 Maren Gesper
Maren Gesper Alena B. H. NonnastNina Kumowski
Alena B. H. NonnastNina Kumowski Robert Stoehr
Robert Stoehr Katharina Schuett
Katharina Schuett Nikolaus Marx
Nikolaus Marx Ben A. Kappel*
Ben A. Kappel*- Department of Internal Medicine 1, University Hospital Aachen, Rheinisch-Westfälische Technische Hochschule (RWTH) Aachen University, Aachen, Germany
Background: The gut microbiome has been linked to the onset of cardiometabolic diseases, in part facilitated through gut microbiota-dependent metabolites such as trimethylamine-N-oxide. However, molecular pathways associated to heart failure mediated by microbial metabolites remain largely elusive. Mitochondria play a pivotal role in cellular energy metabolism and mitochondrial dysfunction has been associated to heart failure pathogenesis. Aim of the current study was to evaluate the impact of gut-derived metabolites on mitochondrial function in cardiomyocytes via an in vitro screening approach.
Methods: Based on a systematic Medline research, 25 microbial metabolites were identified and screened for their metabolic impact with a focus on mitochondrial respiration in HL-1 cardiomyocytes. Oxygen consumption rate in response to different modulators of the respiratory chain were measured by a live-cell metabolic assay platform. For one of the identified metabolites, indole-3-propionic acid, studies on specific mitochondrial complexes, cytochrome c, fatty acid oxidation, mitochondrial membrane potential, and reactive oxygen species production were performed. Mitochondrial function in response to this metabolite was further tested in human hepatic and endothelial cells. Additionally, the effect of indole-3-propionic acid on cardiac function was studied in isolated perfused hearts of C57BL/6J mice.
Results: Among the metabolites examined, microbial tryptophan derivative indole-3-propionic acid could be identified as a modulator of mitochondrial function in cardiomyocytes. While acute treatment induced enhancement of maximal mitochondrial respiration (+21.5 ± 7.8%, p < 0.05), chronic exposure led to mitochondrial dysfunction (−18.9 ± 9.1%; p < 0.001) in cardiomyocytes. The latter effect of indole-3-propionic acids could also be observed in human hepatic and endothelial cells. In isolated perfused mouse hearts, indole-3-propionic acid was dose-dependently able to improve cardiac contractility from +26.8 ± 11.6% (p < 0.05) at 1 μM up to +93.6 ± 14.4% (p < 0.001) at 100 μM. Our mechanistic studies on indole-3-propionic acids suggest potential involvement of fatty acid oxidation in HL-1 cardiomyocytes.
Conclusion: Our data indicate a direct impact of microbial metabolites on cardiac physiology. Gut-derived metabolite indole-3-propionic acid was identified as mitochondrial modulator in cardiomyocytes and altered cardiac function in an ex vivo mouse model.
Introduction
Heart failure (HF) is a multifactorial syndrome characterized by mechanical cardiac dysfunction and inability to provide sufficient perfusion to the body. Despite recent therapeutic advances, morbidity and mortality of HF still remains high. Therefore, there is an urgent need to identify HF-related pathomechanisms to develop novel preventive and therapeutic strategies. During the past decade, gut microbiota have been identified as important contributor to various pathologic conditions including cardiovascular disease (CVD) (1). While gut microbiota are able to influence the host in different ways, technical advances in metabolomics identified gut-derived metabolites as mediators of CVD and other metabolic diseases. These metabolites enter the blood flow and are thereby able to interact with cells of the cardiovascular system (1, 2). One prominent microbial metabolite is trimethylamine-N-oxide (TMAO). As part of bacterial choline metabolism, TMAO has been associated with progression of CVD and cardiovascular mortality (3–5). Besides TMAO, other microbial metabolites, like short-chain fatty acids (SCFA) and tryptophan derivatives, have been linked to cardiovascular disorders (6, 7).
While past studies have focused on gut microbiota-related mechanism associated to atherosclerosis, atherothrombosis and myocardial infarction (4, 8, 9), only few have investigated the role of gut bacteria on HF. Patients with HF exhibit changes in the gut flora (10, 11). However, pathomechanisms between intestinal bacteria and HF remain elusive.
Mitochondria play a pivotal role in metabolism and bioenergetics, particularly in an organ with high energy demand, such as the heart. Mitochondrial dysfunction is a characteristic of HF and one of the first steps during metabolic remodeling in the failing heart (12–15). Although a vast number of circulating microbiota-derived metabolites have been described, their impact on cardiac metabolism is unknown. Therefore, we conducted this study to screen the influence of microbial metabolites on metabolic function in cardiomyocytes. Using a live-cell metabolic assay platform with focus on mitochondrial respiration, 25 circulating gut-derived metabolites of seven different pathways were tested in HL-1 cardiomyocytes. Here, we identified indole-3-propionic acid (IPA), a microbial tryptophan derivative, as modulator of mitochondrial function in vitro. Using an isolated perfused heart mouse model, we were able to show that IPA exhibits direct effect on cardiac function.
Together, our data suggest that gut microbiota are capable to impact cardiac physiology via gut-derived metabolites. Our data may therefore provide implications for further studies investigating the role of gut microbiota on HF.
Materials and Methods
Systematic Medline Search
To identify and select different gut-derived metabolites, a systematic Medline search was performed (July 2017). The following search term was used: [((bacteria[Title] OR bacterial[Title] OR flora[Title] OR microbiota[Title] OR microbiome[Title] OR microbe[Title] OR microflora[Title])) AND (gut[Title] OR intestine[Title] OR intestinal[Title] OR colon[Title] OR colonic[Title] OR cecum[Title] OR caecum[Title] OR cecal[Title] OR caecal[Title] OR stool[Title])) AND (metabolite[Title/Abstract] OR metabolites[Title/Abstract] OR metabolome[Title/Abstract] OR metabolomics[Title/Abstract] OR metabolic[Title/Abstract]]. Based on these results, 25 commercially available metabolites were selected with potential biological function on cardiovascular or other biological systems.
Reagents
If not differently specified, all reagents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Metabolites Used for Cell Experiments
Trimetyhlamine (TMA), trimethylamine-N-oxide (TMAO), phenylacetylglutamine (Bachem; Bubendorf, Switzerland), 3,4-dihydroxyphenylacetic acid, 4-hydroxyphenylacetic acid, sodium taurodeoxycholate hydrate, sodium propionate and sodium butyrate were dissolved in distillated water. Indole-3-propionic acid (IPA) was also dissolved as aqueous solution and adjusted to pH: 7.4. Working solutions were prepared in distillated water. Indole, indole-3-acetic acid, 2-oxindole, phenylacetic acid, p-cresol, trans-3-hydroxycinnamic acid, 3-(4-hydroxyphenyl)propionic acid, benzoic acid, 3-hydroxybenzoic acid, hippuric acid, o-hydroxyhippuric acid, cholic acid, and valeric acid were dissolved in dimethyl sulfoxide (DMSO). Deoxycholic acid and ursodeoxycholic acid were dissolved in 100% ethanol (Carl Roth; Karlsruhe, Germany). It was ensured that final concentrations of vehicles (ethanol/DMSO) did not exceed 0.1% of the total volume.
Cell Culture
For the metabolic screening of the metabolites three different cell types were used. Murine cardiomyocytes (HL-1) (16) were cultured in Claycomb medium supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) penicillin/streptomycin (100 Units/ml penicillin and 100 μg/ml streptomycin), 0.1 mM norepinephrine (in 30 mM ascorbic acid) and 2 mM L-glutamine. Flasks, 96-well and Seahorse XF96 cell culture plates (Agilent; Santa Clara, CA, USA) were pre-coated with 0.02% (v/v) gelatin containing 5 μg/ml fibronectin.
Human hepatoma cell line (Huh7) (17) was cultured in Dulbecco's Modified Eagle's Medium (DMEM—high glucose) supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin (100 Units/ml penicillin and 100 μg/ml streptomycin).
Human umbilical vein endothelial cells (HUVEC) (18) were cultured in Endothelial Cell Growth Medium-2 (EGM-2; Lonza, Basel, Switzerland). HUVECs were used between passage 3 and 8. All cell types were cultured at 37°C in a humidified atmosphere with 5% CO2.
Cell Viability Assay
To analyze the cell viability in response to the metabolites, a resazurin-based assay with the reagent PrestoBlue (ThermoFisher; Walthman, MA, USA) was used (19). The assay was performed according to the manufacture's protocol. Briefly, cells were seeded with a density of 10,000 cells/well in 96-well-plates (Corning 3610; Corning, NY, USA). Cells were treated with different concentrations of the metabolites (1 μM−1 mM) for 24 h at 37°C (with 5% CO2). After the treatment, cells were incubated with media containing 1× PrestoBlue for 1 h at 37°C (with 5% CO2) and fluorescence intensity was measured at Ex/Em 560/590 nm (Tecan infinite M200; Tecan Group Ltd., Männedorf, Switzerland).
Cell Proliferation Assay
Cell proliferation in response to indole-3-propionic acid (IPA) treatment was detected by incorporation of the pyrimidine analog 5-Bromo-2′-deoxyuridine (BrdU) into the DNA (20). The cell proliferation ELISA, BrdU (chemiluminescence) (Roche; Basel, Switzerland) was used according to the manufacture's protocol. Cells were seeded with a density of 2,000 cells/well in 96-well-plates and treated with different concentrations of IPA (10 μM−1 mM). Cells were incubated at 37°C (with 5% CO2) and medium was changed daily. Incorporation of BrdU into the DNA was measured via luminescence with an integration time of 2,500 ms (Tecan infinite M200). Proliferation via BrDU assay was measured after 24 h, 48 h, and 72 h.
Real-Time Extracellular Flux Analysis
Metabolic function in cells in response to the metabolites was characterized by Seahorse XFe96 Flux Analyzer (Agilent). This technique allows real-time measurements of the oxygen consumption rate (OCR) and glycolysis (ECAR) in living cells. Two different Seahorse Kits, Seahorse XF Cell Energy Phenotype Test and Seahorse XF Mito Stress Test, were used to characterize metabolic function of cells in response to gut-derived metabolites.
Cells were seeded with a density of 10,000 cells/well on XF96 cell culture plates and treated with metabolites for 24 h (37°C, 5% CO2). In selected experiments, cells were treated with IPA for shorter periods of time. On the day of the assay, medium was changed to unbuffered XF Assay Medium (Agilent) (supplemented with 10 mM D-glucose, 2 mM L-glutamine and 1 mM sodium pyruvate; adjusted to pH: 7,4) to a final volume of 180 μl/well, after three washing steps with assay medium. For pre-equilibration, cells were incubated for 1 h in a non-CO2 incubator at 37°C. In addition, one day before the assay, XFe96 Sensor Cartridge (Agilent) was hydrated with autoclaved water over night at 37°C in a non-CO2 atmosphere. Before the Sensor Cartridge was loaded with different inhibitors to the respiration chain, water was exchanged with Seahorse XF Calibrant Solution (Agilent) and incubated for 45 min at 37°C (non-CO2).
For the Seahorse XF Cell Energy Phenotype Test, ATP synthase inhibitor oligomycin at a final concentration of 1 μM and the uncoupler carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) at a final concentration of 0.4 μM were injected at the same time point after a general baseline measurement of OCR.
For the Seahorse Mito Stress Kit, oligomycin (1 μM), FCCP (0.4 μM) and a mixture of rotenone/antimycin A (final concentration of mix: 0.5 μM), inhibitors of complex I and II, were injected sequentially after baseline measurements.
The general protocol of the measurements includes three baseline measurements with mix (3 min)/measure (3 min) followed by injection of port A. Afterwards respiration was measured for five (Cell Energy Phenotype) to three times (Mito Stress) with mix (3 min)/measure (3 min). For the Mito Stress, port B and C was injected subsequently with the same measurement cycle of three times mix/measure in between.
