 Iaroslav Semin
Iaroslav Semin Justus Ninnemann
Justus Ninnemann Marina Bondareva
Marina Bondareva Ilia Gimaev
Ilia Gimaev Andrey A. Kruglov
Andrey A. Kruglov- 1German Rheumatism Research Center (DRFZ), a Leibniz Institute, Berlin, Germany
- 2Belozersky Institute of Physico-Chemical Biology and Biological Faculty, M.V. Lomonosov Moscow State University, Moscow, Russia
- 3Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia
The intestinal tract is densely populated by microbiota consisting of various commensal microorganisms that are instrumental for the healthy state of the living organism. Such commensals generate various molecules that can be recognized by the Toll-like receptors of the immune system leading to the inflammation marked by strong upregulation of various proinflammatory cytokines, such as TNF, IL-6, and IL-1β. To prevent excessive inflammation, a single layer of constantly renewing, highly proliferating epithelial cells (IEC) provides proper segregation of such microorganisms from the body cavities. There are various triggers which facilitate the disturbance of the epithelial barrier which often leads to inflammation. However, the nature and duration of the stress may determine the state of the epithelial cells and their responses to cytokines. Here we discuss the role of the microbiota-TLR-cytokine axis in the maintenance of the epithelial tissue integrity. In particular, we highlight discrepancies in the function of TLR and cytokines in IEC barrier during acute or chronic inflammation and we suggest that intervention strategies should be applied based on the type of inflammation.
Introduction
The intestinal barrier represents a complex system of epithelial cells, Paneth cells, goblet cells, infiltrating immune cells, mucus, immunoglobulin A (IgA) antibodies and antimicrobial peptides (1). Underneath the epithelial cells multiple immune cell subsets are localized, which contribute to the maintenance of the border between the host and the microbiota. Disturbance of this barrier by extrinsic and intrinsic factors may result in the influx of various bacterial products inside of the host body leading to chronic inflammatory reactions. Such stimuli include dietary components, commensal microflora or invading pathobionts from the environmental side. Moreover, genetic variability of the host and adaptive immune response toward these stimuli may also influence barrier integrity.
The main component of the intestinal barrier is a layer of epithelial cells that forms the very first physical border between the host organism and its external surroundings, which could be potentially detrimental for the host cells. These epithelial cells are tightly connected with each other to ensure proper control of the molecules that enter the body from the intestinal fluids. The junctional complex of intestinal epithelial cells is composed of the three main different types of connections—tight junction (TJ), adherence junction and desmosome. Tight junctions between epithelial cells are facilitated by a set of proteins [Claudin, ZO1, Occludin, F-actin, Myosin, Myosin light chain kinase (MLCK)], which form together an apical junctional complex in order to seal the paracellular space between epithelial cells. There are two additional zones of cell-to-cell contacts beneath TJ named “Adherence junction” and “Desmosome.” They consist of E-cadherin, α-catenin 1, β-catenin, catenin- δ1 and desmoglein, desmocollin, desmoplakin, respectively (2). Together they provide cell-to-cell and cell-to-matrix connections and create a paracellular space. Normal gut permeability facilitates paracellular transport of nutrients, water and essential solutes. Disruption of such TJ may result in the penetration of various molecules and microorganisms, leading to inflammation.
The whole spectrum of cell types within the gut epithelium develops from the epithelial stem cells located at the base of the crypts. Stem cells give rise to distinct cell types of the intestinal epithelium: absorptive cells (enterocytes) and secretory cells (goblet, Paneth, enteroendocrine, and tuft cells). Fate decision toward the absorptive phenotype is critically dependent on the NOTCH pathway (3). Genetic and pharmacological manipulation of NOTCH signaling also revealed its crucial role in the maintenance of the epithelial stem cell niche (4–6). Apart from NOTCH, wingless and Int-1 (WNT) signaling plays an essential role in epithelial stem cell functions influencing functioning of different transcription factors including Ascl2, sox9, Lgr5 (7–9).
Regulation of The Epithelial Cell Functions During Homeostasis
In steady state, a delicate balance is maintained between bacterial composition, the mucosal immune system and the intact epithelial barrier. Commensal microbiota is transported in a highly controlled manner to be recognized by the immune system in the gut-associated lymphoid tissues (2). Due to the non-pathogenic nature of such microorganisms, the immune system responds with the production of non-inflammatory cytokines, such as TGF-β1, IL-10 and cytokines which are important for the IEC barrier, like IL-22 (Figure 1). Both mutation of IL-10 pathway in humans and the genetic ablation of Il10 resulted in development of intestinal inflammation demonstrating a crucial role for IL-10 in the tolerance maintenance and barrier integrity (10). Although Il10−/− mice are not defective in mucin production, but have its defective loose quality that makes mice suffer from spontaneous colitis (11). Similarly, TGF-β1 directly modulates TJ protein expression (12, 13), significantly decreasing JNK-pathway activation and protects cells from TNF-mediated downregulation of occludin and ZO-1 (14). IL-22 controls not only the expression of TJ proteins (15), but also the expression of various antimicrobial proteins. IL-22 deficient animals exhibited defects in IEC barrier (15) and failed to repair IEC functions in multiple inflammatory models linked to the disruption of the IEC barrier. IL-22 was further reported as a necessary cytokine for TJ formation and mucin production (16). Patients with HIV infections have decreased IL-22 levels and concomitantly impaired IEC barrier and increased bacterial translocation (16). Interestingly, the natural antagonist of IL-22 (IL-22BP; IL-22Ra2) which regulates the biological actions of IL-22 was found to be expressed by various immune cells (17). Recent data suggested that type III innate lymphoid cells (ILC3) instruct a special subset of dendritic cells in the isolated lymphoid follicles to produce IL-22BP via lymphotoxin (LTα1β2)–lymphotoxin β receptor (LTβR) interaction (18), revealing a novel mechanism of the epithelial barrier control in steady state and during inflammation.
