 Jordi Bover1*†
Jordi Bover1*† Armando Aguilar2*†
Armando Aguilar2*† Carolt Arana1
Carolt Arana1 Pablo Molina3
Pablo Molina3 María Jesús Lloret1Jackson Ochoa1Gerson Berná1Yessica G. Gutiérrez-Maza2Natacha Rodrigues4
María Jesús Lloret1Jackson Ochoa1Gerson Berná1Yessica G. Gutiérrez-Maza2Natacha Rodrigues4 Luis D'Marco5
Luis D'Marco5 José L. Górriz5
José L. Górriz5- 1Department of Nephrology, Fundació Puigvert, IIB Sant Pau, Universitat Autònoma, REDinREN, Barcelona, Spain
- 2Department of Nephrology, Instituto Mexicano del Seguro Social, Hospital General de Zona No. 2, Tuxtla Gutiérrez, Mexico
- 3Department of Nephrology, Hospital Universitario Dr Peset, Universidad de Valencia, REDinREN, Valencia, Spain
- 4Division of Nephrology and Renal Transplantation, Department of Medicine, Centro Hospitalar Universitário Lisboa Norte, EPE, Lisboa, Portugal
- 5Servicio de Nefrología, Hospital Clínico Universitario, INCLIVA, Universidad de Valencia, Valencia, Spain
Chronic kidney disease (CKD) is associated with a very high morbimortality, mainly from cardiovascular origin, and CKD is currently considered in the high- or very high risk- cardiovascular risk category. CKD-mineral and bone disorders (CKD-MBDs), including vascular and/or valvular calcifications, are also associated with these poor outcomes. Vascular calcification (VC) is very prevalent (both intimal and medial), even in non-dialysis dependent patients, with a greater severity and more rapid progression. Simple X-ray based-scores such as Adragão's (AS) are useful prognostic tools and AS (even AS based on hand-X-ray only) may be superior to the classic Kauppila's score when evaluating non-dialysis CKD patients. Thus, in this mini-review, we briefly review CKD-MBD-related aspects of VC and its complex pathophysiology including the vast array of contributors and inhibitors. Furthermore, although VC is a surrogate marker and is not yet considered a treatment target, we consider that the presence of VC may be relevant in guiding therapeutic interventions, unless all patients are treated with the mindset of reducing the incidence or progression of VC with the currently available armamentarium. Avoiding phosphate loading, restricting calcium-based phosphate binders and high doses of vitamin D, and avoiding normalizing (within the normal limits for the assay) parathyroid hormone levels seem logical approaches. The availability of new drugs and future studies, including patients in early stages of CKD, may lead to significant improvements not only in patient risk stratification but also in attenuating the accelerated progression of VC in CKD.
Introduction: Chronic Kidney Disease And Cardiovascular Disease
Chronic kidney disease (CKD) has become a major public health problem because of the associated high mortality and elevated cost of treatment for dialysis patients. Worldwide, it is estimated that about 500 million adults suffer from CKD (1), but prevalence varies widely between countries (2). In Spain, CKD prevalence is 15.1% (3), is more common in men, in patients with cardiovascular disease (CVD) (39.8%), and increases with age (37.3% in those ≥65 years) (3). Given this significant link between CKD and CVD, CKD is currently considered an independent cardiovascular risk factor (4). A similar scenario is described for diabetes (5), a leading cause of CKD. Moreover, an independent, graded association was observed between a reduced estimated glomerular filtration rate (eGFR) and the risk of death, CVD, and hospitalization in a huge community-based population (6). Consequently, the European Society of Cardiology and European Atherosclerosis Society (ESC/EAS) recently considered moderate (eGFR 30–59 ml/min/1.73m2) and severe (eGFR <30 ml/min/1.73m2) CKD to be in the high- and the very high risk category of cardiovascular risk factors (7).
Since the seminal report by Foley et al. (8), who noted an extremely high cardiovascular mortality even in young dialysis patients, the existence of non-traditional cardiovascular risk factors, including mineral metabolism parameters, has caught the attention of many. Subclinical atherosclerosis is very frequent in CKD patients, and its evolution is also closely linked with CKD progression and related to several CKD-associated factors (9, 10). Thus, bone was recently described as a new endocrine organ “at the heart” of CKD and mineral-bone disorders (CKD-MBD) (11). Subsequently, the presence (12, 13) and progression (14, 15) of vascular and valvular calcification (16), which are also associated with “normal” aging (17), were clearly linked to prognosis (18–22), including in non-dialysis CKD (ND-CKD) patients, especially if the coronary artery calcification (CAC) score was evaluated (20–24). Therefore, CKD could be viewed conceptually as an accelerator of traditional cardiovascular risk factors and vascular calcification (VC) (17). Thus, in this review we will focus on the CKD-MBD aspects of VC including potentially helpful treatment considerations.
Chronic Kidney Disease-Mineral and Bone Disorders (Ckd-Mbd) and Vascular Calcification
The term CKD-MBD was first coined in 2006 (25). It described a systemic disorder due to CKD, manifested by mineral and bone abnormalities and/or extraskeletal calcifications (25). Several studies have demonstrated the high prevalence of cardiovascular calcifications in CKD, even in ND-CKD patients (20, 21, 26–30), with a greater severity (20, 31) and more rapid progression (32, 33). Furthermore, it has been suggested that these cardiovascular calcifications may be not only a useful prognostic tool but also relevant in guiding therapeutic interventions (19, 34, 35), as it will be reviewed in section Treatment Implications. However, we will first let readers understand the different diagnostic methods and the important contribution of CKD-MBD-related aspects in the pathophysiology of VC in CKD patients (section Pathophysiology of CKD-MBD-related Vascular Calcification).
In clinical studies, VC is frequently detected by multisliced computed tomography (CT) and less often by electron-beam CT (20, 36–38), multiterritorial vascular ultrasound (9), intravascular ultrasound (IVUS) or Virtual Histology® IVUS (39, 40), arteriography, or even positron emission tomography (PET) scans (41, 42). Thus, the prevalence of CAC has been recently systematically reviewed and authors found a variable prevalence ranging from 28 to 93% in predialysis patients (59% pooled prevalence) (20). High heterogeneity was present, at least partially related to ethnic and population differences (such as age or CKD stages) (20). However, simple X-rays may also be used despite their much lower sensitivity and variable associations with CAC (43). In fact, guidelines suggest that a lateral abdominal radiograph can be used to detect VC and an echocardiogram can be used to detect valvular calcifications, as reasonable alternatives to CT-based imaging (35, 44). These guidelines also suggest that patients with CKD G3a–G5D with known vascular or valvular calcification be considered at highest cardiovascular risk (evidence 2A) and that it is reasonable to use this information to guide CKD-MBD management (35, 44).
Lateral abdominal X-ray is helpful not only to semiquantify an aortic risk score such as Kauppila's (KS) (45) (Figure 1) but also to detect unnoticed vertebral fractures, with potential therapeutic renewed interest in CKD-associated osteoporosis guidelines (35, 44, 46, 47). Spanish CKD-MBD guidelines also suggest use of the simpler Adragão's score (AS) (19, 48) (Figure 1). In their original study in hemodialysis patients, an AS≥3 was independently associated with coronary, peripheral, and vascular disease (48). Importantly, patients with an AS≥3 had a 2–4-fold higher risk of cardiovascular mortality, hospitalizations, and fatal or non-fatal cardiovascular events. In another study, it was found that an AS>3 was also significantly associated with pulse wave velocity (PWV), pulse pressure, and lower adjusted cumulative survival[hazard ratio (HR) = 3.31)] (19).
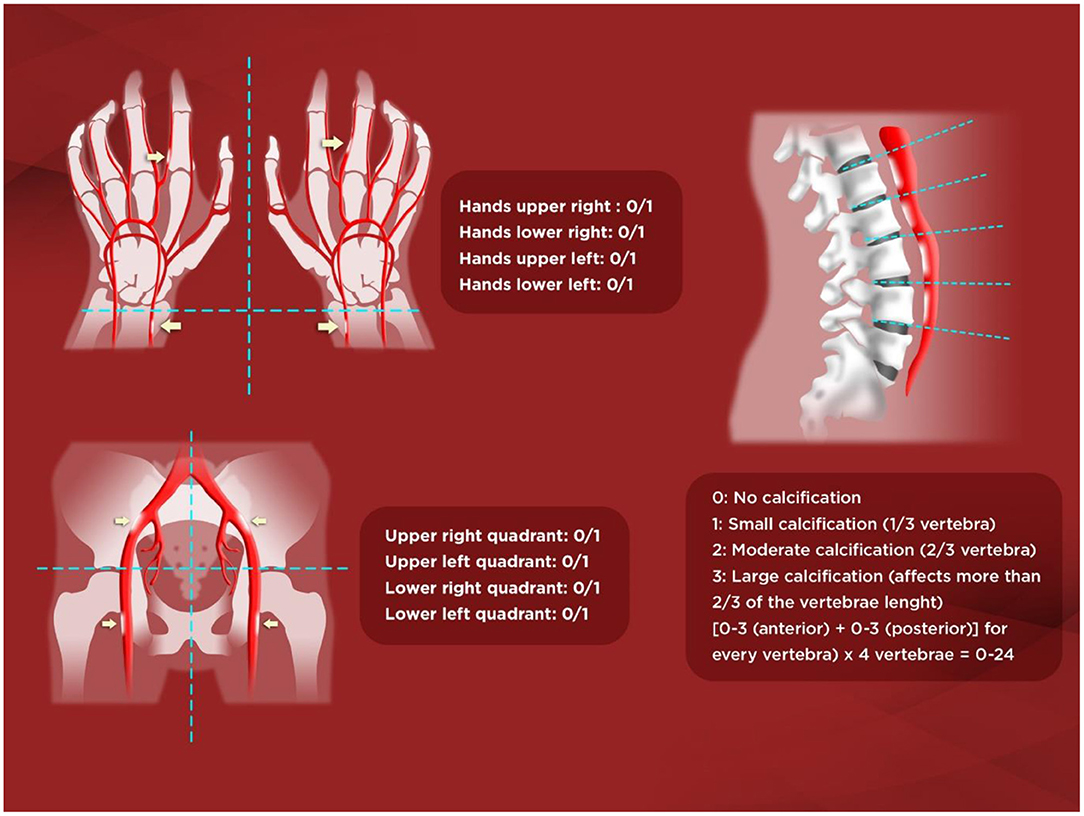
Figure 1. Adragão and Kauppila scores. Kauppila score (KS). The score is assigned from 1 to 3 [1: small calcification (1/3 of the vertebral length), 2: moderate (2/3), 3: large (> 2/3)] depending on the length of each plaque detected. The anterior and posterior part of the aorta shall be taken into consideration, associating them with the place where they are located, in front of the L1, L2, L3, or L4 vertebrae. With KS, a final score is achieved between 0 and 24 points. Adragão score (AS). The score is determined by the sum of the absence of calcification (0 points), or the presence of unilateral (1 point) or bilateral (2 points) linear calcification in each section. AS analyzes the calcification of iliac, femoral, radial, and digital arteries. The final value ranges from 0 to 8 points (0–4 in the pelvis and 0–4 in the hands).
