
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 18 March 2021
Sec. Infectious Diseases – Surveillance, Prevention and Treatment
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.620990
This article is part of the Research Topic COVID-19: Integrating Artificial Intelligence, Data Science, Mathematics, Medicine and Public Health, Epidemiology, Neuroscience, and Biomedical Science in Pandemic Management View all 95 articles
 Hawraa Issa1,2
Hawraa Issa1,2 Ali H. Eid3,4
Ali H. Eid3,4 Bassam Berry5Vahideh Takhviji6Abbas Khosravi6
Bassam Berry5Vahideh Takhviji6Abbas Khosravi6 Sarah Mantash1Rawan Nehme1
Sarah Mantash1Rawan Nehme1 Rawan Hallal1
Rawan Hallal1 Hussein Karaki1
Hussein Karaki1 Kawthar Dhayni1,7
Kawthar Dhayni1,7 Wissam H. Faour8
Wissam H. Faour8 Firas Kobeissy9
Firas Kobeissy9 Ali Nehme10
Ali Nehme10 Kazem Zibara1*
Kazem Zibara1*Coronavirus disease-2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is currently the most concerning health problem worldwide. SARS-CoV-2 infects cells by binding to angiotensin-converting enzyme 2 (ACE2). It is believed that the differential response to SARS-CoV-2 is correlated with the differential expression of ACE2. Several reports proposed the use of ACE2 pharmacological inhibitors and ACE2 antibodies to block viral entry. However, ACE2 inhibition is associated with lung and cardiovascular pathology and would probably increase the pathogenesis of COVID-19. Therefore, utilizing ACE2 soluble analogs to block viral entry while rescuing ACE2 activity has been proposed. Despite their protective effects, such analogs can form a circulating reservoir of the virus, thus accelerating its spread in the body. Levels of ACE2 are reduced following viral infection, possibly due to increased viral entry and lysis of ACE2 positive cells. Downregulation of ACE2/Ang (1-7) axis is associated with Ang II upregulation. Of note, while Ang (1-7) exerts protective effects on the lung and cardiovasculature, Ang II elicits pro-inflammatory and pro-fibrotic detrimental effects by binding to the angiotensin type 1 receptor (AT1R). Indeed, AT1R blockers (ARBs) can alleviate the harmful effects associated with Ang II upregulation while increasing ACE2 expression and thus the risk of viral infection. Therefore, Ang (1-7) agonists seem to be a better treatment option. Another approach is the transfusion of convalescent plasma from recovered patients with deteriorated symptoms. Indeed, this appears to be promising due to the neutralizing capacity of anti-COVID-19 antibodies. In light of these considerations, we encourage the adoption of Ang (1-7) agonists and convalescent plasma conjugated therapy for the treatment of COVID-19 patients. This therapeutic regimen is expected to be a safer choice since it possesses the proven ability to neutralize the virus while ensuring lung and cardiovascular protection through modulation of the inflammatory response.
Starting November 2019, several cases of pneumonia of unknown etiology were reported in Wuhan, China (1). The causal agent was identified as severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). The newly identified betacoronavirus differs from severe acute respiratory syndrome coronavirus (SARS-CoV-1) and Middle East respiratory syndrome coronavirus (MERS-CoV); however, it causes similar symptoms associated with pneumonia (2–4). In contrast to SARS-CoV-1, which caused the 2002 outbreak, SARS-CoV-2 exhibits a higher risk of transmission as evident from the rapid global rise in the number of Coronavirus disease 2019 (COVID-19) cases (2–4). As of mid February 2021, more than 110 million cases have been confirmed, and nearly 2,400,000 deaths were reported globally, with the rapid increase of numbers in many countries.
SARS-CoV-2 is mainly transmitted from person to person through respiratory droplets, contact, aerosol, or oral-fecal transmission (5, 6). While most reported COVID-19 cases present mild to moderate pathology, 20% of infected patients may develop severe disease and need intensive care (7–12). Severe cases progress to acute respiratory distress syndrome (ARDS) after 8–9 days of symptoms onset (1). ARDS seen in severe COVID-19 cases is characterized by difficulty in breathing and low blood oxygen level, leading to respiratory failure, which is the main cause of death in 70% of fatal COVID-19 cases (8–10). Plasma analysis of severe cases showed a massive release of cytokines by the immune system (cytokine storm) in response to the viral infection and/or potential secondary bacterial and fungal infections (8, 9). This uncontrolled inflammation induced by SARS-CoV-2 infection results in multi-organ damage, leading to organ failure (10).
Certain groups of the population are more susceptible to SARS-CoV-2 infection (11–14). The case fatality rate (CFR) seems to be age-dependent, with a higher percentage in the elderly, especially men. SARS-CoV-2 may have a higher transmissibility than SARS-CoV-1 and MERS-CoV (1, 8, 15, 16). Patients with pre-existing comorbidities such as chronic obstructive pulmonary disease (COPD), cardiovascular diseases, hypertension and type 2 diabetes mellitus, are more likely to display a severe course and to have higher mortality rates (13–18).