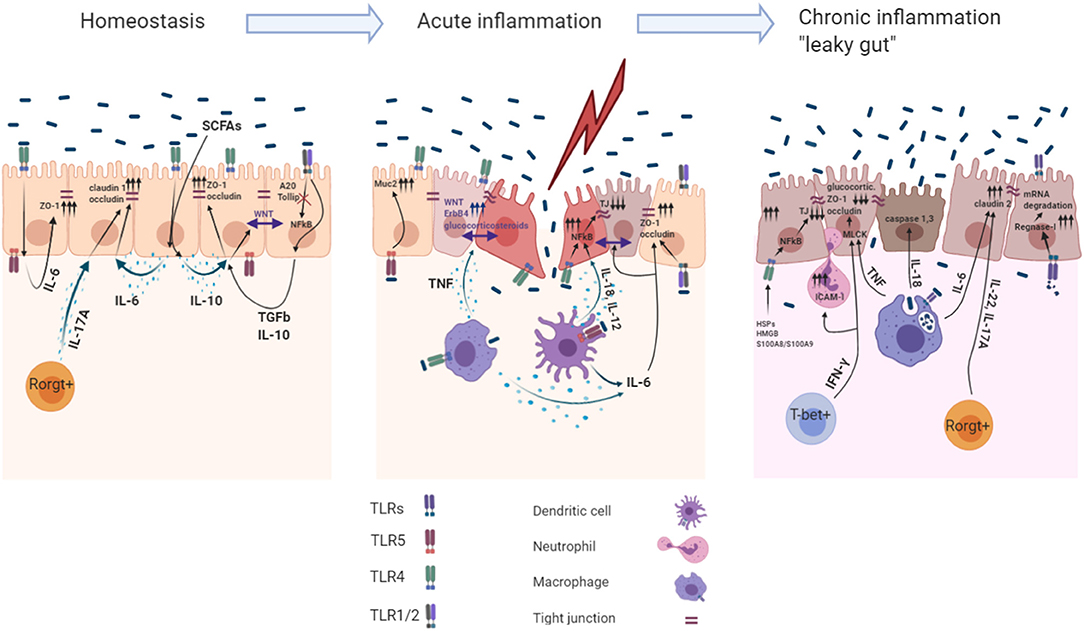
Figure 1. Role of the TLR induced cytokines in acute and chronic intestinal inflammation. The intestinal mucosa is separated from the body immune environment by a single layer of the intestinal epithelial cells (IECs) that provides a physical and functional barrier. Beneath the IECs immune cells reside in the lamina propria, maintaining the intestinal tissue at the hyporesponsive state. Intestinal immune homeostasis: Constant recognition of microbiota by TLR4 and TLR1/2 leads to IL- 6, IL−10, TGF-β1 production within IECs. Autocrine recognition of these cytokines maintains IEC barrier integrity by promoting expression of the TJ proteins (ZO-1, claudin-1, occludin). Moreover, action of the IL-10 induce Wnt signaling within IECs, which maintains their proliferation. A20 and Tollip are the main inhibitors of the TLR1/2 signaling facilitating the avoidance of undesired response toward microbiota. Rorgt+ cells during homeostasis produce IL-17A to maintain constant production of claudin-1 and occluding within IECs. Acute inflammation: Sensing microbiota within the lamina propria induces production of pro-inflammatory cytokines and cytoprotective factors via NFkB dependent mechanism. Basolateral TLR5 mediated recognition of bacteria leads to MUC2 production in IECs. IL-6, TNF production by M?s and DCs during acute inflammation enables barrier repair program within IECs. TNF induced production of the glucocorticoids and ErbB4 receptor tyrosine kinase in IECs induce tissue repair functions and resolve late stages of the acute inflammation. Pro-inflammatory cytokines IL-18, IL-12 involved in IEC barrier dysfunction by downregulating TJ proteins (ZO-1, occludin). TLR1/2 basolateral recognition of bacteria promotes ZO-1/occluding expression in IECs. Chronic inflammation: Chronic TNF sensing by IECs reduces their ability to migrate toward crypts villi, modulates MLCK which decreases claudin-1, ZO-1, and ZO-2 expression and decreases glucocorticoids synthesis, which is indispensable for later inflammation resolution. IFN-γ activates the expression of ICAM-1 which resulted in increased IEC barrier permeability caused by neutrophil migration into subepithelial layers and paracellular space via modulation of MLCK. TLR4 dependent recognition of the HSPs, HMGB1 and S100A8/S100A9 by IECs leads to downregulation of the expression of ZO-1.
Commensal microbiota produces multiple “non-self” ligands and IECs recognize such molecules and tune their transcriptional program to keep the barrier tight. There are several families of receptors sensing various microbial products: Toll-like receptors (TLR), NOD-like receptors (NLR), RIG-like receptors (RLR), and others (19). TLRs are widely expressed on the epithelial cells in the small and large intestine and their expression is tightly regulated in order to ensure the proper innate immune recognition. Mostly, TLRs are expressed among the whole IEC lineage: absorptive enterocytes (20, 21), stem cells (22), enteroendocrine cells (23), goblet cells (24, 25), Paneth cells (26–28), and micro-fold cells (29, 30). The distribution pattern of TLR expression on epithelial cells varies among the intestinal tract. Price et al. recently provided an elegant analysis of TLRs expression in the large and small intestine of mice (27). It was shown that TLR2, TLR5, and TLR9 are more restricted to the small intestine when TLR2, TLR4, and TLR5 are upregulated in the colonic epithelial cells. In addition, TLR signaling is controlled by the polarized expression on the cell surface. For instance, TLR2 and TLR4 are expressed at low levels on basolateral sides of IEC in the small intestine, while TLR5 is expressed mainly on basolateral sides of the colon (31). Furthermore, apical TLR9 recognition of CpG oligonucleotides prevents NFkB translocation into the nucleus and limits inflammatory response.
The tuning of the immune responses via IEC-derived TLRs is achieved by several mechanisms. Epithelial cells modulate TLR receptor-ligand interactions by the downregulation of the receptor expression (32) or by translocating receptors from apical to basolateral sides or to lysosomes (33–36) to avoid excessive sensing of bacterial products. Indeed, overexpression of Tlr4 on epithelial cells resulted in the overactivation of TLR4 pathway in IECs that lead to the increased production of IgA by plasma B cells (37). This loop potentially demonstrates a regulatory mechanism where IgA antibodies after being induced neutralize excessive bacteria-TLR4 interaction (20). Next, expression of molecules downstream of TLRs is modulated in IEC via various posttranslational modifications like glycosylation, phosphorylation, and ubiquitination (38, 39). Finally, IECs were reported to bind and modify immunogenic parts of MAMPs in order to diminish ligands property to induce signals (40, 41).