In the observational multicentre OSERCE-2 study (26), we prospectively analyzed VC in a cohort of 742 ND-CKD G3–G5 patients. VC was present in 79%, and VC was severe (AS≥3 or KS>6) in 47%. Age, phosphate (P) levels, and diabetes were independently associated with AS≥3. After a median follow-up of 35 months, AS≥3 but not KS>6 was independently associated with all-cause (HR = 2.07) and cardiovascular (HR = 3.46) mortality, and a shorter hospitalization event-free period (HR = 1.14). Moreover, only AS based on hand-X-ray only (AS-hands) (Figure 1) showed a significant correlation with parathyroid hormone (PTH) levels and renal function. Interestingly, the group AS≥3 showed more than double the risk of all-cause mortality, whereas the risk increased 5-fold using AS-hands. Moreover, AS≥3 but not KS>6 independently predicted all-cause, cardiovascular mortality and hospitalization, and AS-hands ≥1 was also an independent predictor of cardiovascular mortality and hospitalization-related outcomes. Nevertheless, other authors have described significant associations with hard-outcomes in ND-CKD patients evaluated for aortic, pelvic or even breast artery calcifications, and significant differences were also present among distinct vascular beds and associated factors (43, 49–52).
We therefore confirmed the potentially better predictive ability of simple X-rays with the AS (vs. KS), and extended its value into the ND-CKD population. Moreover, the even simpler AS-hands demonstrated an important predictive ability, probably because the radial and cubital arteries are muscular arteries with a greater tendency to calcification of the medial layer (arteriosclerosis), especially affected in CKD, as opposed to the elastic abdominal aorta, which may be more prone to intimal calcification (atheromatosis) (26, 48, 53).
Medial and intimal VC lead to different comorbidities: medial calcification (Mönckeberg's disease) induces arterial stiffness and consequently an increased PWV, left ventricular hypertrophy and heart failure, whereas intimal calcification is associated with ischemic episodes and is pathophysiologically characterized by endothelial damage, lipid accumulation, and infiltration of inflammatory cells. These data support the concept that VC (especially in CKD patients) is not just a single entity but the consequence of a wide range of different biological processes (54), affecting both intimal and medial layers, and it is difficult to study separately because common radiological techniques do not allow clear distinction between them (55, 56). Reliable differentiation between medial and intimal VC can only be achieved pathomorphologically (57). Moreover, VC is not exclusive to CKD and is especially prevalent in diabetes and during the aging process (also accelerated in CKD patients) (17).
Nevertheless, while the high prevalence and prognostic value of VC in CKD patients are now well-established, the clinical usefulness of early screening is still controversial since it is far from proven that early diagnosis permits beneficial actions that improve outcomes (e.g., regarding different P binders or vitamin D (VD) derivatives).
Pathophysiology of CKD-MBD-Related Vascular Calcification
CKD-MBD-related factors are obviously not the only mechanisms responsible for VC but, as mentioned before, CKD acts as an accelerator of the VC process (17). Moreover, VC is not only a passive process of excess calcium (Ca) and P deposition (enhanced in CKD due to the loss of excretory function) but also an active process where transdifferentiation of vascular smooth muscle cells (VSMCs) from a contractile to a secretory calcific phenotype plays an important role (53, 54). This is probably magnified by CKD-MBD-related factors (11, 17). These VSMCs can also display adipogenic and macrophagic features (58–60). Moreover, adventitial Gli1+ mesenchymal stem-cells serve as progenitors of VSMC which migrate into the media and neointima during arteriosclerosis and atheromatosis (61).
Multiple physiopathological mechanisms and pathways are involved in the process of VC, but a comprehensive review is beyond the goals of this article, and we therefore refer interested readers to other references (62–67). Interestingly, some critical factors might play a double and sometimes opposite roles in terms of vascular health, such as the effects of high and low PTH levels on stem-cells (68, 69), or the roles of fibroblast growth factor-23 (FGF23) (70), VD (71), and calcimimetics (72). Importantly, in addition to a genetic background, the altered balance between calcification contributors (73–80) (Figure 2) and circulating or tissue calcification inhibitors (81–88) (Figure 2) makes CKD patients more or less susceptible to accelerated VC. Different comorbid pathologies associated with intimal atheroma calcification (ischemic events), arteriosclerosis (arterial stiffening), and heart valvular calcification may be overlapping, and CKD predisposes to all these types of VC (54, 64). Finally, the revolution in omics technology (genomics, epigenomics, transcriptomics, proteomics, and metabolomics) has resulted in the accumulation of cellular and molecular-level data (63, 67) which will provide novel approaches on the mechanisms associated with VC (63, 67). Although clinical application is still in its early stages, CKD may become an interesting model to analyse (17, 63), especially considering that as many as 17% of dialysis patients show no calcification (89).
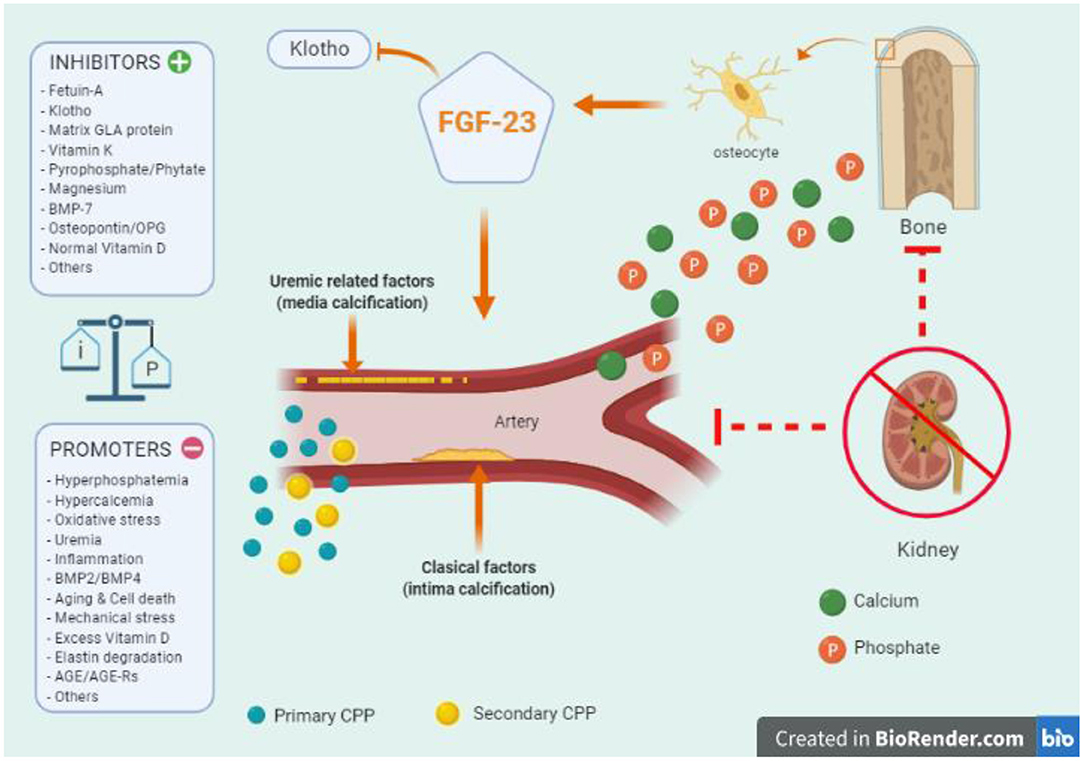
Figure 2. Key aspects of vascular calcification (VC) in chronic kidney disease (CKD): the important balance between calcification contributors and inhibitors (tissue or circulating factors) in the pathophysiology of vascular calcification. The altered balance between calcification contributors and inhibitors makes CKD patients more or less susceptible to accelerated calcifications. Of note, CKD patients exhibit both types of VC, intimal atheroma calcification, and medial arteriosclerosis. A key role in the initiation and propagation are calcium (green) and phosphate (orange) ions. The passive formation of calciprotein nanoparticles (CPP) act as active precursors of micro-calcification. Initially, the small calcium/phosphate complex can be removed by Fetuin-A, which can eventually become saturated leading to primary CPP (light blue) that can develop secondary CPP particles (yellow). Later, phosphate entries into vascular smooth muscle cells producing osteochondrocytic transdifferentiation (vascular “ossification”). Many of the contributors may also promote this phenotypic osteochondrocytic transdifferentiation. Among them, tissue non-specific alkaline phosphatase (TNAP), aldosterone, parathyroid hormone, activin-A, collagen I, osteocalcin, osteonectin, oxidized-low density lipoproteins, have been associated with vascular calcification. On the other hand, parathyroid hormone related-peptide, collagen IV, high density lipoproteins, nitric oxide, and others, have been described as potential inhibitors. At the bone level, the role of FGF-23 and the co-factor Klotho are relevant in all the process, and a genetic background (e.g., proteins derived from Ank, Npps, ENPP1 genes) should also be considered. BMP, Bone Morphogenetic Proteins; TNF-α, Tumor Necrosis Factor- α; IL, Interleukin; AGE, Advanced-Glication End-products; AGE-Rs, AGE-receptors.
