 Ana Ávila
Ana Ávila Eva Gavela
Eva Gavela Asunción Sancho†
Asunción Sancho†- Nephrology Department, University Hospital Dr. Peset, Valencia, Spain
Thrombotic microangiopathy is a rare but serious complication that affects kidney transplant recipients. It appears in 0.8–14% of transplanted patients and negatively affects graft and patient survival. It can appear in a systemic form, with hemolytic microangiopathic anemia, thrombocytopenia, and renal failure, or in a localized form, with progressive renal failure, proteinuria, or arterial hypertension. Post-transplant thrombotic microangiopathy is classified as recurrent atypical hemolytic uremic syndrome or de novo thrombotic microangiopathy. De novo thrombotic microangiopathy accounts for the majority of cases. Distinguishing between the 2 conditions can be difficult, given there is an overlap between them. Complement overactivation is the cornerstone of all post-transplant thrombotic microangiopathies, and has been demonstrated in the context of organ procurement, ischemia-reperfusion phenomena, immunosuppressive drugs, antibody-mediated rejection, viral infections, and post-transplant relapse of antiphospholipid antibody syndrome. Although treatment of the causative agents is usually the first line of treatment, this approach might not be sufficient. Plasma exchange typically resolves hematologic abnormalities but does not improve renal function. Complement blockade with eculizumab has been shown to be an effective therapy in post-transplant thrombotic microangiopathy, but it is necessary to define which patients can benefit from this therapy and when and how eculizumab should be used.
Introduction
Thrombotic microangiopathy (TMA) is a life-threatening disease, characterized by endothelial dysfunction and the presence of thrombi in small blood vessels. As the thrombus forms, there is platelet consumption and mechanical disturbance of red blood cells, leading to thrombocytopenia and microangiopathic hemolytic anemia. Vessel occlusion results in tissue ischemia and organ damage, primarily affecting the kidneys, although other organs can be involved (1–4).
TMA syndromes can be classified according to the pathogenic mechanism (5, 6). Primary TMA syndromes include TMA whose etiology is known: thrombotic thrombocytopenic purpura (TTP), due to deficiency of the von Willebrand factor-cleaving protease ADAMTS13 (7); typical hemolytic uremic syndrome (HUS), caused by Shiga toxin-producing Escherichia coli (8); pneumococcal-associated HUS (9, 10); and atypical HUS (aHUS) caused by inherited or acquired abnormalities in complement proteins leading to unregulated activation of the alternative pathway of the complement system and the formation of the membrane attack complex (MAC) (11, 12). Other genetic causes, such as diacylglycerol kinase ε, an endothelial cell and podocyte protein, and cobalamin C deficiency have been described as causes of primary aHUS, mainly in children (13, 14). Secondary TMA syndromes occur in the context of infections, organ transplantation (solid organ and hematopoietic stem cell transplantation), drugs (cancer chemotherapy, vascular endothelial growth factor [VEGF] inhibitors, immunosuppressants such as calcineurin inhibitors [CNIs] and mammalian target of rapamycin inhibitors [mTORis]), malignancies, pregnancy, malignant hypertension, and autoimmune diseases (systemic lupus erythematosus, antiphospholipid syndrome, scleroderma, and vasculitis) (3, 5, 6). The distinction between primary and secondary TMAs is not absolute because genetic variants have been identified in patients with secondary TMAs. Moreover, secondary TMAs are also called secondary aHUS, because a complement deregulation has been described in some of those conditions, suggesting an overlap between these categories (15).
Post-transplant thrombotic microangiopathy (PT-TMA) is a rare but devastating condition that can lead to poor patient and graft outcomes. It can occur as a de novo disease or as a recurrence of a previous aHUS (sometimes undiagnosed before kidney transplantation). De novo PT-TMA is caused by various pathogenic mechanisms, whereas aHUS recurrence is a consequence of complement system deregulation triggered by several activating conditions (Table 1). The primary aHUS normally requires a second hit for disease to develop. The aHUS triggers match several situations that can produce secondary TMA, making the distinction between the two entities difficult (16).

Table 1. Causes of post-transplant thrombotic microangiopathy.
Epidemiology of Post-Transplant Thrombotic Microangiopathy
Post-transplant TMA is observed in 0.8–14% of kidney transplants (17, 18). (USRDS) de novo PT-TMA is much more frequent than recurrent aHUS (90 vs. 10% of all cases), but the risk associated with the development of PT-TMA is much higher (36.5 times; 29 vs. 0.8%) in patients with a history of aHUS (18).
Clinical Manifestations of Post-Transplant Thrombotic Microangiopathy
The manifestations of PT-TMA are quite variable and can range from a limited form confined to the kidney to a full-blown systemic variant (19, 20, 24). The systemic form is typically acute, consisting of the classic triad of thrombocytopenia, microangiopathic hemolytic anemia with increase in lactate dehydrogenase, reduced haptoglobin, and schistocyte formation, and acute kidney injury (AKI); this has been described in 18–62% of patients with PT-TMA (19, 21, 25, 26). The localized form can manifest as isolated AKI or as a chronic form with slowly progressive graft dysfunction, proteinuria, or difficult-to-control arterial hypertension, and it can only be diagnosed when a kidney biopsy is performed (5, 27).
Although aHUS recurrence and PT-TMA are clinically and pathologically indistinguishable, a personal and family history of aHUS, an abrupt onset, and a complete and systemic TMA are suggestive of aHUS recurrence (28). Extrarenal manifestations of aHUS apart from hemolytic anemia (29–34) are frequent in aHUS recurrence, but they are rarely observed in de novo PT-TMA (35).
PT-TMA and aHUS recurrence can appear at any time in the post-transplant course (17, 36), but they develop primarily in the first 3 months after transplantation (21, 37), in conjunction with the presence of more complement activating events (e.g., ischemia-reperfnusion injury, high immunosuppressive drug levels, higher infectious risk). Systemic TMA usually appears in the early post-transplant period, and the localized form can appear at all stages after transplantation.
Histological Changes
In the kidney biopsy, the active lesions are intraluminal fibrin occlusive thrombi along with endothelial cell activation signs, such as endothelial swelling, fragmented red blood cells in capillaries, mesangiolysis and microaneurisms, myocyte necrosis, and intramural fibrin in arterioli. Immunofluorescence microscopy is negative, except for fibrinogen. Electron microscopy shows subendothelial widening by flocculent material (“subendothelial fluff”). In the chronic phase, the characteristic lesions are double contour formation in peripheral walls with hyaline deposits in arterioles and fibrous intimal thickening with concentric lamination (onion skin). By electron microscopy, new subendothelial basement membrane and widening of the subendothelial zone can be observed (15, 21, 25, 38). The lack of thrombi in the biopsy does not exclude TMA (15). The biopsy does not allow the identification of the etiology, although some changes might suggest certain etiologies, such as C4d deposition, peritubular capilaritis, and glomerulitis in antibody mediated rejection (ABMR) or intimal thickening, reduplication of the elastic lamina, and hyaline degeneration in arterial hypertension. In PT-TMA, histological characteristics can be influenced by the donor's previous injuries. Thus, the interpretation of chronic injuries can be more complex. The findings of any acute injuries in the biopsy, together with clinical and laboratory data, can lead us to suspect TMA activity and the need for active treatment.
