 Michael Stürzl
Michael Stürzl Meik Kunz
Meik Kunz Susanne M. Krug
Susanne M. Krug Elisabeth Naschberger1
Elisabeth Naschberger1- 1Division of Molecular and Experimental Surgery, Department of Surgery, Universitätsklinikum Erlangen, Friedrich-Alexander University (FAU) of Erlangen-Nürnberg, Erlangen, Germany
- 2Chair of Medical Informatics, Friedrich-Alexander-University (FAU) of Erlangen-Nürnberg, Erlangen, and Fraunhofer Institute of Toxicology and Experimental Medicine, Hannover, Germany
- 3Clinical Physiology/Nutritional Medicine, Charité-Universitätsmedizin Berlin, Berlin, Germany
Inflammatory bowel disease describes chronic inflammatory disorders. The incidence of the disease is rising. A major step in disease development is the breakdown of the epithelial cell barrier. Numerous blood vessels are directly located underneath this barrier. Diseased tissues are heavily vascularized and blood vessels significantly contribute to disease progression. The gut-vascular barrier (GVB) is an additional barrier controlling the entry of substances into the portal circulation and to the liver after passing the first epithelial barrier. The presence of the GVB rises the question, whether the vascular and endothelial barriers may communicate bi-directionally in the regulation of selective barrier permeability. Communication from epithelial to endothelial cells is well-accepted. In contrast, little is known on the respective backwards communication. Only recently, perfusion-independent angiocrine functions of endothelial cells were recognized in a way that endothelial cells release specific soluble factors that may directly act on the epithelial barrier. This review discusses the putative involvement of angiocrine inter-barrier communication in the pathogenesis of IBD.
Clinical Presentation and Epidemiology of Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) includes inflammatory diseases of the colon and small intestine with Crohn's disease and ulcerative colitis being the major clinical presentations (1). Crohn's disease affects the small intestine and large intestine, as well as the mouth, esophagus, stomach and the anus, whereas ulcerative colitis primarily affects the colon and the rectum (2). Crohn's disease and ulcerative colitis are different diseases, but commonly present with any of the following symptoms: abdominal pain, diarrhea, rectal bleeding, severe internal cramps/muscle spasms in the region of the pelvis and weight loss. In addition, anemia is a common extra-intestinal complication of IBD.
IBD is classically considered as a disease of Westernized countries but has started to rise worldwide in the beginning of the twenty first century (3). The rise is population-dependent and categorized into four different epidemiological stages: first, the Emergence Stage with sporadic cases of IBD observed in developing countries, second, the Acceleration in Incidence Stage with rising incidence and relatively low prevalence in newly industrialized countries, third, the Compounding Prevalence Stage with stable incidence and steeply rising prevalence in countries of the Western world, and forth, the Prevalence Equilibrium Stage, which represents the opposing forces between an aging IBD population and the incidence of IBD. In Germany at present 620,085 persons are suffering from IBD with a predicted rise up to 815,200 patients in 2030. In the U.S. presently 2,489,362 patients are registered and a rise up to 3,544,480 is expected within the next 10 years (4).
IBD is characterized by a chronically relapsing intestinal inflammation that is thought to result from an exaggerated immune response to the commensal microbiota. However, the specific molecular mechanisms driving IBD pathogenesis are still unclear. Many different putative susceptibility genes for IBD are reported but all of these are associated with only low risk and differ in different countries of the world. At present, it is commonly accepted, that cytokines, such as, tumor necrosis factor (TNF), interleukin (IL)-10, transforming growth factor (TGF)-β, IL-6, IL-12, IL-13, IL-17, IL-21, IL-23, interferon (IFN)-γ and C-X-C motif chemokine ligand (CXCL)10, are drivers of the excessive immune response, leading to leukocyte infiltration and mucosal damage. In addition, there is agreement that IBD pathogenesis is closely associated with a loss of intestinal epithelial barrier functions associated with bacterial translocation, likely representing an initiating or early event in the disease (5–10).
Recently, it became evident that the intestinal barrier involves two sequential physical barriers. The first being the epithelial barrier consisting of a single cell layer of epithelial cells and a mucus layer which physically separates the microbiota in the gut lumen and epithelial cells (11). Directly below the epithelial barrier an additional barrier was identified, the gut-vascular barrier (GVB) controlling the entry of substances into the portal circulation and their access to the liver after passage of the first epithelial barrier (12, 13). The discriminative control of nutrient uptake and tight sealing towards potentially pathological microorganisms requires a profound regulation of the barrier permeability.
Structure and Function of the Epithelial Barrier in IBD
The epithelial barrier allows the co-existence of commensal microbiota and mucosal immune cells in the gut. It consists of a physical barrier established by the epithelial cells situated on a basement membrane. Collagen type IV and laminins are the predominant components of the basement membrane (14). The basement membrane is subject of continuous remodeling. Increased remodeling was observed under inflammatory conditions in association with decreased barrier functions (14). At the cellular level barrier functions are established by (i) densely packed microvilli on the apical side of intestinal epithelial cells termed the brush border (15), (ii) tight cell-cell interactions between the epithelial cells, (iii) the cellular resistance to bacterial transcytosis (16), and (iv) specialized epithelial cells, such as mucus-producing goblet cells and anti-microbial peptide secreting Paneth cells (12). Altogether, the epithelium exerts manifold functions, establishing a physical barrier against pathogen invasion and also performing innate immune functions and nutrient uptake (17). Thereby, the preservation of the epithelial integrity is a major aspect in order to preserve homeostasis and to avoid the progress of inflammation in mucosal tissues (18) [for review see: Lopez-Posadas et al. (11)].
At the molecular level the intercellular barrier of the intestinal epithelium is established by apical junction complexes comprised of tight and adherens junctions. Adherens junctions consist of cadherins and nectins and are mainly important for the cell-cell-adhesion (19, 20). Tight junctions are multiprotein-complexes consisting of several transmembrane proteins: tight junction associated MARVEL proteins (TAMP) like occludin, marvelD3 and tricellulin, junctional adhesion molecules (JAM), angulins and the family of claudins, which has in mammalia 27 members that either possess barrier- or channel-forming properties affecting the overall permeability characteristics of the epithelia [for review see Günzel and Fromm (21)]. Adherens junctions as well as tight junctions establish zipper-like structures, sealing the paracellular space within the epithelial cell layer (22). These intercellular junctions are connected to the actin cytoskeleton via cytoplasmic adaptors, such as zonula occludens proteins, and catenins supporting the mechanical strength of the junctions (23–25). Cell activation with molecules that induce permeability causes actin reorganization into stress fibers. This is associated with increasing traction forces, which lead to the detachment of adherens junctions from the cytoskeleton followed by the formation of gaps between adjacent cells (26, 27). Further mechanisms such as the removal of cell-cell interaction molecules from the cell surface by internalization and/or by proteolytic cleavage can regulate the intestinal barrier permeability (11, 28).
The epithelium is constantly renewed without an effect on its tightness. Within this process stem cells at the crypt bottom proliferate and differentiate into the different intestinal epithelial cell subtypes with specialized biological functions (29). Subsequently, most of the differentiated epithelial cells migrate upwards to the villus tip, where aged cells die and are shed into the lumen (30, 31). The tightness of the epithelial layer is maintained by the intercellular junctions during this process (23). During cell shedding, epithelial integrity is maintained in cytoskeleton and membrane trafficking-dependent processes regulating the redistribution of junctional proteins along lateral membranes (32, 33).