Central to the increasing knowledge on mechanisms involved in VC is the discovery that calcifying extracellular vesicles act as active precursors of cardiovascular microcalcification in diverse vascular beds (90). Novel in vitro assays may quantify the propensity for VC in serum (as a composite measure capturing a global effect) by evaluating the semimaximal transformation time (T50) from the so-called primary to secondary calciprotein particles (CPP) when challenged with additional Ca and P (91, 92). Primary CPP are amorphous accumulations of Ca-P, and their transformation to secondary CPP, composed of crystalline Ca-P, may provide information about the contributor/inhibitor balance (91, 93). T50 has been associated not only with more severe CAC and progression in patients with CKD G2–G4, but also with cardiovascular and all-cause mortality in individuals with ND-CKD, hemodialysis and transplant patients (94–97). T50 could also reflect factors that promote intimal calcification (98) since secondary CPP stimulates inflammation and apoptosis of macrophages, which may promote ectopic calcification (99, 100). Actually, early stages of CKD were associated with local up-regulation of proinflammatory and pro-osteogenic molecules in the vascular wall and calcification of the aortic media layer (101). Moreover, Ca × P product correlated well with markers of inflammation, but not with calcification itself (101). In fact, Takx et al. (102) recently conducted a retrospective study including CKD-G3 patients and matched controls who underwent fluorodeoxyglucose-PET or CT imaging and concluded that moderate CKD is associated with arterial inflammation, independently of subclinical atherosclerosis. On the other hand, Joshi et al. (103) demonstrated the presence of VC at baseline to be associated with progressive calcification and suggested that VC can be the inducer of inflammation and not only its consequence. Of note, hyperphosphatemia, among other mineral metabolism parameters, has also been independently associated with inflammation and VC in CKD (104, 105). Thus, T50 may not directly reflect the initial pathophysiological pathway of VC but could theoretically help in clinical decisions on CKD-MBD-related treatments and/or identifying high-risk patients (97). Similarly, the so-called fetuin-A (VC inhibitor) reduction ratio was also evaluated as a potential parameter to measure extraosseous calcification stress (97, 106, 107).
Treatment Implications
Although VC is a surrogate marker and is not yet considered a treatment target in CKD (108), the presence of VC may have important therapeutic implications (34, 35, 44), unless all patients are treated with the mindset of reducing the incidence or progression of VC (i.e., using all minimization strategies such as reducing exogenous Ca and P).
Phosphate Binders
Several studies have shown that non-Ca-based P binders, mainly sevelamer in dialysis patients, attenuate the progression of valvular and VC as compared to Ca-based P binders (109–112). Non-Ca-based P binders have also been associated with improved survival (109, 113–115), although certainty of evidence is low (116). Russo et al. (117) randomized 90 non-diabetic ND-CKD G3-G5 patients without a history of CVD to three groups: low P-diet only, Ca-based P binders, or sevelamer. They monitored patients for an average of 2 years and found that CAC significantly increased in patients on the low-P diet alone, to a lesser extent in Ca-carbonate-treated patients, and not at all in sevelamer-treated patients (117). Moreover, Di Iorio et al. found in 212 patients with CKD G3-G4 that sevelamer provided benefits in all-cause mortality and in the composite end point of death or dialysis inception (118). Interestingly, considering only patients with CAC>0, a significant regression of CAC was observed in more patients treated with sevelamer than Ca-carbonate (24 vs. 2). Over 24 months, the final cumulative percentage of de novo onset of CAC was 12.8 and 81.8%, respectively (118). Consequently, recent guidelines (35, 44) increased the degree of evidence from 2C to 2B, suggesting that in adult patients with CKD G3a–G5D the dose of Ca-based P binders should be restricted.
In these guidelines, lowering elevated P levels only toward the normal range is also suggested, mainly because of an absence of data supporting the benefit of efforts to maintain P in the normal range (e.g., in G3a–G4 patients) and also some safety concerns (35, 119). Therefore, guidelines now suggest that treatment should only aim at overt hyperphosphatemia and emphasize that early “preventive” P-lowering treatment is currently not supported by data (35). Nevertheless, recent studies have analyzed the potential of “preventive” treatment of early P loading in CKD patients (120, 121). Bouma-de Krijger et al. (120) found that sevelamer carbonate for 8 weeks did not induce a significant reduction in PWV and that serum FGF23 did not decrease despite a decline in 24-h urine P excretion. These findings challenged to some extent the concept that “preventive” P binder therapy is a useful approach, at least over this short period. Interestingly, in a subgroup of patients with absent or limited abdominal VC, treatment did result in a statistically significant reduction in adjusted PWV, suggesting that PWV is amenable to improvement in this subset (120, 122). On the other hand, Toussaint et al. (121) recently reported that lanthanum carbonate did not affect arterial stiffness or VC in CKD G3b-4 patients; however, an absence of a significant decline in 24-h urine P excretion seems difficult to explain. Therefore, interpretation of the scarce and heterogeneous observations described in early CKD remains difficult, and the possibility of beneficial effects of a “preventive” treatment may not yet be completely disregarded (122). Finally, significant beneficial effects of a fixed dose of ferric citrate in advanced ND-CKD patients merit further study (123), since it was not a placebo-controlled randomized clinical trial (RCT), about 1/3 of patients in the “standard of care” arm took Ca-based P binders, and the outcome was a composite end-point.
Treatment for Hyperparathyroidism
Recent guidelines also state that in patients with ND-CKD G3a–G5, the optimal PTH level is not known (35, 44). However, an association between improvement of PTH levels with all-cause mortality has not only been described in dialysis but also in ND-CKD patients (124–126). Guidelines also suggest that patients with intact PTH levels progressively rising or persistently above the assay's upper normal limit should be evaluated for modifiable factors, including hyperphosphatemia, hypocalcemia, high P intake, and VD deficiency (evidence 2C) (35). In fact, low calcidiol levels represent a novel cardiovascular risk marker and have been directly associated with the presence and progression of VC and decreased survival (127, 128). VD may have atheroprotective effects; however, clinical studies do not show improved survival with VD administration (129).
Supplementation with native (nutritional) VD has been shown to have beneficial effects on several biological parameters, even in dialysis patients (130, 131), but a significant RCT with positive hard-outcomes is lacking. On the other hand, in adult patients with ND-CKD G3a–G5, recent guidelines (35) suggest that calcitriol and VD analogs should not now be routinely used (evidence 2C), now considering reasonable to reserve them for patients with CKD G4–G5 with severe and progressive hyperparathyroidism (35). Recent RCTs of VD analogs failed to demonstrate improvements in clinically relevant outcomes and an increased risk of hypercalcemia (132, 133). However, very high doses of paricalcitol and/or Ca-based P binders were frequently used in those studies (132, 133). All guidelines agree that modest increases in PTH may represent an appropriate adaptive response in CKD and “progressively rising” PTH levels (trends) rather than PTH “above the upper normal limit” should be considered for treatment (35, 44). Nevertheless, other guidelines do not consider it acceptable to wait until severe hyperparathyroidism is present, and physicians are then advised to avoid hypercalcemia and/or hyperphosphatemia but not to aim for complete normalization of PTH (44, 134). In experimental animals, low clinically relevant dosages of calcitriol and paricalcitol seem to protect against CKD-related VC (135). Paricalcitol and calcitriol at equipotent doses also showed different effects on VC (136, 137), and distinct positive pleiotropic effects and survival benefits were described in retrospective studies in dialysis patients (138, 139); however, they have not been demonstrated in RCTs.
On the other hand, calcimimetics are important contributors to the achievement of biochemical treatment goals (especially Ca and PTH, but also P) in CKD-MBD (140, 141). Calcimimetics have been shown to decrease VC in experimental uremic animals (142–145) and, in the ADVANCE study, Raggi et al. demonstrated that in hemodialysis patients with secondary hyperparathyroidism, cinacalcet plus low-dose VD may attenuate cardiac valve and VC (37). In fact, calcimimetics do not only correct PTH hypersecretion but also modulate the vascular Ca-sensor activity (72). However, cinacalcet has not been approved in ND-CKD patients, although it can be prescribed in patients with primary hyperparathyroidism (with or without CKD) as an alternative or bridging therapy to treat hypercalcemia (146–148).