Analysis of Respiratory Activity in Permeabilized Cells
Measurement of mitochondrial respiration in permeabilized cells by Seahorse XFe96 Flux Analyzer allows a detail overview on the different complexes of mitochondrial respiratory chain and their activity (21, 22). For this, HL-1 cardiomyocytes were seeded in a density of 10,000 cells/well on XF96 cell culture plates and were treated with IPA for 24 h (37°C, 5% CO2). Before the measurement, cells were washed with 1× mannitol and sucrose (MAS) buffer (220 mM mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, and 1 mM EGTA). After a washing step, 1× MAS buffer was changed to 1× MAS buffer supplemented with 4 mM ADP (ADP sodium salt) and 10 μg/ml saponin, as permeabilizing agent, in a final volume of 180 μl/well. Saponin only permeabilizes the plasma membrane without affecting mitochondrial membrane (21), thus enabling the addition of different substrates to the complexes of the respiratory chain.
To characterize the activity of the different complexes sequentially, 10 mM pyruvate and 1 mM malate were further added in 1× MAS buffer to analyze complex I of the respiratory chain. For inhibition of complex I, 20 μM rotenone (final concentration in well: 2 μM) was injected via port A. Subsequently, 100 mM succinate (final concentration: 10 mM) was injected to analyze complex II activity. To inhibit complex II, 20 μM antimycin A (final concentration: 20 μM) was injected via port C. At the end of the measurement, complex IV was characterized by the injection of 1 mM N,N,N′,N′-Tetrametyhl-p-phenylenediamine (TMPD) and 100 mM ascorbic acid (final concentrations: 0.1/10 mM). Measurement periods were shorter with cycles of two times mix (0.5 min)/wait (0.5 min)/measure (2 min) without equilibration step.
Beside the comprehensive study of the respiratory complexes, respiratory activity based on medium- and long-chain fatty acid oxidation was measured. Therefore, 1× MAS buffer was supplemented with 4 mM ADP, 10 μg/ml saponin and either 40 μM palmitoyl-L-carnitine/1 mM malate or 40 μM octanoyl-L-carnitine/1 mM malate. For these experiments, the Mito Stress Kit was performed as described previously with the adapted protocol for permeabilized cells.
Supplementation of Cytochrome c in Permeabilized Cells
Cytochrome c is an important protein of the respiratory chain and loss can lead to reduction in respiratory activity. Exogenous cytochrome c addition may rescue mitochondrial function (23, 24). Therefore, permeabilized HL-1 cardiomyocytes were measured via Seahorse XFe96 Flux Analyzer after pre-incubation with IPA for 24 h as previously described. 1 mM exogenous cytochrome c (cytochrome c from equine heart; final concentration: 100 μM) was simultaneously injected with saponin (final concentration of 250 μg/ml), 4 mM ADP, 10 mM succinate, and 2 μM rotenone. After two baseline measurements, respiratory activity was measured five times with cycles of mix (0.5 min)/wait (0.5 min)/measure (2 min). At the end of the measurement a mix of 20 μM rotenone/antimycin A (final concentration: 2 μM) was injected to the cells.
Measurement of Mitochondrial Membrane Potential
To detect direct effects on mitochondrial membrane potential, the fluorescence dye tetramethylrhodamine ethyl ester perchlorate (TMRE) was used. TMRE is a cell permeable, cationic dye, which accumulates to active, negatively-charged mitochondrial membrane (25, 26). This allows a live cell analysis of the membrane potential in response to treatment with metabolites. HL-1 cardiomyocytes were seeded with a density of 10,000 cells/well in 96-well-plates and treated with different concentrations of IPA for 24 h. As positive control, 20 μM FCCP was added 10 min before staining. After pre-incubation with IPA/FCCP, cells were then stained with 200 nM TMRE in media for 30 min (37°C, 5% CO2, dark environment). Before measurement of the fluorescence intensity [Ex/Em 540/595 nm (Tecan infinite M200)], cells were washed twice with pre-warmed PBS.
Measurement of Hydroxyl Radicals
Mitochondrial dysfunction can be caused by reactive oxidative species (ROS). To evaluate ROS production in response to IPA treatment, the dye 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) was used. H2DCFDA is a cell permeable, non-fluorescent dye, which become fluorescent after oxidation with hydroxyl radicals (27). HL-1 cardiomyocytes were seeded in a density of 10,000 cells/well in 96-well-plates and treated with IPA for different time periods (30 min and 24 h). As positive control, 1 mM iron(II) sulfate heptahydrate for 30 min before staining was used. After treatment, cells were washed once with pre-warmed PBS and then stained with 25 μM H2DCFDA in PBS for 30 min (37°C, 5% CO2, dark environment). After a second washing step with PBS, fluorescence intensity was measured at Ex/Em 504/529 nm (Tecan infinite M200).
Langendorff Mouse Model
To characterize whether IPA exhibits direct effects on cardiac function, the ex vivo Langendorff mouse model was used (28–30). Animal experiments were approved by the government of North Rhine-Westphalia, Germany. C57BL/6J male mice were purchased from Janvier Labs (Le Genest-Saint-Isle, France). Mice were housed in filtertop cages in a controlled environment (12 h daylight cycle) at the animal facility of the RWTH Aachen University. Mice had free access to water and food. At age of 9–12 weeks (average weight of 22 g) mice were anesthetized with ketamine hydrochloride and xylazine hydrochloride and anticoagulated with 1,000 IE heparin.
The heart was quickly excised and was prepared in ice-cold Krebs–Henseleit buffer. The aorta was cannulated for retrograde perfusion at constant pressure of 90 mmHg using a Langendorff perfusion apparatus (Harvard Apparatus, Massachusetts, USA). The perfusion solution was heated to 37°C and oxygenated with 100% oxygen. A latex balloon filled with distillated water connected to a pressure transducer was inserted into the left ventricle (LV). LV diastolic pressure was set to 12 mmHg. The heart was stimulated resulting in a heart rate of 600 bpm. LV systolic (LVPsys) and LV developed pressure (LVPdp), as well as maximum rate of increase (dLVPmax) and maximum rate of decrease in left ventricular pressure during isovolumic contraction (dLVPmin) were measured continuously (all 2 s) with ISOHEART Isolated Heart Data Acquisition Software (Harvard Apparatus, Massachusetts, USA). Hearts were perfused for a 15 min equilibration period. Following, 200 μL Krebs-Henseleit buffer was administered through the aortic cannula into the heart. After 10 min, dobutamine was administered (1 mg/ml, diluted 1:256). In cycles of 10 min, IPA in different concentrations (1–100 μM) was administered following dobutamine as positive control to ensure proper heart function. Therefore, IPA was dissolved in Krebs-Henseleit buffer and pH-adjusted to 7.4 afterwards. pH-adjusted buffer was used as control to exclude side-effects dependent on buffer administration. In between, washing steps with buffer were performed.
Statistics
Results are presented as mean ± SEM or SD. For statistical analysis GraphPad Prism version 8 (GraphPad software Inc., San Diego, CA, USA) was used. Data were analyzed by one-way ANOVA or two-way ANOVA with post-hoc test. A p < 0.05 was considered to be statistically significant. All data are representatives of at least two independent biological replicates. For every measurement four to five technical replicates per group were performed.
Results
In vitro-Screening of Gut-Derived Metabolites on Metabolic Function in Cardiomyocytes
Based on a detailed Medline research, 25 commercially available gut-derived metabolites with potential effects on cardiovascular system were selected to analyze their impact on metabolic function in HL-1 cardiomyocytes. Metabolites were categorized into seven different pathways, i.e., microbial tryptophan and choline metabolism (Table 1). Cytotoxity and cell viability in response to metabolites were evaluated in all cell types used (data not shown). Only non-toxic concentrations were used for further analyses. To evaluate the effects of gut-derived metabolites on mitochondrial function, we performed mitochondrial stress testing in HL-1 cardiomyocytes. Unexpectedly, we did not observe significant effects on mitochondrial function by most of the tested metabolites (Figures 1A,B). However, we noticed that several microbiota-derived tryptophan metabolites exerted effects on stressed OCR. Particularly, these were indole, indole-3-acetic acid, and indole-3-propionic acid (IPA). IPA exhibited the strongest effect with a clear reduction of −26.8 ± 6.0% of stressed OCR at a concentration of 1 mM (Figure 1B). In contrast to IPA, indole, indole-3-acetic acid and also bile acid ursodeoxycholic acid showed only very moderate effects on stressed OCR (Figure 1B and Supplementary Figure 1).
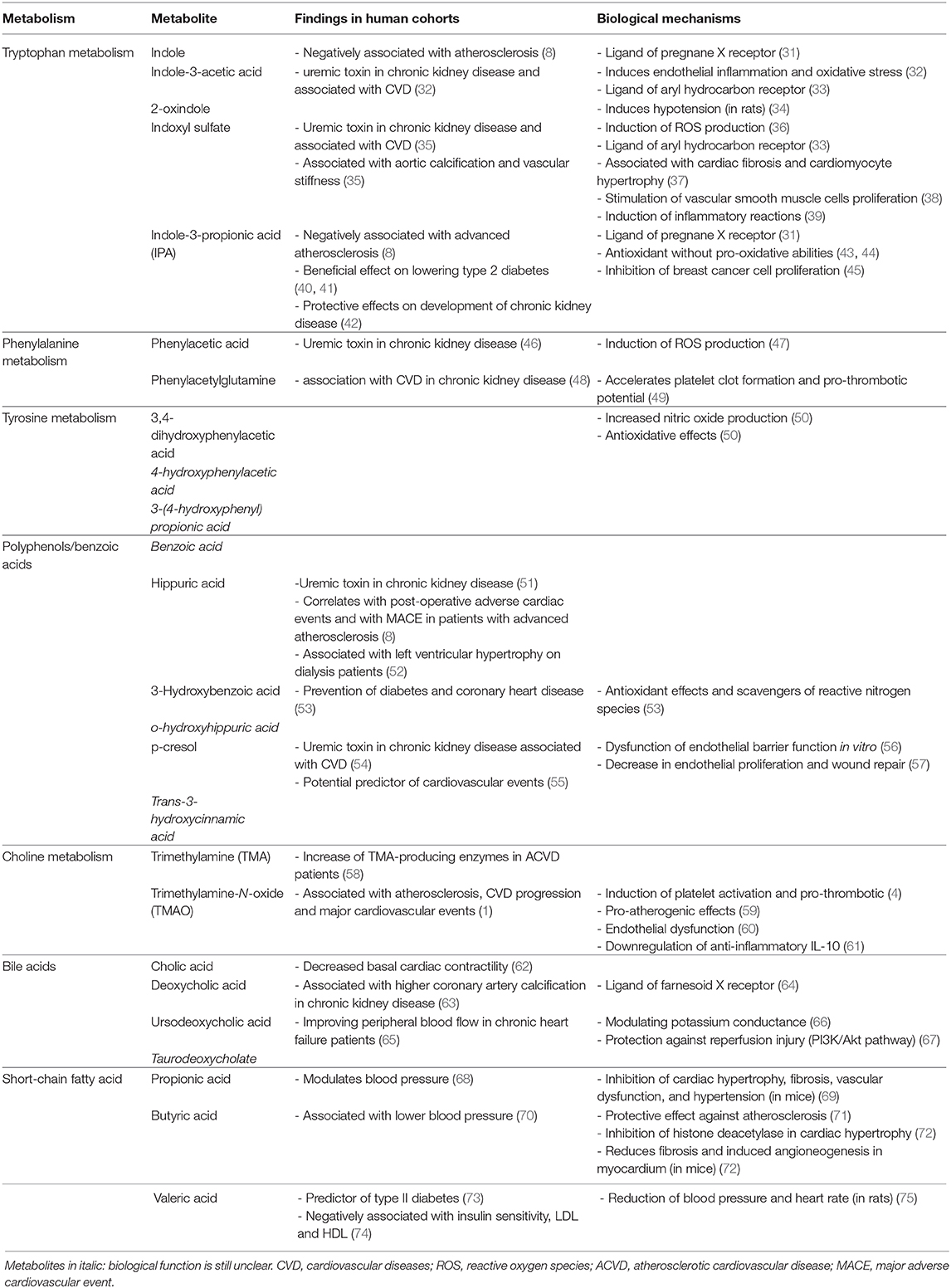
Table 1. Gut-derived metabolites and their impact on host metabolism.