After the diagnosis of TMA, the etiology of the native kidney ESRD should be investigated to rule out previously missed aHUS (38).
Prognosis for Post-Transplant Thrombotic Microangiopathy
The overall prognosis for PT-TMA is quite poor for the allograft and for the patient, resulting in a graft loss rate of 33–40% in the first 2 years (17, 18, 20, 21, 23), and a patient survival of 50% at 3 years after TMA diagnosis in a previous series (18); recently, however, 97% 1-year patient survival has been reported (25). Recurrence of aHUS appeared in 60% of transplant patients with the disease, leading to 89–90% graft loss in the first year in the pre-eculizumab era (39). In case of PT-TMA, although poorer short-term graft survival had been described in patients with the systemic form of PT-TMA, reflecting a more severe disease with a higher incidence of dialysis-dependent AKI and plasma exchange (PE) needs (19), the prognosis in the long term is similar in both forms (19, 25, 26). The prognosis is also similar in early (<3 months) and late (>3 months after transplantation) presentation (25).
Causes of Post-Transplant Thrombotic Microangiopathy
At the time of kidney transplantation, the coincidence of several mechanisms that activate the complement system can trigger the recurrence of aHUS in patients with a genetic background or the development of de novo PT-TMA.
aHUS Recurrence
In aHUS, the recurrence risk is determined by a genetic mutation in complement proteins (11, 15, 39–41, 48). Stratifying the risk according to the mutation is the key to making decisions about the post-transplant management of aHUS. A higher recurrence risk is considered in patients with recurrence in previous transplants and in carriers of pathogenic variants in CFH/CFB/CFH:CFHR1 rearrangements/TBHD; moderate risk in carriers of CFI variants/C3/anti-FH antibodies/homozygous for haplotypes CFH-H3/absence of variants (29, 39–48, 114); and low risk in those with isolated MCP/DGKE variants and with negative anti-FH antibodies at the time of transplantation (13, 40, 48–50) (Table 2). The use of prophylactic eculizumab reduced the post-transplant recurrence rate from 49 to 12% in patients with aHUS, reducing the probability of graft loss, and significantly increasing the number of patients with aHUS who receive a kidney transplant (51).
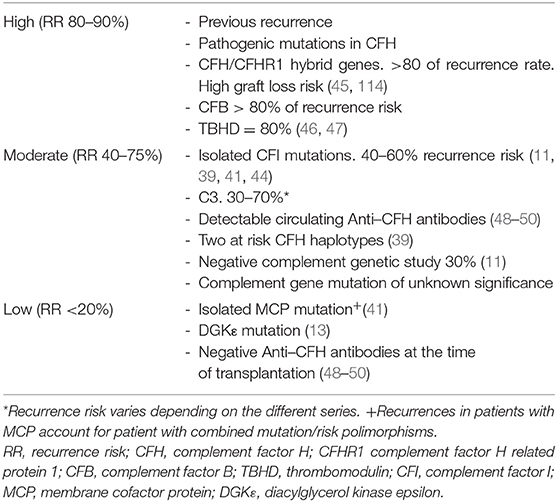
Table 2. Recurrence risk of aHUS after kidney transplantation in the preeculizumab era.
Secondary Thrombotic Microangiopathy
In the absence of previous aHUS, Chua et al. (52) showed activation of the classical and terminal complement in several transplant-associated conditions: in donors after brain death (DBD) or donors after circulatory death (DCD) (53–55), or associated with ischemia reperfusion injury (56), immunosuppressive drugs (mainly CNI or mTORi) (41, 42, 57–61), ABMR (62–68), viral and fungal infection (69–82), and recurrence of antiphospholipid syndrome (APS) (83–85). The laboratory tests needed to evaluate causes of PT-TMA are described in Table 3. De novo aHUS has also been described in patients with C3 glomerulopathy in the native kidneys (86, 87) and in 1 patient with TTP (88).

Table 3. Main laboratory tests to perform in PT-TMA.
All these triggers appear in patients with or without a known genetic background; Le Quintrec reported 30% complement genetic mutations in a series of patients with de novo PT-TMA (29), indicating an overlap between recurrent and de novo TMA.
Complement Activation Related to the Donor and Procurement Process
Activation of the complement system can be observed from earlier stages of transplant. This activation can be related to the type of donor. Naesens et al. (53) showed a relevant increase in expression of complement factors, such as C1, C3, and CFB, in renal allograft pre-implantation and after transplantation biopsies in DBD compared with living donor (LD) biopsies. Ischemia reperfusion damage has been associated with early injury of the renal allograft. Experimental data have suggested that, after ischemia reperfusion, complement is activated by the lectin pathway, and afterward, the alternative complement pathway could amplify the injury through the release of C3, C5, and the MAC. In addition, the damage to endothelial glycocalyx secondary to ischemia would reduce the union of factor H to the endothelial cells. In a similar manner, this complement activation after ischemia reperfusion would be higher in recipients of kidneys from DBDs than from LDs (54–56). The use of kidneys from DCDs has been associated with a higher risk of PT-TMA. The prolonged warm ischemia in this type of donor would aggravate the endothelial lesions in the graft and would increase complement activation and secondary damage (41, 112).
Drug-Induced Thrombotic Microangiopathy
Drug induced TMA (DITMA) is suspected when there is a sudden onset acute kidney injury, usually within hours or a few days after drug exposure, and resolution can be observed when the drug is stopped or reduced (57, 89, 113).
The association between CNIs and de novo PT-TMA has been well documented in the literature, with the risk higher with cyclosporine than with tacrolimus (41). Various mechanisms have been associated with the development of PT-TMA after CNI treatment. The loss of normal equilibrium between vasoactive peptides, with an increase in vasoconstrictor substances, such as angiotensin II, thromboxane A2, and endothelin, and a reduction of vasodilatory molecules, such as prostaglandin (PG) E2, prostacyclin (PGI2), and nitric oxide leads to arteriolar vasoconstriction and endothelial injury secondary to renal ischemia. CNIs also favor platelet aggregation and plasminogen activation, with a higher risk of thrombosis. Cyclosporine causes endothelial cells to release microparticles that activate the alternate complement pathway (58).
The diagnosis of CNI-related TMA is found in the early post-transplant period when the levels of these drugs are high. Recently, new advances in the understanding of TMA and its association with complement abnormalities have questioned the relevance of CNIs in this disease. It has been suggested that there must be some predisposing factors for PT-TMA development in patients taking CNIs, given that more than 95% of renal transplant recipients receive this treatment. Data from USRDS have shown a higher PT-TMA incidence in patients without CNI treatment. In addition, results from a French aHUS registry have not shown the association between CNIs and PT-TMA, and the lack of CNI use did not prevent the recurrence of aHUS in this study group (42). It was thought that mTORi could be a good alternative to CNIs for patients with aHUS. Unfortunately, various studies have not shown this protective effect. Data from USRSD showed a higher incidence of PT-TMA with sirolimus than with CNIs, and the French registry results highlighted a higher risk of recurrence post-transplant in patients with aHUS when mTORis were used. However, the fact that mTORis might have been used as a rescue therapy after diagnosis of PT-TMA limits the interpretation of these results (18, 41, 57). Inhibition of mTOR inhibition leads to the death of endothelial progenitor cells and the decrease in renal expression of vascular endothelial growth factor (VEGF), which also would lead to a reduction in Factor H synthesis. Other factors, such as an increased procoagulant and a reduced fibrinolytic state, are also believed to contribute to the pathogenesis of TMA in patients taking mTORis (59, 60). Recently, the combined use of CNIs with mTORis is increasing as an alternative treatment for renal transplant recipients, but this combination increases the risk of PT-TMA compared with single medication treatment, particularly when the blood levels of both drugs are high (22, 23, 61).