Finally, the fact that a statin paradox exists (statins promoted coronary atheroma calcification but improved clinical outcomes) (149) means that the implication of VC may be reconsidered (150). Nevertheless, many experimental and/or preliminary clinical studies on VC with different compounds are ongoing (151–160). Thus, other factors (directly or indirectly related with CKD-MBD) such as magnesium, vitamin K, inhibitors of intestinal P transporters, bisphosphonates, denosumab, sodium thiosulfate, alkaline phosphatase inhibitors, apabetalone, sotatercept, recombinant BMP-7, as well as metformin, antioxidants, spironolactone, senolytics, zinc, and the recently tested SNF472 (35, 161, 162) have been shown to potentially attenuate VC. However, as it has been mentioned before, prospective RCTs on hard-outcomes are still lacking with any treatment.
Conclusion
CKD-MBD is a common complication and contributes to the high morbimortality (mainly cardiovascular) in CKD patients. The prevalence of VC (intimal and medial), even in early stages of CKD, is very high and progresses rapidly. Although VC may not be a direct cause of CVD, it may reflect underlying pathophysiologic derangements and does have deleterious hemodynamic consequences linked to a negative prognosis. Several common treatments may increase or attenuate its natural progression; therefore, CKD-MBD treatment requires an integral approach, addressing all its components, although clear evidence that this strategy improves hard outcomes is lacking. Meanwhile, it seems logical to avoid P loading, restrict Ca-based P-binders, avoid high doses of VD, and avoid normalizing PTH levels. The availability of new drugs and future studies including early CKD patients may lead to significant improvements not only in patient stratification but also in slowing the natural progression of VC.
Author Contributions
JB was invited as an expert in the field as co-coordinator of the Spanish Nephrology guidelines and experience in the field of chronic kidney disease and mineral and bone disorders (pathophysiology and treatment), contributed to the conceptualization and writing and editing of the manuscript. AA contributed to the conceptualization, search and selection of the most important references, and writing and editing of the manuscript, as well as creation of illustrative Figure 1. CA contributed to the search and selection of the most important references and editing of the manuscript. PM, ML, JO, GB, YG-M, and NR contributed to the conceptualization and review and editing of the manuscript. LD'M contributed to the conceptualization and review and editing of the manuscript, as well as the creation of illustrative Figure 2. JG contributed to the conceptualization, review and editing of the manuscript, and he is the first author of the main contribution to the field of vascular calcification in non-dialysis dependent CKD patients.
Conflict of Interest
JB declares advisory, lecture fees, and/or travel funding from Amgen, Abbvie, Sanofi-Genzyme, Shire, Vifor-Fresenius-Renal Pharma, Rubió, and Sanifit. AA declares lecture fees and/or travel funding from Amgen. PM acknowledges consultation or speaker honoraria from Abbott Nutrition, Amgen, Fresenius-Kabi, Nutricia, Palex, Sanofi-Genzyme, and ViforPharma. ML declares lecture fees from Abbvie and Vifor-Fresenius-Renal Pharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JB belongs to the Spanish Red Nacional RedinRen (RD06/0016/0001 and RD12/0021/0033) and the Red de Biobancos Nacional Española (RD09/0076/00064), as well as to the Catalan Grupo Catalán de Investigación AGAUR (2009 SGR-1116). Collaborations with the Fundation Iñigo Alvarez de Toledo (FRIAT) have also been arranged. We also thank Mr. Ricardo Pellejero for his priceless bibliographic assistance.
References
1. Mills KT, Xu Y, Zhang W, Bundy JD, Chen C-S, Kelly TN, et al. A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int. (2015) 88:950–7. doi: 10.1038/ki.2015.230
2. Brück K, Stel VS, Gambaro G, Hallan S, Völzke H, Ärnlöv J, et al. On behalf of the European CKD Burden Consortium. CKD prevalence varies across the European general population. J Am Soc Nephrol. (2016) 27:2135–47. doi: 10.1681/ASN.2015050542
3. Gorostidi M, Sánchez-Martínez M, Ruilope LM, Graciani A, de la Cruz JJ, Santamaría R, et al. Prevalencia de enfermedad renal crónica en España: impacto de la acumulación de factores de riesgo cardiovascular. Nefrologia. (2018) 38:606–15. doi: 10.1016/j.nefro.2018.04.004
4. Briasoulis A, Bakris GL. Chronic kidney disease as a coronary artery disease risk equivalent. Curr Cardiol Rep. (2013) 15:340. doi: 10.1007/s11886-012-0340-4
5. Hajar R. Diabetes as “Coronary Artery Disease Risk Equivalent”: a historical perspective. Heart Views. (2017) 18:34–7 doi: 10.4103/HEARTVIEWS.HEARTVIEWS_37_17
6. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu C-Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. (2004) 351:1296–305. doi: 10.1056/NEJMoa041031
7. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. (2020) 41:111–88. doi: 10.15829/1560-4071-2020-3826
8. Foley RN, Parfrey PS, Sarnak MJ. Epidemiology of cardiovascular disease in chronic renal disease. J Am Soc Nephrol. (1998) 9:S16–23.
9. Valdivielso JM, Rodríguez-Puyol D, Pascual J, Barrios C, Bermúdez-López M, Sánchez-Niño MD, et al. Atherosclerosis in chronic kidney disease: more, less, or just different? Arterioscler Thromb Vasc Biol. (2019) 39:1938–66. doi: 10.1161/ATVBAHA.119.312705
10. Palanca A, Castelblanco E, Betriu À, Perpiñán H, Soldevila B, Valdivielso JM, et al. Subclinical atherosclerosis burden predicts cardiovascular events in individuals with diabetes and chronic kidney disease. Cardiovasc Diabetol. (2019) 18:93. doi: 10.1186/s12933-019-0897-y
11. Vervloet MG, Massy ZA, Brandenburg VM, Mazzaferro S, Cozzolino M, Ureña-Torres P, et al. Bone: a new endocrine organ at the heart of chronic kidney disease and mineral and bone disorders. Lancet Diabetes Endocrinol. (2014) 2:427–36. doi: 10.1016/S2213-8587(14)70059-2
12. Detrano R, Guerci AD, Carr JJ, Bild DE, Burke G, Folsom AR, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. (2008) 358:1336–45. doi: 10.1056/NEJMoa072100
13. Rennenberg R, Kessels Schurgers, Engelshoven V, de Leeuw P, Kroon. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. (2009) 5:185–97. doi: 10.2147/VHRM.S4822
14. Budoff MJ, Hokanson JE, Nasir K, Shaw LJ, Kinney GL, Chow D, et al. Progression of coronary artery calcium predicts all-cause mortality. JACC Cardiovasc Imaging. (2010) 3:1229–36. doi: 10.1016/j.jcmg.2010.08.018
15. Budoff MJ, Young R, Lopez VA, Kronmal RA, Nasir K, Blumenthal RS, et al. Progression of coronary calcium and incident coronary heart disease events: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol. (2013) 61:1231–9. doi: 10.1016/j.jacc.2012.12.035
16. Ureña-Torres P, D'Marco L, Raggi P, García-Moll X, Brandenburg V, Mazzaferro S, et al. Valvular heart disease and calcification in CKD: more common than appreciated. Nephrol Dial Transplant. (2020) 35:2046–53. doi: 10.1093/ndt/gfz133
17. Covic A, Vervloet M, Massy ZA, Torres PU, Goldsmith D, Brandenburg V, et al. Bone and mineral disorders in chronic kidney disease: implications for cardiovascular health and ageing in the general population. Lancet Diabetes Endocrinol. (2018) 6:319–31. doi: 10.1016/S2213-8587(17)30310-8
18. Bover J, Evenepoel P, Urena-Torres P, Vervloet MG, Brandenburg V, Mazzaferro S, et al. Pro: cardiovascular calcifications are clinically relevant. Nephrol Dial Transplant. (2015) 30:345–51. doi: 10.1093/ndt/gfv020
19. Adragao T, Pires A, Birne R, Curto JD, Lucas C, Goncalves M, et al. A plain X-ray vascular calcification score is associated with arterial stiffness and mortality in dialysis patients. Nephrol Dial Transplant. (2008) 24:997–1002 doi: 10.1093/ndt/gfn584