Apart from this, TLRs are involved in crypt dynamics control. For instance the depletion of MyD88 or TLR2 was associated with an abrogation of trefoil factor 3 (TFF3) expression, which is required for goblet cells maturation (24). Furthermore, TLR4 was shown to mediate NOTCH expression implying that TLRs may interfere with processes of stemness and differentiation in the stem cell niche (1). However, the role of TLR4 on stem cell differentiation remains controversial (42, 43). Deletion of TLR1 or TLR5 in mice induced the loss of the mucus layer integrity via impaired MUC2 production in goblet cells (1). Moreover, ablation of the TLR recognition by MyD88 deletion abrogated the production of antimicrobial peptides RegIIIβ and RegIIIγ by goblet cells in mice (20). Thus, sensing of bacterial products via TLRs modulates mucus layer permeability that limits the direct interaction of commensals with the epithelium and induction of spontaneous inflammation (33, 44).
Altogether, IEC barrier exerts multiple strategies to avoid activation of inflammatory pathways in normal conditions via cytokine production and regulation of TLR signaling to maintain its integrity.
Regulation of The Epithelial Cell Functions During Acute Inflammation
Disruption of the cell-to-cell contacts at the epithelial layer leads to increased bacterial products penetration, which triggers inflammatory immune responses. The nature of the damage may further define the type of immune response and subsequent immune reactions driving IEC repair (Figure 1). Epithelial barrier disruption may be induced by acute stimuli, such as ingestion of toxic substances (oxazolone, dextran sodium sulfate etc.) (45), by physical force or by the invasion of various pathogens, such as Clostridium difficles, Citrobacter rodentium, Salmonella enterica etc. These acute stimuli result in IEC layer erosion, the influx of commensal bacteria and activation of the innate arm of the immune system (46), while the chronic reduction of the barrier leads to the mobilization of both arms of the immune system as well as the genomic instability of epithelial cells (47). In case of the acute damage of the epithelium caused by pathogens, the immune system should eliminate the causing agent or pathogen, while ensuring the proper restoration of the barrier. Thus, the gut immune system is determined to restore the barrier functions in both acute and chronic settings, but triggers are different and, thereby, advocate for different intervention strategies.
Eliciting a protective immune response is required for the successful restoration of the barrier during bacteria-induced colitis. Here TLR-proinflammatory cytokine module is instrumental for the clearance of the inflammatory triggers and it is also involved in further tissue repair processes. Indeed, there are multiple examples of protective functions of TLR receptors in this setting. For instance, TLR1 is found to be crucial for the protection during acute intestinal inflammation induced by Yersinia enterocolitica in mice and the maintenance of the increased IEC barrier permeability (48). TLR5 was reported to limit intestinal colonization with vancomycin-resistant Enterococcus (VRE) by the induction of RegIIIγ expression (49) and IEC-derived TLR5 mediates production of IL-6 and IL-12 by CD11c+ in response to Salmonella enterica infection (50). The significance of TLR/MyD88 signaling pathway for the recovery of IECs was also shown during acute colitis induced by Helicobacter hepaticus or Citrobacter rodentium (51). Furthermore, Myd88−/−, Tlr1−/−, Tlr2−/− mice were characterized by the early loss of tight junctions and diminished transepithelial resistance during acute intestinal inflammation (52).
Apart from IEC barrier disruption by pathogens, there is a significant amount of the research directed toward the dissection of the pathways which are crucial for IEC barrier restoration during injury caused by chemical agents, such as DSS, oxazolone and others. Herein the inflammation is caused by the influx of commensal microbiota in the intestinal tissue. Thus, TLR signaling pathways and pro-inflammatory cytokines facilitate the inflammation that is needed for the clearance of the bacteria and may possess protective functions. Consistently, seminal work from Medzhitov's lab showed the crucial role of TLR4/MyD88 signaling for the maintenance of the intestinal homeostasis and barrier repair during acute DSS colitis in microbiota dependent manner (53). Activation of TLR4 signaling pathways was crucial for the clearance of commensal bacteria by infiltrating innate immune cells (54). In contrast, several other studies highlighted the pathogenic function of TLR4 signaling in DSS colitis. In particular, an increase of E. Coli in the microbiota was associated with less severe colitis in TLR4 deficient mice (55). LPS, main TLR4 agonist, also may induce epithelial damage in vitro and in vivo via excessive phosphorylation of the focal actin kinase (FAK) in TLR4/MyD88 dependent pathway in epithelial cells (56). Using an ileal cell line, LPS was further reported to be instrumental in the induction of paracellular permeability via ZO-1 and occludin downregulation via TLR4 (57). Interestingly, LPS serotypes differentially affect inflammatory cytokines expression in vitro. Among others, LPS from S. marcescens has the most pronounced effect on the reduction of transepithelial electrical resistance. That correlated with an increase in NFkB activation, IL-8 production as well as TNF (58). Furthermore, E. coli LPS, but not LPS from B. dorei, influenced the incidence of autoimmune diabetes in non-obese diabetic mice and correlated with the development of autoimmunity in humans (59). Therefore, the role of TLR4 during acute IEC disruption is determined by the microbiota composition and therapeutic strategies targeting TLR4 should be considered given the prevalence of various microorganisms and pathogens in individual contexts.