Antibody-Mediated Rejection-Associated Thrombotic Microangiopathy
The complement system plays an important role in ABMR. The donor-specific antibodies bind to human leukocyte antigens on the allograft endothelium and activate the classical complement pathway through C1q, leading to activation of C4 and C3. The deposit of C3b on the membrane of endothelial cells triggers the activation of the alternate complement pathway with the generation of the MAC, which produces cell lysis and an inflammatory infiltration (62). The histological finding of TMA in patients with ABMR has wide variability, between 4 and 46%, most likely as a consequence of the focal presentation of the disease (63, 64). A negative impact of TMA on ABMR has been described. Wu et al. (65) found lower graft survival in patients with ABMR and TMA compared with ABMR without TMA. Diagnosis of TMA in the sensitized kidney transplant recipient is predominantly confirmed by histological findings.
Infection-Related Thrombotic Microangiopathy
Viral infections can trigger PT-TMA due to the endothelial trophism of the virus, which induces the expression of adhesion molecules and the release of von Willebrand factor, causing platelet adhesion and microvascular thrombosis (71). CMV is the most frequently involved virus (69–73). In all CMV-related TMA cases, treatment with intravenous ganciclovir and plasma exchange (PE) resolved hemolysis; however, a TMA recurrence occurred in one case, which was resolved with valganciclovir and eculizumab (71). Parvovirus (74–76), hepatitis C virus, and its treatment (77–79) and fungal infections such as histoplasmosis PT-TMA have also been described (80, 81). Recently a case of ABMR and TMA associated with Nile Fever has been reported (82).
Other Causes of Post-transplant Thrombotic Microangiopathy
APS can cause ESRD due to large and small kidney vessel thrombosis and TMA. These symptoms can recur after transplantation, and eculizumab has shown a beneficial effect in patients with this disease, preventing and treating the recurrence of APS (83–85).
Treatment of Post-transplant Thrombotic Microangiopathy
Treatment of aHUS Recurrence
Classically, transplantation of patients with aHUS has shown low graft survival with a high rate of loss due to recurrence of the disease. Prophylactic treatment or recurrence treatment are the two alternatives. Prophylactic treatment based on plasmapheresis does not effectively prevent recurrence in patients with high or moderate risk mutations, and subclinical complement activation have been reported in these patients. Excellent results with eculizumab as the first-line therapy have been reported; the usefulness of a first dose 1 h before reperfusion and a second dose 24 h after transplantation has been considered, both to reduce secondary complement activation due to ischemia-reperfusion. In the treatment of recurrence, little success has been achieved after plasmapheresis; nevertheless, good results have been observed with an early introduction of eculizumab after recurrence (42).
Treatment of de novo Post-transplant Thrombotic Microangiopathy
Treatment of de novo PT-TMA should be based on correcting the potential cause of the disease and varies depending on the time of onset. Given the extreme heterogenicity of the mechanisms related to the appearance of TMA, therapeutic maneuvers must be individualized. The first step is to avoid complement over-activation before donation, preventing renal hypoperfusion during organ procurement, and reducing cold ischemia time.
In cases of PT-TMA secondary to immunosuppressive medications, the first step is to reduce or stop the offending agent, by switching from a CNI to another CNI or to an mTORi. This approach can resolve the TMA, but the effectiveness of this strategy is controversial. Satoskar showed no difference in outcomes between changing immunosuppression or not (20). Belatacept, a cytotoxic T lymphocyte antigen 4-immunoglobulin fusion protein that inhibits T cell function, allows the minimization or discontinuation of endothelial toxic immunosuppressants such as CNIs and mTORis (90, 91). However, a higher risk of acute kidney transplant rejection compared with current standard immunosuppressive therapy has been observed after conversion to belatacept in kidney transplant rejection (92).
In ABMR-associated TMA the mainstay of treatment is plasmapheresis (PP), with or without IVIg and additional immunosuppression (65–68). Despite the implication of complement activation in ABMR, the efficacy of complement inhibitors for the prophylaxis or treatment of ABMR is difficult to assess with current clinical data and is yet to be established. Instead, eculizumab use is currently recommended as a rescue therapy in AMR-associated TMA when hemolysis persists despite maximal management including plasma exchange (PLEX) and in those with PLEX dependency (66–68).
PT-TMA that is unresponsive to the previous measures has typically been treated with PE. PE has been shown to reduce mortality in TTP patients (93, 94), and it was the first-line therapy for aHUS in the pre-eculizumab era. PE can remove vasoconstrictor molecules such as thromboxane A2 and mutant complement proteins and provides deficient factors such as PGI2-stimulating factor and normally functioning complement components (27, 36). Due to these effects in primary TMA, the use of PE was extrapolated to PT-TMA. In 2003, Karthikeyan et al. (36) reported a graft salvage rate of 80% with PE in addition to CNI withdrawal in 29 patients with biopsy-proven TMA. Epperla et al. (95) showed a 100% response rate in 5 patients using withdrawal of the suspected offending agent associated with PE in 4, eculizumab in 2, and rituximab in 1 patient. However, the use of PE in a large proportion of patients does not improve kidney function despite correcting the hematological abnormalities, and it is associated with a 20–42% risk of graft loss (97, 98). Schwimmer showed similar graft outcomes in a series of 742 PT-TMA transplants, regardless of whether they had received PE (19).
Eculizumab, a recombinant, fully humanized monoclonal antibody targeted against human complement protein C5, blocks generation of the lytic C5b-9 MAC. It has been shown to be effective in the treatment and prevention of recurrent aHUS after transplantation (99–103). In post-transplant TMA, a complement over-activation has been demonstrated (51–55, 104), even in patients without pathogenic complement proteins variants. Therefore, the inhibition of the complement system could be a suitable PT-TMA approach (Figure 1) (96).