20. Wang XR, Zhang JJ, Xu XX, WU YG. Prevalence of coronary artery calcification and its association with mortality, cardiovascular events in patients with chronic kidney disease: a systematic review and meta-analysis. Ren Fail. (2019) 41:244–56. doi: 10.1080/0886022X.2019.1595646
21. Cano-Megías M, Guisado-Vasco P, Bouarich H, de Arriba-de la Fuente G, de Sequera-Ortiz P, Álvarez-Sanz C, et al. Coronary calcification as a predictor of cardiovascular mortality in advanced chronic kidney disease: a prospective long-term follow-up study. BMC Nephrol. (2019) 20:188. doi: 10.1186/s12882-019-1367-1
22. Chen J, Budoff MJ, Reilly MP, Yang W, Rosas SE, Rahman M, et al. Coronary artery calcification and risk of cardiovascular disease and death among patients with chronic kidney disease. JAMA Cardiol. (2017) 2:635–43. doi: 10.1001/jamacardio.2017.0363
23. Watanabe R, Lemos MM, Manfredi SR, Draibe SA, Canziani MEF. Impact of cardiovascular calcification in nondialyzed patients after 24 months of follow-up. Clin J Am Soc Nephrol. (2010) 5:189–94. doi: 10.2215/CJN.06240909
24. Matsushita K, Sang Y, Ballew SH, Shlipak M, Katz R, Rosas SE, et al. Subclinical atherosclerosis measures for cardiovascular prediction in CKD. J Am Soc Nephrol. (2015) 26:439–47. doi: 10.1681/ASN.2014020173
25. Moe S, Drüeke T, Cunningham J, Goodman W, Martin K, Olgaard K, et al. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. (2006) 69:1945–53. doi: 10.1038/sj.ki.5000414
26. Górriz JL, Molina P, Cerverón MJ, Vila R, Bover J, Nieto J, et al. Vascular calcification in patients with nondialysis CKD over 3 years. Clin J Am Soc Nephrol. (2015) 10:654–66. doi: 10.2215/CJN.07450714
27. García-Canton C, Bosch E, Ramírez A, Gonzalez Y, Auyanet I, Guerra R, et al. Vascular calcification and 25-hydroxyvitamin D levels in non-dialysis patients with chronic kidney disease stages 4 and 5. Nephrol Dial Transplant. (2011) 26:2250–6. doi: 10.1093/ndt/gfq650
28. Sigrist MK, Taal MW, Bungay P, McIntyre CW. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin J Am Soc Nephrol. (2007) 2:1241–8. doi: 10.2215/CJN.02190507
29. Russo D, Palmiero G, De Blasio AP, Balletta MM, Andreucci VE. Coronary artery calcification in patients with CRF not undergoing dialysis. Am J Kidney Dis. (2004) 44:1024–30. doi: 10.1053/j.ajkd.2004.07.022
30. Garland JS, Holden RM, Groome PA, Lam M, Nolan RL, Ross Morton A, et al. Prevalence and associations of coronary artery calcification in patients with stages 3 to 5 CKD without cardiovascular disease. Am J Kidney Dis. (2008) 52:849–58. doi: 10.1053/j.ajkd.2008.04.012
31. Budoff MJ, Rader DJ, Reilly MP, Mohler ER 3rd, Lash J, Yang W, et al. Relationship of estimated GFR and coronary artery calcification in the CRIC (Chronic Renal Insufficiency Cohort) Study. Am J Kidney Dis. (2011) 58:519–26. doi: 10.1053/j.ajkd.2011.04.024
32. Kestenbaum BR, Adeney KL, de Boer IH, Ix JH, Shlipak MG, Siscovick DS. Incidence and progression of coronary calcification in chronic kidney disease: the Multi-Ethnic Study of Atherosclerosis. Kidney Int. (2009) 76:991–8. doi: 10.1038/ki.2009.298
33. Kronmal RA, McClelland RL, Detrano R, Shea S, Lima JA, Cushman M, et al. Risk factors for the progression of coronary artery calcification in asymptomatic subjects: results from the Multi-Ethnic Study of Atherosclerosis (MESA). Circulation. (2007) 115:2722–30. doi: 10.1161/CIRCULATIONAHA.106.674143
34. Bover J, Ureña-Torres P, Lloret MJ, Ruiz-García C, DaSilva I, Diaz-Encarnacion MM, et al. Integral pharmacological management of bone mineral disorders in chronic kidney disease (part I): from treatment of P imbalance to control of PTH and prevention of progression of cardiovascular calcification. Expert Opin Pharmacother. (2016) 17:1247–58. doi: 10.1080/14656566.2016.1182155
35. Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017. Clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. (2017) 7:1–59. doi: 10.1016/j.kisu.2017.04.001
36. Callister TQ, Cooil B, Raya SP, Lippolis NJ, Russo DJ, Raggi P. Coronary artery disease: improved reproducibility of calcium scoring with an electron-beam CT volumetric method. Radiology. (1998) 208:807–14. doi: 10.1148/radiology.208.3.9722864
37. Raggi P, Chertow GM, Torres PU, Csiky B, Naso A, Nossuli K, et al. The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant. (2011) 26:1327–39. doi: 10.1093/ndt/gfq725
38. Rumberger JA, Simons DB, Fitzpatrick LA, Sheedy PF, Schwartz RS. Coronary artery calcium area by electron-beam computed tomography and coronary atherosclerotic plaque area. a histopathologic correlative study. Circulation. (1995) 92:2157–62. doi: 10.1161/01.CIR.92.8.2157
39. Tuzcu EM, Murat Tuzcu E, Berkalp B, De Franco AC, Ellis SG, Goormastic M, et al. The dilemma of diagnosing coronary calcification: angiography versus intravascular ultrasound. J Am Coll Cardiol. (1996) 27:832–8. doi: 10.1016/0735-1097(95)00537-4
40. Nair A, Kuban BD, Tuzcu EM, Schoenhagen P, Nissen SE, Vince DG. Coronary plaque classification with intravascular ultrasound radiofrequency data analysis. Circulation. (2002) 106:2200–6. doi: 10.1161/01.CIR.0000035654.18341.5E
41. Dweck MR, Chow MWL, Joshi NV, Williams MC, Jones C, Fletcher AM, et al. Coronary arterial 18F-sodium fluoride uptake: a novel marker of plaque biology. J Am Coll Cardiol. (2012) 59:1539–48. doi: 10.1016/j.jacc.2011.12.037
42. Joshi NV, Vesey AT, Williams MC, Shah ASV, Calvert PA, Craighead FHM, et al. 18F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: a prospective clinical trial. Lancet. (2014) 383:705–13. doi: 10.1016/S0140-6736(13)61754-7
43. Mizuiri S, Nishizawa Y, Yamashita K, Mizuno K, Ishine M, Doi S, et al. Coronary artery calcification score and common iliac artery calcification score in non-dialysis CKD patients. Nephrology. (2018) 23:837–45. doi: 10.1111/nep.13113
44. Torregrosa V, Bover J, Rodriguez M, Gonzalez-Parra E, Arenas MD, Caravaca F, et al. Spanish Society of Nephrology recommendations for controlling mineral and bone disorder in chronic kidney disease patients (S.E.N.-M.B.D.). Nefrologia. (2021).
45. Kauppila LI, Polak JF, Cupples LA, Hannan MT, Kiel DP, Wilson PW. New indices to classify location, severity and progression of calcific lesions in the abdominal aorta: a 25-year follow-up study. Atherosclerosis. (1997) 132:245–50. doi: 10.1016/S0021-9150(97)00106-8
46. Evenepoel P, Cunningham J, Ferrari S, Haarhaus M, Javaid MK, Lafage-Proust M-H, et al. European Consensus Statement on the diagnosis and management of osteoporosis in chronic kidney disease stages G4-G5D. Nephrol Dial Transplant. (2021) 36:42–59. doi: 10.1093/ndt/gfaa192
47. Bover J, Ureña-Torres P, Torregrosa J-V, Rodríguez-García M, Castro-Alonso C, Górriz JL, et al. Osteoporosis, bone mineral density and CKD-MBD complex (I): diagnostic considerations. Nefrologia. (2018) 38:476–90. doi: 10.1016/j.nefroe.2018.10.005
48. Adragao T, Pires A, Lucas C, Birne R, Magalhaes L, Goncalves M, et al. A simple vascular calcification score predicts cardiovascular risk in haemodialysis patients. Nephrol Dial Transplant. (2004) 19:1480–8. doi: 10.1093/ndt/gfh217
49. Hwang HS, Kim SY, Hong YA, Cho WK, Chang YK, Shin SJ, et al. Clinical impact of coexisting retinopathy and vascular calcification on chronic kidney disease progression and cardiovascular events. Nutr Metab Cardiovasc Dis. (2016) 26:590–96. doi: 10.1016/j.numecd.2016.02.005
50. Disthabanchong S, Vipattawat K, Phakdeekitcharoen B, Kitiyakara C, Sumethkul V. Abdominal aorta and pelvic artery calcifications on plain radiographs may predict mortality in chronic kidney disease, hemodialysis and renal transplantation. Int Urol Nephrol. (2018) 50:355–64. doi: 10.1007/s11255-017-1758-9