Increased epithelial tight junction permeability is a hallmark in the gut of IBD patients (34–38). It is believed that the disruption of intercellular junctions and cytoskeleton rearrangements in the context of infection or inflammation lead to a breakdown of epithelial integrity (39–41). Although a correlation between epithelial barrier permeability and disease activity has been observed in patients with Crohn's disease, the cause of this barrier collapse is still a matter of controversy (42, 43). Experimental animal studies demonstrated that a deficiency of single tight junction proteins is not sufficient to cause pathology due to compensatory mechanisms (44, 45) with the exception of claudin-15 (46). However, agreement exists that inflammation-derived soluble mediators such as IL-6 (47), IL-13 (48, 49), TNF (50), and IFN-γ (51, 52) affect tight junctions and may increase intestinal permeability in experimental colitis models and IBD (53–55). These observations suggested that the epithelial barrier breakdown occurs as a consequence of proinflammatory cytokine stimulation. In contrast, recent studies in IBD patients demonstrated that an increase of epithelial permeability precedes flares of inflammatory bowel pointing towards a causative role of epithelial barrier breakdown in the development of intestinal inflammation (35, 56–58). The latter is supported by reports that a decrease of epithelial permeability by application of vitamin D (59, 60), probiotics (61–63), IL-22-triggered mucus production (64), butyrate (65, 66), or an anti-TNF antibody caused clinical amelioration of chronic colitis (67, 68). Moreover, alternative portals for gut leakiness such as brush border functions and intestinal bacterial endocytosis by epithelial cells have to be considered and may play important pathogenic roles providing putative targets for therapy of inflammatory bowel disease (15). Altogether, these results suggest that the epithelial barrier function is important and its maintenance can counteract the development of inflammatory bowel disease.
The Impact of Blood Vessels on IBD Pathogenesis
Capillaries are located in close proximity to the intestinal epithelial cell barrier (Figure 1A). Blood vessels in adult tissues evolve through sprouting from preexisting vessels, a process termed angiogenesis (69). Angiogenic activity correlates with disease severity in IBD suggesting that blood vessels may contribute to pathogenesis (70–73). Moreover, elevated levels of angiogenic growth factors including vascular endothelial growth factor (VEGF)-A and basic fibroblast growth factor (bFGF), that synergize in angiogenesis activation, have been detected in the inflamed mucosa and in the blood during active IBD (74, 75). However, experimental colitis models provided conflicting results on the contribution of angiogenesis to disease activity. Neutralization of VEGF-A resulted in a decreased vessel density and improvement of the disease in dextran sulfate sodium (DSS)–induced and 2,4,6-trinitrobenzenesulfonic acid (TNBS)–induced colitis (73, 76). In contrast, reduced angiogenic activity induced by deficiency of placental growth factor failed to ameliorate colitis in the same experimental models (77). These results indicated that besides vessel density additional parameters such as vessel quality are of relevance in IBD pathogenesis. In fact, newly formed vessels in IBD tissues are strongly disorganized and leaky as evident by associated edema (78).
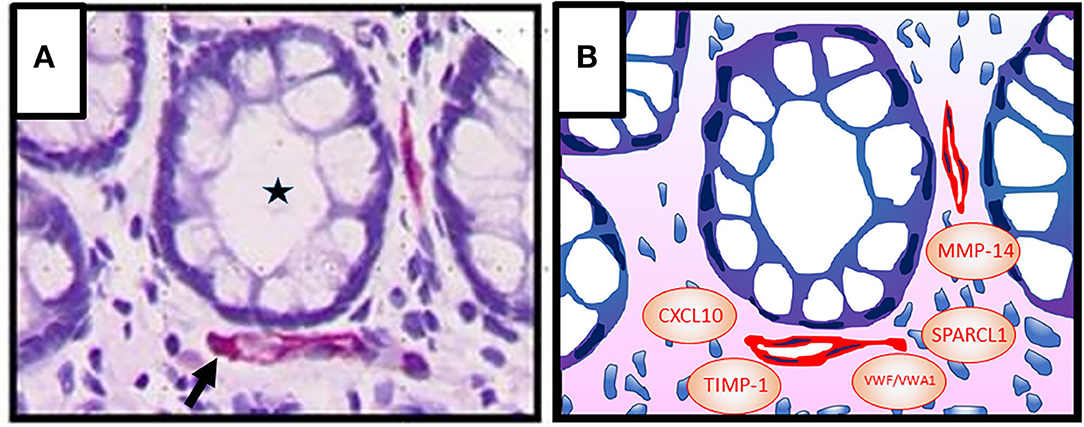
Figure 1. (A) Colonic crypt (intestinal gland, asterisk) with vessels (red, arrow) in the lamina propria. Epithelial cells (1st barrier) and endothelial cells (2nd barrier) are directly adjacent, indicating active inter-barrier communication. Vascular endothelial cells were stained immunohistochemically using an anti-CD31 antibody. Cell nuclei (blue) were stained by haematoxylin. (B) Graphic presentation of (A) indicating possible factors that may be involved in angiocrine regulation of epithelial barrier functions in IBD [von Willebrand factor A domain containing 1 (VWA1), von Willebrand factor (VWF), matrix metalloproteinase (MMP)-14, tissue inhibitor of metalloproteinases (TIMP)-1, C-X-C motif chemokine ligand (CXCL) 10, secreted protein, acidic and rich in cysteine–like 1 (SPARCL1)].
The difficulties in determining the precise role of blood vessel function in IBD may be due to the fact that the intestinal endothelial cells are both, targets and regulators of inflammation (78). In this framework, IBD-associated inflammatory cytokines such as TNF-α, IL-1β and IFN-γ can activate endothelial cells by inducing the expression of adhesion molecules for leukocytes such as E-selectin, intercellular adhesion molecule (ICAM)-1 or vascular cell adhesion molecule (VCAM)-1 (79). Macrophages are important drivers of IBD and are characteristically expressing high amounts of TNF-α and IL-1β, which may amplify the extravasation of these cells being responsible for the high numbers of macrophages present in IBD tissues (80). In addition, inflammation is associated with increased angiogenesis supporting immune cell recruitment by increase of blood flow and endothelial surface (81). As mentioned above the intestinal endothelium also establishes an additional barrier in the gut, the GVB (12, 13). The GVB constitutes a semipermeable barrier between the blood stream and the interstitium regulating the transport of nutrients, tissue fluid homeostasis and the transmigration of immune cells but is non-permissive to bacterial penetration (13, 28, 78, 82). The latter is in agreement with the observation that bacterial lipopolysaccharides (LPS) in low concentrations are stabilizing the vascular barrier (83). In contrast, high concentrations of LPS (>10 μg/ml) inhibit endothelial cell migration, down-regulate intercellular junction molecules and increase the permeability of the vascular barrier (83). Paracellular (i.e., in between the cells) or transcellular (i.e., across the cells) routes are available to cross the endothelial cell monolayer. Transcellular exchange is accomplished via either solute transporters, or transcytosis via vesicular carriers (e.g., caveolae), or pore-like subcellular structures (i.e., fenestrae and transendothelial channels) (84, 85). The paracellular route is controlled by adherens junctions and tight junction proteins similar as in the epithelial barrier. In intestinal endothelial cells, tight junctions are composed mainly of occludin, junctional adhesion molecule (JAM)-A, zonula occludens (ZO)-1, and cingulin (13). Claudin-3, -5, and -12 from the claudin family are known to be mainly expressed in endothelia (86, 87).
Adherens junctions are formed by vascular endothelial (VE)-cadherin and β-catenin (13). Of note, the same cytokines regulating immune cell extravasation can also deregulate adherens and tight junction formation in endothelial cells supporting translocation of bacteria thereby further amplifying the inflammatory process [for review see: Lopez-Posadas et al. (11)].