TLR signaling mediates the production of multiple pro-inflammatory cytokines, among them TNF, IL-6, and IL-1β. TNF a cytokine with pleiotropic functions in the body is of particular significance in this context. On the one hand, TNF is crucial for the host defense against intracellular pathogens (60) but on the other hand it drives multiple autoimmune pathologies associated with a reduction of the epithelial barrier, such as inflammatory bowel disease (IBD), ankylosing spondylitis and rheumatoid arthritis. Importantly, anti-TNF therapy is highly effective in the treatment aforementioned autoimmune pathologies (61). Despite the tremendous success of the TNF blockade, a significant proportion of patients do not respond to this type of biological interventions further highlighting the heterogeneity of given autoimmune conditions and pleiotropy of TNF itself. It is worth mentioning that TNF exerts its functions via two receptors, TNFR1 and TNFR2 (62) inducing distinct transcriptional programs. TNF plays a protective role during acute colitis induced by DSS, as TNF deficient mice and anti-TNF therapy in wild type mice during colitis resulted in severe inflammation (63). Short acute IEC exposure to TNF induced glucocorticoid synthesis and, thereby, ameliorated the late stages of DSS colitis (64). Furthermore, TNFR1 mediated protective functions, while TNFR2 was deleterious upon acute disruption of epithelium (65). Apart from the induction of anti-inflammatory mediators that are crucial for the barrier restoration, TNF also contributes to the restoration of the epithelial barrier via modulation of Wnt (66). TNF administration during acute DSS colitis promoted the intestinal cell survival and restitution via elevating expression the ErbB4 receptor tyrosine kinase (67). In addition, another study conducted on the IL-10 deficient mice colitis model suggested that the binding of TNF by TNFR1 and following Il1b upregulation is essential for the early defensive response within colonic epithelial cells (68, 69). Kuhn et al. showed that Bacteroidales spp. induced IL-6 secretion by IECs in a MyD88-dependent manner, while Il6−/− mice were more susceptible to Citrobacter rodentium infection and had a thinner mucus layer, as well as decreased claudin-1 expression (70). Finally, IL-6 also activated NOTCH dependent program of IEC barrier restoration during acute DSS colitis (71).
Thus, proinflammatory cytokines exert its protective functions during acute barrier injury to facilitate efficient clearance of invading microorganisms.
Regulation Of the Epithelial Cell Functions During Chronic Inflammation
Various extrinsic factors, such as the environment, particular diet, and exposure to hazardous chemicals, may result in the chronic elevation of pro-inflammatory cytokines and the reduction of the gut permeability for a long period of time (Figure 1). The state of an increased gut permeability and the perturbation of local immunity in the gut is called “leaky gut.” This phenomenon has been described not only in IBD patients, but also in many metabolic and autoimmune disorders. “Leaky gut” syndrome is characterized by an impaired mucin synthesis, a decreased expression of junctional proteins and epithelial cell death. Importantly, increased permeability of the epithelium is often found before the development of clinical symptoms (72).
Taking into account the fundamentally different nature of IEC barrier reduction during acute and chronic stress, it is plausible that TLR and cytokines may have distinct, and even opposing functions depending on the duration of inflammation. Consistently, deep analysis of the mutational landscape from inflamed IBD tissue and corresponding non-inflamed parts revealed mutations in several genes, such as NFKBIZ, ZC3H12A (Regnase-1) and PIGR. Interestingly, Regnase-1 is activated in response to TLR stimulation and degrades mRNA of many downstream immune signaling genes (47), including PIGR (73), NFKBIZ (74), and members of the IL-17 pathway (75). Furthermore, DNA methylation patterns and transcriptional program in IECs differed between healthy and IBD patients (76). Chronic exposure of IECs to TNF exclusively affected their migration from the crypt to the villus (77). In addition, chronic inflammation modeled by long-term culture of colonic organoids in the presence of TLR agonists and pro-inflammatory cytokines resulted in chronic NFkB activation and the transformation of epithelial cells. Finally, organoid cultures from IBD patients showed an inflammatory phenotype with decreased size and budding capacity and inverted polarization (78). Altogether, these data suggested that chronic inflammation might transform the genetic program and the functions of IECs and their ability to maintain the epithelial barrier.
Chronic subclinical inflammation is characterized by an increase in cytokine production and in release of endogenous TLR4 ligands. In particular, high mobility group box 1 (HMGB1) protein, the heat shock proteins and calcium binding protein A8 and A9 (S100A8/S100A9) (79) are released during an inflammation and chronic conditions, like metabolic disorders (80). Their binding to TLR4 leads to the secretion of the pro-inflammatory cytokines IL-1β, TNF, IL-6, IL-17A, IL-18, and IL-12 in the intestine (31, 81). Furthermore, TLR4 activation within the gut epithelium is associated with the activation of myosin light chain kinase (MLCK), which reduces the tight junction of IEC barrier and may lead to the development of “leaky gut” (82–84).
As mentioned earlier, increased gut permeability may be induced by extrinsic factors, like diet, environmental factors but also by intrinsic factors, such as elevated levels of pro-inflammatory cytokines (85, 86). In particular, TNF, IL-6 and IFN-γ are associated with the epithelial barrier impairment and increased gut permeability (31, 87–89). These cytokines once produced chronically may significantly reduce IEC barrier. So IFN-γ was found to modulate the expression of the neutrophil adhesion molecule ICAM-1, which resulted in increased permeability and the migration of neutrophils into the subepithelial layers and paracellular space (90). Apart from this, IFN-γ enhanced Th1 immune responses and also increased CD14 and TLR4 expression, as well as LPS uptake by IECs (86). For instance, IL-6 increased permeability-promoting tight junction protein (claudin-2) in colonic cell culture via activation of c-Jun N-terminal kinase (JNK) pathway (91). IEC stimulation with TNF lead to the upregulation of the MLCK, phosphorylation of myosin II light chain (MLC) and the subsequent decrease in barrier integrity. Furthermore, TNF induced the loss of ZO-1 and occludin expression and decreased trans-epithelial electrical resistance (92). In immune-mediated colitis model, it was further shown that TNFR2 pathway, but not TNFR1 signaling, increases MLCK expression resulting in tight junction dysregulation, barrier loss, and more severe disease (93). Chronic exposure to TNF, in contrast to acute stimuli, actually decreased glucocorticosteroid production and perpetuated inflammation (94). Given multiple effects of TNF on the intestinal biology, it is predicted that anti-TNF therapy restores the intestinal barrier in many autoimmune diseases (95). It has been shown in several reports that anti-TNF therapy directly influenced tight junction protein expression (96), while others showed the restoration of EC survival rate (97). In vitro experiments also indicated that sera from IBD patients directly regulates ZO-1 and occludin expression in IECs via TNF. Moreover, TNF was further shown to downregulate claudin-1, claudin-2, claudin-4, and occludin expression in IECs layer (95). Interestingly, IL-6 promoted crypt organoid proliferation stem cell numbers (98). Furthermore, anti-IL-6 therapy in IBD patients ameliorated the disease, but increased the risk of developing GI abscesses and perforation (99), suggesting that IL-6 contribute to inflammatory processes, but also may maintain epithelial barrier. Thus, upon chronic inflammatory stimuli epithelial cells modify their transcriptional program, expression patterns of receptors and, thereby, may respond differently toward pro-inflammatory cytokines.