51. Zhou Y, Hellberg M, Kouidi E, Deligiannis A, Höglund P, Clyne N. Relationships between abdominal aortic calcification, glomerular filtration rate, and cardiovascular risk factors in patients with non-dialysis dependent chronic kidney disease. Clin Nephrol. (2018) 90:380–9. doi: 10.5414/CN109441
52. Hassan NA, D'Orsi ET, D'Orsi CJ, O'Neill WC. The risk for medial arterial calcification in CKD. Clin J Am Soc Nephrol. (2012) 7:275–9. doi: 10.2215/CJN.06490711
53. Coll B, Betriu A, Martínez-Alonso M, Amoedo ML, Arcidiacono MV, Borras M, et al. Large artery calcification on dialysis patients is located in the intima and related to atherosclerosis. Clin J Am Soc Nephrol. (2011) 6:303–10. doi: 10.2215/CJN.04290510
54. London GM. Cardiovascular calcifications in uremic patients: clinical impact on cardiovascular function. J Am Soc Nephrol. (2003) 14(9 Suppl. 4):S305–9. doi: 10.1097/01.ASN.0000081664.65772.EB
55. Drüeke TB, Massy ZA. Chronic kidney disease: medial or intimal calcification in CKD-does it matter? Nat Rev Nephrol. (2011) 7:250–1. doi: 10.1038/nrneph.2011.41
56. Zwakenberg SR, de Jong PA, Hendriks EJ, Westerink J, Spiering W, de Borst GJ, et al. Intimal and medial calcification in relation to cardiovascular risk factors. PLoS ONE. (2020) 15:e0235228. doi: 10.1371/journal.pone.0235228
57. Amann K. Media calcification and intima calcification are distinct entities in chronic kidney disease. Clin J Am Soc Nephrol. (2008) 3:1599–605. doi: 10.2215/CJN.02120508
58. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
59. Metz RP, Patterson JL, Wilson E. Vascular smooth muscle cells: isolation, culture, and characterization. Methods Mol Biol. (2012) 843:169–76. doi: 10.1007/978-1-61779-523-7_16
60. Sinha S, Iyer D, Granata A. Embryonic origins of human vascular smooth muscle cells: implications for in vitro modeling and clinical application. Cell Mol Life Sci. (2014) 71:2271–88. doi: 10.1007/s00018-013-1554-3
61. Kramann R, Goettsch C, Wongboonsin J, Iwata H, Schneider RK, Kuppe C, et al. Adventitial MSC-like cells are progenitors of vascular smooth muscle cells and drive vascular calcification in chronic kidney disease. Cell Stem Cell. (2016) 19:628–42. doi: 10.1016/j.stem.2016.08.001
62. Jono S, Shioi A, Ikari Y, Nishizawa Y. Vascular calcification in chronic kidney disease. J Bone Miner Metab. (2006) 24:176–81. doi: 10.1007/s00774-005-0668-6
63. Duan M, Zhao W-L, Zhou L, Novák P, Zhu X, Yin K. Omics research in vascular calcification. Clin Chim Acta. (2020) 511:319–28. doi: 10.1016/j.cca.2020.10.022
64. Zaker B, Ardalan M. Vascular calcification; stony bridge between kidney and heart. J Cardiovasc Thorac Res. (2020) 12:165–71. doi: 10.34172/jcvtr.2020.29
65. Disthabanchong S, Srisuwarn P. mechanisms of vascular calcification in kidney disease. Adv Chronic Kidney Dis. (2019) 26:417–26. doi: 10.1053/j.ackd.2019.08.014
66. Voelkl J, Lang F, Eckardt K-U, Amann K, Kuro-o M, Pasch A, et al. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci. (2019) 76:2077–91. doi: 10.1007/s00018-019-03054-z
67. Lee SJ, Lee IJ, Jeon JH. Vascular calcification- new insights into its mechanism. Int J Mol Sci. (2020) 21:2685. doi: 10.3390/ijms21082685
68. Brunner S, Weinberger T, Huber BC, Segeth A, Zaruba MM, Theiss HD, et al. The cardioprotective effects of parathyroid hormone are independent of endogenous granulocyte-colony stimulating factor release. Cardiovasc Res. (2012) 93:330–9.68. doi: 10.1093/cvr/cvr303
69. Cianciolo G, Capelli I, Cappuccilli M, Schillaci R, Cozzolino M, La Manna G. Calcifying circulating cells: an uncharted area in the setting of vascular calcification in CKD patients. Clin Kidney J. (2016) 9:280–6. doi: 10.1093/ckj/sfv145
70. Cianciolo G, Galassi A, Capelli I, Schillaci R, La Manna G, Cozzolino M. Klotho-FGF23, cardiovascular disease, and vascular calcification: black or white?. Curr Vasc Pharmacol. (2018) 16:143–56. doi: 10.2174/1570161115666170310092202
71. Razzaque MS. The dualistic role of vitamin D in vascular calcifications. Kidney Int. (2011) 79:708–14. doi: 10.1038/ki.2010.432
72. Hénaut L, Boudot C, Massy ZA, Lopez-Fernandez I, Dupont S, Mary A, et al. Calcimimetics increase CaSR expression and reduce mineralization in vascular smooth muscle cells: mechanisms of action. Cardiovasc Res. (2014) 101:256–65. doi: 10.1093/cvr/cvt249
73. Moe SM, Chen NX. Mechanisms of vascular calcification in chronic kidney disease. J Am Soc Nephrol. (2008) 19:213–6. doi: 10.1681/ASN.2007080854
74. Lau WL, Pai A, Moe SM, Giachelli CM. Direct effects of phosphate on vascular cell function. Adv Chronic Kidney Dis. (2011) 18:105–12. doi: 10.1053/j.ackd.2010.12.002
75. Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol. (2009) 297:H1673–84. doi: 10.1152/ajpheart.00455.2009
76. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. (2008) 283:15319–27. doi: 10.1074/jbc.M800021200
77. Coscas R, Bensussan M, Jacob M-P, Louedec L, Massy Z, Sadoine J, et al. Free DNA precipitates calcium phosphate apatite crystals in the arterial wall in vivo. Atherosclerosis. (2017) 259:60–7. doi: 10.1016/j.atherosclerosis.2017.03.005
78. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. (2015) 21:628–37. doi: 10.1038/nm.3866
79. Pai A, Leaf EM, El-Abbadi M, Giachelli CM. Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am J Pathol. (2011) 178:764–73. doi: 10.1016/j.ajpath.2010.10.006
80. Neves KR, Graciolli FG, dos Reis LM, Graciolli RG, Neves CL, Magalhães AO, et al. Vascular calcification: contribution of parathyroid hormone in renal failure. Kidney Int. (2007) 71:1262–70. doi: 10.1038/sj.ki.5002241
81. Schurgers LJ, Cranenburg ECM, Vermeer C. Matrix Gla-protein: the calcification inhibitor in need of vitamin K. Thromb Haemost. (2008) 100:593–603. doi: 10.1160/TH08-02-0087
82. Paloian NJ, Leaf EM, Giachelli CM. Osteopontin protects against high phosphate-induced nephrocalcinosis and vascular calcification. Kidney Int. (2016) 89:1027–36. doi: 10.1016/j.kint.2015.12.046
83. Davies MR, Lund RJ, Hruska KA. BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J Am Soc Nephrol. (2003) 14:1559–67 doi: 10.1097/01.ASN.0000068404.57780.DD
84. Lomashvili KA, Narisawa S, Millán JL, O'Neill WC. Vascular calcification is dependent on plasma levels of pyrophosphate. Kidney Int. (2014) 85:1351–6. doi: 10.1038/ki.2013.521
85. Chang JR, Guo J, Wang Y, Hou YL, Lu WW, Zhang JS, et al. Intermedin1-53 attenuates vascular calcification in rats with chronic kidney disease by upregulation of?-Klotho. Kidney Int. (2016) 89:586–600. doi: 10.1016/j.kint.2015.12.029
86. Bressendorff I, Hansen D, Schou M, Pasch A, Brandi L. The effect of increasing dialysate magnesium on serum calcification propensity in subjects with end stage kidney disease: a randomized, controlled clinical trial. Clin J Am Soc Nephrol. (2018) 13:1373–80. doi: 10.2215/CJN.13921217
87. Gungor O, Kocyigit I, Yilmaz MI, Sezer S. Role of vascular calcification inhibitors in preventing vascular dysfunction and mortality in hemodialysis patients. Semin Dial. (2018) 31:72–81. doi: 10.1111/sdi.12616
88. Villa-Bellosta R, Egido J. Phosphate, pyrophosphate, and vascular calcification: a question of balance. Eur Heart J. (2015) 38:1801–4. doi: 10.1093/eurheartj/ehv605
89. Hujairi NMA, Afzali B, Goldsmith DJA. Cardiac calcification in renal patients: what we do and don't know. Am J Kidney Dis. (2004) 43:234–43. doi: 10.1053/j.ajkd.2003.10.014
90. Aikawa E, Blaser MC. 2020 Jeffrey M. Hoeg Award Lecture: calcifying extracellular vesicles as building blocks of microcalcifications in cardiovascular disorders. Arterioscler Thromb Vasc Biol. (2021) 41:117–27. doi: 10.1161/ATVBAHA.120.314704