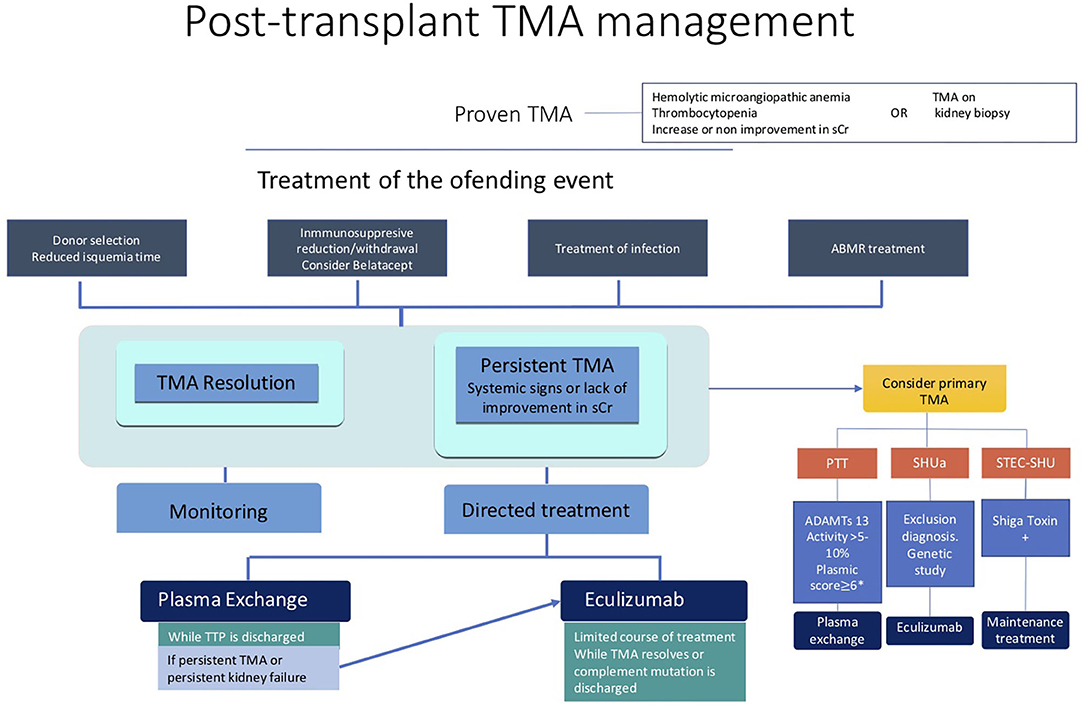
Figure 1. Post-transplant TMA management.
The main risk associated with the eculizumab use is meningococcal infection; thus, vaccination against Neisseria meningitidis serogroups B, A, C, W135, and Y is mandatory. Vaccination should be administered 2 weeks before initiation of eculizumab treatment. But in PT TMA, usually it is not possible to delay eculizumab treatment two weeks, and prophylactic antibiotic should be used in the meantime. Given neither vaccines nor prophylaxis guarantee full protection against meningitis, patients and their families should be taught to recognize the alarm signs and symptoms of the infection (48).
Several case reports and small case series have been published documenting the efficacy of eculizumab in de novo PT-TMA refractory to previously mentioned treatments (Table 4) (53–61, 71, 83–85, 97, 98). Recently, the efficacy of eculizumab has been described in larger series. Cavero et al. (97) showed 15 kidney transplanted patients with PT-TMA; 14 patients were using tacrolimus, 4 of them combined with an mTORi. All but one withdrew the offending drug. Twelve patients also received PE (a mean of 5.46 sessions per patient, range 2–12) without improvements in serum creatinine (mean 4 mg/dl, 3.4–5.6). Eculizumab was started from 4 to 53 days after TMA detection, and a mean of 6.4 doses from 2 to 17 per patient were used. At the end of follow-up, mean serum creatinine was 1.7 mg/dl, with no graft losses or recurrences of TMA after eculizumab discontinuation. Portolés et al. (98) reported 22 patients with PT-TMA, 16 with early- (<1 month) and 6 with late- (>1 month post-transplant) onset TMA. All patients presented hematological TMA and most had a confirmatory biopsy. Some 77% of early and 100% of late patients with PT-TMA received PE, achieving a complete hematological response, but with an incomplete or an absent renal response in most cases. Eculizumab was then added, for an average of 21 days in the early TMA group and 83.5 days in the late TMA group. In the early group, 8 complete and 2 partial responses were observed, whereas in the late group, one patient had a complete response, two had a partial renal response, and the remaining patients lost their grafts. The patients with better responses had a shorter time lapse between diagnosis and the beginning of the treatment. Eculizumab was withdrawn in all cases, without relapses.
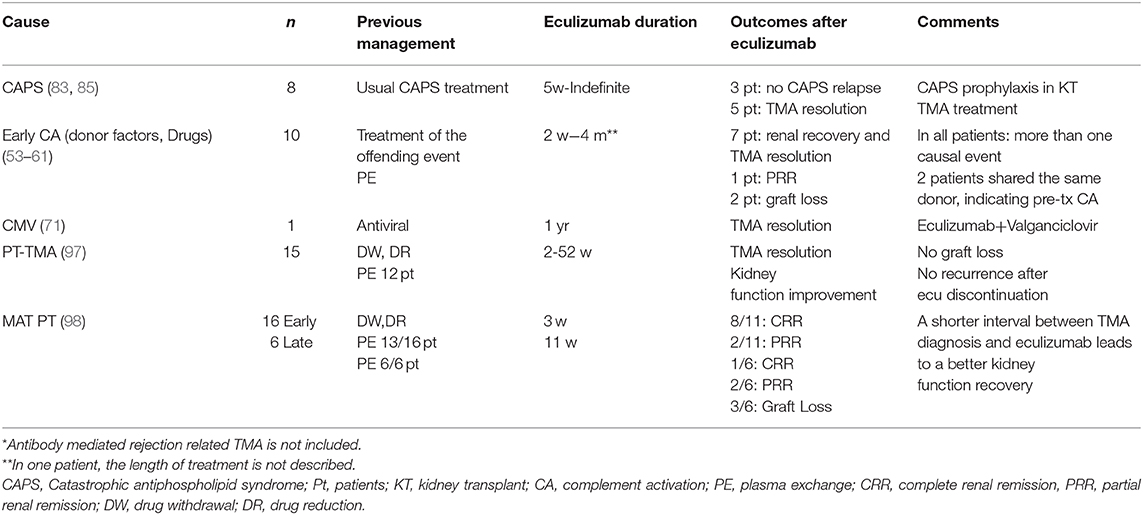
Table 4. Eculizumab in kidney transplant associated TMA*.
Eculizumab has been used more frequently in cases of lack of renal recovery after a short course of PE, obtaining an improvement of kidney function with few adverse events.
The duration of eculizumab treatment in cases of aHUS recurrence can be lifelong (depending on the genetic mutation); in secondary TMA, however, it is not clearly addressed. In previous reports, a short course of eculizumab was shown to be an efficient therapy to control TMA and to reduce the risk of graft loss due to this disease. Discontinuation of the therapy can be considered when complications of TMA have completely resolved, and on a case-by-case basis. It can also be considered when the kidney function has not improved after 3 to 6 months or when a lack of viability of the kidney graft is documented by means of a kidney biopsy or imaging tests (computed tomography, magnetic resonance imaging).
The current data are not strong enough to expand the recommendation for the use of eculizumab in all patients with PT-TMA, but in cases resistant to treatment of the offending event, eculizumab is a good therapeutic option. Although PE has been used, its efficacy is limited and it is important to note that in patients with PT-TMA, the sooner eculizumab starts, the better the renal function will be at the end of the follow-up (97, 98).
Conclusion
The incidence and the impact of PT-TMA, either de novo or recurrent, on allograft survival is underestimated. Several factors related to the donor and procurement process and to the recipient and events during the post-transplant period (immunosuppressive drugs, rejection, and infections), trigger the development of this disease. The treatment of PT-TMA includes management of the offending event, PE, and recently, eculizumab, with promising results. However, further studies are needed to establish which PT-TMA kidney transplant recipients are most likely to benefit from eculizumab therapy, including when and how to use it given the high economic burden associated with this approach.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
The authors have received funding for publication fees from the Valencian Society of Nephrology.
