P, et al. Human recombinant lysozyme downregulates advanced glycation endproduct-induced interleukin-6 production and release in an in-vitro model of human proximal tubular epithelial cells. Exp Biol Med. (2014) 239:337–46. doi: 10.1177/1535370213518281
73. Schaefer L, Gröne HJ, Raslik I, Robenek H, Ugorcakova J, Budny S, et al. Small proteoglycans of normal adult human kidney: distinct expression patterns of decorin, biglycan, fibromodulin, and lumican. Kidney Int. (2000) 58:1557–68. doi: 10.1046/j.1523-1755.2000.00317.x
74. Chakravarti S, Magnuson T, Lass JH, Jepsen KJ, LaMantia C, Carroll H. Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J Cell Biol. (1998) 141:1277–86. doi: 10.1083/jcb.141.5.1277
75. Schaefer L, Raslik I, Grone HJ, Schonherr E, Macakova K, Ugorcakova J, et al. Small proteoglycans in human diabetic nephropathy: discrepancy between glomerular expression and protein accumulation of decorin, biglycan, lumican, and fibromodulin. FASEB J. (2001) 15:559–61. doi: 10.1096/fj.00-0493fje
76. O'Rourke KM, Laherty CD, Dixit VM. Thrombospondin 1 and thrombospondin 2 are expressed as both homo- and heterotrimers. J Biol Chem. (1992) 267:24921–4. doi: 10.1016/S0021-9258(19)73983-0
77. Shen Y, Yu J, Jing Y, Zhang J. MiR-106a aggravates sepsis-induced acute kidney injury by targeting THBS2 in mice model. Acta Cirurgica Brasil. (2019) 34:e201900602. doi: 10.1590/s0102-865020190060000002
78. Yeh SH, Chang WC, Chuang H, Huang HC, Liu RT, Yang KD. Differentiation of type 2 diabetes mellitus with different complications by proteomic analysis of plasma low abundance proteins. J Diabetes Metab Disord. (2015) 15:24. doi: 10.1186/s40200-016-0246-6
79. Elmarakby AA, Sullivan JC. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc Ther. (2012) 30:49–59. doi: 10.1111/j.1755-5922.2010.00218.x
80. Yamagishi S, Imaizumi T. Diabetic vascular complications: pathophysiology, biochemical basis and potential therapeutic strategy. Curr Pharmaceut Design. (2005) 11:2279–99. doi: 10.2174/1381612054367300
81. Srivastava Adams-Huet B, Vega GL, Toto RD. Effect of losartan and spironolactone on triglyceride-rich lipoproteins in diabetic nephropathy. J Investig Med. (2016) 64:1102–8. doi: 10.1136/jim-2016-000102
82. Zheng Z, Zheng F. Immune cells and inflammation in diabetic nephropathy. J Diabetes Res. (2016) 2016:1841690. doi: 10.1155/2016/1841690
83. Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol. (2006) 17:368–77. doi: 10.1681/ASN.2005080859
84. Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA, et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. (1999) 398:718–23. doi: 10.1038/19546
85. P. von Hundelshausen, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, Weber C. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. (2001) 103:1772–7. doi: 10.1161/01.CIR.103.13.1772
86. Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, et al. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med. (2001) 194:1361–73. doi: 10.1084/jem.194.9.1361
87. Okada S, Shikata K, Matsuda M, Ogawa D, Usui H, Kido Y, et al. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes. (2003) 52:2586–93. doi: 10.2337/diabetes.52.10.2586
88. Lane TA, Lamkin GE, Wancewicz E. Modulation of endothelial cell expression of intercellular adhesion molecule 1 by protein kinase C activation. Biochem Biophys Res Commun. (1989) 161:945–52. doi: 10.1016/0006-291X(89)91334-X
89. Heidland Sebekova K, Schinzel R. Advanced glycation end products and the progressive course of renal disease. Am J Kidney Dis. (2001) 38:S100–6. doi: 10.1053/ajkd.2001.27414
90. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. (2014) 26:192–7. doi: 10.1016/j.cellsig.2013.11.004
91. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
92. Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: molecular insights and therapeutic strategies. Cardiovasc Diabetol. (2018) 17:121. doi: 10.1186/s12933-018-0763-3
93. Tarnow Michelson AD, Barnard MR, Frelinger AL III, Aasted B, Jensen BR, Parving HH, et al. Nephropathy in type 1 diabetes is associated with increased circulating activated platelets and platelet hyperreactivity. Platelets. (2009) 20:513–9. doi: 10.3109/09537100903221001
94. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soci Nephrol(2017) 12:2032–045. doi: 10.2215/CJN.11491116
95. Yang YL, Hu F, Xue M, Jia YJ, Zheng ZJ, Li Y, et al. Early growth response protein-1 upregulates long noncoding RNA Arid2-IR to promote extracellular matrix production in diabetic kidney disease. Am J Physiol Cell Physiol. (2019) 316:C340–52. doi: 10.1152/ajpcell.00167.2018
96. Estevez B, Du X. New concepts and mechanisms of platelet activation signaling. Physiology. (2017) 32:162–77. doi: 10.1152/physiol.00020.2016
97. Li Z, Delaney MK, O'Brien KA, Du X. Signaling during platelet adhesion and activation. Arteriosclerosis Thrombosis Vasc Biol. (2010) 30:2341–9. doi: 10.1161/ATVBAHA.110.207522
98. Gawlowski T, Stratmann B, Ruetter R, Buenting CE, Menart B, Weiss J, et al. Advanced glycation end products strongly activate platelets. Eur J Nutr. (2009) 48:475–81. doi: 10.1007/s00394-009-0038-6
99. Liu D, Cao Y, Zhang X, Peng C, Tian X, Yan C, et al. Chemokine CC-motif ligand 2 participates in platelet function and arterial thrombosis by regulating PKCα-P38MAPK-HSP27 pathway. Biochim Biophys Acta. (2018) 1864:2901–12. doi: 10.1016/j.bbadis.2018.05.025
100. Gear AR, Camerini D. Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, host defense. Microcirculation. (2003) 10:335–50. doi: 10.1080/713773647
101. Chang TT, Chen JW. The role of chemokines and chemokine receptors in diabetic nephropathy. Int J Mol Sci. (2020) 21:3172. doi: 10.3390/ijms21093172
102. de Man Nieuwland FH, van der Laarse RA, Romijn F, Smelt AH, Gevers Leuven JA, Sturk A. Activated platelets in patients with severe hypertriglyceridemia: effects of triglyceride-lowering therapy. Atherosclerosis. (2000) 152:407–14. doi: 10.1016/S0021-9150(99)00485-2
103. Trachtman Futterweit S, Crimmins DL. High glucose inhibits nitric oxide production in cultured rat mesangial cells. J Am Soc Nephrol. (1997) 8:1276–82. doi: 10.1681/ASN.V881276
104. Sokolovska Dekante A, Baumane L, Pahirko L, Valeinis J, Dislere K, Rovite V, et al. Nitric oxide metabolism is impaired by type 1 diabetes and diabetic nephropathy. Biomed Rep. (2020) 12:251–8. doi: 10.3892/br.2020.1288
105. Singh Mok M, Christensen AM, Turner AH, Hawley JA. The effects of polyphenols in olive leaves on platelet function. Nutr Metab Cardiovasc Dis. (2008) 18:127–32. doi: 10.1016/j.numecd.2006.09.001
106. Nikolic-Paterson DJ, Atkins RC. The role of macrophages in glomerulonephritis. Nephrol Dial Transplant. (2001) 16(Suppl. 5):3–7. doi: 10.1093/ndt/16.suppl_5.3
107. Wu YG, Lin H, Qi XM, Wu GZ, Qian H, Zhao M, et al. Prevention of early renal injury by mycophenolate mofetil and its mechanism in experimental diabetes. Int Immunopharmacol. (2006) 6:445–53. doi: 10.1016/j.intimp.2005.09.006
108. Zhang Y, Ma KL, Gong YX, Wang GH, Hu ZB, Liu L, et al. Platelet microparticles mediate glomerular endothelial injury in early diabetic nephropathy. J Am Soc Nephrol. (2018) 29:2671–95. doi: 10.1681/ASN.2018040368
109. De Filippo Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. (2013) 121:4930–7. doi: 10.1182/blood-2013-02-486217
110. Wu CC, Sytwu HK, Lin YF. Cytokines in diabetic nephropathy. Adv Clin Chem. (2012) 56:55–74. doi: 10.1016/B978-0-12-394317-0.00014-5
111. Flodström Shi FD, Sarvetnick N, Ljunggren HG. The natural killer cell – friend or foe in autoimmune disease? Scandinavian J of immunology. (2002) 55:432–1. doi: 10.1046/j.1365-3083.2002.01084.x