Initial research on SARS-CoV-2 demonstrated that it binds to host cells using the angiotensin-converting enzyme 2 (ACE2), similar to SARS-CoV-1. SARS-CoV-2 binds to ACE2 proteins as a receptor in bats, civet cats, swine, cats, ferrets, non-human primates, and humans (16–18).
The binding affinity of SARS-CoV-2 RBD to ACE2 seems stronger than that of SARS-CoV-1. Besides, SARS-CoV-2 has evolved to use a wide array of host proteases (transmembrane protease serine 2 (TMPRSS2), cathepsin L/B, furin, trypsin, etc.) for S-protein priming, thus facilitating cell entry following receptor binding. This may explain the considerably larger global influence of COVID-19 than the initial SARS epidemic of 2003 (19–21). It is worth noting that new SARS-CoV-2 strains, namely the British (B.1.1.7) and South African variants (B.1.351), that emerged toward the end of 2020 show increased transmission capacity, associated with increased interaction force between Spike and ACE2 proteins due to viral mutations (22, 23). In addition, the RBD/ACE2 mediated SARS-CoV-2 entry into cells is followed by subsequent downregulation of surface ACE2 expression (24). Several reports indicate that the reduction in ACE2 function influences blood pressure, perturbs fluid/electrolyte balance, enhances inflammation, and vascular permeability in the airways, and facilitates the development of multiorgan damage from SARS-CoV-2 infections (25–29). Consequently, ACE2 appears as a critical factor in understanding COVID-19 pathology and a potential target for COVID-19 treatment.
ACE2 is a membrane-bound glycoprotein of 805 amino acids that exhibits 40% identity and 61% similarity to human angiotensin converting enzyme (ACE). Full-length ACE2 consists of a heavily glycosylated N-terminal signal sequence containing the active site, a hydrophobic transmembrane sequence, and a short C-terminal cytoplasmic tail. A soluble and catalytically active form of ACE2 can be also produced by several mechanisms, including the action of ADAMs family members of zinc metalloproteases (30–33). ACE2 is the main enzyme involved in the production of the Ang (1-7) peptide of the renin-angiotensin system (RAS) (Figure 1). The latter comprises successive enzymatic reactions that regulate multiple biological processes, including cellular growth, proliferation, migration, extracellular matrix remodeling, and inflammation. While RAS includes multiple enzymatic axes leading to the production of different bioactive peptides, local tissue effects of RAS are driven mostly by the balance between the pro-inflammatory/pro-fibrotic and anti-inflammatory/anti-fibrotic actions of Ang-II and Ang (1–7), respectively (34, 35) (Figure 1).
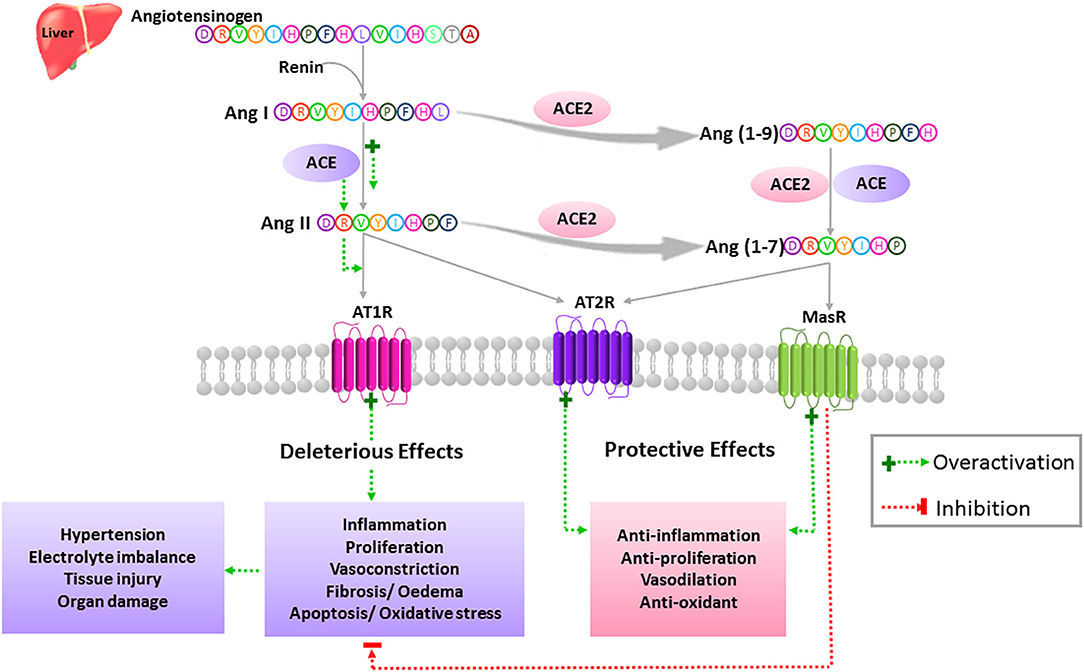
Figure 1. Simplified view of the extended RAS. In the classically described RAS, the inactive zymogen angiotensinogen secreted mainly by the liver, is converted into Ang I by the action of the renal aspartyl protease, renin. Ang I is then cleaved by ACE to generate the Ang II octapeptide. Ang II is a multifunctional hormone that regulates blood pressure and fluid homeostasis. This peptide exerts its actions through binding to two main receptors, AT1R and AT2R, which are typical seven transmembrane GPCRs. More specifically, Ang II mediates its vasoconstrictor effects by stimulating AT1Rs while AT2Rs are known to balance the actions of AT1Rs via activation of vasodilatory pathways. Dysregulation of the RAS in favor of the ACE/Ang II/AT1R axis leads to the pathogenesis of hypertension as well as tissue injury and multi-organ damage through activation of oxidative stress, proliferation, inflammation, fibrosis, edema, and apoptosis. Ang II can either bind to its receptors or is further cleaved to yield degradation products such as Ang (1-7). This bioactive peptide is produced mainly by means of ACE2. Ang (1-7) is obtained directly by the action of ACE2 on Ang II or indirectly by generating Ang (1-9) as an intermediate product. Ang (1-7) exerts its protective effects through activation of the AT2R and MasR and opposes the described detrimental effects of the ACE/Ang II/AT1R axis.