91. Pasch A, Farese S, Gräber S, Wald J, Richtering W, Floege J, et al. Nanoparticle-based test measures overall propensity for calcification in serum. J Am Soc Nephrol. (2012) 23:1744–52. doi: 10.1681/ASN.2012030240
92. Pasch A. Novel assessments of systemic calcification propensity. Curr Opin Nephrol Hypertens. (2016) 25:278–84. doi: 10.1097/MNH.0000000000000237
93. Miura Y, Iwazu Y, Shiizaki K, Akimoto T, Kotani K, Kurabayashi M, et al. Identification and quantification of plasma calciprotein particles with distinct physical properties in patients with chronic kidney disease. Sci Rep. (2018) 8:1256. doi: 10.1038/s41598-018-19677-4
94. Smith ER, Ford ML, Tomlinson LA, Bodenham E, McMahon LP, Farese S, et al. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J Am Soc Nephrol. (2014) 25:339–48. doi: 10.1681/ASN.2013060635
95. Pasch A, Block GA, Bachtler M, Smith ER, Jahnen-Dechent W, Arampatzis S, et al. Blood calcification propensity, cardiovascular events, and survival in patients receiving hemodialysis in the EVOLVE Trial. Clin J Am Soc Nephrol. (2017) 12:315–22. doi: 10.2215/CJN.04720416
96. Dahle DO, Åsberg A, Hartmann A, Holdaas H, Bachtler M, Jenssen TG, Dionisi M, et al. Serum calcification propensity is a strong and independent determinant of cardiac and all?Cause mortality in kidney transplant recipients. Am J Transplant. (2019) 16:204–12. doi: 10.1111/ajt.13443
97. Bundy JD, Cai X, Scialla JJ, Dobre MA, Chen J, Hsu C-Y, et al. Serum calcification propensity and coronary artery calcification among patients with CKD: the CRIC (Chronic Renal Insufficiency Cohort) study. Am J Kidney Dis. (2019) 73:806–14. doi: 10.1053/j.ajkd.2019.01.024
98. Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation. (2008) 117:2938–48. doi: 10.1161/CIRCULATIONAHA.107.743161
99. Smith ER, Hanssen E, McMahon LP, Holt SG. Fetuin-A-containing calciprotein particles reduce mineral stress in the macrophage. PLoS ONE. (2013) 8:e60904. doi: 10.1371/journal.pone.0060904
100. Kutikhin AG, Velikanova EA, Mukhamadiyarov RA, Glushkova TV, Borisov VV, Matveeva VG, et al. Apoptosis-mediated endothelial toxicity but not direct calcification or functional changes in anti-calcification proteins defines pathogenic effects of calcium phosphate bions. Sci Rep. (2016) 6:27255. doi: 10.1038/srep27255
101. Benz K, Varga I, Neureiter D, Campean V, Daniel C, Heim C, et al. Vascular inflammation and media calcification are already present in early stages of chronic kidney disease. Cardiovasc Pathol. (2017) 27:57–7. doi: 10.1016/j.carpath.2017.01.004
102. Takx RAP, MacNabb MH, Emami H, Abdelbaky A, Singh P, Lavender ZR, et al. Increased arterial inflammation in individuals with stage 3 chronic kidney disease. Eur J Nucl Med Mol Imaging. (2016) 43:333–9. doi: 10.1007/s00259-015-3203-6
103. Joshi FR, Rajani NK, Abt M, Woodward M, Bucerius J, Mani V, et al. Does vascular calcification accelerate inflammation?: A substudy of the dal-PLAQUE trial. J Am Coll Cardiol. (2016) 67:69–78. doi: 10.1016/j.jacc.2015.10.050
104. Navarro-González JF, Mora-Fernández C, Muros M, Herrera H, García J. Mineral metabolism and inflammation in chronic kidney disease patients: a cross-sectional study. Clin J Am Soc Nephrol. (2009) 4:1646–54. doi: 10.2215/CJN.02420409
105. Yamada S, Tokumoto M, Tatsumoto N, Taniguchi M, Noguchi H, Nakano T, et al. Phosphate overload directly induces systemic inflammation and malnutrition as well as vascular calcification in uremia. Am J Physiol Renal Physiol. (2014) 306:F1418–28. doi: 10.1152/ajprenal.00633.2013
106. Hamano T, Matsui I, Mikami S, Tomida K, Fujii N, Imai E, et al. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol. (2010) 21:1998–2007. doi: 10.1681/ASN.2009090944
107. Smith ER. The isolation and quantification of fetuin-A containing calciprotein particles from biological fluids. Methods Mol Biol. (2016) 1397:221–40. doi: 10.1007/978-1-4939-3353-2_15
108. Zoccali C, London G. Con: vascular calcification is a surrogate marker, but not the cause of ongoing vascular disease, and it is not a treatment target in chronic kidney disease. Nephrol Dial Transplant. (2015) 30:352–7. doi: 10.1093/ndt/gfv021
109. Komaba H, Wang M, Taniguchi M, Yamamoto S, Nomura T, Schaubel DE, et al. Initiation of sevelamer and mortality among hemodialysis patients treated with calcium-based phosphate binders. Clin J Am Soc Nephrol. (2017) 12:1489–97. doi: 10.2215/CJN.13091216
110. Raggi P, Bommer J, Chertow GM. Valvular calcification in hemodialysis patients randomized to calcium-based phosphorus binders or sevelamer. J Heart Valve Dis. (2004) 13:134–41.
111. Kakuta T, Tanaka R, Hyodo T, Suzuki H, Kanai G, Nagaoka M, et al. Effect of sevelamer and calcium-based phosphate binders on coronary artery calcification and accumulation of circulating advanced glycation end products in hemodialysis patients. Am J Kidney Dis. (2011) 57:422–31. doi: 10.1053/j.ajkd.2010.10.055
112. Chertow GM, Burke SK, Raggi P, for the Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. (2002) 62:245–52. doi: 10.1046/j.1523-1755.2002.00434.x
113. Jamal SA, Vandermeer B, Raggi P, Mendelssohn DC, Chatterley T, Dorgan M, et al. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. Lancet. (2013) 382:1268–77. doi: 10.1016/S0140-6736(13)60897-1
114. Patel L, Bernard LM, Elder GJ. Sevelamer versus calcium-based binders for treatment of hyperphosphatemia in CKD: a meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol. (2016) 11:232–44. doi: 10.2215/CJN.06800615
115. Sekercioglu N, Thabane L, Díaz Martínez JP, Nesrallah G, Longo CJ, Busse JW, et al. Comparative effectiveness of phosphate binders in patients with chronic kidney disease: a systematic review and network meta-analysis. PLoS ONE. (2016) 11:e0156891. doi: 10.1371/journal.pone.0156891
116. Bravo-Soto GA, Madrid T. Sevelamer versus calcium-based phosphate binders for chronic kidney disease. Medwave. (2017) 17:e6942. doi: 10.5867/medwave.2017.6942
117. Russo D, Miranda I, Ruocco C, Battaglia Y, Buonanno E, Manzi S, et al. The progression of coronary artery calcification in predialysis patients on calcium carbonate or sevelamer. Kidney Int. (2007) 72:1255–61. doi: 10.1038/sj.ki.5002518
118. Di Iorio B, Bellasi A, Russo D; INDEPENDENT Study Investigators. Mortality in kidney disease patients treated with phosphate binders: a randomized study. Clin J Am Soc Nephrol. (2012) 7:487–93. doi: 10.2215/CJN.03820411
119. Block GA, Wheeler DC, Persky MS, Kestenbaum B, Ketteler M, Spiegel DM, et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol. (2012) 23:1407–15. doi: 10.1681/ASN.2012030223
120. Bouma-de Krijger A, van Ittersum FJ, Hoekstra T, Ter Wee PM, Vervloet MG. Short-term effects of sevelamer-carbonate on fibroblast growth factor 23 and pulse wave velocity in patients with normophosphataemic chronic kidney disease Stage 3. Clin Kidney J. (2019) 12:678–85. doi: 10.1093/ckj/sfz027
121. Toussaint ND, Pedagogos E, Lioufas NM, Elder GJ, Pascoe EM, Badve SV, et al. A randomized trial on the effect of phosphate reduction on vascular end points in CKD (IMPROVE-CKD). J Am Soc Nephrol. (2020) 31:2653–66. doi: 10.1681/ASN.2020040411
122. Bover J, Cozzolino M. Small steps towards the potential of “preventive” treatment of early phosphate loading in chronic kidney disease patients. Clin Kidney J. (2019) 12:673–7. doi: 10.1093/ckj/sfz082
123. Block GA, Block MS, Smits G, Mehta R, Isakova T, Wolf M, et al. A pilot randomized trial of ferric citrate coordination complex for the treatment of advanced CKD. J Am Soc Nephrol. (2019) 30:1495–504. doi: 10.1681/ASN.2018101016
124. Molina P, Pallardó L, Torralba J, Escudero V, Álvarez L, Peris A, et al. Disorders in bone-mineral parameters and the risk of death in persons with chronic kidney disease stages 4 and 5: the PECERA study. J Nephrol. (2021). doi: 10.1007/s40620-020-00916-9. [Epub ahead of print].