Conflict of Interest
AA have received fees for conferences from Alexion pharmaceutical.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Zhang K, Lu Y, Harley KT, Tran MH. Atypical hemolytic uremic syndrome: a brief review. Hematol Rep. (2017) 9:7053. doi: 10.4081/hr.2017.7053
2. Sheerin NS, Glover E. Haemolytic uremic syndrome: diagnosis and management. F1000Res. (2019) 8:8. doi: 10.12688/f1000research.19957.1
3. Bayer G, von Tokarski F, Thoreau B, Bauvois A, Barbet C, Cloarec S, et al. Etiology and outcomes of thrombotic microangiopathies. Clin J Am Soc Nephrol. (2019) 14:557–66. doi: 10.2215/CJN.11470918
4. Dixon BP, Gruppo RA. Atypical hemolytic uremic syndrome. Pediatr Clin North Am. (2018) 65:509–25. doi: 10.1016/j.pcl.2018.02.003
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. (2014) 371:654–66. doi: 10.1056/NEJMra1312353
6. Avila Bernabeu AI, Cavero Escribano T, Cao Vilarino M. Atypical hemolytic uremic syndrome: new challenges in the complement blockage era. Nephron. (2020) 144:537–49. doi: 10.1159/000508920
7. De la Rubia J, Contreras E, Del Río-Garma J. Thrombotic thrombocytopenic purpura. Med Clin (Barc). (2011) 136:534–40. doi: 10.1016/j.medcli.2010.02.011
8. Freedman SB, Xie J, Neufeld MS, Hamilton WL, Hartling L, Tarr PI, et al. Shiga toxin-producing Escherichia coli infection, antibiotics, and risk of developing hemolytic uremic syndrome: a meta-analysis. Clin Infect Dis. (2016) 62:1251–8. doi: 10.1093/cid/ciw099
9. Constantinescu AR, Bitzan M, Weiss LS, Christen E, Kaplan BS, Cnaan A, et al. Nonenteropathic hemolytic uremic syndrome:causes and short-term course. Am J Kidney Dis. (2004) 43:976–82. doi: 10.1053/j.ajkd.2004.02.010
10. Jokiranta TS. HUS and atypical HUS. Blood. (2017) 129:2847–56. doi: 10.1182/blood-2016-11-709865
11. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. (2009) 361:1676–87. doi: 10.1056/NEJMra0902814
12. Loirat C, Frémeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. (2011) 6:60. doi: 10.1186/17570-1172-6-60
13. Azukaitis K, Simkova E, Majid MA, Galiano M, Benz K, Amann K, et al. The phenotypic spectrum of nephropathies associated with mutations in diacylglycerol kinase. J Am Soc Nephrol. (2017) 28:3066–75. doi: 10.1681/ASN.2017010031
14. Cornec-Le Gall E, Delmas Y, De Parscau L, Doucet L, Ogier H, Benoist JF, et al. Adult-onset eculizumab-resistant hemolytic uremic syndrome associated with cobalamin C deficiency. Am J Kidney Dis. (2014) 63:119–23. doi: 10.1053/j.ajkd.2013.08.031
15. Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. (2017) 91:539–51. doi: 10.1016/j.kint.2016.10.005
16. Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: core curriculum 2015. Am J Kidney Dis. (2015) 66:359–75. doi: 10.1053/j.ajkd.2015.03.040
17. Zarifian A, Meleg-Smith S, O'donovan R, Tesi RJ, Batuman V. Cyclosporine-associated thrombotic microangiopathy in renal allografts. Kidney Int. (1999) 55:2457–66. doi: 10.1046/j.1523-1755.1999.00492.x
18. Reynolds JC, Agodoa LY, Yuan CM, Abbot KC. Thrombotic microangiopathy after renal transplantation in the United States. Am J Kidney Dis. (2003) 42:1058–68. doi: 10.5500/wjt.v8.i5.122
19. Schwimmer J, Nadasdy TA, Spitalnik PF, Kaplan KL, Zand MS. De novo thrombotic microangiopathy in renal transplant recipients: a comparison of hemolytic uremic syndrome with localized renal thrombotic microangiopathy. Am J Kidney Dis. (2003) 41:471–9. doi: 10.1053/ajkd.2003.50058
20. Satoskar AA, Pelletier R, Adams P, Nadasdy GM, Brodsky S, Pesavento T, et al. De novo thrombotic microangiopathy in re-nal allograft biopsies-role of antibody-mediated rejection. Am J Transplant. (2010) 10:1804–11. doi: 10.1111/j.1600-6143.2010.03178.x
21. Broecker V, Bardsley V, Torpey N, Perera R, Montero R, Dorling A, et al. Clinical-pathological correlations in post-transplant thrombotic microangiopathy. Histopathology. (2019) 75:88–103. doi: 10.1111/his.13855
22. Langer RM, Van Buren CT, Katz SM, Kahan BD. De novo hemolytic uremic syndrome after kidney transplantation in patients treated with cyclosporine-sirolimus combination. Transplantation. (2002) 73:756–60. doi: 10.1097/00007890-200203150-00017
23. Fortin MC, Raymond MA, Madore F, Fugère JA, Pâquet M, St-Louis G, et al. Increased risk of thrombotic microangiopathy in patients receiving a cyclosporin-sirolimus combination. Am J Transplant. (2004) 4:946–52. doi: 10.1111/j.1600-6143.2004.00428
24. Nadasdy T. Thrombotic microangiopathy in renal allografts: the diagnostic challenge. Curr Opin Organ Transplant. (2014) 19:283–92. doi: 10.1097/MOT.0000000000000074
25. Iglesias Teixeira C, Gonçalves Mota R, Vieira Afonso BG, Vieira Carneiro T, Gonçalves Meira GS, Urias Mendonça D. Use of Eculizumab in atypical hemolytic uremic syndrome after renal transplantation. J Bras Nefrol. (2015) 37:127–30. doi: 10.5935/0101-2800.20150018
26. Saikumar Doradla LP, Lal H, Kaul A, Bhaduaria D, Jain M, Prasad N, et al. Clinical profile and outcomes of De novo posttransplant thrombotic microangiopathy. Saudi J Kidney Dis Transpl. (2020) 31:160–8. doi: 10.4103/1319-2442.279936
27. Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. (2008) 358:1129–36. doi: 10.1056/NEJMoa0707330
28. Campistol JM, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. (2015) 35:421–7. doi: 10.1016/j.nefro.2015.07.005
29. Le Quintrec M, Lionet A, Kamar N, Karras A, Barbier S, Buchler S, et al. Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation. Am J Transplant. (2008) 8:1694–701. doi: 10.1111/j.1600-6143.2008.02297.x
30. Noris M, Remuzzi G. Thrombotic microangiopathy after kidney transplantation. Am J Transplant. (2010) 10:1517–23. doi: 10.1111/j.1600-6143.2010.03156
31. Formeck C, Swiatecka-Urban A. Extra-renal manifestations of atypical hemolytic uremic syndrome. Pediatr Nephrol. (2019) 34:1337–48. doi: 10.1007/s00467-018-4039-7
32. Ávila A, Vizcaíno B, Molina P, Gavela E, Perez-Ebri ML, Pallardó L, et al. Remission of aHUS neurological damage with eculizumab. Clin Kidney J. (2015) 8:232–6. doi: 10.1093/ckj/sfu144
33. Noris M, Remuzzi G. Cardiovascular complications in atypical haemolytic uraemic syndrome. Nat Rev Nephrol. (2014) 10:174–80. doi: 10.1038/nrneph.2013.280
34. Sampedro López A, Domínguez Moro B, Baltar Martin JM, Garcia Monteavaro C, Barbón García JJ, et al. Ocular involvement in atypical haemolytic uraemic syndrome. Arch Soc Esp Oftalmol. (2017) 92:594–7. doi: 10.1016/j.oftal.2017.02.007
35. Haghikia A, Heeren M, Bockmeyer C, Haubitz B, Gwinner W. Progressive multifocal cerebral infarction in a young kidney transplant recipient due to thrombotic microangiopathy. BMC Nephrol. (2014) 15:59. doi: 10.1186/1471-2369-15-59
36. Karthikeyan V, Parasuraman R, Shah V, Vera E, Venkat KK. Outcome of plasma exchange therapy in thrombotic microangiopathy after renal transplantation. Am J Transplant. (2003) 3:1289–1294. doi: 10.1046/j.1600-6143.2003.00222.x
37. Ardalan MR. Review of thrombotic microangiopathy (TMA), and post-renal transplant TMA. Saudi J Kidney Dis Transpl. (2006) 17:235–44.