The impact of the GVB in intestinal inflammation is substantiated by mouse models of acute and chronic DSS-colitis. In these models intestinal vessel perfusion remained constant during colitis whereas vessel permeability strongly increased (5). Using experimental animal models with an endothelial cell specific knockout of the interferon-γ-receptor 2 (IFNγR2) it was shown that the IBD-associated cytokine IFN-γ induces a breakdown of the vascular barrier based on the disruption of the adherens junction protein VE-cadherin and this was significantly increasing DSS-induced experimental colitis. Importantly, the disease-associated vascular barrier dysfunction could be confirmed in human IBD patients indicating the clinical relevance of the findings. Imatinib (brand name Gleevec) is a kinase inhibitor acting against Abelson tyrosine kinase BCR–ABL, the KIT and PDGF receptors and is used for therapy of chronic myeloid leukemia (CML), gastrointestinal stromal tumors (GIST) and several other malignancies (88). Interestingly, treatment with imatinib restored adherens junctions, inhibited vascular permeability, and significantly reduced colonic inflammation in experimental colitis. Altogether, these results highlighted the pathogenic impact of inflammation–associated vascular barrier defects in IBD and opens new avenues for vascular-directed treatment of the disease (81).
The detection of an additional intestinal barrier rises the question whether the epithelial and the vascular barriers may communicate in prevention or progression of the disease. Epithelial to endothelial cell communication is commonly accepted. For example, the nutrient composition of the chyme (partially digested food) and not simply gut distension modulates blood flow. Specialized subsets of intestinal epithelial cells transport nutrients through the epithelial monolayer into the lamina propria from where they are transported through the fenestrated blood endothelium to be distributed systemically (89, 90). Moreover, in response to pathogen invasion or loss of barrier integrity, both intestinal epithelial cells and tissue-resident leukocytes secrete cytokines, chemokines, reactive oxygen species, and lipid mediators that activate endothelial cells to modulate the number and structure of vessels and to promote immune cell extravasation. For example, intestinal epithelial cells in IBD were shown to secret the chemokines CXCL8/IL-8 and CCL20 (91, 92), both of which can activate angiogenesis (93, 94). In addition, these cells secrete the cytokine TNF-α (91), which regulates vessel remodeling and by directly acting on endothelial cells may inhibit angiogenesis (95, 96). In addition, vascular permeability is increased by inflammatory mediators released from epithelial cells fostering both, inter- and trans-cellular diapedesis (90, 97).
Angiocrine Functions of Blood Vessels in Organ Development and Diseases
The endothelium is not a passive response organ for nutrient supply, tissue entry of immune cells, and metabolite removal, but actively regulates the tissue microenvironment in organ development and diseases as indicated by novel results. These perfusion-independent functions of endothelial cells were recognized in experimental tumor models in mice for the first time, where the inhibition of angiogenesis in certain instances did not abrogate tumor growth but instead enhanced tumor invasiveness (98). Based on this the hypothesis arose that endothelial cells release specific soluble factors that may directly regulate tumor growth in a perfusion-independent manner. This respective mechanism was termed as “angiocrine” regulation of tumorigenesis (98).
Subsequent studies confirmed that endothelial cells may activate tumorigenesis by secreted factors (98, 99). For example, angiocrine factors were reported to stimulate growth and migration of lymphoma tumor cells (100), to maintain stem cell like properties in colorectal carcinoma and glioblastoma cells (101–103), to inhibit anoikis in head and neck cancer stem cells (104) and, to activate proliferation, survival and epithelial to mesenchymal transition of lung carcinoma cells (103) [for review see: Lee et al. (105)].
Vice versa, it was noted that endothelial cells can also suppress cancer growth through angiocrine signaling. In this framework contact-dependent interactions between the endothelial cell surface receptor duffy antigen/receptor for chemokines and the carcinoma cell surface receptor kang ai-1 were shown to suppress metastasis (106). In addition, in breast cancer endothelial cell-released slit homolog 2 protein (Slit 2), perlecan and additional as yet unknown factors were reported to inhibit proliferation, invasion and pro-tumorigenic signaling of the cancer cells (107, 108). In addition, thrombospondin is regarded as a putative anti-angiogenic factor secreted from endothelial cells (98). Angiocrine factors also exert key functions in physiologic condition such as kidney development (109, 110), liver bud (111) and pancreatic bud formation (112), in neuronal development (113), lung regeneration (114), osteogenesis (115) and hematopoiesis (113).
Of note, a specific impact of angiocrine signaling on epithelial barrier functions was observed in retina development (116). Endothelial cells secrete factors that remodel the retinal pigment epithelium (RPE) basement membrane and integrin receptors sense these changes by triggering GTPase signals that modulate RPE tight junctions and enhance RPE barrier function (116). Similar parenchymal cell barrier regulatory mechanisms may be active in other organs.
Altogether, angiocrine factors are involved in tumorigenic, homeostatic, regenerative and morphogenetic processes in a paracrine or juxtacrine manner. The term “angiocrine” factors meanwhile includes secreted and membrane-bound inhibitory or stimulatory growth factors, trophogens, chemokines, cytokines, extracellular matrix components, exosomes and other cellular products (117). The angiocrine profile of endothelial cells can differ between tissues, reflecting the diversity of cell types found adjacent to endothelial cells in organs (113, 117).
The Impact of Angiocrine Signaling on Epithelial Barrier Function in IBD
Angiocrine functions in IBD have not been investigated extensively as yet, despite the manifold effects of angiocrine signaling on epithelial cell functions in cancer, organ development and tissue regeneration. However, first results indicating angiocrine activities in the colon have emerged. For example, endothelial cells release jagged 1, generated by proteolytic activity of ADAM metallopeptidase domain 17 (ADAM17) activating Notch in human colorectal cancer cells and thereby promoting a cancer stem cell phenotype and chemo-resistance (103, 118). Moreover, it was shown that selectively endothelial cells isolated from colorectal carcinomas with a prognostically favorable Th-1-like immune environment released the matricellular protein secreted protein, acidic and rich in cysteine–like 1 (SPARCL1), which autocrinely and paracrinely inhibited angiogenesis and proliferation of different cancer cell lines (119, 120). The latter indicated that angiocrine activities in the colon may trigger the course of diseases in a microenvironment–dependent manner. A recent single cell RNAseq approach of intestinal cells and subsequent bioinformatics interaction analyses supported the molecular interaction between endothelial cells and epithelial cells in the colon (121).
Specific support for angiocrine functions in IBD was obtained from a recent report on an increased susceptibility for acute and chronic DSS-induced colonic inflammation in mice lacking the angiocrinely active SPARCL1 protein (122). SPARCL1 is almost exclusively expressed in vascular cells in the colon (119, 123, 124). In SPARCL1 (Sc1) KO animals colonic inflammation and colon vessel permeability were significantly increased and colon length was shorter as compared to wildtype animals. Exaggerated inflammation in Sc1 KO animals was further supported by an increased detection of fibrosis and the presence of tertiary lymphoid structures similar to the human chronic disease. Altogether, these results indicated that intestinal angiocrine functions may establish a chemical barrier affecting both, epithelial and endothelial cell barrier functions in IBD (122).