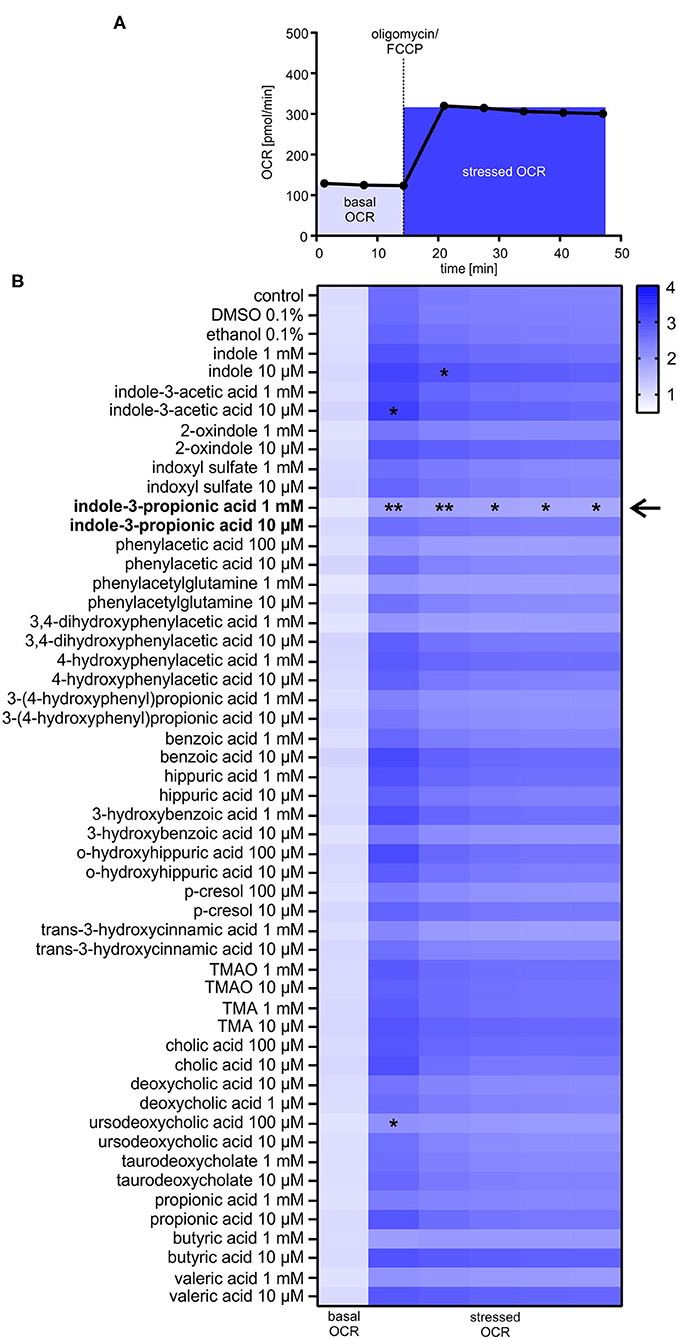
Figure 1. Effects of gut-derived metabolites on mitochondrial respiration in HL-1 cardiomyocytes. (A) Flowchart of mitochondrial stress testing by Seahorse Flux Analyzer. Oligomycin and FCCP were simultaneously added to the cells (stressed OCR). (B) Effects of gut-derived metabolites on mitochondrial function in HL-1 cardiomyocytes after 24 h incubation with substances. Basal oxygen consumption rate (OCR) of control cells was defined as 1.0. Basal OCR is the sum of three baseline measurements. All metabolites were displayed in relation to control or their vehicle (DMSO/ethanol). All results were verified in two independent replicates with N = 5/group. Data were shown as mean. *p < 0.05 and **p < 0.01 by 2-way ANOVA with Dunnett post-hoc test. The black arrow indicates the most prominent effect on mitochondrial function of tested metabolites.
IPA Inhibits Cell Proliferation and Mitochondrial Function in HL-1 Cardiomyocytes
Dependent on the previous findings in our screening approach, we further explored the effects of IPA on HL-1 cardiomyocytes. Cell viability and cytotoxity of HL-1 cardiomyocytes was not affected by IPA at concentrations used from 1 μM to 1 mM (Figure 2A). Based on the reduced mitochondrial function under stress conditions, we determined the effect of IPA on cell proliferation. After 72 h, proliferation was decreased −59.6 ± 4.1% by 1 mM IPA (control: 1,487,379.6 rlu/s ± 34,096.4 vs. 1 mM IPA: 601,400.0 rlu/s ± 60,458.6; p < 0.0001), whereas no growth deprivation was observed at 10 μM IPA (Figure 2B).
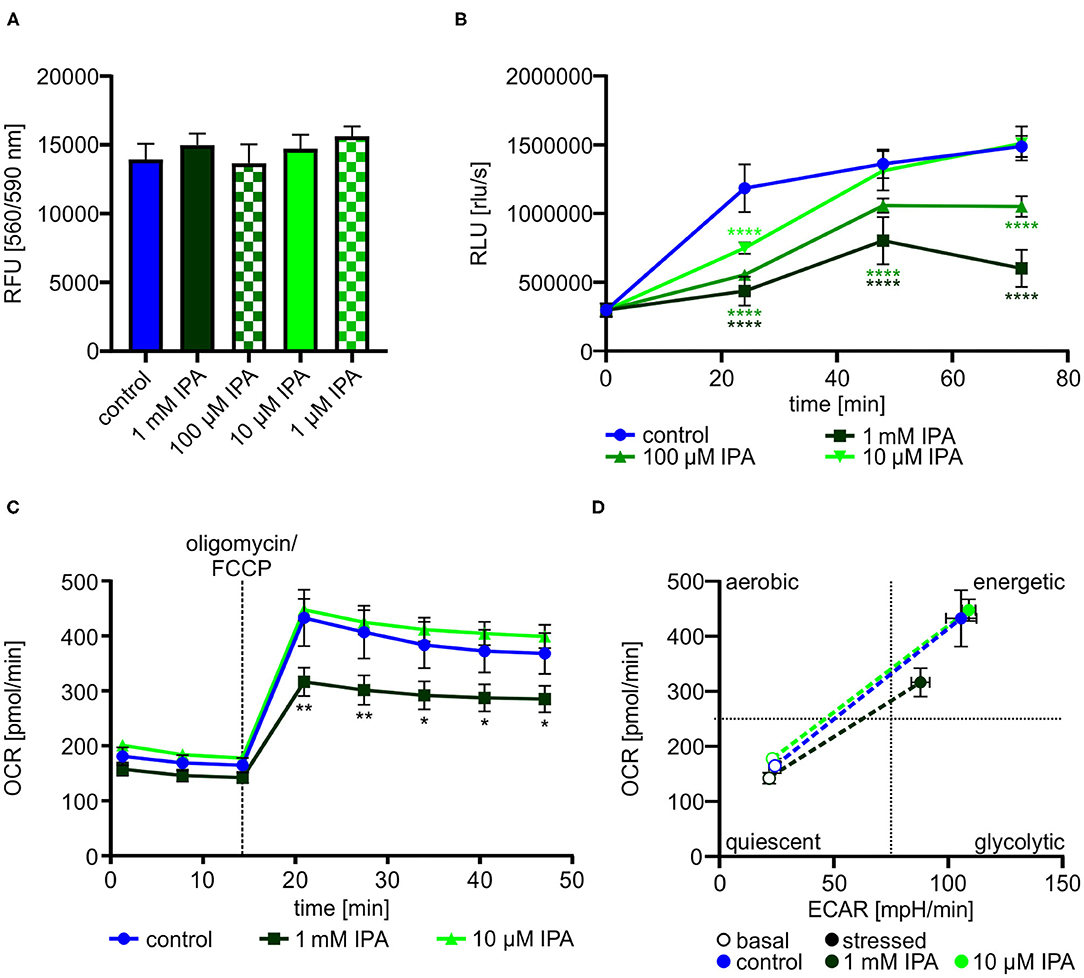
Figure 2. Effects of indole-3-propionic acid (IPA) on HL-1 cardiomyocytes. (A) Cell viability and cytotoxic effects of IPA after 24 h of treatment in HL-1 cardiomyocytes by resazurin-based assay. RFU, relative fluorescence units. N = 4/group, mean ± SD, 1-way ANOVA with post-hoc test. (B) Cell proliferation was measured by BrdU assay after 24, 48, and 72 h treatment with IPA. RLU, relative light units. N = 5/group, mean ± SEM, 1-way ANOVA with post-hoc test. (C,D) Measurement of the metabolic function by Seahorse Flux Analyzer in HL-1 cardiomyocytes after incubation with IPA for 24 h. (C) Changes in oxygen consumption rate (OCR) over time. N = 5/group. Two-way ANOVA with Dunnett post-hoc test. Data are mean ± SEM. (D) Metabolic phenotypes including OCR and extracellular acidification rate (ECAR) under basal (opened cycle) and stressed (filled cycle) conditions. Data were shown as mean ± SEM with N = 5/group. *p < 0.05, **p < 0.01, and ****p < 0.0001.
We next performed a detailed analysis of IPA on mitochondrial respiration in HL-1 cardiomyocytes. As previously shown in our screening approach, 1 mM IPA after 24 h incubation showed a significant reduction of stressed OCR (control: 432.6 pmol/min ± 51.5 vs. 1 mM IPA: 316.3 pmol/min ± 26.1; p < 0.001) (Figure 2C). Basal OCR remained unchanged in response to any concentration of IPA (Figure 2C). In accordance to our findings of reduced proliferation by IPA, evaluation of the metabolic phenotype revealed a shift from an energetic to a more quiescent phenotype in response to 1 mM IPA (Figure 2D).
IPA Affects Maximal Respiration and Respiratory Spare Capacity in Cardiomyocytes
To gain deeper insights into the impact of IPA on mitochondrial respiration, we next evaluated mitochondrial function in response to serial injections different modulators of the respiratory chain (Figures 3A,B).