Ang II is produced by the cleavage of Ang I by the angiotensin-converting enzyme (ACE), and it exerts its pro-inflammatory/pro-fibrotic effects by binding to its angiotensin type 1 receptor (AT1R) (Figure 1). On the other hand, ACE2 is the most potent Ang (1-7) generating enzyme. By its single catalytic domain, ACE2 can produce Ang (1-7) directly by cleaving one amino acid from the C-terminal domain of Ang II or indirectly through two successive cleavage reactions from Ang I (36) (Figure 1). Of note, Ang II is the preferred substrate for ACE2, with an affinity of 400-fold higher than that of Ang I (37). Hence, Ang-(1–7) production is based on both ACE2 levels and the ACE/ACE2 ratio.
Ang (1-7) exerts its effects by binding to and activating the G-coupled receptor Mas (MasR), which opposes the pro-inflammatory and pro-fibrotic actions of Ang II through AT1R activation (38, 39). Moreover, MasR can inhibit Ang II-mediated actions by hetero-oligomerization and inhibition of AT1R. Ang (1-7) can also exert its protective effects by binding directly to the angiotensin type 2 receptor (AT2R) (40, 41). Interestingly, the AT2R can exert similar protective effects when bound by Ang II (34, 35). Thus, the crucial role of ACE2 in RAS stems from the fact that it cleaves and opposes the action of Ang II (Figure 1). Consequently, it has a beneficial role in many diseases such as diabetes and cardiovascular diseases (CVD) (33), as well as in COVID-19 (42).
RAS gained an increased complexity and appreciation with the identification of local RAS in different tissues, including the brain, kidneys, heart, ovary, pancreas, and the vascular wall, independent of the well-known traditional circulatory RAS. Also, the discovery of additional RAS components (alternative enzymes, receptors, and bioactive angiotensin peptides) has extended the system's role far beyond blood pressure regulation and body electrolyte balance. Indeed, novel actions for each member of the RAS are continuously discovered in physiology and diseases (43).
ACE2 is expressed in human airway epithelia and lung parenchyma, suggesting a role in the regulation of pulmonary physiology (44, 45). A large body of evidence has shown the protective role of the ACE2/Ang (1-7)/MasR axis in several models of lung injury, including SARS-CoV-1 mediated injury (25, 46). In fact, to protect from severe lung failure, ACE2 is known to inhibit Ang II production, ACE activity, and AT1R activation. Of note, the anti-inflammatory and anti-fibrotic responses of ACE2 are mediated by the production of Ang (1-7) bioactive peptide, which protects against acute lung failure via activation of MaSR and AT2R. More specifically, Ang (1-7)/MasR exerts its beneficial effects by inhibiting ERK1/2 and NF-κB signaling pathways in a rat model of ARDS (47) and a mouse model of chronic allergic lung inflammation (44, 48). Magalhaes et al. also showed that MasR knockout mice failed to attenuate inflammation and pathological lung remodeling and presented aggravated asthma due to disruption of the protective arm of the RAS (49). The same research group confirmed that Ang (1-7) infusion resolved inflammation through correction of eosinophile defective apoptosis leading to lung damage (48). Ang (1-7) drug was also demonstrated to prevent bronchial responsiveness, a hallmark sign of chronic asthma (44).
Dysregulation of ACE/ACE2 balance leads to impaired lung function due to inflammation, fibrosis, and lung edema. The latter phenomenon is most probably induced via increasing pulmonary blood vessels' permeability (25, 46, 47). Elevated ACE concentrations have been detected in many potentially fibrotic lung diseases, including idiopathic pulmonary fibrosis (50) and ARDS (51, 52). Similar pro-fibrotic effects were also observed in a mouse model of ARDS using the MasR antagonist A779 (53). Moreover, Ang II has been shown to stimulate lung fibroblast proliferation and procollagen production by stimulating AT1R and the autocrine action of TGFβ. Of interest, losartan (AT1R blocker) and ramipril (ACE inhibitor), and Ang (1-7) were shown to reduce lung collagen deposition in the same study (53, 54). Thus, the protective effects of ACE2 on the lungs can be attributed to the inactivation of the ACE/Ang II/AT1R axis in favor of the ACE2/Ang (1-7)/MasR-AT2R axis (25).