In a next step, we applied a meta-analysis to further investigate whether angiocrine signaling may impact barrier functions. To this goal, an in silico secretome screening against the human proteome was performed using the VerSeDa database [Vertebrate Secretome Database (125)]. Transcripts with a prediction cut-off value > 0.8 (SignalP 4.1, TargetP 1.1, SecretomeP) were considered as secreted proteins. The resulting 1,050 genes (1,959 proteins; 1,959 gene transcripts) were used for a functional gene and phenotype annotation using the Ensembl BioMart database (http://www.ensembl.org/index.html). Next, candidates were selected based on data mining (inflammatory, angiocrine, epithel, extracellular, endothelial, barrier, cytokine, bowel, secreted). Subsequently, the resulting 257 genes were mapped to profiles from human endothelial cells of different origin, including human umbilical vein endothelial cells (HUVEC) exposed to shear stress (126), under LPS-stimulation (127), overexpressing γ-interferon-inducible protein (IFI) 16 (128) and unstimulated (129), as well as endothelial cells from brain, lung, heart (130) and colorectal carcinoma (119, 131). This analysis identified in total 28 genes (Table 1). Six of these may be of specific interest as candidates of angiocrine barrier effects in IBD (Figure 1B). This includes components of the von Willebrand factor domain superfamily (VWA1, VWF) and tissue inhibitor of metalloproteinases (TIMP)-1, which were retrieved from three different studies, respectively. vWF is a classical endothelial cell marker protein, that promotes adhesion of platelets to the sites of vascular injury by forming a molecular bridge between sub-endothelial collagen matrix and the platelet-surface receptor complex (132). Its impact on the epithelial barrier warrants further investigation. TIMP-1 is an inhibitor of the matrix metalloproteinases (MMPs). It is able to promote cell proliferation in a wide range of cell types, has an anti-apoptotic function and can modulate the vascular barrier (133, 134). TIMP-1 may impact the epithelial cell barrier activity in the gut through these activities. In this framework, it is interesting that MMP-14 was also identified by our meta-analyses as angiocrine mediator. MMP-14 was reported as an angiocrine factor in lung regeneration and as a member of the membrane-type matrix metalloproteinases that are not inhibited by TIMP-1 (114, 135). In addition, CXCL10, regarded as a major driver in IBD pathogenesis (6), was also identified as angiocrine mediator in our meta analyses. In the DSS-model blockade of CXCL10 enhanced crypt cell survival (136) and mice with a knock out of the CXCL10 receptor CXCR3 showed considerably lower crypt damage (137). Based on these findings it was suggested that CXCL10 may exert direct effects on epithelial cells in the gut (138).
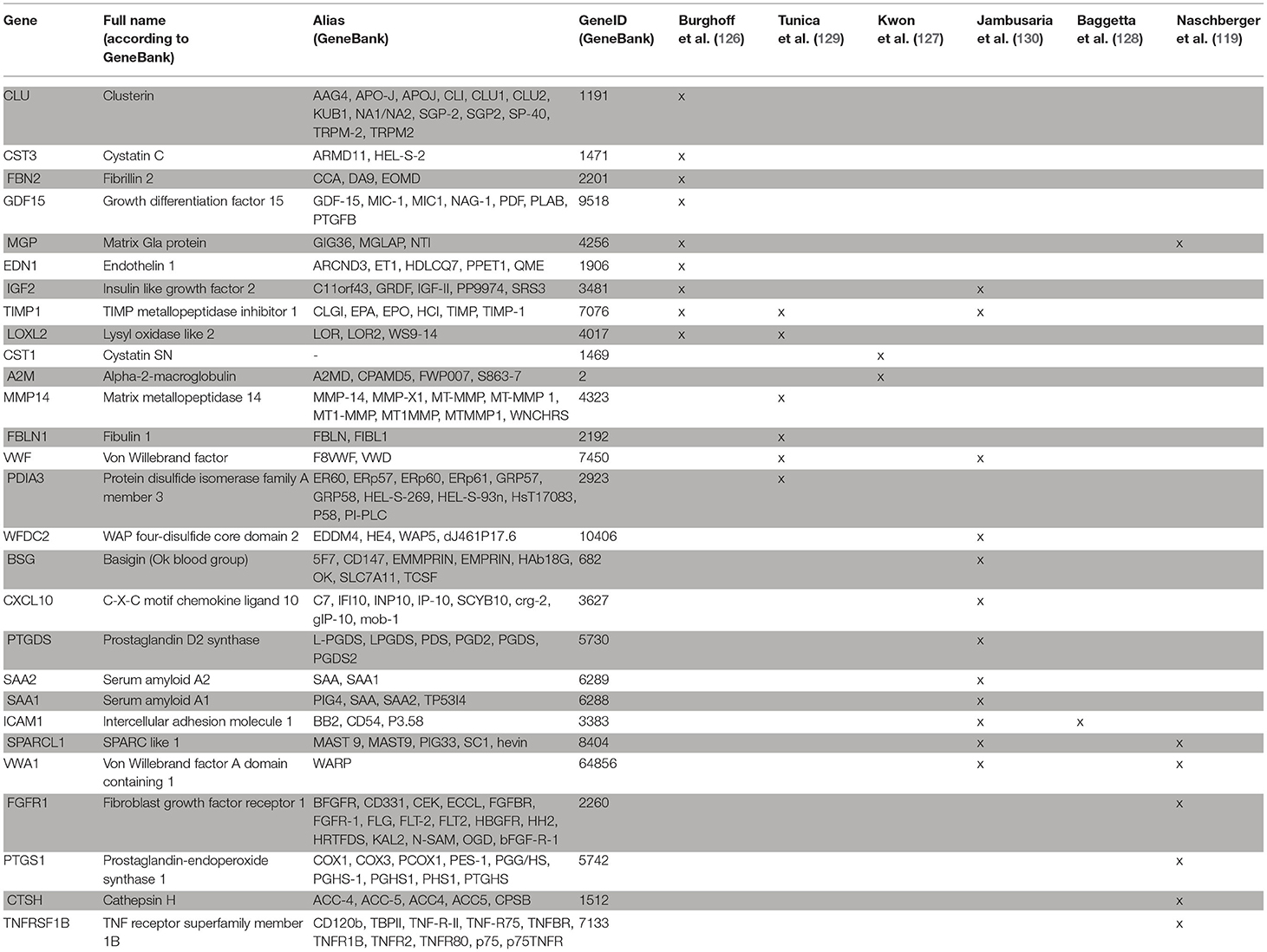
Table 1. Angiocrine barrier-modulating candidate genes in inflammatory bowel disease.
The bioinformatical analysis showed that the overlap of genes retrieved from the different studies was low. This is well in agreement with the high variation of activation and organ-dependent plasticity of endothelial cells. In this framework, the six genes identified in endothelial cells from colorectal carcinoma may exhibit the highest relevance for IBD (see Table 1). Interestingly, SPARCL1, which has been shown to affect susceptibility to experimental colitis in mice was part of this group (122). In summary, this analysis identified several interesting candidates, which may participate in the angiocrine inter-barrier communication in IBD. These factors may provide putative new targets for treatment of the disease. The specific impact of most of these factors on the epithelial barrier functions has to be determined in future studies.
Conclusion
First evidence exists that the gut-vascular barrier (GVB) communicates via angiocrine signals with the epithelial barrier during IBD. The molecules involved in this communication may provide new targets for clinical monitoring and treatment of the disease. In-depth elucidation of the underlying effects and the specific mechanisms warrants further studies.
Author Contributions
MS, SK, and EN: analyzed the literature and wrote the manuscript. MK performed the bioinformatical analysis. All authors approved the final version of the manuscript.
Funding
The work of the authors was supported by grants from the German Research Foundation (DFG) FOR 2438 (subproject 2 to EN and MS); SFB/TRR 241 (subproject A06 to MS and B06 to SK), STU 238/10-1 (to MS), TRR 305 (subproject B08 to EN); the Interdisciplinary Center for Clinical Research (IZKF) of the Clinical Center Erlangen (to MS); the Programm zur Förderung von Corona-Forschungsprojekten, StMWK, München (to MS); W. Lutz Stiftung (to MS); the Forschungsstiftung Medizin am Universitätsklinikum Erlangen (to MS); the German Federal Ministry of Education and Research (BMBF) Era-Net grant 01KT1801 (to MK); and the CompLS program grant 031L0262C (to MK).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all colleagues cited in this review for their inspiring work and we apologize at those colleagues whose work was not integrated into this manuscript. We also thank the reviewers for their very thoughtful and helpful comments.