38. Garg N, Rennke HG, Pavlakis M, Zandi-Nejad K. De novo thrombotic microangiopathy after kidney transplantation. Transplant Rev. (2018) 32:58–68. doi: 10.1016/j.trre.2017.10.001
39. Bresin E, Daina E, Noris M, Castelletti F, Stefanov R, Hill P, et al. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. (2006) 50:88–99. doi: 10.2215/CJN.00050505
40. Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, et al. European Working Party on Complement Genetics in Renal, Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. (2013) 24:475–86. doi: 10.1681/ASN.2012090884
41. Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant. (2013) 13:663–75. doi: 10.1111/ajt.12077
42. Zuber J, Le Quintrec M, Morris H, Frémeaux-Bacchi V, Loirat C, Legendre C. Targeted strategies in the prevention and management of atypical HUS recurrence after kidney transplantation. Transplant Rev (Orlando). (2013) 27:117– 25. doi: 10.1016/j.trre.2013.07.003
43. Bienaime F, Dragon-Durey MA, Regnier CH, Nilsson SC, Kwan WH, Blouin J, et al. Mutations in components of complement influence the outcome of Factor I-associated atypical hemolytic uremic syndrome. Kidney Int. (2010) 77:339–49. doi: 10.1038/ki.2009.472
44. Chan MR, Thomas CP, Torrealba JR, Djamali A, Fernandez LA, Nishimura CJ, et al. Recurrent atypical hemolytic uremic syndrome associated with factor I mutation in a living related renal transplant recipient. Am J Kidney Dis. (2009) 53:321–6. doi: 10.1053/j.ajkd.2008.06.027
45. Román-Ortiz E, Mendizabal Oteiza S, Pinto S, López-Trascasa M, Sánchez-Corral P, Rodríguez de Cordoba S. Eculizumab long-term therapy for pediatric renal transplant in aHUS with CFH/CFHR1 hybrid gene. Pediatr Nephrol. (2014) 29:149–53. doi: 10.1007/s00467-013-2591-8
46. Caroti L, Di Maria L, Carta P, Moscarelli L, Cirami C, Minetti EE. Posttransplant outcome of atypical haemolytic uraemic syndrome in a patient with thrombomodulin mutation: a case without recurrence. Clin Kidney J. (2015) 8:329–1. doi: 10.1093/ckj/sfv025
47. Sinibaldi S, Guzzo I, Piras R, Bresin E, Emma F, Dello Strologo L. Post-transplant recurrence of atypical hemolytic uremic syndrome in a patient with thrombomodulin mutation. Pediatr Transplant. (2013) 17:E177–81. doi: 10.1111/petr.12151
48. Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. (2016) 31:15–39. doi: 10.1007/s00467-015-3076-8
49. Khandelwal P, Sinha A, Hari P, Bansal VK, Dinda AK, Bagga A. Outcomes of renal transplant in patients with anti-complement factor H antibody-associated hemolytic uremic syndrome. Pediatr Transplant. (2014) 18:E134–9. doi: 10.1111/petr.12273
50. Bagga A, Sinha A, Dragon-Durey MA. Therapy for patients with antibodies to complement factor H associated HUS. Pediatr Nephrol. (2014) 29:939–40. doi: 10.1038/ki.2013.373
51. Zuber J, Frimat M, Caillard S, Kamar N, Gatault P, Petitprez F, et al. Use of highly individualized complement blockade has revolutionized clinical outcomes after kidney transplantation and renal epidemiology of atypical hemolytic uremic syndrome. J Am Soc Nephrol. (2019) 30:2449–63. doi: 10.1681/ASN.2019040331
52. Chua JS, Baelde HJ, Zandbergen M, Wilhelmus S, van Es LA, de Fijter JW, et al. Complement factor C4d is a common denominator in thrombotic microangiopathy. J Am Soc Nephrol. (2015) 26:2239–47. doi: 10.1681/ASN.2014050429
53. Naesens M, Li L, Ying L, Sansanwal P, Sigdel TK, Hsieh SC, et al. Expression of complement components differs between kidney allografts from living and deceased donors. JASN August. (2009) 20:1839–51. doi: 10.1681/ASN.2008111145
54. Damman J, Nijboer WN, Schuurs TA. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol Dial Transplant. (2011) 26:2345–54. doi: 10.1093/ndt/gfq717
55. Van Werkhoven MB, Damman J, van Dijk MCRF, Leuvenink HG, Morariu AM, Tullius SG, et al. Complement mediated renal inflammation induced by donor brain death: role of renal C5a-C5aR interaction. Am J Transplant. (2013) 13:875–82. doi: 10.1111/ajt.12130
56. de Vries DK, van der Pol P, van Anken GE, van Gijlswijk DJ, Damman J, Lindeman JH, et al. Acute but transient release of terminal complement complex after reperfusion in clinical kidney transplantation. Transplantation. (2013) 95:816–20. doi: 10.1097/TP.0b013e31827e31c9
57. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug induced thrombotic microangiopathy: a systematic review of published reports. Blood. (2015) 125:616–8. doi: 10.1182/blood-2014-11-611335
58. Renner B, Klawitter J, Goldberg R, McCullough JW, Ferreira VP, Cooper JE, et al. Cyclosporine induces endothelial cell release of complement-activating microparticles. J Am Soc Nephrol. (2013) 24:1849–62. doi: 10.1681/ASN.2012111064
59. Miriuka SG, Rao V, Peterson M, Tumiati L, Delgado DH, Mohan R, et al. mTOR inhibition induces endothelial progenitor cell death. Am J Transplant. (2006) 6:2069–79. doi: 10.1111/j.1600-6143.2006.01433
60. Keir LS, Firth R, Aponik L, Feitelberg D, Sakimoto S, Aguilar E, et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. J Clin Invest. (2017) 127:199–214. doi: 10.1172/JCI86418
61. Nava F, Cappelli G, Mori G, Granito M, Magnoni G, Botta C, et al. Everolimus, cyclosporine, and thrombotic microangiopathy: clinical role and preventive tools in renal transplantation. Transplant Proc. (2014) 46:2263–8. doi: 10.1016/j.transproceed.2014.07.062
62. Stegall MD, Chedid MF, Cornell LD. The role of complement in antibody-mediated rejection in kidney transplantation. Nat Rev Nephrol. (2012) 8:670–8. doi: 10.1038/nrneph.2012.212
63. Jeong HJ. Diagnosis of renal transplant rejection: Banff classification and beyond. Kid Res Clin Prac. (2020) 39:17–31. doi: 10.23876/j.krcp.20.003
64. Riedl M, Orth-Höller D, Würzner R. An update on the thrombotic microangiopathies hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP). Semin Thromb Hemost. (2014) 40:413–5. doi: 10.1055/s-0034-1376521