The expression of ACE2 in human airways and lung tissues highlights its role in respiratory infections, including SARS-CoV-1 and the related human respiratory coronavirus NL63 (55). Although ACE2 is the main door for virus entry, the total ACE2 activity seems to be protective. In fact, several reports mentioned that ACE2 could be downregulated after virus entry and/or host cell lysis, as in SARS-CoV-1. The latter is reported to reduce ACE2 expression at the cell surface as well as the release of active ACE2 ectodomains (56, 57). This fact may further accentuate the pathogenesis of COVID-19, as ACE2 is shown to be protective in several models of lung injury, including SARS-CoV-1 mediated injury (25, 46).
Both SARS-CoV-1 and SARS-CoV-2 use the same receptor, ACE2, to infect cells. Interestingly, SARS-CoV-2 was shown to have a higher affinity for ACE2 than SARS-CoV-1 (58–60). Higher affinity values could be related to the dynamic of infection and the rapid spread that characterize this virus (61). For instance, mutations that increase the infectivity on RBD could explain why SARS-CoV-2 is more infectious than SARS-CoV-1 (62). Notably, mutations affecting SARS-CoV-2 have also been reported. In fact, by the end of August 2020, the C.1 lineage of SARS-CoV-2 presenting one amino acid substitution, D614G, on the spike protein, among 16 other nucleotide mutations, became the predominant lineage in South Africa (63). Analyses of over 28,000 SARS-CoV-2 spike protein gene sequences revealed that the D614G amino acid substitution facilitates the binding to ACE2 receptor and thus enhances viral replication in human lung epithelial cells and primary human airway tissues. This might account for its increased virulence to the respiratory system (64, 65). In addition, the 501Y.V2 variant that appeared in South Africa in December 2020 showed three important mutations in RBD (K417N, E484K, and N501Y) that are most probably correlated with functional significance (66). Another study on the B.1.1.7 British lineage revealed that the N501Y mutation of the SARS-CoV-2 spike protein is linked with increased interaction with ACE2 receptor, which explains its high infectivity rate (23).
The SARS-CoV-2 entry into target cells is initiated by the binding of the surface unit, S1, of the spike (S) protein to the ACE2 cellular receptor (Figure 2). The entry then requires S protein priming by TMPRSS2 serine proteases, which entails S protein cleavage and allows the fusion of viral and cellular membranes (67). Of note, several studies highlighted TMPRSS2 implication as a critical factor for the spread of clinically relevant viruses, including influenza A and other coronaviruses (68–70). One study conducted on a cohort of Italian patients announced that COVID-19 susceptibility is determined by genetic variability of TMPRSS2 known to be involved in SARS-CoV-2 entry into target cells. In this context, the data showed that in comparison to other European populations, Italians might have a higher level of TMPRSS2 or activity since they show a significant decrease in the deleterious variants of this protein. This can be considered as a risk factor for a more severe illness course (71). TMPRSS2 mediated activation of S protein priming enables viral infection of ACE2 positive cells. This initial phase is associated with viral replication and leads to pyroptosis, an inflammatory form of apoptosis, inducing lung injury. Of note, the formation of new viruses is also correlated with the induction of a cytokine storm via activation of various immune cells (56, 72).
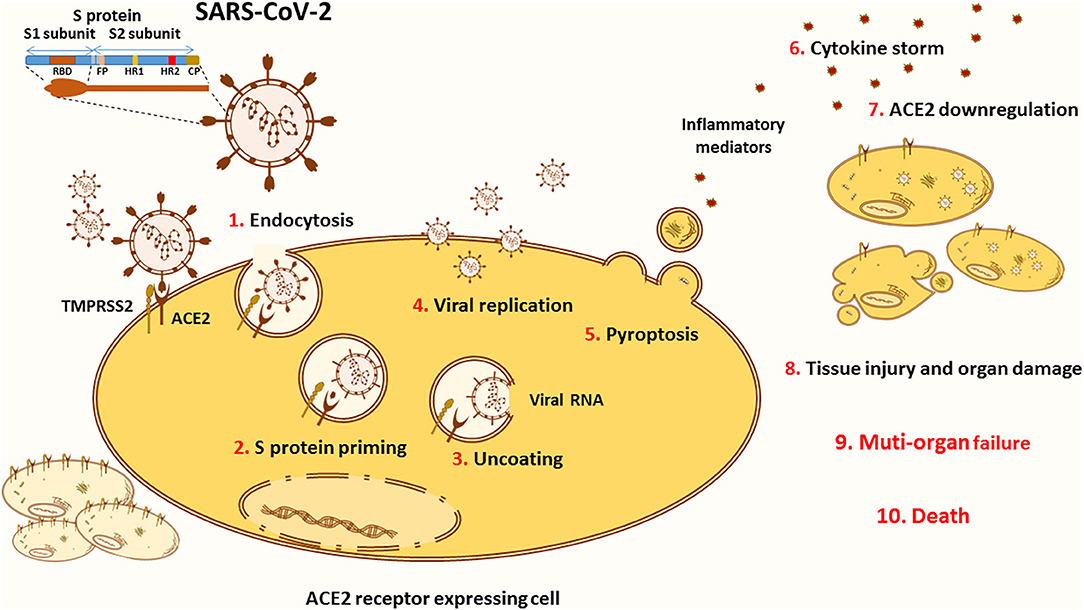
Figure 2. ACE2 in the pathology of COVID-19. The novel SARS-CoV-2 infects the cells through binding to its main receptor ACE2. The latter recognizes the RBD of the S1 subunit and allows the endocytosis of the virus (1). Once exposed to the action of proteases, such as the cellular TMPRSS2, the S1 subunit is cleaved away to ensure S protein priming (2). The fusion peptide (FP) of the S2 subunit is thus exposed to the cellular membrane. The FP initiates the fusion of the viral coat to the endosomal membrane enabling the uncoating of the virus (3). Released into the cytoplasm of the host cell, the viral RNA hijacks the cellular machinery to produce novel viral particles (4). Massive viral replication is thought to be linked with pyroptosis (5), an inflammatory form of apoptosis associated with the release of inflammatory mediators that activate various immune cells in order to create a cytokine storm (6) contributing to the pathogenesis of COVID-19. Viral entry and cellular apoptosis lead to ACE2 downregulation (7), thus stimulating the harmful effects of the ACE/Ang II/AT1R axis. Altogether, these processes are translated into tissue injury and multi-organ damage (8) that can evolve into respiratory, cardiac, hepatic, and/or renal failure (9), causing death (10).