References
1. Rutgeerts P, Vermeire S, Van Assche G. Biological therapies for inflammatory bowel diseases. Gastroenterology. (2009) 136:1182–97. doi: 10.1053/j.gastro.2009.02.001
2. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. (2007) 448:427–34. doi: 10.1038/nature06005
3. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. (2018) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
4. Kaplan GG, Windsor JW. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. (2020) 18:56–66. doi: 10.1038/s41575-020-00360-x
5. Haep L, Britzen-Laurent N, Weber TG, Naschberger E, Schaefer A, Kremmer E, et al. Interferon gamma counteracts the angiogenic switch and induces vascular permeability in dextran sulfate sodium colitis in mice. Inflamm Bowel Dis. (2015) 21:2360–71. doi: 10.1097/MIB.0000000000000490
6. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. (2014) 14:329–42. doi: 10.1038/nri3661
7. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. (2014) 14:141–53. doi: 10.1038/nri3608
8. Uhlig HH, Powrie F. Dendritic cells and the intestinal bacterial flora: a role for localized mucosal immune responses. J Clin Invest. (2003) 112:648–51. doi: 10.1172/JCI19545
9. Yamamoto-Furusho JK. Inflammatory bowel disease therapy: blockade of cytokines and cytokine signaling pathways. Curr Opin Gastroenterol. (2018) 34:187–93. doi: 10.1097/MOG.0000000000000444
10. Pai YC, Weng LT, Wei SC, Wu LL, Shih DQ, Targan SR, et al. Gut microbial transcytosis induced by tumor necrosis factor-like 1A-dependent activation of a myosin light chain kinase splice variant contributes to IBD. J Crohns Colitis. (2020) 15:258–72. doi: 10.1093/ecco-jcc/jjaa165
11. Lopez-Posadas R, Stürzl M, Atreya I, Neurath MF, Britzen-Laurent N. Interplay of GTPases and cytoskeleton in cellular barrier defects during gut inflammation. Front Immunol. (2017) 8:1240. doi: 10.3389/fimmu.2017.01240
12. Spadoni I, Fornasa G, Rescigno M. Organ-specific protection mediated by cooperation between vascular and epithelial barriers. Nat Rev Immunol. (2017) 17:761–73. doi: 10.1038/nri.2017.100
13. Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science. (2015) 350:830–4. doi: 10.1126/science.aad0135
14. Lindholm M, Manon-Jensen T, Madsen GI, Krag A, Karsdal MA, Kjeldsen J, et al. Extracellular matrix fragments of the basement membrane and the interstitial matrix are serological markers of intestinal tissue remodeling and disease activity in dextran sulfate sodium colitis. Dig Dis Sci. (2019) 64:3134–42. doi: 10.1007/s10620-019-05676-6
15. Yu LC. Commensal bacterial internalization by epithelial cells: an alternative portal for gut leakiness. Tissue Barriers. (2015) 3:e1008895. doi: 10.1080/21688370.2015.1008895
16. Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. (2002) 122:44–54. doi: 10.1053/gast.2002.30294
17. Zhang K, Hornef MW, Dupont A. The intestinal epithelium as guardian of gut barrier integrity. Cell Microbiol. (2015) 17:1561–9. doi: 10.1111/cmi.12501
18. Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. (2013) 4:280. doi: 10.3389/fimmu.2013.00280
19. Gates J, Peifer M. Can 1000 reviews be wrong? Actin, alpha-Catenin, and adherens junctions. Cell. (2005) 123:769–72. doi: 10.1016/j.cell.2005.11.009
20. Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. (2003) 112:535–48. doi: 10.1016/S0092-8674(03)00108-9
21. Gunzel D, Fromm M. Claudins and other tight junction proteins. Compr Physiol. (2012) 2:1819–52. doi: 10.1002/cphy.c110045
22. Fries W, Belvedere A, Vetrano S. Sealing the broken barrier in IBD: intestinal permeability, epithelial cells and junctions. Curr Drug Targets. (2013) 14:1460–70. doi: 10.2174/1389450111314120011
23. Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. (2007) 127:2525–32. doi: 10.1038/sj.jid.5700865
24. Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. (2010) 11:502–14. doi: 10.1038/nrm2927
25. Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. (2008) 1778:660–9. doi: 10.1016/j.bbamem.2007.07.012
26. Mangold S, Wu SK, Norwood SJ, Collins BM, Hamilton NA, Thorn P, et al. Hepatocyte growth factor acutely perturbs actin filament anchorage at the epithelial zonula adherens. Curr Biol. (2011) 21:503–7. doi: 10.1016/j.cub.2011.02.018
27. Prasain N, Alexeyev M, Balczon R, Stevens T. Soluble adenylyl cyclase-dependent microtubule disassembly reveals a novel mechanism of endothelial cell retraction. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L73–83. doi: 10.1152/ajplung.90577.2008
28. Komarova YA, Kruse K, Mehta D, Malik AB. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ Res. (2017) 120:179–206. doi: 10.1161/CIRCRESAHA.116.306534
29. van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. (2009) 71:241–60. doi: 10.1146/annurev.physiol.010908.163145
30. Günther C, Neumann H, Neurath MF, Becker C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. (2013) 62:1062–71. doi: 10.1136/gutjnl-2011-301364
31. Watson AJ, Duckworth CA, Guan Y, Montrose MH. Mechanisms of epithelial cell shedding in the mammalian intestine and maintenance of barrier function. Ann N Y Acad Sci. (2009) 1165:135–42. doi: 10.1111/j.1749-6632.2009.04027.x
32. Guan Y, Watson AJ, Marchiando AM, Bradford E, Shen L, Turner JR, et al. Redistribution of the tight junction protein ZO-1 during physiological shedding of mouse intestinal epithelial cells. Am J Physiol Cell Physiol. (2011) 300:C1404–14. doi: 10.1152/ajpcell.00270.2010
33. Marchiando AM, Shen L, Graham WV, Edelblum KL, Duckworth CA, Guan Y, et al. The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology. (2011) 140:1208–18.e1–2. doi: 10.1053/j.gastro.2011.01.004
34. Gitter AH, Wullstein F, Fromm M, Schulzke JD. Epithelial barrier defects in ulcerative colitis: characterization and quantification by electrophysiological imaging. Gastroenterology. (2001) 121:1320–8. doi: 10.1053/gast.2001.29694
35. Hollander D. Permeability in Crohn's disease: altered barrier functions in healthy relatives? Gastroenterology. (1993) 104:1848–51. doi: 10.1016/0016-5085(93)90668-3
36. Mankertz J, Schulzke JD. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr Opin Gastroenterol. (2007) 23:379–83. doi: 10.1097/MOG.0b013e32816aa392
37. Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ, et al. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. (1999) 116:301–9. doi: 10.1016/S0016-5085(99)70126-5
38. Ukabam SO, Clamp JR, Cooper BT. Abnormal small intestinal permeability to sugars in patients with crohn's disease of the terminal ileum and colon. Digestion. (1983) 27:70–4. doi: 10.1159/000198932
39. Betanzos A, Javier-Reyna R, Garcia-Rivera G, Banuelos C, Gonzalez-Mariscal L, Schnoor M, et al. The ehCPADH112 complex of Entamoeba histolytica interacts with tight junction proteins occludin and claudin-1 to produce epithelial damage. PLoS ONE. (2013) 8:e65100. doi: 10.1371/journal.pone.0065100
40. Nusrat A, von Eichel-Streiber C, Turner JR, Verkade P, Madara JL, Parkos CA. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect Immun. (2001) 69:1329–36. doi: 10.1128/IAI.69.3.1329-1336.2001