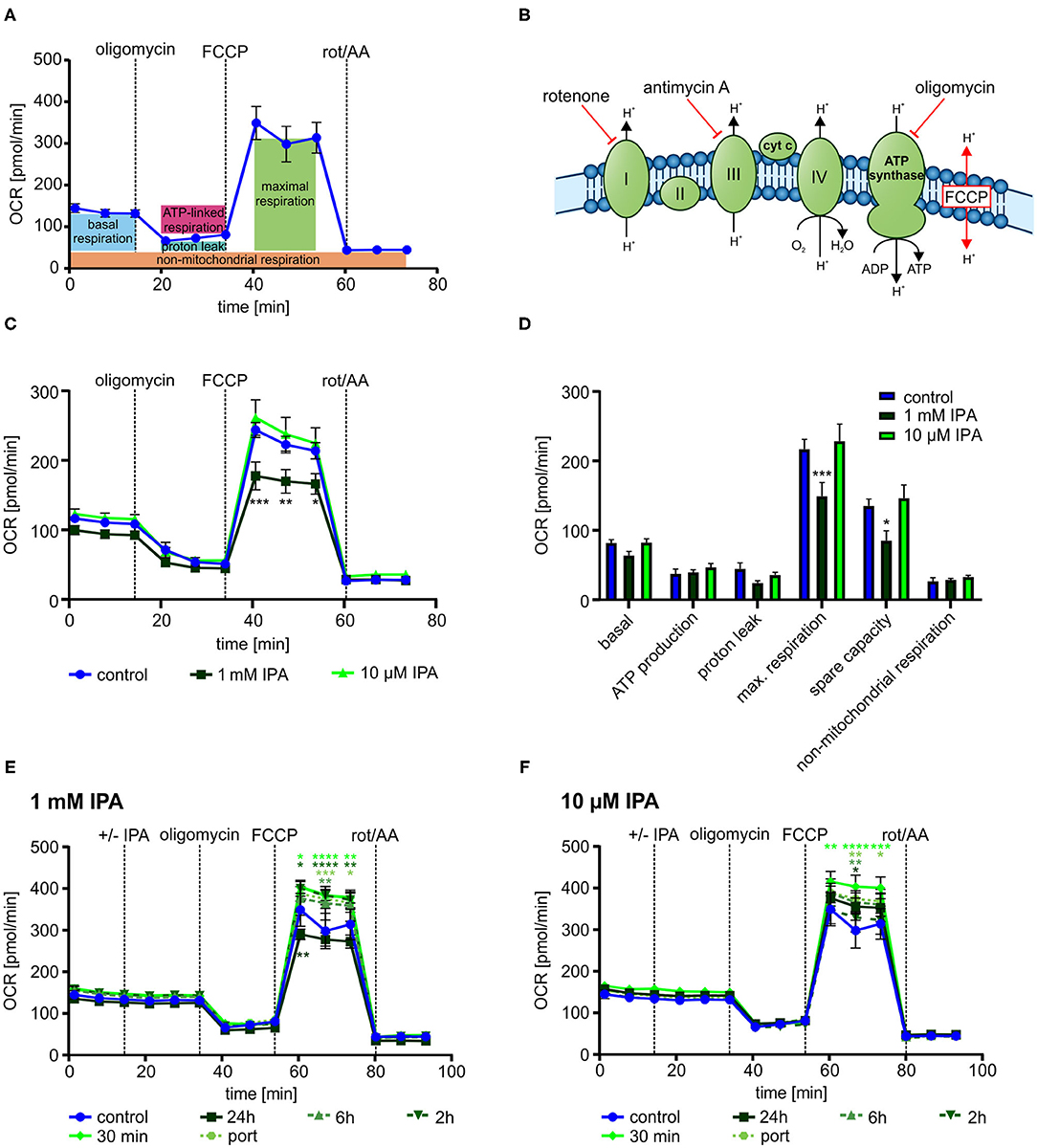
Figure 3. Mitochondrial characterization of HL-1 cardiomyocytes in response to treatment with indole-3-propionic acid (IPA). (A) Schematic representation of real-time measurement of mitochondrial function by Seahorse Flux Analyzer. Oligomycin, FCCP and rotenone/antimycin A (rot/AA) were injected sequentially with determination of basal respiration, ATP production, proton leak, maximum respiration including spare capacity and non-mitochondrial respiration. (B) Schematic overview on the mitochondrial respiratory chain with points of action of different inhibitors. (C,D) Mitochondrial stress testing of HL-1 cardiomyocytes after 24 h incubation with IPA. (C) Changes in oxygen consumption rate (OCR) over time in response to different inhibitors. Data were shown as mean ± SEM with N = 5/group. (D) Changes in basal respiration, ATP production, proton leak, maximal respiration, spare capacity, and non-mitochondrial respiration after 24 h incubation with IPA. For calculation, the last measurement points at basal and the first measurement points after injection of the respiratory chain inhibitors were used. Data were shown as mean ± SEM with N = 5/group. (E,F) Mitochondrial stress testing of HL-1 cardiomyocytes after incubation of IPA at different time points (24 h, 6 h, 2 h, 30 min and direct injection). (E) Changes in OCR over time in response to different inhibitors and IPA 1 mM. Data were shown as mean ± SEM with N = 5/group. (F) Changes in OCR over time in response to different inhibitors and IPA 10 μM. Data were shown as mean ± SEM with N = 5/group. For all data, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, 2-way ANOVA with Dunnett post-hoc test.
After 24 h of incubation, 1 mM IPA showed a significant decrease of OCR after FCCP injection in HL-1 cardiomyocytes (control: 243.5 pmol/min ± 10.8 vs. 1 mM IPA: 177.5 pmol/min ± 20.0; p < 0.001) (Figure 3C). However, no changes were observed at baseline as well as after injections of oligomycin and combination of rotenone/antimycin A, thus indicating a specific effect of IPA on maximal respiration (−18.9 ± 9.1%; p < 0.001) including respiratory spare capacity (−37.0 ± 10.6%; p < 0.05) (Figures 3C,D).
While the previous effects by IPA were observed after a 24 h incubation period, we next asked whether IPA exerts acute effects on mitochondrial function. Therefore, we set up an experiment with different incubation times of IPA. Cells were treated for 24 h, 6 h, 2 h, and 30 min with IPA prior to the assay or by direct port-based injection of IPA during the real-time assay. While the previously observed reduction of maximum respiration by 1 mM IPA after 24 h incubation was confirmed, we observed in both tested concentrations, 1 mM and 10 μM IPA, an increase of maximum respiration and spare capacity particularly at the shorter incubation times with a maximum at 30 min of incubation (Figures 3E,F). Remarkably, the greatest enhancement of maximal respiration of +21.5 ± 7.8% was observed at a concentration of 10 μM IPA after 30 min (control: 305.2 pmol/min ± 40.6 vs. 10 μM IPA: 370.8 pmol/min ± 23.7; p < 0.05). However, also an increase of OCR in response to FCCP was noticed in cells treated with direct injections of IPA during the assay (Figures 3E,F).
IPA Exhibits Direct Effects on Cardiac Function in an Isolated Perfused Heart Model
Our previous experiments revealed that IPA modulates mitochondrial function in cardiac cells. Therefore, we asked whether IPA also affects cardiac function. An isolated perfused mouse heart model was used to evaluate the effects of IPA on heart function ex vivo (Figure 4A). In agreement with our previous findings of increased maximum respiration after short-term administration of IPA in vitro, we observed in the Langendorff model that IPA dose-dependently increased left ventricular developed pressure (LVdp), a validated parameter of cardiac function (Figures 4B,C). A percentage enhancement of LVdp from +26.4 ± 11.6% at 1 μM IPA (p < 0.05), +51.6 ± 11.7% at 10 μM (p < 0.001) and up to +93.6 ± 14.4% at 100 μM IPA (p < 0.0001) was observed. As expected, IPA also increased other cardiac performance indices such as LV pressure, dLVPmax, and dLVPmin (Supplementary Table 1). Serial injections of buffer did not have any effects on cardiac performance, e.g., shown by LVdp (Supplementary Figure 2).
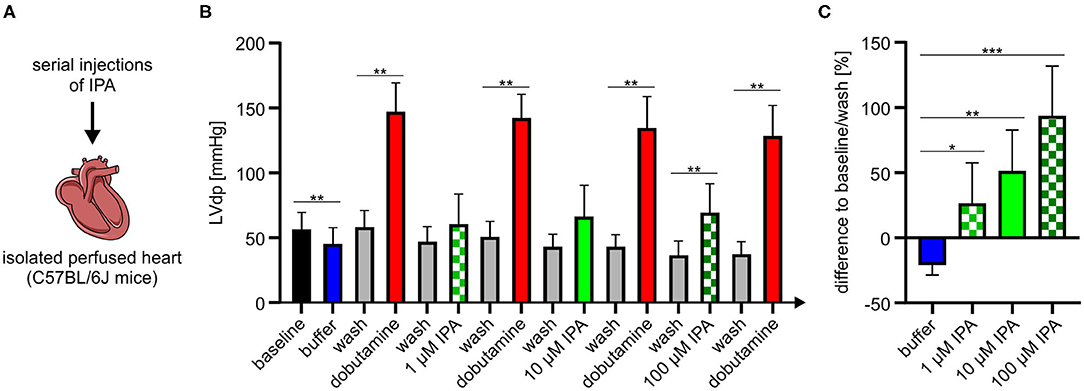
Figure 4. (A) Schematic overview of experimental procedure of Langendorff model. (B) Serial injections of buffer, dobutamine, and IPA with intermittent washing steps in the Langendorff mouse model show dose-depended effects of IPA on left ventricle developed pressure (LVdp). Dobutamine (1 mg/ml, 1:256) was administered as positive control. (C) Percentage difference of LVdp in relation to baseline/wash between buffer, IPA, and wash. All data were shown as mean ± SD with N = 7/group. *p < 0.05, **p < 0.01, and ***p < 0.001 by 1-way ANOVA.
Impact of IPA on Mitochondrial Function in Human Hepatic and Endothelial Cells
While the previous effects were observed in the murine heart, we wanted to know whether IPA affects mitochondrial function also in other tissues as well as in human cells. Therefore, we evaluated the effect of IPA in human hepatic cell line (Huh7) and Human Umbilical Vein Endothelial Cells (HUVEC). Both, Huh7 and HUVEC, showed a reduction of the maximal respiration after IPA treatment similar to the effects in HL-1 cardiomyocytes (Huh7: control: 98.4 pmol/min ± 17.6 vs. 1 mM IPA: 59.8 pmol/min ± 6.9; p < 0.01; HUVEC: control: 22.4 pmol/min ± 3.1 vs. 1 mM IPA: 16.5 pmol/min ± 2.1; p = 0.0575) (Figure 5). However, although toxicity of IPA was excluded in these cells, basal respiration (Huh7: −46.4 ± 5.0% and HUVEC: −38.5 ± 7.7%; both p < 0.05) was also significantly decreased (Figures 5B,D). Together, these findings indicate that effects of IPA on mitochondrial function is not specific for cardiac tissue and mouse cells.