Severe COVID-19 cases are mostly observed among elderly patients, with males suffering from chronic comorbidities such as cardiovascular and cerebrovascular diseases, diabetes, and others. Recently, it was established that risk factors including age, male sex, and hypertension provide a convenient tool to identify high-risk individuals. Hypertension elucidates the involvement of RAS in the pathogenesis of COVID-19 due to the interplay between SARS-CoV-2 and ACE2 (73–75). It is believed that ACE2 expression pattern in different organs, tissues, and cell types is permissive for the susceptibility of SARS-CoV-2 infection since ACE2 receptor permits coronavirus entry, replication, spread, and pathogenesis (76).
In order to identify the initial reservoirs of SARS-CoV-2, ACE2 expression levels were assessed within the lung and the upper/lower airway epithelium. ACE2 was expressed at low levels in the respiratory tract, and it was expressed in multiple epithelial cell types across the lower airway, with the highest expression being observed in club epithelial cells in comparison with basal and ciliated epithelial cells (77). ACE2 expression was also detected in alveolar type II cells in the lung parenchyma (77). In fact, alveolar type II cells, which account for only 5% of the alveoli, are essential to maintain lung elasticity and act as progenitors for alveolar type I cells responsible for gas exchange. Thus, SARS-CoV-2 might be responsible for depletion of the alveolar stem cells leading to the development of irreversible lung injury (56, 76). Remarkably, in the upper airway, nasal epithelial cells, including goblet and ciliated cells, showed the highest expression of ACE2 among all investigated cells in the respiratory tree; thus, highlighting their role in facilitating initial viral entry, transmission, and clearance (45, 77). Moreover, ACE2 expression is dynamic and depends on the differentiation status of epithelial cells. For instance, it is worth noting that differentiated epithelial cells expressing a higher level of ACE2 are readily infected in comparison to undifferentiated cells with low ACE2 expression (55). These findings may raise the theoretical assumption that the differential response to COVID-19 in patients could be in some aspects attributed to changes in ACE2 expression.
In this context, it has been reported that long term smokers express high levels of ACE2 receptor (78), namely in type-2 pneumocytes and alveolar macrophages (79), making them at high risk of infection by SARS-CoV-2 (78). This elevated expression occurs through activation of the α7 subtype of the nicotine acetylcholine receptor (α7-nAChR) (80). This is further highlighted in a recent meta-analysis revealing that patients with a history of smoking, as well as active smokers, recorded a significant severity of COVID-19 (81). In fact, cigarette smoking promotes alterations in the respiratory tract that might increase the risk of viral infections through multiple mechanisms such as impairment of mucociliary clearance, mucus hypersecretion, fibrosis, and dysfunction of the epithelial barrier. These mechanisms are accompanied by alterations in the immune response, eventually harming the function of the lungs, including gas exchange (82–86).
Although different studies have reported the upregulation of ACE2 expression in the lungs of cigarette smokers (78, 87), surprisingly, a very recent study revealed a decrease in the levels of ACE2 receptor in both alveolar and bronchial epithelial cells of mice exposed to cigarette smoking. Additionally, an in vitro study on Calu3 human lung cancer cell line treated with cigarette smoke showed no effect on ACE2 levels but effectively inhibited SARS-CoV-2 replication (88). These conflicting results urge for more work to clarify the role of cigarette smoking on ACE2 expression, SARS-CoV-2 infection and its severe respiratory complications.