41. Shifflett DE, Clayburgh DR, Koutsouris A, Turner JR, Hecht GA. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab Invest. (2005) 85:1308–24. doi: 10.1038/labinvest.3700330
42. D'Inca R, Di Leo V, Corrao G, Martines D, D'Odorico A, Mestriner C, et al. Intestinal permeability test as a predictor of clinical course in Crohn's disease. Am J Gastroenterol. (1999) 94:2956–60. doi: 10.1111/j.1572-0241.1999.01444.x
43. Yacyshyn BR, Meddings JB. CD45RO expression on circulating CD19+ b cells in Crohn's disease correlates with intestinal permeability. Gastroenterology. (1995) 108:132–7. doi: 10.1016/0016-5085(95)90017-9
44. Khounlotham M, Kim W, Peatman E, Nava P, Medina-Contreras O, Addis C, et al. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity. (2012) 37:563–73. doi: 10.1016/j.immuni.2012.06.017
45. Pope JL, Bhat AA, Sharma A, Ahmad R, Krishnan M, Washington MK, et al. Claudin-1 regulates intestinal epithelial homeostasis through the modulation of notch-signalling. Gut. (2014) 63:622–34. doi: 10.1136/gutjnl-2012-304241
46. Tamura A, Kitano Y, Hata M, Katsuno T, Moriwaki K, Sasaki H, et al. Megaintestine in claudin-15-deficient mice. Gastroenterology. (2008) 134:523–34. doi: 10.1053/j.gastro.2007.11.040
47. Al-Sadi R, Ye D, Boivin M, Guo S, Hashimi M, Ereifej L, et al. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS ONE. (2014) 9:e85345. doi: 10.1371/journal.pone.0085345
48. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. (2005) 129:550–64. doi: 10.1053/j.gastro.2005.05.002
49. Kawashima R, Kawamura YI, Oshio T, Son A, Yamazaki M, Hagiwara T, et al. Interleukin-13 damages intestinal mucosa via TWEAK and Fn14 in mice-a pathway associated with ulcerative colitis. Gastroenterology. (2011) 141:2119–29.e8. doi: 10.1053/j.gastro.2011.08.040
50. Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, et al. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. (2004) 286:G367–76. doi: 10.1152/ajpgi.00173.2003
51. Bardenbacher M, Ruder B, Britzen-Laurent N, Naschberger E, Becker C, Palmisano R, et al. Investigating intestinal barrier breakdown in living organoids. J Vis Exp. (2020) 157:1–9. doi: 10.3791/60546
52. Bardenbacher M, Ruder B, Britzen-Laurent N, Schmid B, Waldner M, Naschberger E, et al. Permeability analyses and three dimensional imaging of interferon gamma-induced barrier disintegration in intestinal organoids. Stem Cell Res. (2019) 35:101383. doi: 10.1016/j.scr.2019.101383
53. Blum MS, Toninelli E, Anderson JM, Balda MS, Zhou J, O'Donnell L, et al. Cytoskeletal rearrangement mediates human microvascular endothelial tight junction modulation by cytokines. Am J Physiol. (1997) 273:H286–94. doi: 10.1152/ajpheart.1997.273.1.H286
54. Capaldo CT, Farkas AE, Hilgarth RS, Krug SM, Wolf MF, Benedik JK, et al. Proinflammatory cytokine-induced tight junction remodeling through dynamic self-assembly of claudins. Mol Biol Cell. (2014) 25:2710–9. doi: 10.1091/mbc.e14-02-0773
55. Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. (2009) 1788:864–71. doi: 10.1016/j.bbamem.2008.08.027
56. Kiesslich R, Duckworth CA, Moussata D, Gloeckner A, Lim LG, Goetz M, et al. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut. (2012) 61:1146–53. doi: 10.1136/gutjnl-2011-300695
57. Kiesslich R, Goetz M, Angus EM, Hu Q, Guan Y, Potten C, et al. Identification of epithelial gaps in human small and large intestine by confocal endomicroscopy. Gastroenterology. (2007) 133:1769–78. doi: 10.1053/j.gastro.2007.09.011
58. Lim LG, Neumann J, Hansen T, Goetz M, Hoffman A, Neurath MF, et al. Confocal endomicroscopy identifies loss of local barrier function in the duodenum of patients with Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. (2014) 20:892–900. doi: 10.1097/MIB.0000000000000027
59. Martinesi M, Ambrosini S, Treves C, Zuegel U, Steinmeyer A, Vito A, et al. Role of vitamin D derivatives in intestinal tissue of patients with inflammatory bowel diseases. J Crohns Colitis. (2014) 8:1062–71. doi: 10.1016/j.crohns.2014.02.005
60. Zhao H, Zhang H, Wu H, Li H, Liu L, Guo J, et al. Protective role of 1,25(OH)2 vitamin D3 in the mucosal injury and epithelial barrier disruption in DSS-induced acute colitis in mice. BMC Gastroenterol. (2012) 12:57. doi: 10.1186/1471-230X-12-57
61. Corridoni D, Pastorelli L, Mattioli B, Locovei S, Ishikawa D, Arseneau KO, et al. Probiotic bacteria regulate intestinal epithelial permeability in experimental ileitis by a TNF-dependent mechanism. PLoS ONE. (2012) 7:e42067. doi: 10.1371/journal.pone.0042067
62. Mennigen R, Nolte K, Rijcken E, Utech M, Loeffler B, Senninger N, et al. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol. (2009) 296:G1140–9. doi: 10.1152/ajpgi.90534.2008
63. Pagnini C, Saeed R, Bamias G, Arseneau KO, Pizarro TT, Cominelli F. Probiotics promote gut health through stimulation of epithelial innate immunity. Proc Natl Acad Sci USA. (2010) 107:454–9. doi: 10.1073/pnas.0910307107
64. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. (2008) 118:534–44. doi: 10.1172/JCI33194
65. Steinhart AH, Hiruki T, Brzezinski A, Baker JP. Treatment of left-sided ulcerative colitis with butyrate enemas: a controlled trial. Aliment Pharmacol Ther. (1996) 10:729–36. doi: 10.1046/j.1365-2036.1996.d01-509.x
66. Vernia P, Annese V, Bresci G, d'Albasio G, D'Inca R, Giaccari S, et al. Topical butyrate improves efficacy of 5-ASA in refractory distal ulcerative colitis: results of a multicentre trial. Eur J Clin Invest. (2003) 33:244–8. doi: 10.1046/j.1365-2362.2003.01130.x
67. Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol. (2002) 97:2000–4. doi: 10.1111/j.1572-0241.2002.05914.x
68. Suenaert P, Bulteel V, Vermeire S, Noman M, Van Assche G, Rutgeerts P. Hyperresponsiveness of the mucosal barrier in Crohn's disease is not tumor necrosis factor-dependent. Inflamm Bowel Dis. (2005) 11:667–73. doi: 10.1097/01.MIB.0000168371.87283.4b
69. Cavallo T, Sade R, Folkman J, Cotran RS. Ultrastructural autoradiographic studies of the early vasoproliferative response in tumor angiogenesis. Am J Pathol. (1973) 70:345–62.
70. Alkim C, Alkim H, Koksal AR, Boga S, Sen I. Angiogenesis in inflammatory bowel disease. Int J Inflam. (2015) 2015:970890. doi: 10.1155/2015/970890
71. Danese S. VEGF in inflammatory bowel disease: a master regulator of mucosal immune-driven angiogenesis. Dig Liver Dis. (2008) 40:680–3. doi: 10.1016/j.dld.2008.02.036
72. Danese S, Sans M, de la Motte C, Graziani C, West G, Phillips MH, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology. (2006) 130:2060–73. doi: 10.1053/j.gastro.2006.03.054
73. Scaldaferri F, Vetrano S, Sans M, Arena V, Straface G, Stigliano E, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology. (2009) 136:585–95e5. doi: 10.1053/j.gastro.2008.09.064
74. Cornali E, Zietz C, Benelli R, Weninger W, Masiello L, Breier G, et al. Vascular endothelial growth factor regulates angiogenesis and vascular permeability in Kaposi's sarcoma. Am J Pathol. (1996) 149:1851–69.