Conclusions
IEC barrier integrity is maintained not only by a complex system of tight junction proteins and strict compartment-dependent distribution of TLRs on apical and basolateral sides of IECs but also by a network of immune cells that mediate cell proliferation and epithelial permeability via cytokines. In a healthy state IECs exhibit multiple mechanisms that dampen TLR-dependent recognition of the microbiota. During acute injury of IEC barrier by chemical agents or pathogens the TLR-TNF axis is triggered toward the clearance of the pro-inflammatory stimuli and further drives IEC layer restoration via activation of the glucocorticosteroid synthesis, WNT pathway and ErbB4 kinase. In contrast to acute damage, chronic inflammation induces genetic instability, changes of methylome, transcriptome and the polarity of TLRs expression in IECs. This results in their modified response toward TLR agonists and TNF. Thus, the character and duration of inflammation should be considered for the modeling of studies aiming to dissect the mechanisms of IEC barrier integrity during various injury.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
IS, MB, JN, IG, and AK analyzed the literature and studies and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by DFG (TRR241 A04, AK) and Russian Science Foundation (#21-14-00223, AK).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Burgueño JF, Abreu MT. Epithelial Toll-like receptors and their role in gut homeostasis and disease. Nat Rev Gastroenterol Hepatol. (2020) 17:263–78. doi: 10.1038/s41575-019-0261-4
2. Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. (2009) 9:799–809. doi: 10.1038/nri2653
3. Noah TK, Shroyer NF. Notch in the intestine: regulation of homeostasis and pathogenesis. Annu Rev Physiol. (2013) 75:263–88. doi: 10.1146/annurev-physiol-030212-183741
4. Riccio O, van Gijn ME, Bezdek AC, Pellegrinet L, van Es JH, Zimber-Strobl U, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. (2008) 9:377–83. doi: 10.1038/embor.2008.7
5. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. (2010) 464:1052–7. doi: 10.1038/nature08878
6. Tran IT, Sandy AR, Carulli AJ, Ebens C, Chung J, Shan GT. Blockade of individual Notch ligands and receptors controls graft-versus-host disease. J Clin Invest. (2013) 123:1590–604. doi: 10.1172/JCI65477
7. Chin AM, Hill DR, Aurora M, Spence JR. Morphogenesis and maturation of the embryonic and postnatal intestine. Semin Cell Dev Biol. (2017) 66:81–93. doi: 10.1016/j.semcdb.2017.01.011
8. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. (1999) 96:5522–7. doi: 10.1073/pnas.96.10.5522
9. Flanagan DJ, Austin CR, Vincan E, Phesse TJ. Wnt signalling in gastrointestinal epithelial stem cells. Genes (Basel). (2018) 9:178. doi: 10.3390/genes9040178
10. Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. (1993) 75:263–74. doi: 10.1016/0092-8674(93)80068-P
11. Johansson ME, Gustafsson JK, Holmen-Larsson J, Jabbar KS, Xia L, Xu H, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. (2014) 63:281–91. doi: 10.1136/gutjnl-2012-303207
12. Hahm KB, Im YH, Parks TW, Park SH, Markowitz S, Jung HY, et al. Loss of transforming growth factor beta signalling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut. (2001) 49:190–8. doi: 10.1136/gut.49.2.190
13. Howe KL, Reardon C, Wang A, Nazli A, McKay DM. Transforming growth factor-beta regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am J Pathol. (2005) 167:1587–97. doi: 10.1016/S0002-9440(10)61243-6
14. Xiao K, Cao S, Jiao L, Song Z, Lu J, Hu C. TGF-β1 protects intestinal integrity and influences Smads and MAPK signal pathways in IPEC-J2 after TNF-α challenge. Innate Immun. (2017) 23:276–84. doi: 10.1177/1753425917690815
15. Keir M, Yi T, Lu T, Ghilardi N. The role of IL-22 in intestinal health and disease. J Exp Med. (2020) 217:e20192195. doi: 10.1084/jem.20192195
16. Kim C, Nazli A, Chege D, Alidina Z, Huibner S, Mujib S, et al. A role for mucosal IL-22 production and Th22 cells in HIV-associated mucosal immunopathogenesis. Mucosal Immunol. (2012) 5:670–80. doi: 10.1038/mi.2012.72
17. Xu W, Presnell SR, Parrish-Novak J, Kindsvogel W, Jaspers S, Chen Z, et al. A soluble class II cytokine receptor, IL-22RA2, is a naturally occurring IL-22 antagonist. Proc Natl Acad Sci USA. (2001) 98:9511–6. doi: 10.1073/pnas.171303198
18. Kempski J, Giannou AD, Riecken K, Zhao L, Steglich B, Lücke J, et al. IL22BP mediates the antitumor effects of lymphotoxin against colorectal tumors in mice and humans. Gastroenterology. (2020) 159:1417–30.e3. doi: 10.1053/j.gastro.2020.06.033
19. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
20. Lee J, Mo JH, Katakura K, Alkalay I, Rucker AN, Liu YT, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. (2006) 8:1327–36. doi: 10.1038/ncb1500
21. Cario E, Rosenberg IM, Brandwein SL, Beck PL, Reinecker HC, Podolsky DK. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J Immunol. (2000) 164:966–72. doi: 10.4049/jimmunol.164.2.966
22. Neal MD, Sodhi CP, Jia H, Dyer M, Egan CE, Yazji I, et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J Biol Chem. (2012) 287:37296–308. doi: 10.1074/jbc.M112.375881
23. Palazzo M, Balsari A, Rossini A, Selleri S, Calcaterra C, Gariboldi S, et al. Activation of enteroendocrine cells via TLRs induces hormone, chemokine, and defensin secretion. J Immunol. (2007) 178:4296–303. doi: 10.4049/jimmunol.178.7.4296