RAS dysregulation, highlighted by Ang II upregulation, is associated with the pathogenesis of CVD. RAS inhibitors, such as ACE inhibitors (ACEI) and AT1R blockers (ARBs), are commonly used for the treatment of CVD patients. ACEI are known to downregulate the expression of Ang II, whereas ARBs are known to block Ang II-mediated detrimental effects (41). Due to the beneficial effects for the activation of the ACE2/Ang (1-7) axis, there has been substantial interest in considering the effect of RAS inhibitors on ACE2 expression in patients with CVD. Previous evidence in several animal models indicated that certain ARBs and ACEIs exhibit the ability to increase ACE2 mRNA and protein expression levels in the heart (89, 90). More importantly, ARBs were shown to alter the expression of ACE2 more consistently than ACEI (91). Although some animal studies displayed an elevation in ACE2 expression under the effect of RAS inhibitors (92), other studies did not. In this context, it has been reported that ACEI did not alter the activity of ACE2 in vitro (93). In another study, the use of the ACEI ramipril or of the ARB valsartan did not increase cardiac ACE2 expression in a rat model of myocardial infarction (94).
In contrast to animal models, limited investigation has been conducted in humans to consider the influence of RAS inhibition on the expression of ACE2 (95). In fact, a human study involving patients with hypertension showed higher urinary ACE2 levels in patients treated with the ARB olmesartan, but not with other ARBs or the ACEI enalapril (96). Importantly, ACE2 upregulation has been mostly noticed in renal vasculature and in cardiac tissue. However, the outcomes differed depending on the RAS inhibitors used (97). Furthermore, most human studies relied on measuring the soluble ACE2 levels in the blood. It's worth noting that measuring the membrane-bound ACE2 expression in vivo is technically challenging. In this regard, an increase in soluble ACE2 levels may refer to a decrease in the membrane-bound form of ACE2. Therefore, the distinction between soluble and membrane-bound ACE2 must be clear (97).
Overall, upregulation of ACE2 expression in CVD patients under ARBs or ACEIs raised several theoretical assumptions that these treatment regimens might put them at a greater risk of infection by SARS-CoV-2 (60, 95).
ACE2 appears as a potential target for COVID-19 treatment based on the fact that it is an entry receptor critically involved in mediating SARS-CoV-2 infection and on its central role in cardiac pathology as well as in lung damage (98). Some reports suggested introducing ACE2 blockers, such as the MLN-4760 chemical inhibitor, or targeted antibodies to disrupt the viral entry into cells (99, 100). This approach could be detrimental to the risk of reducing ACE2 protective and anti-inflammatory activity, which further increases the susceptibility of lungs for more damage. Instead, viral entry could be impaired by protease inhibitors targeting TMPRSS2 protease implicated in SARS-CoV-2 cell entry, without risking the endogenous ACE2 activity (98) (Figure 3).
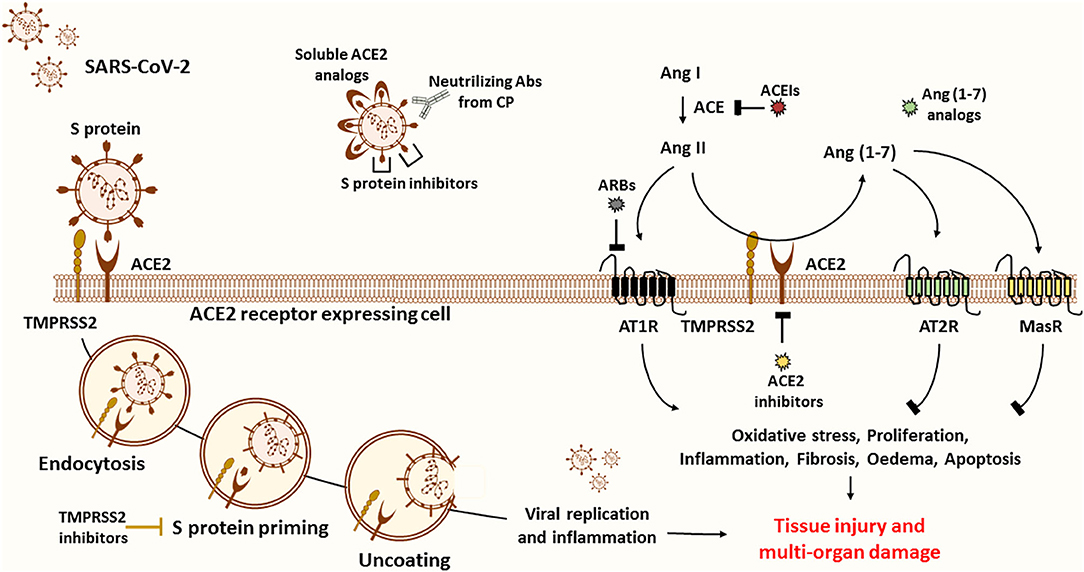
Figure 3. Possible treatment strategies for COVID-19. ACE2 is believed to be the main entry door for the SARS-CoV-2. ACE2 interaction with the RBD of the S1 subunit mediates viral entry into the host cell. To inhibit viral entry, researchers suggest the use of several drugs, including ACE2 inhibitors, soluble ACE2 analogs, S protein inhibitors, and transfusion of convalescent plasma from recovered patients. First, ACE2 inhibitors (pharmacological inhibitors and Abs) are more harmful than protective since ACE2 is known to be the primary source of the anti-inflammatory Ang (1-7) peptide. ACE2 inhibition and upregulation of Ang II expression stimulate the pathogenesis of many diseases through activation of AT1R. The latter stimulates oxidative stress, proliferation, inflammation, fibrosis, edema, and apoptosis, thus leading to tissue injury and multi-organ damage. Of note, Ang (1-7) counteract the ACE/Ang II/AT1R axis by activating the MasR and the AT2R. Second, soluble ACE2 analogs act as a trap competitively binding the virus to prevent cellular entry while rescuing ACE2 activity. Despite their beneficial effects, they can form a circulating reservoir of the virus. Third, spike protein inhibitors appear to be more promising to reduce disease severity. Fourth, another