75. Di Sabatino A, Ciccocioppo R, Armellini E, Morera R, Ricevuti L, Cazzola P, et al. Serum bFGF and vEGF correlate respectively with bowel wall thickness and intramural blood flow in Crohn's disease. Inflamm Bowel Dis. (2004) 10:573–7. doi: 10.1097/00054725-200409000-00011
76. Cromer WE, Ganta CV, Patel M, Traylor J, Kevil CG, Alexander JS, et al. VEGF-A isoform modulation in an preclinical TNBS model of ulcerative colitis: protective effects of a VEGF164b therapy. J Transl Med. (2013) 11:207. doi: 10.1186/1479-5876-11-207
77. Hindryckx P, Waeytens A, Laukens D, Peeters H, Van Huysse J, Ferdinande L, et al. Absence of placental growth factor blocks dextran sodium sulfate-induced colonic mucosal angiogenesis, increases mucosal hypoxia and aggravates acute colonic injury. Lab Invest. (2010) 90:566–76. doi: 10.1038/labinvest.2010.37
78. Cromer WE, Mathis JM, Granger DN, Chaitanya GV, Alexander JS. Role of the endothelium in inflammatory bowel diseases. World J Gastroenterol. (2011) 17:578–93. doi: 10.3748/wjg.v17.i5.578
79. Pober JS Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15. doi: 10.1038/nri2171
80. Ruder B, Becker C. At the forefront of the mucosal barrier: the role of macrophages in the intestine. Cells. (2020) 9:2162. doi: 10.3390/cells9102162
81. Langer V, Vivi E, Regensburger D, Winkler TH, Waldner MJ, Rath T, et al. IFN-gamma drives inflammatory bowel disease pathogenesis through VE-cadherin-directed vascular barrier disruption. J Clin Invest. (2019) 129:4691–707. doi: 10.1172/JCI124884
82. Habtezion A, Nguyen LP, Hadeiba H, Butcher EC. Leukocyte trafficking to the small intestine and colon. Gastroenterology. (2016) 150:340–54. doi: 10.1053/j.gastro.2015.10.046
83. Zheng X, Zhang W, Hu X. Different concentrations of lipopolysaccharide regulate barrier function through the PI3K/Akt signalling pathway in human pulmonary microvascular endothelial cells. Sci Rep. (2018) 8:9963. doi: 10.1038/s41598-018-28089-3
84. Aird WC. Phenotypic heterogeneity of the endothelium: i. Structure, function, and mechanisms. Circ Res. (2007) 100:158–73. doi: 10.1161/01.RES.0000255691.76142.4a
85. Tse D, Stan RV. Morphological heterogeneity of endothelium. Semin Thromb Hemost. (2010) 36:236–45. doi: 10.1055/s-0030-1253447
86. Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, et al. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. (2003) 105:586–92. doi: 10.1007/s00401-003-0688-z
87. Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. (1999) 147:185–94. doi: 10.1083/jcb.147.1.185
88. Cohen P, Cross D, Janne PA. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov. (2021) 20(7):551–69. doi: 10.1038/s41573-021-00195-4
89. Stan RV, Tse D, Deharvengt SJ, Smits NC, Xu Y, Luciano MR, et al. The diaphragms of fenestrated endothelia: gatekeepers of vascular permeability and blood composition. Developmental Cell. (2012) 23:1203–18. doi: 10.1016/j.devcel.2012.11.003
90. Gentile ME, King IL. Blood and guts: the intestinal vasculature during health and helminth infection. PLoS Pathog. (2018) 14:e1007045. doi: 10.1371/journal.ppat.1007045
91. Ferrari D, Cimino F, Fratantonio D, Molonia MS, Bashllari R, Busa R, et al. Cyanidin-3-O-Glucoside modulates the in vitro inflammatory crosstalk between intestinal epithelial and endothelial cells. Mediators Inflamm. (2017) 2017:3454023. doi: 10.1155/2017/3454023
92. Franze E, Marafini I, De Simone V, Monteleone I, Caprioli F, Colantoni A, et al. Interleukin-34 induces cc-chemokine ligand 20 in gut epithelial cells. J Crohns Colitis. (2016) 10:87–94. doi: 10.1093/ecco-jcc/jjv181
93. Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. (2003) 170:3369–76. doi: 10.4049/jimmunol.170.6.3369
94. Benkheil M, Van Haele M, Roskams T, Laporte M, Noppen S, Abbasi K, et al. CCL20, a direct-acting pro-angiogenic chemokine induced by hepatitis C virus (HCV): potential role in HCV-related liver cancer. Exp Cell Res. (2018) 372:168–77. doi: 10.1016/j.yexcr.2018.09.023
95. Baluk P, Yao LC, Feng J, Romano T, Jung SS, Schreiter JL, et al. TNF-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J Clin Invest. (2009) 119:2954–64. doi: 10.1172/JCI37626
96. Guenzi E, Töpolt K, Cornali E, Lubeseder-Martellato C, Jörg A, Matzen K, et al. The helical domain of GBP-1 mediates the inhibition of endothelial cell proliferation by inflammatory cytokines. EMBO J. (2001) 20:5568–77. doi: 10.1093/emboj/20.20.5568
97. Boueiz A, Hassoun PM. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc Res. (2009) 77:26–34. doi: 10.1016/j.mvr.2008.10.005
98. Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. (2010) 10:138–46. doi: 10.1038/nrc2791
99. Franses JW, Drosu NC, Gibson WJ, Chitalia VC, Edelman ER. Dysfunctional endothelial cells directly stimulate cancer inflammation and metastasis. Int J Cancer. (2013) 133:1334–44. doi: 10.1002/ijc.28146
100. Hamada J, Cavanaugh PG, Lotan O, Nicolson GL. Separable growth and migration factors for large-cell lymphoma cells secreted by microvascular endothelial cells derived from target organs for metastasis. Br J Cancer. (1992) 66:349–54. doi: 10.1038/bjc.1992.269
101. Pedrosa AR, Trindade A, Carvalho C, Graca J, Carvalho S, Peleteiro MC, et al. Endothelial jagged1 promotes solid tumor growth through both pro-angiogenic and angiocrine functions. Oncotarget. (2015) 6:24404–23. doi: 10.18632/oncotarget.4380
102. Galan-Moya EM, Le Guelte A, Lima Fernandes E, Thirant C, Dwyer J, Bidere N, et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. (2011) 12:470–6. doi: 10.1038/embor.2011.39
103. Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of jagged-1. Cancer Cell. (2013) 23:171–85. doi: 10.1016/j.ccr.2012.12.021
104. Campos MS, Neiva KG, Meyers KA, Krishnamurthy S, Nor JE. Endothelial derived factors inhibit anoikis of head and neck cancer stem cells. Oral Oncol. (2012) 48:26–32. doi: 10.1016/j.oraloncology.2011.09.010
105. Lee E, Pandey NB, Popel AS. Crosstalk between cancer cells and blood endothelial and lymphatic endothelial cells in tumour and organ microenvironment. Expert Rev Mol Med. (2015) 17:e3. doi: 10.1017/erm.2015.2
106. Bandyopadhyay S, Zhan R, Chaudhuri A, Watabe M, Pai SK, Hirota S, et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat Med. (2006) 12:933–8. doi: 10.1038/nm1444
107. Brantley-Sieders DM, Dunaway CM, Rao M, Short S, Hwang Y, Gao Y, et al. Angiocrine factors modulate tumor proliferation and motility through EphA2 repression of Slit2 tumor suppressor function in endothelium. Cancer Res. (2011) 71:976–87. doi: 10.1158/0008-5472.CAN-10-3396
108. Franses JW, Baker AB, Chitalia VC, Edelman ER. Stromal endothelial cells directly influence cancer progression. Sci Transl Med. (2011) 3:66ra5. doi: 10.1126/scitranslmed.3001542