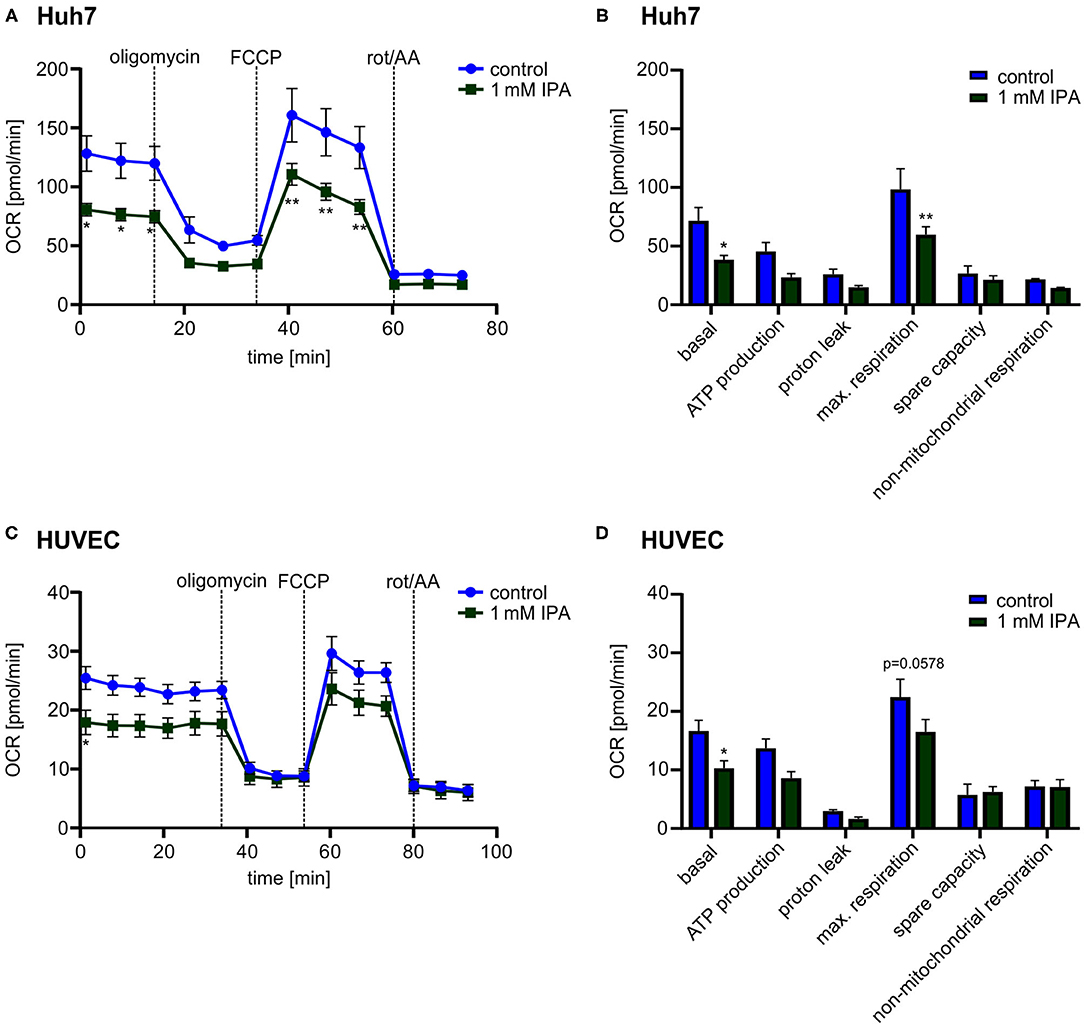
Figure 5. Mitochondrial characterization in human hepatic cell line (Huh7) and Human Umbilical Vein Endothelial Cells (HUVEC) in response to 1 mM indole-3-propionic acid (IPA) after 24 h. (A,B) Mitochondrial stress testing in Huh7 after 1 mM IPA treatment for 24 h. (A) Changes of oxygen consumption rate (OCR) over time in response to respiratory chain inhibitors. Data were shown as mean ± SEM with N = 5/group. (B) Changes in basal respiration, ATP production, proton leak, maximal respiration, spare capacity and non-mitochondrial respiration after 24 h incubation of 1 mM IPA. For calculation, the last measurement points at basal and the first measurement points after injection of the respiratory chain inhibitors were used. Data were shown as mean ± SEM with N = 5/group. (C,D) Mitochondrial stress testing in HUVEC after 1 mM IPA treatment for 24 h. (C) Changes of OCR over time in response to respiratory chain inhibitors. Data were shown as mean ± SEM with N = 5/group. (D) Changes in basal respiration, ATP production, proton leak, maximal respiration, spare capacity and non-mitochondrial respiration after 24 h incubation of 1 mM IPA. Data were shown as mean ± SEM with N = 5/group. Rot/AA, combination of the inhibitors rotenone and antimycin A. *p < 0.05 and **p < 0.01, 2-way ANOVA with Dunnett post-hoc test.
Evaluation of Mechanisms Associated Mitochondrial Modulation by IPA
We next explored mechanisms associated to modulated metabolic function by IPA. While a clear impact of IPA on mitochondrial respiration was observed, no effects of IPA on glycolysis were noticed (Supplementary Figure 3). These findings indicate that IPA may pre-dominantly modulate the mitochondrial respiratory chain. Therefore, single complexes of the respiratory chain were analyzed in the next steps (Figure 6A).
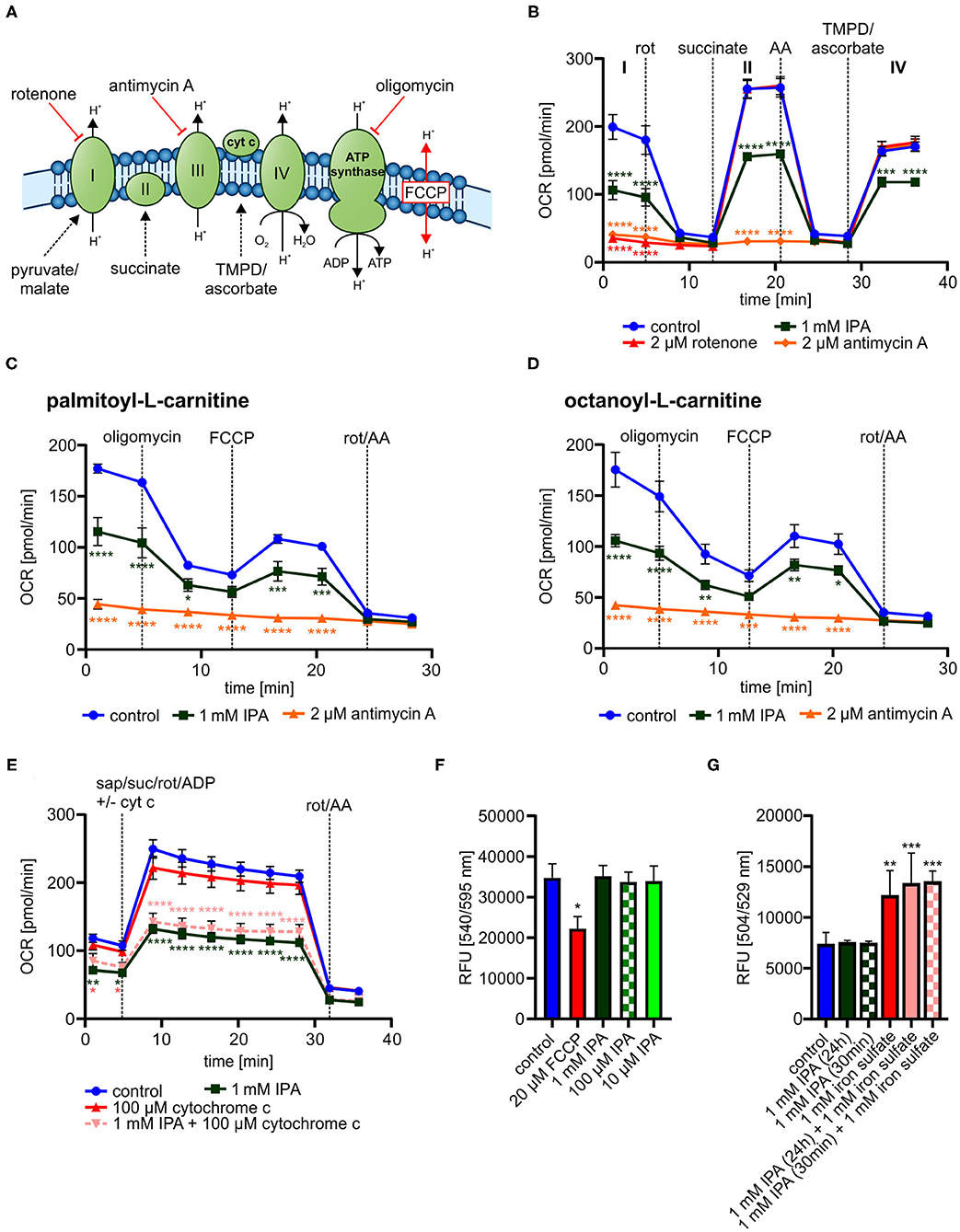
Figure 6. Detailed characterization of mitochondrial function in response to indole-3-propionic acid (IPA) in HL-1 cardiomyocytes. (A) Schematic representation of respiratory chain including substrates for detailed analysis of respiratory chain complexes. TMPD: N,N,N′,N′-Tetrametyhl-p-phenylenediamine. (B) Measurement of oxygen consumption rate (OCR) over time in response to substrates of respiratory chain complexes injected sequentially. Rot, rotenone; AA, antimycin A. (C) Mitochondrial stress testing in permeabilized HL-1 cardiomyocytes including palmitoyl-L-carnitine in response to 1 mM IPA (24 h). (D) Mitochondrial stress testing in permeabilized HL-1 cardiomyocytes including octanoyl-L-carnitine in response to 1 mM IPA (24 h). (E) Addition of exogenous cytochrome c to permeabilized HL-cardiomyocytes after treatment with IPA (24 h). Measurement of OCR over time. Sap, saponin; suc, succinate; rot, rotenone; cyt c, cytochrome c. (B–E) Data were shown as mean ± SEM with N = 5/group by 2-way ANOVA with Dunnett post-hoc test. (F) Mitochondrial membrane potential was measured by TMRE staining in HL-1 cardiomyocytes after treatment with IPA (24 h). 20 μM FCCP for 10 min was used as positive control. RFU: relative fluorescence units. Data were shown as mean ± SD with N = 4–5/group by 1-way ANOVA with post-hoc test. (G) Measurement of ROS production and hydroxyl radicals by H2DCFDA assay in HL-1 cardiomyocytes. HL-1 cardiomyocytes were treated with 1 mM IPA for 24 h and 30 min., 1 mM iron(II) sulfate for 30 min was used as positive control. Data were shown as mean ± SD with N = 4/group by 1-way ANOVA with post-hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
In all tested complexes, 1 mM IPA significantly reduced OCR approximately from 50 to 30% in a similar way (p < 0.001–0.0001) (Figure 6B), while rotenone (2 μM for 30 min) (complex I) and antimycin A (2 μM for 30 min) (complex II) did not show respiratory activity at the specific complex (Figure 6B). This finding indicates that IPA has no specific activity on complex I, II, or IV.
While the entire respiratory chain seemed to be affected by IPA without a complete blocking, we next tested whether IPA modulated mitochondrial fatty acid oxidation (FAO). FAO is an important energy source for mitochondrial respiration and defective FAO may eventually lead to mitochondrial dysfunction (21). In both analyzed FAO pathways, 1 mM IPA significantly reduced OCR (basal: palmitoyl-L-carnitine: −34.8 ± 7.8% and octanoyl-L-carnitine: −39.8 ± 3.5% (both p < 0.0001); max. respiration: palmitoyl-L-carnitine: −29.2 ± 8.9%; p < 0.001 and octanoyl-L-carnitine: −25.6 ± 5.2%; p < 0.01) (Figures 6C,D). As seen in our previous analysis of complex I, II, and IV, respiratory activity was reduced by IPA, but not completely inhibited as by the positive control antimycin A (Figures 6C,D).
While our previous data indicate modulation of FAO by IPA, involvement of cytochrome c release as well as changes in the mitochondrial membrane potential by IPA may also explain a general reduction of mitochondrial respiratory function. As shown in Figure 6E, cytochrome c supplementation was not able to restore the effect of IPA. Additionally, mitochondrial membrane potential was significantly affected by the positive control FCCP (20 μM for 10 min, p < 0.05), but no effect was observed by IPA (Figure 6F). Oxidative stress is another strong modulator of mitochondrial function. IPA has been described as antioxidant without pro-oxidative ability in neurons with pre-dominately protective effects against hydroxyl radicals (43, 76). Therefore, the impact of IPA on ROS production with focus on hydroxyl radicals was further analyzed. Neither alone nor in combination with the positive control iron(II) sulfate (1 mM), IPA modulated hydroxyl radical production in HL-1 cardiomyocytes (Figure 6G).