effective plan is based on the use of COVID-19 Abs from CP of recovered patients to neutralize the virus. This alternative has been proved to be safe and efficient in critically ill patients. Fifth, other therapeutic approaches encourage targeting S protein priming by means of protease inhibitors such as TMPRSS2 inhibitors to prevent the release of viral RNA into the cytoplasm of host cells, thus blocking subsequent viral replication and inflammation. In fact, virus entry and apoptosis are associated with ACE2 downregulation and consequently Ang II overproduction. Sixth, recent reports propose the use of Ang (1-7) analogs to block excessive inflammation through stimulation of the protective arm of the RAS. Importantly, Ang (1-7) drug formulation is useful in the management of several diseases, including cancer. Seventh, others suggest that ARBs and ACEIs might be useful in blunting the detrimental effects of ACE/Ang II/AT1R axis. Of note, these could also upregulate the expression of ACE2 and thus the risk of viral entry. Based on the above, we encourage the adoption of CP and Ang (1-7) conjugated therapy to neutralize the virus while controlling the inflammatory process to ensure organ protection.
A better alternative to attenuate the viral load and infection, in comparison with ACE2 antibodies and pharmacological inhibitors, would be to deliver excessive soluble viral receptor analogs in order to intercept the viral binding to the membrane-bound ACE2. The conceptual principle is that soluble ACE2 may act as a trap, competitively binding and neutralizing the virus while rescuing cellular ACE2 activity and protecting the lungs from injury (99). In fact, a fusion protein of recombinant human ACE2 (rhACE2) was reported to show high affinity to SARS-CoV-2 binding domain and to neutralize the virus in vitro (101). Furthermore, Monteil et al. recently demonstrated that a clinical-grade rhACE2 is capable of reducing the viral load of SARS-CoV-2 infection in Vero-E6 cells and of blocking its entry into the cells. This study revealed that rhACE2 could inhibit SARS-CoV-2 infections in human organoids, such as the kidneys, during early stages of viral infection (102). Moreover, a clinical pilot study was planned (NCT number: NCT04287686) to deliver soluble rhACE2 infusions in a small COVID-19 patient cohort in China. Nevertheless, this study was withdrawn for non-stated reasons. Regardless, a large phase II clinical trial has been initiated at the beginning of April by the Austrian pharmaceutical company APEIRON to treat COVID-19 patients with APN01-rhACE2, in Austria, Germany, and Denmark (98). On the other hand, soluble ACE2 appears to have a short half-life and may lack the capacity to overcome massive virus infection. This agent could form a reservoir of circulating viruses, thus increasing its propagation (103–105) (Figure 3).
Altogether, clinical trials using ACE2-based treatments are eagerly awaited to exclude possible adverse effects and to prove their promising potential to enhance the positive outcomes in patients infected with COVID-19.
It is believed that SARS-CoV-2 infection may cause an increase in lung injury and severe acute respiratory failure due to ACE2/Ang (1-7) downregulation. In addition, since Ang II is the major substrate of ACE2, inhibition of ACE2 following viral entry is generally associated with Ang II upregulation as well as activation of the AT1R. In fact, elevated levels of Ang II were reported in the plasma of SARS-CoV-2 infected patients, which were positively correlated with viral load and lung injury. Thus, the harmful effects of COVID-19 could be achieved by inhibiting Ang II-mediated harmful effects (25, 26, 47, 56, 106). Due to its anti-fibrotic and anti-inflammatory properties, ARBs effectively block Ang II-mediated AT1R activation and reduce acute lung injury in patients diagnosed with pneumonia, sepsis, and influenza (107–109). Therefore, ARBs blocking Ang II/AT1R pathway could overcome the adverse effects of ACE2 downregulation by SARS-CoV-2. On the other hand, several studies reported that ARBs beneficial effects could be correlated to the increase of ACE2 expression and activity in patients. Despite the beneficial properties of ACE2/Ang (1-7) axis (96, 110, 111), ACE2 upregulation after ARB treatment may open the door for viral entry, thus increasing the susceptibility of patients to SARS-CoV-2 infection.