112. Imani F, Horii Y, Suthanthiran M, Skolnik EY, Makita Z, Sharma V, et al. Advanced glycosylation endproduct-specific receptors on human and rat T-lymphocytes mediate synthesis of interferon gamma: role in tissue remodeling. J Exp Med. (1993) 178:2165–72. doi: 10.1084/jem.178.6.2165
113. Peng X, Xiao Z, Zhang J, Li Y, Dong Y, Du J. IL-17A produced by both γδ T and Th17 cells promotes renal fibrosis via RANTES-mediated leukocyte infiltration after renal obstruction. J Pathol. (2015) 235:79–89. doi: 10.1002/path.4430
114. Smith MJ, Simmons KM, Cambier JC. B cells in type 1 diabetes mellitus and diabetic kidney disease. Nat Rev Nephrol. (2017) 13:712–20. doi: 10.1038/nrneph.2017.138
115. Violi F, Pignatelli P. Platelet NOX, a novel target for anti-thrombotic treatment. Thromb Haemost. (2014) 111:817–23. doi: 10.1160/TH13-10-0818
116. Nergiz-Unal R, Rademakers T, Cosemans JM, Heemskerk JW. CD36 as a multiple-ligand signaling receptor in atherothrombosis. Cardiovasc Hematol Agents Med Chem. (2011) 9:42–55. doi: 10.2174/187152511794182855
117. Englyst NA, Taube JM, Aitman TJ, Baglin TP, Byrne CD. A novel role for CD36 in VLDL-enhanced platelet activation. Diabetes. (2003) 52:1248–55. doi: 10.2337/diabetes.52.5.1248
118. Wilhelmsen P, Kjær J, Thomsen KL, Nielsen CT, Dige A, Maniecki MB, et al. Elevated platelet expression of CD36 may contribute to increased risk of thrombo-embolism in active inflammatory bowel disease. Arch Physiol Biochem. (2013) 119:202–8. doi: 10.3109/13813455.2013.808671
119. Pasterk Lemesch S, Leber B, Trieb M, Curcic S, Stadlbauer V, Schuligoi R, et al. Oxidized plasma albumin promotes platelet-endothelial crosstalk and endothelial tissue factor expression. Sci Rep. (2016) 6:22104. doi: 10.1038/srep22104
120. Hänzelmann S, Wang J, Güney E, Tang Y, Zhang E, Axelsson AS, et al. Thrombin stimulates insulin secretion via protease-activated receptor-3. Islets. (2015) 7:e1118195. doi: 10.1080/19382014.2015.1118195
121. McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci USA. (2007) 104:5662–7. doi: 10.1073/pnas.0700763104
122. Nakanishi-Matsui Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. (2000) 404:609–13. doi: 10.1038/35007085
123. Bynagari YS, Nagy B, Jr Tuluc F, Bhavaraju K, Kim S, Vijayan KV, et al. Mechanism of activation and functional role of protein kinase Ceta in human platelets. J Biol Chem. (2009) 284:13413–21. doi: 10.1074/jbc.M808970200
124. Rayes J, Bourne JH, Brill A, Watson SP. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res Pract Thromb Haemost. (2020) 4:23–35. doi: 10.1002/rth2.12266
125. Reeves WB, Andreoli TE. Transforming growth factor beta contributes to progressive diabetic nephropathy. Proc Natl Acad Sci USA. (2000) 97:7667–9. doi: 10.1073/pnas.97.14.7667
126. Kalantarinia K, Awad AS, Siragy HM. Urinary and renal interstitial concentrations of TNF-alpha increase prior to the rise in albuminuria in diabetic rats. Kidney Int. (2003) 64:1208–13. doi: 10.1046/j.1523-1755.2003.00237.x
Keywords: diabetic nephropathy, pathogenesis, bioinformatics, platelet, glomerulus, immune cell
Citation: Yao X, Shen H, Cao F, He H, Li B, Zhang H, Zhang X and Li Z (2021) Bioinformatics Analysis Reveals Crosstalk Among Platelets, Immune Cells, and the Glomerulus That May Play an Important Role in the Development of Diabetic Nephropathy. Front. Med. 8:657918. doi: 10.3389/fmed.2021.657918
Received: 24 January 2021; Accepted: 28 April 2021;
Published: 24 June 2021.
Edited by:
Xu-Jie Zhou, Peking University First Hospital, ChinaReviewed by:
Tao Huang, Shanghai Institute of Nutrition and Health (CAS), ChinaCopyright © 2021 Yao, Shen, Cao, He, Li, Zhang, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiguo Li, bGl6aGlndW9AbmNzdC5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.