Independently of mitochondrial function, IPA has been described as agonist to human and rodent pregnane X receptor (PXR) (31). Therefore, we tested the effects of PXR agonists on mitochondrial function not only in murine HL-1 cardiomyocytes, but also in human Huh7 hepatic cells. Rifampicin was used as human PXR agonist and 5-Pregnen-3β-ol-20-one-16α-carbonitrile (PCN) as murine PXR agonist, as previously described (77, 78). In Huh7, 1 mM IPA and rifampicin revealed similar effects on OCR with no additional reduction by combination of both substances (control: 160.8 pmol/min ± 22.7; 1 mM IPA: 110.6 pmol/min ± 9.2; 10 μM rifampicin: 111.1 ± 18.5 pmol/min; 1 mM IPA and 10 μM rifampicin: 112.5 pmol/min ± 8.9; all p < 0.001) (Supplementary Figure 4A). While these results in Huh7 may indicate a PXR-dependent effect of IPA, murine PXR agonist PCN alone or in combination with IPA did not modify OCR in HL-1 cardiomyocytes (Supplementary Figure 4B). Neither did rifampicin (as weak agonist of murine PXR) modulate respiratory function in HL-1 cardiomyocytes (Supplementary Figure 4B). Together with the fact that PXR is only weakly expressed in the heart, a PXR-dependent effect of IPA on cardiac mitochondrial function seems unlikely.
Discussion
Gut microbiota play an important role in the onset and progression of cardiometabolic diseases (79). The interaction between host and intestinal bacteria is complex and involves many different mechanisms. Particularly, these include microbiota dysbiosis (11, 80), modulation of energy harvest, induction of systemic low-grade inflammation, immunological reactions as well as microbiome-related metabolites (1, 81–83). Gut microbiota produce a vast number of metabolites, which enter blood stream and may exert biological functions in the host (1, 79). Several gut-derived metabolites have been identified to be associated with cardiovascular disease (CVD). While serum levels of TMAO have been linked to increased cardiovascular mortality and enhanced atherothrombotic risk (3, 4, 84), other gut-derived metabolites exert positive effects in the cardiovascular system. Particularly, SCFA have been described as positive modulators of CVD (71, 80, 85). Recently, we and others found beneficial effects of microbiota-derived tryptophan metabolites in relation to CVD (7, 83).
While many studies revealed the association between gut bacteria and cardiovascular diseases such as hypertension and atherosclerosis, little is known about the impact of the gut microbiome on heart failure (HF). Patients with HF show changes in their gut microbiome, but a direct link between onset of HF and microbiota could not been established so far (11). HF is a heterogenous disease with different pathomechanism involved. However, a molecular hallmark of HF is mitochondrial dysfunction in cardiomyocytes (12, 13). Metabolomics analyses have identified a large number of circulating metabolites that are derived from gut bacteria metabolism. To date, the impact of those metabolites on mitochondrial function in cardiomyocytes has not been evaluated. Therefore, we selected 25 different gut-derived metabolites with potential biological activity and screened their impact on metabolic function with focus on mitochondrial respiration in cardiomyocytes.
Unexpectedly, our screening approach did not discover many metabolites that affect metabolic function in cardiomyocytes. Choline derivatives and SCFA, substances that have been associated to CVD, did not modify metabolic activity in cardiomyocytes in our study. Beneficial effects of SCFA on mitochondrial function have been described in Akt2-knockout cardiomyocytes before, but no effects were detected in wildtype cardiomyocytes, as seen in our study (86). In a mouse model of myocardial infarction, administration of antibiotics decreased cardiac function by reducing levels of SCFA. The authors suggested a mechanism of modulation of myeloid cells by SCFA (80). Therefore, SCFA might not affect cardiomyocyte function. Biological effects of TMAO have been associated to atherosclerosis and atherothrombosis (4, 84). In agreement with these findings, TMAO has been predictive for mortality in ischemic HF, but not dilative HF or HF with preserved ejection fraction (5, 87). Thus, TMAO might not directly modulate mitochondrial function in cardiomyocytes.
However, we found that several gut-derived tryptophan derivatives, which exerted effects on respiration of cardiomyocytes under stress conditions. Of all analyzed metabolites, tryptophan derivative indole-3-propionic acid (IPA) showed the largest impact on stressed OCR. IPA is a circulating metabolite that is exclusively produced by gut bacteria, predominantly by Clostridium sporogenes (2, 88). Under physiological conditions, serum concentrations of IPA are around 1 μM in humans and 5 μM in rodents (40, 89). In contrast to other microbial tryptophan derivatives, like indoxyl sulfate or indole-3-acetic acid, IPA does not accumulate in chronic kidney disease (CKD) (35, 42, 90). Higher levels of IPA were even correlated with a lower risk of rapid renal decline in CKD patients (42). Effects of IPA on cardiometabolic diseases have been described as well. In human and mice, levels of IPA were negatively associated to the burden of atherosclerosis (7, 8). Further, low IPA levels were predictive for the incidence of type 2 diabetes mellitus (40, 41). In rodent models, administration of IPA reduced microbial dysbiosis and inflammation in steatohepatitis (91), reduced weight gain (89) and decreased inflammation in a model of colitis (31). Together, these studies indicate beneficial effects of IPA in vivo.
Based on the previous results, we investigated the effects of IPA on cardiomyocyte function more deeply. High, supraphysiological concentrations of IPA induced a more quiescent phenotype, which was linked to a specific reduction of maximal respiration and respiratory spare capacity in mitochondria. Reduced maximal respiration and spare capacity are both indicators of mitochondrial dysfunction. Spare capacity represent the reverse capacity of ATP, which can be used in response to cellular stress without a following ATP crisis (92). Mitochondria are the main cellular energy producers. In organs with high demands of energy, such as the heart, a defective mitochondrial respiration could be fatal. Mitochondrial dysfunction in cardiomyocytes is one of the first steps in molecular cardiac remodeling, eventually leading to clinical apparent HF (13, 15). In agreement with reduced mitochondrial function, we found that cardiomyocytes treated with IPA were less metabolic active revealing lower proliferation rates.
However, when we explored acute effects of IPA on mitochondrial respiration in a time series experiment, we observed the opposite effect of IPA on mitochondrial function. Both, low and high, concentrations of IPA increased maximal respiration with a maximum of 30 min after administration. This ambivalent characteristic of IPA leads to the speculation that mitochondrial capacity is first increased by IPA, but eventually induces mitochondrial damage by higher, supraphysiological concentrations of IPA in a later stage. However, 24 h treatment of high concentrations of IPA showed only reduced maximal respiration and spare capacity, but did not alter basal respiration or ATP production. Therefore, mitochondrial dysfunction of high-dose IPA might only apparent under cellular stress conditions.
The acute effects of IPA on mitochondrial function are in agreement with the observed effects of IPA on cardiac function in the isolated perfused mouse heart model. IPA immediately increased cardiac function, shown by increased contractility, already at low, physiological concentrations of IPA after acute administration. This effect might be explained by the acute modulation of mitochondrial function, as seen in our experiments with cardiomyocytes. While these data suggest positive cardiac effects of acute IPA treatment, further in vivo mouse experiments must address whether chronic administration of IPA exerts positive or negative effects on cardiac function and HF.
Based on these findings, we explored mechanisms that could be associated to modulation of mitochondria by IPA. IPA had no effect on glycolysis, thus indicating a pre-dominant role on mitochondrial respiration. Our experiments testing the impact of IPA on specific complexes of the respiratory chain did not point to a single complex. A possible target with impact on mitochondrial respiration, however, is oxidation of fatty acids by mitochondrial beta-oxidation. In cardiomyocytes, fatty acids are the pre-dominant fuel accounting for ~60–90% ATP production (15, 93). IPA equally reduced, but not completely inhibited long- and medium-chain fatty acid oxidation. This finding may indicate that IPA has an impact on fatty acid oxidation. Our experiments on cytochrome c involvement, mitochondrial membrane potential change or modulation of ROS production by IPA did not explain the observed mitochondrial effects of IPA. Therefore, modulation of FAO could also be a possible reason for a general decrease in respiration due to lower beta-oxidation. These data may also provide a possible explanation for reduced weight gain in rats upon treatment with IPA, as described by Konopelski et al. (89).
IPA has been described as antioxidant without pro-oxidative ability predominantly scavenging hydroxyl radicals in neurons (43, 76) and prevented against iron-induced oxidative damage in hepatic cells (44). Increase in reactive oxygen species (ROS) has closely been linked to mitochondrial dysfunction. However, we did not observe an effect of IPA on ROS production.
Our experiments in a human hepatic cell line as well as in primary human endothelial cells showed a similar decrease in mitochondrial respiration compared to our experiments in cardiomyocytes. Effects of IPA in other cell types have been described. The inhibitory effect of IPA on cell proliferation was, for example, observed in breast cancer cells, but mitochondrial function was not investigated (45). Besides an inhibition of maximal respiration, we observed in both cell types that also basal respiration was significantly reduced. This could indicate that these cell types are more sensitive to IPA compared to cardiomyocytes. A further explanation for this finding could be activation of pregnane X receptor (PXR) by IPA, as previously described (31, 45). PXR is a xenobiotic receptor, which is predominantly expressed in gastrointestinal and liver tissue, but has also been described in HUVEC before (94, 95). No or very low levels of PXR are expressed in cardiac tissue and HL-1 cardiomyocytes (94, 96). We observed that human PXR agonist rifampicin induced the same effect in Huh7 as IPA alone or in combination. These findings suggest that a reduction of mitochondrial respiration could also be caused by an activation of PXR due to IPA. However, rodent PXR agonist PCN did not show those effects on mitochondrial respiration in murine HL-1 cardiomyocytes. While in human hepatic cells our data are in agreement with PXR activation by IPA, the findings in cardiomyocyte make a general effect of IPA on mitochondria via PXR unlikely. An explanation for our results could be low expression of PXR in HL-1 cardiomyocytes, but also species differences among PXR activation in human and mice (97).
Our study has several limitations. In cardiovascular research, HL-1 cardiomyocytes are an established in vitro model to determine effects on cardiac tissue (96, 98). Therefore, we chose these murine instead of human or primary cardiomyocytes for our screening approach. Further, acute cardiac function was measured in isolated perfused hearts ex vivo. This model has the advantage to test direct cardiac effects of a substance without hormonal and neural influence. Nonetheless, in vivo studies are needed to verify our observed effects, especially to determine chronic effects of IPA on cardiovascular system. Acute and chronic administration of IPA in mouse models of HF, such as transverse aortic constriction and ligation of the left anterior descending artery, will allow to get deeper insights into the relationship between IPA and HF progression.
Mitochondrial dysfunction in our in vitro approach was observed only at supraphysiological conditions. However, stimulating effects of IPA on mitochondrial function in cardiomyocytes and cardiac function in the Langendorff model were observed at low, physiological concentrations (1–10 μM). Further, we could not fully elucidate the specific mechanism of IPA on mitochondrial function, although our data suggest involvement of FAO in HL-1 cardiomyocytes.
In conclusion, our cell-based screening approach identified that gut-derived metabolites are capable to affect cardiac physiology. Particularly, we could show that gut-derived tryptophan derivative IPA impacted mitochondrial respiration in cardiomyocytes as well as cardiac function in an isolated perfused heart model. Further studies will reveal whether IPA modulates cardiovascular function and HF in vivo.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on reasonable request.
Ethics Statement
The animal study was reviewed and approved by Government of North Rhine-Westphalia, Germany.