65. Wu K, Budde K, Schmidt D, Neumayer HH, Lehner L, Bamoulid J, et al. The inferior impact of antibody-mediated rejection on the clinical outcome of kidney allografts that develop de novo thrombotic microangiopathy. Clin Transplant. (2016) 30:105–17. doi: 10.1111/ctr.12645
66. Roberts DM, Jiang SH, Chadban SJ. The treatment of acute antibody-mediated rejection in kidney transplant recipients-a systematic review. Transplantation. (2012) 94:775–83. doi: 10.1097/TP.0b013e31825d1587
67. Johnson CK, Leca N. Eculizumab use in kidney transplantation. Curr Opin Organ Transplant. (2015) 20:643–51, doi: 10.1097/MOT.0000000000000249
68. Loupy A, Lefaucheur C. Antibody-mediated rejection of solid-organ allografts. N Engl J Med. (2018) 379:1150–60. doi: 10.1056/NEJMra1802677
69. De Keyzer K, van Laecke S, Peeters P, Vanholder R. De novo thrombotic microangiopathy induced by cytomegalovirus infection leading to renal allograft loss. Am J Nephrol. (2010) 32:491–6. doi: 10.1159/000321328
70. Olie KH, Goodship TH, Verlaak R, Florquin S, Groothoff JW, Strain L, et al. Posttransplantation cytomegalovirus-induced recurrence of atypical hemolytic uremic syndrome associated with a factor H mutation: Successful treatment with intensive plasma exchanges and ganciclovir. Am J Kidney Dis. (2005) 45:e12–e15. doi: 10.1053/j.ajkd.2004.09.012
71. Java A, Edwards A, Rossi A, Pandey R, Gaut J, De los Santos R„, et al. Cytomegalovirus-induced thrombotic microangiopathy after renal transplant successfully treated with eculizumab: Case report and review of the literature. Transpl Int. (2015) 28:1121–5. doi: 10.1111/tri.12582
72. Jeejeebhoy FM, Zaltzman JS. Thrombotic microangiopathy in association with cytomegalovirus infection in a renal transplant patient: A new treatment strategy. Transplantation. (1998) 65:1645–8. doi: 10.1097/00007890-199806270-00018
73. Rane S, Nada R, Minz M, Joshi K. Spectrum of cytomegalovirus-induced renal pathology in renal allograft recipients. Transplant Proc. (2012) 44:713–6. doi: 10.1016/j.transproceed.2011.11.052
74. Ardalan MR, Shoja MM, Tubbs RS, Jayne D. Parvovirus B19 microepidemic in renal transplant recipients with thrombotic microangiopathy and allograft vasculitis. Exp Clin Transplant. (2008) 6:137–43.
75. Waldman M, Kopp JB. Parvovirus-B19-associated complications in renal transplant recipients. Nat Clin Pract Nephrol. (2007) 3:540–50. doi: 10.1038/ncpneph0609
76. Prasad B, St Onge J. Parvovirus leading to thrombotic microangiopathy in a healthy adult. British Med J Case Rep. (2016) 2016:bcr2015213492. doi: 10.1136/bcr-2015-213492
77. Baid S, Pascual M, Williams WW Jr, Tolkoff-Rubin N, Johnson SM, Collins B, et al. Renal thrombotic microangiopathy associated with anticardiolipin antibodies in hepatitis C-positive renal allograft recipients. J Am Soc Nephrol. (1999) 10:146–53.
78. Baid-Agrawal S, Farris AB III, Pascual M, Mauiyyedi S, Farrell ML, Tolkoff-Rubin N, et al. Overlapping pathways to transplant glomerulopathy: chronic humoral rejection, hepatitis C infection, and thrombotic microangiopathy. Kidney Int. (2011) 80:879–85. doi: 10.1038/ki.2011.194
79. Yamazaki S, Takayama T, Inoue K, Higaki T, Makuuchi M, et al. Transplantation-related thrombotic microangiopathy triggered by preemptive therapy for hepatitis C virus infection. Transplantation. (2008) 86:1010–1. doi: 10.1097/TP.0b013e31818747d8
80. Dwyre DM, Bell AM, Siechen K, Sethi S, Raife TJ, et al. Disseminated histoplasmosis presenting as thrombotic microangiopathy. Transfusion. (2006) 46:1221–5. doi: 10.1111/j.1537-2995.2006.00873
81. Sethi S. Acute renal failure in a renal allograft: an unusual infectious cause of thrombotic microangiopathy. Am J Kidney Dis. (2005) 46:159–62. doi: 10.1053/j.ajkd.2004.11.026
82. Zanoni F, Alfieri C, Moroni G, Passerini P, Regalia A, Meneghini M, et al. Delayed diagnosis of West Nile virus infection in a kidney transplant patient due to inaccuracies in commonly available diagnostic tests. Exp Clin Transplant. (2020) 18:385–9. doi: 10.6002/ect.2018.0107
83. Canaud G, Kamar N, Anglicheau D, Esposito L, Rabant M, Noël LH, et al. Eculizumab improves postransplant thrombotic microangiopathy due to antiphospholipid syndrome recurrence but fails to prevent chronic vascular changes. Am J Transplant. (2013) 13:2179–85. doi: 10.1111/ajt.12319
84. Lonze BE, Zachary AA, Magro CM, Desai NM, Orandi BJ, Dagher NN, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. (2014) 14:459–65. doi: 10.1111/ajt.12540
85. Bakhtar O, Thajudeen B, Braunhut BL, Yost SE, Bracamonte ER, Sussman AN, et al. A case of thrombotic microangiopathy associated with antiphospholipid antibody syndrome successfully treated with eculizumab. Transplantation. (2014) 98:e17–e18. doi: 10.1097/TP.0000000000000267
86. Lorcy N, Rioux-Leclercq N, Lombard ML, Le Pogamp P, Vigneau C. Three kidneys, two diseases, one antibody? Nephrol Dial Transplant. (2011) 26:3811–13. doi: 10.1093/ndt/gfr436
87. Bouatou Y, Bacchi VF, Villard J, Moll S, Martin PY, Hadaya K, et al. Atypical Hemolytic Uremic Syndrome Recurrence after Renal Transplantation: C3-Glomerulonephritis as an Initial Presentation. Transplant Direct. (2015) 1: e9. doi: 10.1097/TXD.0000000000000518
88. Pham PT, Danovitch GM, Wilkinson AH, Gritsch HA, Pham PC, Eric TM, et al. Inhibitors of ADAMTS13: a potential factor in the cause of thrombotic microangiopathy in a renal allograft recipient. Transplantation. (2002) 74:1077–80. doi: 10.1097/00007890-200210270-00003
89. Palma LMP, Sridharan M, Sethi S. Complement in secondary thrombotic microangiopathy. Kidney Int Rep. (2021) 6:11–23. doi: 10.1016/j.ekir.2020.10.009
90. Dedhia P, Govil A, Mogilishetty G, Alloway RR, Woodle ES, Abu Jawdeh BG, et al. Eculizumab and belatacept for de novo atypical hemolytic uremic syndrome associated with CFHR3-CFHR1 deletion in a kidney transplant recipient: a case report. Transplant Proc. (2017) 49:188–92. doi: 10.1016/j.transproceed.2016.11.008