109. Bjarnegard M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A, et al. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. (2004) 131:1847–57. doi: 10.1242/dev.01080
110. Serluca FC, Drummond IA, Fishman MC. Endothelial signaling in kidney morphogenesis: a role for hemodynamic forces. Curr Biol. (2002) 12:492–7. doi: 10.1016/S0960-9822(02)00694-2
111. Hilscher MB, Sehrawat T, Arab JP, Zeng Z, Gao J, Liu M, et al. Mechanical stretch increases expression of CXCL1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology. (2019) 157:193–209.e9. doi: 10.1053/j.gastro.2019.03.013
112. Edsbagge J, Johansson JK, Esni F, Luo Y, Radice GL, Semb H. Vascular function and sphingosine-1-phosphate regulate development of the dorsal pancreatic mesenchyme. Development. (2005) 132:1085–92. doi: 10.1242/dev.01643
113. Ramasamy SK, Kusumbe AP, Adams RH. Regulation of tissue morphogenesis by endothelial cell-derived signals. Trends Cell Biol. (2015) 25:148–57. doi: 10.1016/j.tcb.2014.11.007
114. Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. (2011) 147:539–53. doi: 10.1016/j.cell.2011.10.003
115. Ramasamy SK, Kusumbe AP, Wang L, Adams RH. Endothelial notch activity promotes angiogenesis and osteogenesis in bone. Nature. (2014) 507:376–80. doi: 10.1038/nature13146
116. Benedicto I, Lehmann GL, Ginsberg M, Nolan DJ, Bareja R, Elemento O, et al. Concerted regulation of retinal pigment epithelium basement membrane and barrier function by angiocrine factors. Nat Commun. (2017) 8:15374. doi: 10.1038/ncomms15374
117. Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature. (2016) 529:316–25. doi: 10.1038/nature17040
118. Wang R, Bhattacharya R, Ye X, Fan F, Boulbes DR, Xia L, et al. Endothelial cells activate the cancer stem cell-associated NANOGP8 pathway in colorectal cancer cells in a paracrine fashion. Mol Oncol. (2017) 11:1023–34. doi: 10.1002/1878-0261.12071
119. Naschberger E, Liebl A, Schellerer VS, Schutz M, Britzen-Laurent N, Kölbel P, et al. Matricellular protein SPARCL1 regulates tumor microenvironment-dependent endothelial cell heterogeneity in colorectal carcinoma. J Clin Invest. (2016) 126:4187–204. doi: 10.1172/JCI78260
120. Hu H, Zhang H, Ge W, Liu X, Loera S, Chu P, et al. Secreted protein acidic and rich in cysteines-like 1 suppresses aggressiveness and predicts better survival in colorectal cancers. Clin Cancer Res. (2012) 18:5438–48. doi: 10.1158/1078-0432.CCR-12-0124
121. Uhlitz F, Bischoff P, Peidli S, Sieber A, Obermayer B, Blanc E, et al. Mitogen-activated protein kinase activity drives cell trajectories in colorectal cancer. BioRxiv [Preprint]. (2020). doi: 10.1101/2020.01.10.901579
122. Regensburger D, Tenkerian C, Pürzer V, Schmid B, Wohlfahrt T, Stolzer I, et al. Matricellular protein SPARCL1 regulates blood vessel integrity and antagonizes inflammatory bowel disease. Inflamm Bowel Dis. (2021). doi: 10.1093/ibd/izaa346. [Epub ahead of print].
123. Klingler A, Regensburger D, Tenkerian C, Britzen-Laurent N, Hartmann A, Stürzl M, et al. Species-, organ- and cell-type-dependent expression of SPARCL1 in human and mouse tissues. PLoS ONE. (2020) 15:e0233422. doi: 10.1371/journal.pone.0233422
124. St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, et al. Genes expressed in human tumor endothelium. Science. (2000) 289:1197–202. doi: 10.1126/science.289.5482.1197
125. Cortazar AR, Oguiza JA, Aransay AM, Lavin JL. VerSeDa: vertebrate secretome database. Database (Oxford). (2017) 2017:baw171. doi: 10.1093/database/baw171
126. Burghoff S, Schrader J. Secretome of human endothelial cells under shear stress. J Proteome Res. (2011) 10:1160–9. doi: 10.1021/pr100937a
127. Kwon OK, Lee W, Kim SJ, Lee YM, Lee JY, Kim JY, et al. In-depth proteomics approach of secretome to identify novel biomarker for sepsis in lPS-stimulated endothelial cells. Electrophoresis. (2015) 36:2851–8. doi: 10.1002/elps.201500198
128. Baggetta R, De Andrea M, Gariano GR, Mondini M, Ritta M, Caposio P, et al. The interferon-inducible gene IFI16 secretome of endothelial cells drives the early steps of the inflammatory response. Eur J Immunol. (2010) 40:2182–9. doi: 10.1002/eji.200939995
129. Tunica DG, Yin X, Sidibe A, Stegemann C, Nissum M, Zeng L, et al. Proteomic analysis of the secretome of human umbilical vein endothelial cells using a combination of free-flow electrophoresis and nanoflow lC-MS/MS. Proteomics. (2009) 9:4991–6. doi: 10.1002/pmic.200900065
130. Jambusaria A, Hong Z, Zhang L, Srivastava S, Jana A, Toth PT, et al. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife. (2020) 9:e51413. doi: 10.7554/eLife.51413
131. Naschberger E, Regensburger D, Tenkerian C, Langheinrich M, Engel FB, Geppert C, et al. Isolation of human endothelial cells from normal colon and colorectal carcinoma - an improved protocol. J Vis Exp. (2018) 134:57400. doi: 10.3791/57400
132. Rand JH, Glanville RW, Wu XX, Ross JM, Zangari M, Gordon RE, et al. The significance of subendothelial von Willebrand factor. Thromb Haemost. (1997) 78:445–50. doi: 10.1055/s-0038-1657567
133. Nalluri S, Ghoshal-Gupta S, Kutiyanawalla A, Gayatri S, Lee BR, Jiwani S, et al. TIMP-1 inhibits apoptosis in lung adenocarcinoma cells via interaction with bcl-2. PLoS ONE. (2015) 10:e0137673. doi: 10.1371/journal.pone.0137673
134. Duarte S, Hamada T, Kuriyama N, Busuttil RW, Coito AJ. TIMP-1 deficiency leads to lethal partial hepatic ischemia and reperfusion injury. Hepatology. (2012) 56:1074–85. doi: 10.1002/hep.25710
135. Arpino V, Brock M, Gill SE. The role of tIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. (2015) 44–46:247–54. doi: 10.1016/j.matbio.2015.03.005
136. Sasaki S, Yoneyama H, Suzuki K, Suriki H, Aiba T, Watanabe S, et al. Blockade of cXCL10 protects mice from acute colitis and enhances crypt cell survival. Eur J Immunol. (2002) 32:3197–205. doi: 10.1002/1521-4141(200211)32:11<3197::AID-IMMU3197>3.0.CO;2-1
137. Chami B, Yeung AW, van Vreden C, King NJ, Bao S. The role of CXCR3 in DSS-induced colitis. PLoS ONE. (2014) 9:e101622. doi: 10.1371/journal.pone.0101622
Keywords: endothelial, angiocrine, barrier, inflammatory bowel disease, inflammation, angiogenesis, epithelial
Citation: Stürzl M, Kunz M, Krug SM and Naschberger E (2021) Angiocrine Regulation of Epithelial Barrier Integrity in Inflammatory Bowel Disease. Front. Med. 8:643607. doi: 10.3389/fmed.2021.643607
Received: 18 December 2020; Accepted: 07 July 2021;
Published: 02 August 2021.
Edited by:
Roberto Gramignoli, Karolinska Institutet (KI), SwedenReviewed by:
Prashant Nighot, The Pennsylvania State University, United StatesLinda Chia-Hui Yu, National Taiwan University, Taiwan
Copyright © 2021 Stürzl, Kunz, Krug and Naschberger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Stürzl, bWljaGFlbC5zdHVlcnpsQHVrLWVybGFuZ2VuLmRl