Author Contributions
BK: conceptualization. MG and BK: methodology. MG and BK: development or design of methodology. MG: software. RS, KS, NM, and BK: formal analysis. MG, AN, and NK: investigation. KS, NM, and BK: resources. MG: data curation and writing–original draft. KS, RS, NM, and BK: writing–review and editing. MG, AN, and NK: visualization. RS, KS, NM, and BK: supervision. NM and BK: project administration. KS, NM, and BK: funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
BK was supported by grants from the Deutsche Stiftung für Herzforschung (DSHF) [F-43-16] and RWTH Aachen University (START). KS was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation; SFB/TRR219 C-07). NM was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation; SFB/TRR 219; Project-ID 322900939 [M03, M05]). KS and NM were further supported by a CORONA Foundation grant. NK was supported by the Clinician Scientist program of the Faculty of Medicine of the RWTH Aachen University. AN was supported by the Otto-Hess-Scholarship of the German Cardiac Society.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.648259/full#supplementary-material
References
1. Brown JM, Hazen SL. Microbial modulation of cardiovascular disease. Nat Rev Microbiol. (2018) 16:171–81. doi: 10.1038/nrmicro.2017149
2. Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. (2009) 106:3698–703. doi: 10.1073/pnas0812874106
3. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B., et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. (2011) 472:57–65. doi: 10.1038/nature09922
4. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut microbial metabolite TMAO enhance platelet hyperreactivity and thrombosis risk. Cell. (2016) 165:111–24. doi: 10.1016/j.cell.2016.02011
5. Schuett K, Kleber ME, Scharnagl H, Lorkowski S, März W, Niessner A, et al. Trimethylamine-N-oxide and heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol. (2017) 70:3202–4. doi: 10.1016/j.jacc.2017.10064
6. Kurilshikov A, van den Munckhof ICL, Chen L, Bonder MJ, Schraa K, Rutten JHW, et al. Gut microbial associations to plasma metabolites linked to cardiovascular phenotypes and risk. Circ Res. (2019) 124:1808–20. doi: 10.1161/CIRCRESAHA.118314642
7. Kappel BA, De A.ngelis L, Heiser M, Ballanti M, Stoehr R, Goettsch C, et al. Cross-omics analysis revealed gut microbiome-related metabolic pathways underlying atherosclerosis development after antibiotics treatment. Mol Metab. (2020) 36:100976. doi: 10.1016/j.molmet.2020100976
8. Cason CA, Dolan KT, Sharma G, Tao M, Kulkarni R, Helenowski IB, et al. Plasma microbiome-modulated indole- and phenyl-derived metabolites associate with advanced atherosclerosis and postoperative outcomes. J Vasc Surg. (2018) 68:1552–562.e7. doi: 10.1016/j.jvs.2017.09029
9. Jäckel S, Kiouptsi K, Lillich M, Hendrikx T, Khandagale A, Kollar B, et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood. (2017) 130:542–53. doi: 10.1182/blood-2016-11-754416
10. Cui X, Ye L, Li J, Jin L, Wang W, Li S, et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci Rep. (2018) 8:1–15. doi: 10.1038/s41598-017-18756-2
11. Luedde M, Winkler T, Heinsen F-A, Rühlemann MC, Spehlmann ME, Bajrovic A, et al. Heart failure is associated with depletion of core intestinal microbiota. ESC Hear Fail. (2017) 4:282–90. doi: 10.1002/ehf212155
12. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. (2018) 128:3716–26. doi: 10.1172/JCI120849
13. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko A. V, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. (2018) 50:121–7. doi: 10.1080/07853890.20171417631
14. Buchwald A, Till H, Unterberg C, Oberschmidt R, Figulla HR, Wiegand V. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur Heart J. (1990) 11:509–16. doi: 10.1093/oxfordjournals.eurheartja059743
15. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. (2018) 15:457–70. doi: 10.1038/s41569-018-0044-6
16. Claycomb WC, Lanson NA, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. (1998) 95:2979–84. doi: 10.1073/pnas.95.62979
17. Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. (1982) 42:3858–63.
18. Jaffe EA, Nachman RL, Becker CG, Miinick CR. Culture of human endothelial cells derived from umbilical veins. J Clin Invest. (1973) 52:2745–56. doi: 10.1172/JCI107470
19. Lall N, Henley-Smith CJ, De Canha MN, Oosthuizen CB, Berrington D. Viability reagent, prestoblue, in comparison with other available reagents, utilized in cytotoxicity and antimicrobial assays. Int J Microbiol. (2013) 2013:1–5. doi: 10.1155/2013/420601
20. Gratzner HG. Monoclonal Antibody to 5-Bromo- and 5-iododeoxyuridine: a new reagent for detection of DNA replication. Science. (1982) 218:474–5. doi: 10.1126/science7123245
21. Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc. (2014) 9:421–38. doi: 10.1038/nprot.2014018
22. Divakaruni AS, Rogers GW, Murphy AN. Measuring mitochondrial function in permeabilized cells using the seahorse XF analyzer or a clark-type oxygen electrode. Curr Protoc Toxicol. (2014) 2014:25.2.1–25.2.16. doi: 10.1002/0471140856tx2502s60
23. Jaber SM, Yadava N, Polster BM. Mapping mitochondrial respiratory chain deficiencies by respirometry: beyond the Mito Stress Test. Exp Neurol. (2020) 328:113282. doi: 10.1016/j.expneurol.2020113282
24. Clerc P, Polster BM. Investigation of mitochondrial dysfunction by sequential microplate-based respiration measurements from intact and permeabilized neurons. PLoS One. (2012) 7:e034465. doi: 10.1371/journal.pone0034465
25. Scaduto RC, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J. (1999) 76:469–77. doi: 10.1016/S0006-3495(99)77214-0
26. Yuan C, Gao J, Guo J, Bai L, Marshall C, Cai Z, et al. Dimethyl sulfoxide damages mitochondrial integrity and membrane potential in cultured astrocytes. PLoS One. (2014) 9:e107447. doi: 10.1371/journal.pone0107447
27. LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. (1992) 5:227–31. doi: 10.1021/tx00026a012
28. Langendorff O. Untersuchungen am überlebenden Säugethierherzen. Pflüger Arch Gesammte Physiol Menschen Thiere. (1895) 61:291–332. doi: 10.1007/BF01812150
29. Liao R, Podesser BK, Lim CC. The continuing evolution of the Langendorff and ejecting murine heart: New advances in cardiac phenotyping. Am J Physiol Hear Circ Physiol. (2012) 303:H156. doi: 10.1152/ajpheart.003332012
30. Sutherland FJ, Shattock MJ, Baker KE, Hearse DJ. Mouse isolated perfused heart: characteristics and cautions. Clin Exp Pharmacol Physiol. (2003) 30:867–78. doi: 10.1046/j.1440-1681.2003.03925x
31. Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and toll-like receptor 4. Immunity. (2014) 41:296–310. doi: 10.1016/j.immuni.2014.06014
32. Dou L, Sallée M, Cerini C, Poitevin S, Gondouin B, Jourde-Chiche N, et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J Am Soc Nephrol. (2015) 26:876–87. doi: 10.1681/ASN2013121283
33. Gondouin B, Cerini C, Dou L, Sallée M, Duval-Sabatier A, Pletinck A, et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. (2013) 84:733–44. doi: 10.1038/ki.2013133
34. Carpenedo R, Mannaioni G, Moroni F. Oxindole, a sedative tryptophan metabolite, accumulates in blood and brain of rats with acute hepatic failure. J Neurochem. (2002) 70:1998–2003. doi: 10.1046/j.1471-4159.1998.70051998x
35. Barreto FC, Barreto D. V, Liabeuf S, Meert N, Glorieux G, Temmar M, et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. (2009) 4:1551–8. doi: 10.2215/CJN03980609
36. Muteliefu G, Enomoto A, Jiang P, Takahashi M, Niwa T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol Dial Transplant. (2009) 24:2051–8. doi: 10.1093/ndt/gfn757
37. Lekawanvijit S, Kompa AR, Manabe M, Wang BH, Langham RG, Nishijima F, et al. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS One. (2012) 7:e0041281. doi: 10.1371/journal.pone0041281
38. Yamamoto H, Tsuruoka S, Ioka T, Ando H, Ito C, Akimoto T, et al. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. (2006) 69:1780–5. doi: 10.1038/sj.ki5000340
39. Liu WC, Tomino Y, Lu KC. Impacts of indoxyl sulfate and p-Cresol sulfate on chronic kidney disease and mitigating effects of AST-120. Toxins (Basel). (2018) 10:1–22. doi: 10.3390/toxins10090367
40. Tuomainen M, Lindström J, Lehtonen M, Auriola S, Pihlajamäki J, Peltonen M, et al. Associations of serum indolepropionic acid, a gut microbiota metabolite, with type 2 diabetes and low-grade inflammation in high-risk individuals. Nutr Diabetes. (2018) 8:35. doi: 10.1038/s41387-018-0046-9
41. De M.ello VD, Paananen J, Lindström J, Lankinen MA, Shi L, Kuusisto J, et al. Indolepropionic acid and novel lipid metabolites are associated with a lower risk of type 2 diabetes in the Finnish Diabetes Prevention Study. Sci Rep. (2017) 7:1–2. doi: 10.1038/srep46337
42. Sun CY, Lin CJ, Pan HC, Lee CC, Lu SC, Hsieh YT, et al. Clinical association between the metabolite of healthy gut microbiota, 3-indolepropionic acid and chronic kidney disease. Clin Nutr. (2019) 38:2945–8. doi: 10.1016/j.clnu.2018.11029
43. Chyan YJ, Poeggeler B, Omar RA, Chain DG, Frangione B, Ghiso J, et al. Potent neuroprotective properties against the Alzheimer β-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J Biol Chem. (1999) 274:21937–42. doi: 10.1074/jbc.274.3121937
44. Karbownik M, Reiter RJ, Garcia JJ, Cabrera J, Burkhardt S, Osuna C, et al. Indole-3-propionic acid, a melatonin-related molecule, protects hepatic microsomal membranes from iron-induced oxidative damage: Relevance to cancer reduction. J Cell Biochem. (2001) 81:507–13. doi: 10.1002/1097-4644(20010601)81:3<507::AID-JCB1064>3.0CO;2-M
45. Sári Z, Mikó E, Kovács T, Jankó L, Csonka T, Lente G, et al. Indolepropionic acid, a metabolite of the microbiome, has cytostatic properties in breast cancer by activating ahr and pxr receptors and inducing oxidative stress. Cancers (Basel). (2020) 12:1–27. doi: 10.3390/cancers12092411
46. Jankowski J, Van D.er Giet M, Jankowski V, Schmidt S, Hemeier M, Mahn B, et al. Increased plasma phenylacetic acid in patients with end-stage renal failure inhibits iNOS expression. J Clin Invest. (2003) 112:256–64. doi: 10.1172/JCI200315524
47. Morita M, Yano S, Yamaguchi T, Yamauchi M, Sugimoto T. Phenylacetic acid stimulates reactive oxygen species generation and tumor necrosis factor-α secretion in vascular endothelial cells. Ther Apher Dial. (2011) 15:147–50. doi: 10.1111/j.1744-9987.2010.00887x
48. Poesen R, Claes K, Evenepoel P, de Loor H, Augustijns P, Kuypers D, et al. Microbiota-derived phenylacetylglutamine associates with overall mortality and cardiovascular disease in patients with CKD. J Am Soc Nephrol. (2016) 27:3479–87. doi: 10.1681/ASN2015121302