24. Podolsky DK, Gerken G, Eyking A, Cario E. Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology. (2009) 137:209–20. doi: 10.1053/j.gastro.2009.03.007
25. Birchenough GMH, Nyström EEL, Johansson MEV, Hansson GC. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science. (2016) 352:1535–42. doi: 10.1126/science.aaf7419
26. Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci USA. (2008) 105:20858–63. doi: 10.1073/pnas.0808723105
27. Price AE, Shamardani K, Lugo KA, Deguine J, Roberts AW, Lee BL, et al. A Map of Toll-like Receptor Expression in the Intestinal Epithelium Reveals Distinct Spatial, Cell Type-Specific, and Temporal Patterns. Immunity. (2018) 49:560–575.e6. doi: 10.1016/j.immuni.2018.07.016
28. Rumio C, Sommariva M, Sfondrini L, Palazzo M, Morelli D, Vigano L, et al. Induction of Paneth cell degranulation by orally administered Toll-like receptor ligands. J Cell Physiol. (2012) 227:1107–13. doi: 10.1002/jcp.22830
29. Chabot S, Wagner JS, Farrant S, Neutra MR. TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J Immunol. (2006) 176:4275–83. doi: 10.4049/jimmunol.176.7.4275
30. Cashman SB, Morgan JG. Transcriptional analysis of Toll-like receptors expression in M cells. Mol Immunol. (2009) 47:365–72. doi: 10.1016/j.molimm.2009.09.007
31. Hug H, Mohajeri MH, La Fata G. Toll-like receptors: regulators of the immune response in the human gut. Nutrients. (2018) 10:203. doi: 10.3390/nu10020203
32. Fulde M, Sommer F, Chassaing B, van Vorst K, Dupont A, Hensel M, et al. Neonatal selection by Toll-like receptor 5 influences long-term gut microbiota composition. Nature. (2018) 560:489–93. doi: 10.1038/s41586-018-0395-5
33. Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. (2007) 117:3909–21. doi: 10.1172/JCI33084
34. Cario E, Brown D, McKee M, Lynch-Devaney K, Gerken G, Podolsky DK. Commensal-associated molecular patterns induce selective toll-like receptor-trafficking from apical membrane to cytoplasmic compartments in polarized intestinal epithelium. Am J Pathol. (2002) 160:165–73. doi: 10.1016/S0002-9440(10)64360-X
35. Melmed G, Thomas LS, Lee N, Tesfay SY, Lukasek K, Michelsen KS, et al. Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host-microbial interactions in the gut. J Immunol. (2003) 170:1406–15. doi: 10.4049/jimmunol.170.3.1406
36. Yu S, Gao N. Compartmentalizing intestinal epithelial cell toll-like receptors for immune surveillance. Cell Mol Life Sci. (2015) 72:3343–53. doi: 10.1007/s00018-015-1931-1
37. Shang L, Fukata M, Thirunarayanan N, Martin AP, Arnaboldi P, Maussang D, et al. Toll-like receptor signaling in small intestinal epithelium promotes B-cell recruitment and IgA production in lamina propria. Gastroenterology. (2008) 135:529–38. doi: 10.1053/j.gastro.2008.04.020
38. Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. (2005) 5:446–58. doi: 10.1038/nri1630
39. Leifer CA, Medvedev AE. Molecular mechanisms of regulation of Toll-like receptor signaling. J Leukoc Biol. (2016) 100:927–41. doi: 10.1189/jlb.2MR0316-117RR
40. Candia E, Diaz-Jimenez D, Langjahr P, Nunez LE, de la Fuente M, Farfan N, et al. Increased production of soluble TLR2 by lamina propria mononuclear cells from ulcerative colitis patients. Immunobiology. (2012) 217:634–42. doi: 10.1016/j.imbio.2011.10.023
41. Bentala H, Verweij WR, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, Poelstra K. Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock. (2002) 18:561–6. doi: 10.1097/00024382-200212000-00013
42. Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology. (2012) 143:708–18 e5. doi: 10.1053/j.gastro.2012.05.053
43. Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. (2013) 340:1190–4. doi: 10.1126/science.1234852
44. Carvalho FA, Koren O, Goodrich JK, Johansson MEV, Nalbantoglu I, Aitken JD, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. (2012) 12:139–52. doi: 10.1016/j.chom.2012.07.004
45. Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. (2007) 2:541–6. doi: 10.1038/nprot.2007.41
46. Lechuga S, Ivanov AI. Disruption of the epithelial barrier during intestinal inflammation: quest for new molecules and mechanisms. Biochim Biophys Acta Mol Cell Res. (2017) 1864:1183–94. doi: 10.1016/j.bbamcr.2017.03.007
47. Olafsson S, McIntyre RE, Coorens T, Butler T, Jung H, Robinson PS, et al. Somatic evolution in non-neoplastic IBD-affected colon. Cell. (2020) 182:672–84.e11. doi: 10.1016/j.cell.2020.06.036
48. Kamdar K, Khakpour S, Chen J, Leone V, Brulc J, Mangatu T, et al. Genetic and metabolic signals during acute enteric bacterial infection alter the microbiota and drive progression to chronic inflammatory disease. Cell Host Microbe. (2016) 19:21–31. doi: 10.1016/j.chom.2015.12.006
49. Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest. (2010) 120:4332–41. doi: 10.1172/JCI43918
50. Uematsu S, Jang MH, Chevrier N, Guo Z, Kumagai Y, Yamamoto M, et al. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat Immunol. (2006) 7:868–74. doi: 10.1038/ni1362
51. Jobin C. MyD88 signaling in the intestine: Dr Jekyll and Mr Hyde? Gastroenterology. (2010) 139:383–5. doi: 10.1053/j.gastro.2010.06.027