125. Fernández-Martín J, Martínez-Camblor P, Dionisi M, Floege J, Ketteler M, London G, et al. Improvement of mineral and bone metabolism markers is associated with better survival in haemodialysis patients: the COSMOS study. Nephrol Dial Transplant. (2015) 30:1542–51. doi: 10.1093/ndt/gfv099
126. Lamina C, Kronenberg F, Stenvinkel P, Froissart M, Forer L, Schönherr S, et al. Association of changes in bone mineral parameters with mortality in haemodialysis patients: insights from the ARO cohort. Nephrol Dial Transplant. (2020) 35:478–87. doi: 10.1093/ndt/gfz060
127. Naves-Díaz M, Cabezas-Rodríguez I, Barrio-Vázquez S, Fernández E, Díaz-López JB, Cannata-Andía JB. Low calcidiol levels and risk of progression of aortic calcification. Osteoporos Int. (2012) 23:1177–82. doi: 10.1007/s00198-011-1550-0
128. Pilz S, Iodice S, ZIttermann A, Grant WB, Gandini S. Vitamin D status and mortality risk in CKD: a meta-analysis of prospective studies. Am J Kidney Dis. (2011) 58:374–82. doi: 10.1053/j.ajkd.2011.03.020
129. Reiss AB, Miyawaki N, Moon J, Kasselman LJ, Voloshyna I, D'Avino R Jr, et al. CKD, arterial calcification, atherosclerosis and bone health: Inter-relationships and controversies. Atherosclerosis. (2018) 278:49–59. doi: 10.1016/j.atherosclerosis.2018.08.046
130. Vahdat S, Naini A, Hedaiati Z, Shahzeidi S, Pezeshki A, Nasri H. The effect of vitamin D administration on serum leptin and adiponectin levels in end-stage renal disease patients on hemodialysis with vitamin D deficiency: a placebo-controlled double-blind clinical trial. J Res Med Sci. (2016) 21:1. doi: 10.4103/1735-1995.175144
131. Matias PJ, Jorge C, Ferreira C, Borges M, Aires I, Amaral T, et al. Cholecalciferol supplementation in hemodialysis patients: effects on mineral metabolism, inflammation, and cardiac dimension parameters. Clin J Am Soc Nephrol. (2010) 5:905–11. doi: 10.2215/CJN.06510909
132. Thadhani R, Appelbaum E, Pritchett Y, Chang Y, Wenger J, Tamez H, et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease. JAMA. (2012) 307:674. doi: 10.1001/jama.2012.120
133. Wang AY-M, Fang F, Chan J, Wen Y-Y, Qing S, Chan IH-S, et al. Effect of paricalcitol on left ventricular mass and function in CKD-The OPERA trial. J Am Soc Nephrol. (2014) 25:175–86. doi: 10.1681/ASN.2013010103
134. Torregrosa J-V, Bover J, Cannata, Andía J, Lorenzo V, de Francisco ALM, Martínez I. Spanish Society of Nephrology recommendations for controlling mineral and bone disorder in chronic kidney disease patients (S.E.N.-M.B.D.). Nefrologia. (2011) 31(Suppl. 1):3–32. doi: 10.3265/Nefrologia.pre2011.Jan.10816
135. Mathew S, Lund RJ, Chaudhary LR, Geurs T, Hruska KA. Vitamin D receptor activators can protect against vascular calcification. J Am Soc Nephrol. (2008) 19:1509–19. doi: 10.1681/ASN.2007080902
136. Mizobuchi M, Finch JL, Martin DR, Slatopolsky E. Differential effects of vitamin D receptor activators on vascular calcification in uremic rats. Kidney Int. (2007) 72:709–15. doi: 10.1038/sj.ki.5002406
137. Cardús A, Panizo S, Parisi E, Fernandez E, Valdivielso JM. Differential effects of vitamin D analogs on vascular calcification. J Bone Miner Res. (2007) 22:860–6. doi: 10.1359/jbmr.070305
138. Teng M, Wolf M, Lowrie E, Ofsthun N, Michael Lazarus J, Thadhani R. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. (2003) 349:446–56. doi: 10.1056/NEJMoa022536
139. Teng M, Wolf M, Ofsthun MN, Lazarus JM, Hernán MA, Camargo CA Jr, et al. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol. (2005) 16:1115–25. doi: 10.1681/ASN.2004070573
140. EVOLVE Trial Investigators, Chertow GM, Block GA, Correa-Rotter R, Drüeke TB, Floege J, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. (2012) 367:2482–94. doi: 10.1056/NEJMoa1205624
141. Bover J, Ureña P, Ruiz-García C, daSilva I, Lescano P, del Carpio J, et al. Clinical and practical use of calcimimetics in dialysis patients with secondary hyperparathyroidism. Clin J Am Soc Nephrol. (2016) 11:161–74. doi: 10.2215/CJN.01760215
142. Sakai M, Tokunaga S, Kawai M, Murai M, Kobayashi M, Kitayama T, et al. Evocalcet prevents ectopic calcification and parathyroid hyperplasia in rats with secondary hyperparathyroidism. PLoS ONE. (2020) 15:e0232428. doi: 10.1371/journal.pone.0232428
143. Yu L, Tomlinson JE, Alexander ST, Hensley K, Han C-Y, Dwyer D, et al. Etelcalcetide, a novel calcimimetic, prevents vascular calcification in a rat model of renal insufficiency with secondary hyperparathyroidism. Calcif Tissue Int. (2017) 101:641–53. doi: 10.1007/s00223-017-0319-7
144. Lopez I, Aguilera-Tejero E, Mendoza FJ, Almaden Y, Perez J, Martin D, et al. Calcimimetic R-568 decreases extraosseous calcifications in uremic rats treated with calcitriol. J Am Soc Nephrol. (2006) 17:795–804. doi: 10.1681/ASN.2005040342
145. Kawata T, Nagano N, Obi M, Miyata S, Koyama C, Kobayashi N, et al. Cinacalcet suppresses calcification of the aorta and heart in uremic rats. Kidney Int. (2008) 74:1270–7. doi: 10.1038/ki.2008.407
146. Ng CH, Chin YH, Tan MHQ, Ng JX, Yang SP, Kiew JJ, et al. Cinacalcet and primary hyperparathyroidism: systematic review and meta regression. Endocr Connect. (2020) 9:724–35. doi: 10.1530/EC-20-0221
147. DaSilva-Santos I, Furlano MM, Ruiz-García CE, Lloret-Cora MJ, Guevara MdC, Barreiro-Delgado YB, et al. Hiperparatiroidismo autónomo en paciente renal crónico estadio 3: valoración clínica a propósito de un caso. Dial Traspl. (2013) 34:171–7. doi: 10.1016/j.dialis.2013.06.002
148. Reyes J, DaSilva I, Furlano M, Calero F, Montañés R, Ayasreh N, et al. Autonomous hyperparathyroidism in CKD stages 3-4 treated with cinacalcet. Nephrol Dial Transplant. (2012) 27 (Suppl. 2):ii146–58. doi: 10.1093/ndt/gfs217
149. Puri R, Nicholls SJ, Shao M, Kataoka Y, Uno K, Kapadia SR, et al. Impact of statins on serial coronary calcification during atheroma progression and regression. J Am Coll Cardiol. (2015) 65:1273–82. doi: 10.1016/j.jacc.2015.01.036
150. Vervloet M, Cozzolino M. Vascular calcification in chronic kidney disease: different bricks in the wall?. Kidney Int. (2017) 91:808–17. doi: 10.1016/j.kint.2016.09.024
151. Massy ZA, Nistor I, Apetrii M, Brandenburg VM, Bover J, Evenepoel P, et al. Magnesium-based interventions for normal kidney function and chronic kidney disease. Magnes Res. (2016) 29:126–40 doi: 10.1684/mrh.2016.0412
152. Caluwé C, Pyfferoen L, De Boeck K, De Vriese AS. The effects of vitamin K supplementation and vitamin K antagonists on progression of vascular calcification: ongoing randomized controlled trials. Clin Kidney J. (2016) 9:273–9. doi: 10.1093/ckj/sfv146
153. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. (2001) 108:1167–74. doi: 10.1172/JCI13505
154. Yamada S, Taniguchi M, Tokumoto M, Toyonaga J, Fujisaki K, Suehiro T, et al. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J Bone Miner Res. (2012) 27:474–85. doi: 10.1002/jbmr.539
155. Lomashvili KA, Monier-Faugere MC, Wang X, Malluche HH, O'Neill WC. Effect of bisphosphonates on vascular calcification and bone metabolism in experimental renal failure. Kidney Int. (2009) 75:617–25. doi: 10.1038/ki.2008.646
156. Chen CL, Chen NC, Wu FZ, Wu MT. Impact of denosumab on cardiovascular calcification in patients with secondary hyperparathyroidism undergoing dialysis: a pilot study. Osteoporos Int. (2020) 31:1507–16. doi: 10.1007/s00198-020-05391-3
157. Adirekkiat S, Sumethkul V, Ingsathit A, Domrongkitchaiporn S, Phakdeekitcharoen B, Kantachuvesiri S, et al. Sodium thiosulfate delays the progression of coronary artery calcification in haemodialysis patients. Nephrol Dial Transplant. (2010) 25:1923–9. doi: 10.1093/ndt/gfp755
158. Coyne DW, Singh HN, Smith WT, Giuseppi AC, Connarn JN, Sherman ML, et al. Sotatercept safety and effects on hemoglobin, bone, and vascular calcification. Kidney Int Rep. (2019) 4:1585–97. doi: 10.1016/j.ekir.2019.08.001
159. Hénaut L, Chillon J-M, Kamel S, Massy ZA. Updates on the mechanisms and the care of cardiovascular calcification in chronic kidney disease. Semin Nephrol. (2018) 38:233–50. doi: 10.1016/j.semnephrol.2018.02.004
160. Ruderman I, Holt SG, Hewitson TD, Smith ER, Toussaint ND. Current and potential therapeutic strategies for the management of vascular calcification in patients with chronic kidney disease including those on dialysis. Semin Dial. (2018) 31:487–99. doi: 10.1111/sdi.12710
161. Raggi P, Bellasi A, Bushinsky D, Bover J, Rodriguez M, Ketteler M, et al. Slowing progression of cardiovascular calcification with SNF472 in patients on hemodialysis. Circulation. (2020) 141:728–39. doi: 10.1161/CIRCULATIONAHA.119.044195
Keywords: CKD, CKD-MBD, calcification, vascular calcification, phosphate, calciprotein particles
Citation: Bover J, Aguilar A, Arana C, Molina P, Lloret MJ, Ochoa J, Berná G, Gutiérrez-Maza YG, Rodrigues N, D'Marco L and Górriz JL (2021) Clinical Approach to Vascular Calcification in Patients With Non-dialysis Dependent Chronic Kidney Disease: Mineral-Bone Disorder-Related Aspects. Front. Med. 8:642718. doi: 10.3389/fmed.2021.642718
Received: 16 December 2020; Accepted: 12 April 2021;
Published: 19 May 2021.
Edited by:
Chia-Ter Chao, National Taiwan University Hospital Bei-Hu Branch, TaiwanReviewed by:
Giuseppe Cianciolo, Sant'Orsola-Malpighi Polyclinic, ItalyMaria-Eleni Roumelioti, University of New Mexico, United States
Nigel Toussaint, Royal Melbourne Hospital, Australia
Copyright © 2021 Bover, Aguilar, Arana, Molina, Lloret, Ochoa, Berná, Gutiérrez-Maza, Rodrigues, D'Marco and Górriz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordi Bover, amJvdmVyQGZ1bmRhY2lvLXB1aWd2ZXJ0LmVz; Armando Aguilar, YWd1aWxhcmJldGFAaG90bWFpbC5jb20=
†These authors have contributed equally to this work