49. Nemet I, Saha PP, Gupta N, Zhu W, Romano KA, Skye SM, et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell. (2020) 180:862–77.e22. doi: 10.1016/j.cell.2020.02016
50. Álvarez-Cilleros D, Ramos S, Goya L, Martín MÁ. Colonic metabolites from flavanols stimulate nitric oxide production in human endothelial cells and protect against oxidative stress-induced toxicity and endothelial dysfunction. Food Chem Toxicol. (2018) 115:88–97. doi: 10.1016/j.fct.2018.03006
51. Nallu A, Sharma S, Ramezani A, Muralidharan J, Raj D. Gut microbiome in chronic kidney disease: challenges and opportunities. Transl Res. (2017) 179:24–37. doi: 10.1016/j.trsl.2016.04007
52. Yu TH, Tang WH, Lu YC, Wang CP, Hung WC, Wu CC, et al. Association between hippuric acid and left ventricular hypertrophy in maintenance hemodialysis patients. Clin Chim Acta. (2018) 484:47–51. doi: 10.1016/j.cca.2018.05022
53. Hubková B, Veliká B, Birková A, Guzy J, Mareková M. Hydroxybenzoic acids and their derivatives as peroxynitrite scavengers. Free Radic Biol Med. (2014) 75:S33–S4. doi: 10.1016/j.freeradbiomed.2014.10770
54. Meijers BKI, Bammens B, De M.oor B, Verbeke K, Vanrenterghem Y, Evenepoel P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. (2008) 73:1174–80. doi: 10.1038/ki.200831
55. Meijers BKI, Claes K, Bammens B, De L.oor H, Viaene L, Verbeke K, et al. p-Cresol and cardiovascular risk in mild-to-moderate kidney disease. Clin J Am Soc Nephrol. (2010) 5:1182–9. doi: 10.2215/CJN07971109
56. Cerini C, Dou L, Anfosso F, Sabatier F, Moal V, Glorieux G, et al. P-cresol, a uremic retention solute, alters the endothelial barrier function in vitro. Thromb Haemost. (2004) 92:140–50. doi: 10.1160/TH03-07-0491
57. Dou L, Bertrand E, Cerini C, Faure V, Sampol J, Vanholder R, et al. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. (2004) 65:442–51. doi: 10.1111/j.1523-1755.2004.00399x
58. Jie Z, Xia H, Zhong S-L, Feng Q, Li S, Liang S, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. (2017) 8:845. doi: 10.1038/s41467-017-00900-1
59. Geng J, Yang C, Wang B, Zhang X, Hu T, Gu Y, et al. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed Pharmacother. (2018) 97:941–7. doi: 10.1016/j.biopha.2017.11016
60. Chou RH, Chen CY, Chen IC, Huang HL, Lu YW, Kuo CS, et al. Trimethylamine N-Oxide, Circulating Endothelial Progenitor Cells, and Endothelial Function in Patients with Stable Angina. Sci Rep. (2019) 9:4249. doi: 10.1038/s41598-019-40638-y
61. Chen H, Li J, Li N, Liu H, Tang J. Increased circulating trimethylamine N-oxide plays a contributory role in the development of endothelial dysfunction and hypertension in the RUPP rat model of preeclampsia. Hypertens Pregnancy. (2019) 38:96–104. doi: 10.1080/10641955.20191584630
62. Zavecz JH, Battarbee HD. The role of lipophilic bile acids in the development of cirrhotic cardiomyopathy. Cardiovasc Toxicol. (2010) 10:117–29. doi: 10.1007/s12012-010-9069-8
63. Jovanovich A, Isakova T, Block G, Stubbs J, Smits G, Chonchol M, et al. Deoxycholic acid, a metabolite of circulating bile acids, and coronary artery vascular calcification in CKD. Am J Kidney Dis. (2018) 71:27–34. doi: 10.1053/j.ajkd.2017.06017
64. Khurana S, Raufman JP, Pallone TL. Bile acids regulate cardiovascular function. Clin Transl Sci. (2011) 4:210–8. doi: 10.1111/j.1752-8062.2011.00272x
65. Hanafi NI, Mohamed AS, Kadir SHS.A, Othman MHD Overview of bile acids signaling and perspective on the signal of ursodeoxycholic acid, the most hydrophilic bile acid, in the heart. Biomolecules. (2018) 8:1–9. doi: 10.3390/biom8040159
66. Voiosu A, Wiese S, Voiosu T, Bendtsen F, Møller S Bile acids and cardiovascular function in cirrhosis. Liver Int. (2017) 37:1420–30. doi: 10.1111/liv13394
67. Rajesh KG, Suzuki R, Maeda H, Yamamoto M, Yutong X, Sasaguri S. Hydrophilic bile salt ursodeoxycholic acid protects myocardium against reperfusion injury in a PI3K/Akt dependent pathway. J Mol Cell Cardiol. (2005) 39:766–76. doi: 10.1016/j.yjmcc.2005.07014
68. Miyamoto J, Kasubuchi M, Nakajima A, Irie J, Itoh H, Kimura I. The role of short-chain fatty acid on blood pressure regulation. Curr Opin Nephrol Hypertens. (2016) 25:379–83. doi: 10.1097/MNH0000000000000246
69. Bartolomaeus H, Balogh A, Yakoub M, Homann S, Markó L, Höges S, et al. Short-chain fatty acid propionate protects from hypertensive cardiovascular damage. Circulation. (2019) 139:1407–21. doi: 10.1161/CIRCULATIONAHA.118036652
70. Kim S, Goel R, Kumar A, Qi Y, Lobaton G, Hosaka K, et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci. (2018) 132:701–18. doi: 10.1042/CS20180087
71. Kasahara K, Krautkramer KA, Org E, Romano KA, Kerby RL, Vivas EI, et al. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat Microbiol. (2018) 3:1461–71. doi: 10.1038/s41564-018-0272-x
72. Chen Y, Du J, Zhao YT, Zhang L, Lv G, Zhuang S, et al. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol. (2015) 14:99. doi: 10.1186/s12933-015-0262-8
73. O'Sullivan JF, Morningstar JE, Yang Q, Zheng B, Gao Y, Jeanfavre S, et al. Dimethylguanidino valeric acid is a marker of liver fat and predicts diabetes. J Clin Invest. (2017) 127:4394–402. doi: 10.1172/JCI95995
74. Robbins JM, Herzig M, Morningstar J, Sarzynski MA, Cruz DE, Wang TJ, et al. Association of dimethylguanidino valeric acid with partial resistance to metabolic health benefits of regular exercise. JAMA Cardiol. (2019) 4:636–43. doi: 10.1001/jamacardio.20191573
75. Onyszkiewicz M, Gawrys-Kopczynska M, Sałagaj M, Aleksandrowicz M, Sawicka A, Kozniewska E, et al. Valeric acid lowers arterial blood pressure in rats. Eur J Pharmacol. (2020) 877:173086. doi: 10.1016/j.ejphar.2020173086
76. Poeggeler B, Pappolla MA, Hardeland R, Rassoulpour A, Hodgkins PS, Guidetti P, et al. Indole-3-propionate: a potent hydroxyl radical scavenger in rat brain. Brain Res. (1999) 815:382–8. doi: 10.1016/S0006-8993(98)01027-0
77. Kocarek TA, Schuetz EG, Strom SC, Fisher RA, Guzelian PS. Comparative analysis of cytochrome P4503A induction in primary cultures of rat, rabbit, and human hepatocytes. Drug Metab Dispos. (1995) 23:415–21.
78. Bertilsson G, Heidrich J, Svensson K, Åsman M, Jendeberg L, Sydow-Bäckman M, Ohlsson R, et al. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci U S A. (1998) 95:12208–13. doi: 10.1073/pnas.95.2112208
79. Kappel BA, Federici M. Gut microbiome and cardiometabolic risk. Rev Endocine Metab Disord. (2019) 20:399–406. doi: 10.1007/s11154-019-09533-9
80. Tang TWH, Chen H-C, Chen C-Y, Yen CYT, Lin C-J, Prajnamitra RP, et al. Loss of gut microbiota alters immune system composition and cripples postinfarction cardiac repair. Circulation. (2019) 139:647–59. doi: 10.1161/CIRCULATIONAHA.118035235
81. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. (2006) 444:1027–31. doi: 10.1038/nature05414
82. Fatkhullina AR, Peshkova IO, Dzutsev A, Aghayev T, McCulloch JA, Thovarai V, et al. An interleukin-23-interleukin-22 axis regulates intestinal microbial homeostasis to protect from diet-induced atherosclerosis. Immunity. (2018) 49:943–57.e9. doi: 10.1016/j.immuni.2018.09011
83. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. (2017) 551:585–9. doi: 10.1038/nature24628
84. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. (2013) 19:576–85. doi: 10.1038/nm3145
85. Aguilar EC, Leonel AJ, Teixeira LG, Silva AR, Silva JF, Pelaez JMN, et al. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr Metab Cardiovasc Dis. (2014) 24:606–13. doi: 10.1016/j.numecd.2014.01002
86. Li L, Hua Y, Ren J. Short-chain fatty acid propionate alleviates Akt2 knockout-induced myocardial contractile dysfunction. Exp Diabetes Res. (2012) 2012:851717. doi: 10.1155/2012/851717
87. Trøseid M, Ueland T, Hov JR, Svardal A, Gregersen I, Dahl CP, et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med. (2015) 277:717–26. doi: 10.1111/joim12328
88. Dodd D, Spitzer MH, Van T.reuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature. (2017) 551:648–52. doi: 10.1038/nature24661
89. Konopelski P, Konop M, Gawrys-Kopczynska M, Podsadni P, Szczepanska A, Ufnal M. Indole-3-propionic acid, a tryptophan-derived bacterial metabolite, reduces weight gain in rats. Nutrients. (2019) 11:1–13. doi: 10.3390/nu11030591
90. Evenepoel P, Meijers BKI, Bammens BRM, Verbeke K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. (2009) 76:S12–S9. doi: 10.1038/ki.2009402
91. Zhao ZH, Xin FZ, Xue Y, Hu Z, Han Y, Ma F, et al. Indole-3-propionic acid inhibits gut dysbiosis and endotoxin leakage to attenuate steatohepatitis in rats. Exp Mol Med. (2019) 51:1–14. doi: 10.1038/s12276-019-0304-5
92. Marchetti P, Fovez Q, Germain N, Khamari R, Kluza J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. (2020) 34:13106–24. doi: 10.1096/fj202000767R
93. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. (2005) 85:1093–129. doi: 10.1152/physrev.000062004
94. Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. (2006) 126:789–99. doi: 10.1016/j.cell.2006.06049
95. Zhou T, You WT, Ma ZC, Liang Q, De, Tan HL, Xiao CR, et al. Ginkgolide B protects human umbilical vein endothelial cells against xenobiotic injuries via PXR activation. Acta Pharmacol Sin. (2016) 37:177–86. doi: 10.1038/aps.2015124
96. Elshenawy OH, Anwar-Mohamed A, Abdelhamid G, El-Kadi AOS. Murine atrial HL-1 cell line is a reliable model to study drug metabolizing enzymes in the heart. Vascul Pharmacol. (2013) 58:326–33. doi: 10.1016/j.vph.2012.12002
97. Kojima H, Sata F, Takeuchi S, Sueyoshi T, Nagai T. Comparative study of human and mouse pregnane X receptor agonistic activity in 200 pesticides using in vitro reporter gene assays. Toxicology. (2011) 280:77–87. doi: 10.1016/j.tox.2010.11008
Keywords: gut microbiota, microbial metabolites, heart failure, mitochondrial dysfunction, cardiomyocyte physiology
Citation: Gesper M, Nonnast ABH, Kumowski N, Stoehr R, Schuett K, Marx N and Kappel BA (2021) Gut-Derived Metabolite Indole-3-Propionic Acid Modulates Mitochondrial Function in Cardiomyocytes and Alters Cardiac Function. Front. Med. 8:648259. doi: 10.3389/fmed.2021.648259
Received: 31 December 2020; Accepted: 19 February 2021;
Published: 22 March 2021.
Edited by:
Christoph Reinhardt, University Medical Centre, Johannes Gutenberg University Mainz, GermanyReviewed by:
Antonio Molinaro, University of Gothenburg, SwedenLong-Sheng Lu, Taipei Medical University, Taiwan
Copyright © 2021 Gesper, Nonnast, Kumowski, Stoehr, Schuett, Marx and Kappel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben A. Kappel, YmthcHBlbEB1a2FhY2hlbi5kZQ==