52. Karczewski J, Troost FJ, Konings I, Dekker J, Kleerebezem M, Brummer RJ, et al. Regulation of human epithelial tight junction proteins by Lactobacillus plantarum in vivo and protective effects on the epithelial barrier. Am J Physiol Gastrointest Liver Physiol. (2010) 298:G851–9. doi: 10.1152/ajpgi.00327.2009
53. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. (2004) 118:229–41. doi: 10.1016/j.cell.2004.07.002
54. Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. (2005) 288:G1055–65. doi: 10.1152/ajpgi.00328.2004
55. Heimesaat MM, Fischer A, Siegmund B, Kupz A, Niebergall J, Fuchs D, et al. Shift towards pro-inflammatory intestinal bacteria aggravates acute murine colitis via Toll-like receptors 2 and 4. PLoS ONE. (2007) 2:e662. doi: 10.1371/journal.pone.0000662
56. Guo S, Nighot M, Al-Sadi R, Alhmoud T, Nighot P, Ma TY. Lipopolysaccharide regulation of intestinal tight junction permeability is mediated by TLR4 signal transduction pathway activation of FAK and MyD88. J Immunol. (2015) 195:4999–5010. doi: 10.4049/jimmunol.1402598
57. Bein A, Zilbershtein A, Golosovsky M, Davidov D, Schwartz B. LPS induces hyper-permeability of intestinal epithelial cells. J Cell Physiol. (2017) 232:381–90. doi: 10.1002/jcp.25435
58. Ochieng JB, Boisen N, Lindsay B, Santiago A, Ouma C, Ombok M, et al. Serratia marcescens is injurious to intestinal epithelial cells. Gut Microbes. (2014) 5:729–36. doi: 10.4161/19490976.2014.972223
59. Vatanen T, Kostic AD, d'Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. (2016) 165:842–53. doi: 10.1016/j.cell.2016.04.007
60. Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, et al. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: protective and deleterious effects. Immunity. (2005) 22:93–104. doi: 10.1016/S1074-7613(04)00379-6
61. Kruglov A, Drutskaya M, Schlienz D, Gorshkova E, Kurz K, Morawietz L, et al. Contrasting contributions of TNF from distinct cellular sources in arthritis. Ann Rheum Dis. (2020) 79:1453–9. doi: 10.1136/annrheumdis-2019-216068
62. Atretkhany K-SN, Mufazalov IA, Dunst J, Kuchmiy A, Gogoleva VS, Andruszewski D, et al. Intrinsic TNFR2 signaling in T regulatory cells provides protection in CNS autoimmunity. Proc Natl Acad Sci USA. (2018) 115:13051–6. doi: 10.1073/pnas.1807499115
63. Kojouharoff G, Hans W, Obermeier F, Männel DN, Andus T, Schölmerich J, et al. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin Exp Immunol. (1997) 107:353–8. doi: 10.1111/j.1365-2249.1997.291-ce1184.x
64. Noti M, Corazza N, Mueller C, Berger B, Brunner T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med. (2010) 207:1057–66. doi: 10.1084/jem.20090849
65. Stillie R, Stadnyk AW. Role of TNF receptors, TNFR1 and TNFR2, in dextran sodium sulfate-induced colitis. Inflamm Bowel Dis. (2009) 15:1515–25. doi: 10.1002/ibd.20951
66. Bradford EM, Ryu SH, Singh AP, Lee G, Goretsky T, Sinh P, et al. Epithelial TNF receptor signaling promotes mucosal repair in inflammatory bowel disease. J Immunol. (2017) 199:1886–97. doi: 10.4049/jimmunol.1601066
67. Frey MR, Edelblum KL, Mullane MT, Liang D, Polk DB. The ErbB4 growth factor receptor is required for colon epithelial cell survival in the presence of TNF. Gastroenterology. (2009) 136:217–26. doi: 10.1053/j.gastro.2008.09.023
68. Liu CY, Tam SS, Huang Y, Dubé PE, Alhosh R, Girish N, et al. TNF receptor 1 promotes early-life immunity and protects against colitis in mice. Cell Rep. (2020) 33:108275. doi: 10.1016/j.celrep.2020.108275
69. Pié S, Lallès JP, Blazy F, Laffitte J, Sève B, Oswald IP. Weaning is associated with an upregulation of expression of inflammatory cytokines in the intestine of piglets. J Nutr. (2004) 134:641–7. doi: 10.1093/jn/134.3.641
70. Kuhn KA, Schulz HM, Regner EH, Severs EL, Hendrickson JD, Mehta G, et al. Bacteroidales recruit IL-6-producing intraepithelial lymphocytes in the colon to promote barrier integrity. Mucosal Immunol. (2018) 11:357–68. doi: 10.1038/mi.2017.55
71. Taniguchi K, Wu L-W, Grivennikov SI, de Jong PR, Lian I, Yu F-X, et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. (2015) 519:57–62. doi: 10.1038/nature14228
72. Turpin W, Lee S-H, Raygoza Garay JA, Madsen KL, Meddings JB, Bedrani L, et al. Increased intestinal permeability is associated with later development of Crohn's disease. Gastroenterology. (2020) 159:2092–100.e5. doi: 10.1053/j.gastro.2020.08.005
73. Nakatsuka Y, Vandenbon A, Mino T, Yoshinaga M, Uehata T, Cui X, et al. Pulmonary Regnase-1 orchestrates the interplay of epithelium and adaptive immune systems to protect against pneumonia. Mucosal Immunol. (2018) 11:1203–18. doi: 10.1038/s41385-018-0024-5
74. Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels H-H, Ori D, et al. Regnase-1 and roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell. (2015) 161:1058–73. doi: 10.1016/j.cell.2015.04.029
75. Garg AV, Amatya N, Chen K, Cruz JA, Grover P, Whibley N, et al. MCPIP1 endoribonuclease activity negatively regulates interleukin-17-mediated signaling and inflammation. Immunity. (2015) 43:475–87. doi: 10.1016/j.immuni.2015.07.021
76. Howell KJ, Kraiczy J, Nayak KM, Gasparetto M, Ross A, Lee C, et al. DNA methylation and transcription patterns in intestinal epithelial cells from pediatric patients with inflammatory bowel diseases differentiate disease subtypes and associate with outcome. Gastroenterology. (2018) 154:585–98. doi: 10.1053/j.gastro.2017.10.007