91. Merola J, Yoo PS, Schaub J, Smith JD, Rodriguez-Davalos MI, Tichy E, et al. Belatacept and eculizumab for treatment of calcineurin inhibitor-induced thrombotic microangiopathy after kidney transplantation: case report. Transplant Proc. (2016) 48:3106–8. doi: 10.1016/j.transproceed.2016.04.005
92. van der Zwan M, Hesselink DA, van den Hoogen MWF, Baan CC. Costimulation blockade in kidney transplant recipients. Drugs. (2020) 80:33–46. doi: 10.1007/s40265-019-01226-6
93. Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. (1991) 325:393–7. doi: 10.1056/NEJM199108083250604
94. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. (1991) 325:398–403. doi: 10.1056/NEJM199108083250605
95. Epperla N, Hemauer K, Hamadani M, Friedman KD, Baumann Kreuziger L. Impact of treatment and outcomes for patients with posttransplant drug-associated thrombotic microangiopathy. Transfusion. (2017) 57:2775–81. doi: 10.1111/trf.14263
96. Román E, Mendizábal S, Jarque I, de la Rubia J, Sempere A, Morales E, et al. Secondary thrombotic microangiopathy and eculizumab: A reasonable therapeutic option. Nefrologia. (2017) 37:478–91. doi: 10.1016/j.nefro.2017.01.006
97. Cavero T, Rabasco C, López A, Román E, Ávila A, Sevillano A, et al. Eculizumab in secondary atypical haemolytic uraemic syndrome. Nephrol Dial Transplant. (2017) 32:466–74. doi: 10.1093/ndt/gfw453
98. Portolés J, Huerta A, Arjona E, Gavela E, Agüera M, Jiménez C, et al. Characteristics, management and outcomes of atypical haemolytic uraemic syndrome in kidney transplant patients: a retrospective national study. Clin Kidney J. (2020) 93:1–8. doi: 10.1093/ckj/sfaa096
99. Matar D, Naqvi F, Racusen LC, Carter-Monroe N, Montgomery RA, Alachkar N. Atypical hemolytic uremic syndrome recurrence after kidney transplantation. Transplantation. (2014) 98:1205–12. doi: 10.1097/TP.0000000000000200
100. Legendre C, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolyticuremic syndrome. N Engl J Med. (2013) 368:2169–81. doi: 10.1056/NEJMoa1208981
101. Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. (2015) 87:1061–73. doi: 10.1038/ki.2014.42
102. Noris M, Remuzzi G. Managing and preventing atypical hemolytic uremic syndrome recurrence after kidney transplantation. Curr Opin Nephrol Hypertens. (2013) 22:704–12. doi: 10.1097/MNH.0b013e328365b3fe
103. Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V, French Study Group for aHUS/C3G. French Study Group for aHUS/C3G. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. (2012) 8:643–57. doi: 10.1038/nrneph.2012.214
104. Palomo M, Blasco M, Molina P, Lozano M, Praga M, Torramade-Moix S, et al. Complement activation and thrombotic microangiopathies. Clin J Am Soc Nephrol. (2019) 14:1719–32. doi: 10.2215/CJN.05830519
105. Shochet L, Kanellis J, Simpson I, Ta J, Mulley W, et al. De novo thrombotic microangiopathy following simultaneous pancreas and kidney transplantation managed with eculizumab. Nephrology. (2017) 22 Suppl 1:23–7. doi: 10.1111/nep.12936
106. Ikeda T, Okumi M, Unagami K, Kanzawa T, Sawada A, Kawanishi K, et al. Two cases of kidney transplantation associated thrombotic microangiopathy successfully treated with eculizumab. Nephrology. (2016) 21 Suppl 1:35–4. doi: 10.1111/nep.12768
107. Safa K, Logan MS, Batal I, Gabardi S, Rennke HG, Abdi R. Eculizumab for drug-induced de novo posttransplantation thrombotic microangiopathy: a case report. Clin Nephrol. (2015) 83:125–9.
108. Chandran S, Baxter-Lowe L, Olson JL, Tomlanovich SJ, Webber A. Eculizumab for the treatment of de novo thrombotic microangiopathy post simultaneous pancreas-kidney transplantation–a case report. Transplant Proc. (2011) 43:2097–101. doi: 10.1016/j.transproceed.2011.02.064
109. Wilson CH, Brown AL, White SA, Goodship THJ, Sheerin NS, Manas DM. Successful treatment of de novo posttransplant thrombotic microangiopathy with eculizumab. Transplantation. (2011) 92:e42–e3. doi: 10.1097/TP.0b013e318230c0bd
110. Hadaya K, Ferrari-Lacraz S, Fumeaux D, Boehlen F, Toso C, Moll S, et al. Eculizumab in acute recurrence of thrombotic microangiopathy after renal transplantation. Am J Transplant. (2011) 11:2523–7. doi: 10.1111/j.1600-6143.2011.03696
111. Godara A, Migliozzi DR, Pilichowska M, Goyal N, Varga C, Gordon CE. Use of eculizumab in transplant-associated thrombotic microangiopathy in a patient with polycystic kidney disease immediately post-kidney transplant: a case report. Kidney Med. (2020) 2:652–6. doi: 10.1016/j.xkme.2020.06.007
112. Roberts D, Siegman I, Andeen N, Woodland D, Deloughery T, Rueda J, et al. De novo thrombotic microangiopathy in two kidney transplant recipients from the same deceased donor: A case series. J. Clin Transplant. (2020) 34:e13885. doi: 10.1111/c
113. Reese JA, Bougie DW, Curtis BR, Terrell DR, Vesely SK, Aster RH, et al. Drug induced thrombotic microangiopathy: experience of the oklahoma registry and the bloodcenter of wisconsin. Am J Hematol. (2015) 90:406–10. doi: 10.1002/ajh.23960
114. Valotti E, Alberti M, Tortajada A, Garcia-Fernandez J, Gastoldi S, Besso L, et al. A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. Am J Soc Nephrol. (2015) 26:209–19. doi: 10.1681/ASN.2013121339
Keywords: thrombotic microangiopathy, kidney transplantation, atypical hemolytic uremic syndrome, eculizumab, complement system activation
Citation: Ávila A, Gavela E and Sancho A (2021) Thrombotic Microangiopathy After Kidney Transplantation: An Underdiagnosed and Potentially Reversible Entity. Front. Med. 8:642864. doi: 10.3389/fmed.2021.642864
Received: 16 December 2020; Accepted: 22 February 2021;
Published: 08 April 2021.
Edited by:
Carlo Garofalo, Università della Campania Luigi Vanvitelli, ItalyReviewed by:
Gaetano Alfano, University of Modena and Reggio Emilia, ItalyGaurav Gupta, Virginia Commonwealth University, United States
Copyright © 2021 Ávila, Gavela and Sancho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Ávila, YWFhYXZpbGFiQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work