77. Muraro D, Parker A, Vaux L, Filippi S, Almet AA, Fletcher AG, et al. Chronic TNFα-driven injury delays cell migration to villi in the intestinal epithelium. J R Soc Interface. (2018) 15:20180037. doi: 10.1098/rsif.2018.0037
78. d'Aldebert E, Quaranta M, Sébert M, Bonnet D, Kirzin S, Portier G, et al. Characterization of human colon organoids from inflammatory bowel disease patients. Front Cell Dev Biol. (2020) 8:363. doi: 10.3389/fcell.2020.00363
79. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MAD, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. (2007) 13:1042–9. doi: 10.1038/nm1638
80. Vogl T, Eisenblätter M, Völler T, Zenker S, Hermann S, van Lent P, et al. Alarmin S100A8/S100A9 as a biomarker for molecular imaging of local inflammatory activity. Nat Commun. (2014) 5:4593. doi: 10.1038/ncomms5593
81. Tang H, Pang S, Wang M, Xiao X, Rong Y, Wang H, et al. TLR4 activation is required for IL-17–induced multiple tissue inflammation and wasting in mice. J Immunol. (2010) 185:2563–9. doi: 10.4049/jimmunol.0903664
82. Dheer R, Santaolalla R, Davies JM, Lang JK, Phillips MC, Pastorini C, et al. Intestinal epithelial toll-like receptor 4 signaling affects epithelial function and colonic microbiota and promotes a risk for transmissible colitis. Infect Immun. (2016) 84:798–810. doi: 10.1128/IAI.01374-15
83. Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. (2013) 182:375–87. doi: 10.1016/j.ajpath.2012.10.014
84. Nighot M, Al-Sadi R, Guo S, Rawat M, Nighot P, Watterson MD, et al. Lipopolysaccharide-induced increase in intestinal epithelial tight permeability is mediated by toll-like receptor 4/myeloid differentiation primary response 88 (MyD88) activation of myosin light chain kinase expression. Am J Pathol. (2017) 187:2698–710. doi: 10.1016/j.ajpath.2017.08.005
85. Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol. (2001) 167:1609–16. doi: 10.4049/jimmunol.167.3.1609
86. Suzuki M, Hisamatsu T, Podolsky DK. Gamma interferon augments the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular toll-like receptor 4-MD-2 complex. Infect Immun. (2003) 71:3503–11. doi: 10.1128/IAI.71.6.3503-3511.2003
87. Xiao YT, Yan WH, Cao Y, Yan JK, Cai W. Neutralization of IL-6 and TNF-α ameliorates intestinal permeability in DSS-induced colitis. Cytokine. (2016) 83:189–92. doi: 10.1016/j.cyto.2016.04.012
88. Yang J-Y, Kim M-S, Kim E, Cheon JH, Lee Y-S, Kim Y, et al. Enteric viruses ameliorate gut inflammation via toll-like receptor 3 and toll-like receptor 7-mediated interferon-β production. Immunity. (2016) 44:889–900. doi: 10.1016/j.immuni.2016.03.009
89. Bhutiani N, Li Q, Anderson CD, Gallagher HC, Jesus MD, Singh R, et al. Enhanced gut barrier integrity sensitizes colon cancer to immune therapy. OncoImmunology. (2018) 7:e1498438. doi: 10.1080/2162402X.2018.1498438
90. Sumagin R, Robin AZ, Nusrat A, Parkos CA. Transmigrated neutrophils in the intestinal lumen engage ICAM-1 to regulate the epithelial barrier and neutrophil recruitment. Mucosal Immunol. (2014) 7:905–15. doi: 10.1038/mi.2013.106
91. Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem. (2011) 286:31263–71. doi: 10.1074/jbc.M111.238147
92. Ye X, Sun M. AGR2 ameliorates tumor necrosis factor-α-induced epithelial barrier dysfunction via suppression of NF-κB p65-mediated MLCK/p-MLC pathway activation. Int J Mol Med. (2017) 39:1206–14. doi: 10.3892/ijmm.2017.2928
93. Su L, Nalle SC, Shen L, Turner ES, Singh G, Breskin LA, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. (2013) 145:407–15. doi: 10.1053/j.gastro.2013.04.011
94. Touhami S, Beguier F, Augustin S, Charles-Messance H, Vignaud L, Nandrot EF, et al. Chronic exposure to tumor necrosis factor alpha induces retinal pigment epithelium cell dedifferentiation. J Neuroinflammation. (2018) 15:85. doi: 10.1186/s12974-018-1106-8
95. Fischer A, Gluth M, Pape UF, Wiedenmann B, Theuring F, Baumgart DC. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. (2013) 304:G970–9. doi: 10.1152/ajpgi.00183.2012
96. Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol. (2002) 97:2000–4. doi: 10.1111/j.1572-0241.2002.05914.x
97. Zeissig S, Bojarski C, Buergel N, Mankertz J, Zeitz M, Fromm M, et al. Downregulation of epithelial apoptosis and barrier repair in active Crohn's disease by tumour necrosis factor alpha antibody treatment. Gut. (2004) 53:1295–302. doi: 10.1136/gut.2003.036632
98. Jeffery V, Goldson AJ, Dainty JR, Chieppa M, Sobolewski A. IL-6 Signaling Regulates Small Intestinal Crypt Homeostasis. J Immunol. (2017) 199:304–11. doi: 10.4049/jimmunol.1600960
Keywords: TNF, TLR4, cytokine, intestinal barrier, inflammation
Citation: Semin I, Ninnemann J, Bondareva M, Gimaev I and Kruglov AA (2021) Interplay Between Microbiota, Toll-Like Receptors and Cytokines for the Maintenance of Epithelial Barrier Integrity. Front. Med. 8:644333. doi: 10.3389/fmed.2021.644333
Received: 20 December 2020; Accepted: 06 May 2021;
Published: 28 May 2021.
Reviewed by:
Hiroshi Nakase, Sapporo Medical University, JapanJoana Inês Almeida, Fundação para a Ciência e Tecnologia, Portugal
Approved by:
Pedro M. Baptista, Health Research Institute of Aragon (IIS Aragon), SpainCopyright © 2021 Semin, Ninnemann, Bondareva, Gimaev and Kruglov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrey A. Kruglov, YW5kcmV5X2tydWdsb2ZmQG1haWwucnU=