Since the treatment of CVD patients is based on ARBs, their discontinuation in COVID-19 patients has been proposed to reduce ACE2 expression and, thus, the risk of a more severe infection associated with the increased viral entry. In fact, interruption of treatment may be more harmful than protective. For instance, discontinuing RAS inhibitors, including ARBs, in patients with an unstable clinical state (hypertension, heart failure, myocardial infarction), may result in a decline in clinical status and higher risks of mortality (60, 95). In fact, ARBs are reported to block Ang II/AT1R axis and to reduce acute lung injury pathogenesis. Interruption of ARBs might increase lung injury since Ang II-mediated AT1R activation is associated with vasoconstriction, oxidative stress, increased fibroproliferative, and inflammatory responses as well as lung oedema (54, 60, 112, 113). Importantly, studies have shown that these drugs can be protective against lung injury in SARS-CoV-1 patients by enhancing the protective arm of RAS (114). Also, retrospective studies showed that patients using ACEIs and ARBs present a lower risk of mortality and develop less severe cases as compared with those using other hypertensive drugs. On the other hand, these treatment regimens do not increase the risk of SARS-CoV-2 infection nor disease severity, as summarized in Table 1.
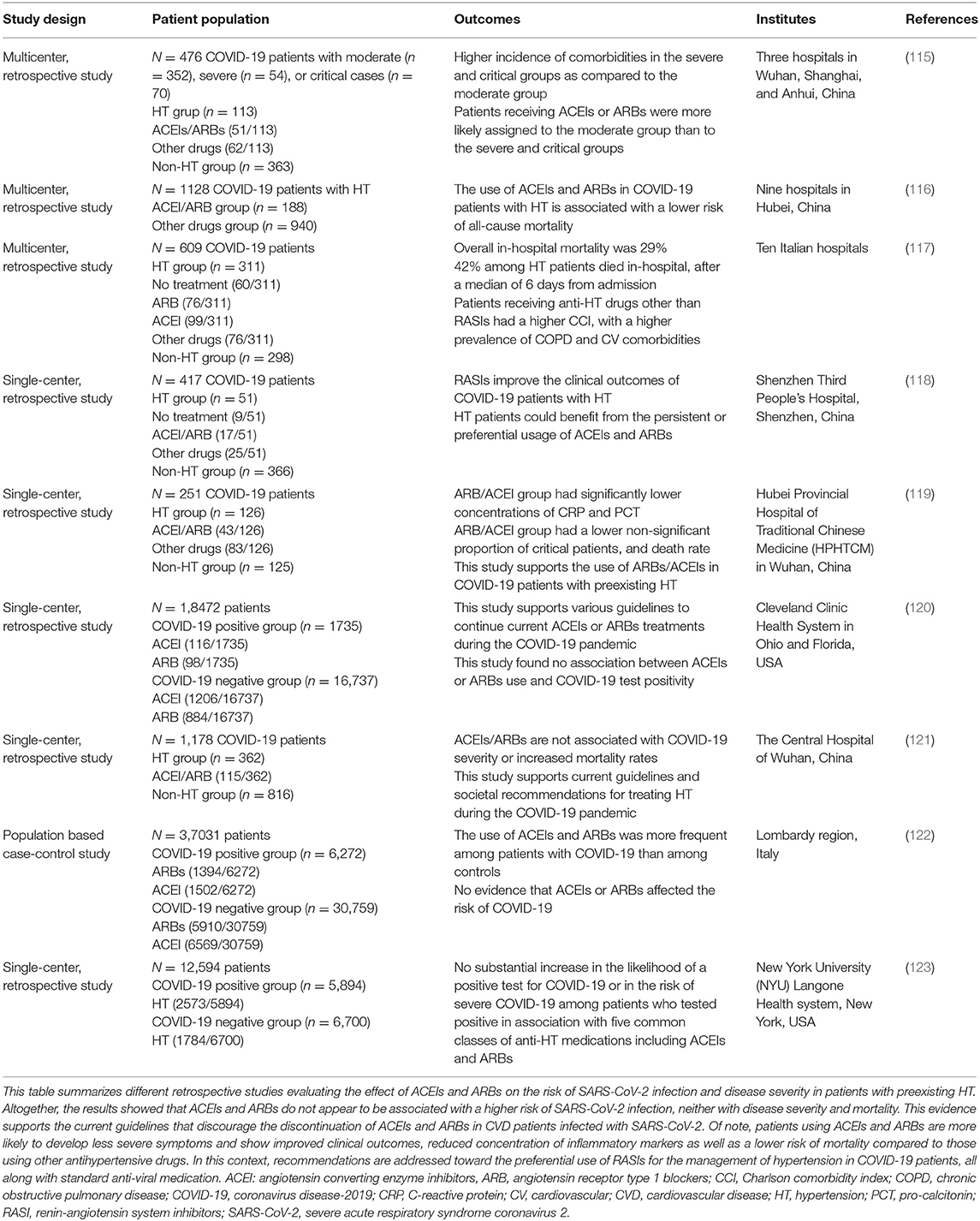
Table 1. The effect of ACEIs and ARBs on COVID-19 patients with CVD.
Taken together, more evidence is needed to support the proper use of ARBs for the treatment of COVID-19 and to exclude the risk of an increased viral entry. ARBs play a protective role in CVD patients by reducing the harmful effects of Ang II/AT1R axis while enhancing ACE2/Ang (1-7) protective axis. Therefore, ARB withdrawal can be potentially harmful rather than protective.
Several therapies are being investigated for the treatment of COVID-19 (124). Passive immunotherapy has also been reported as a treatment option to reduce mortality in many infectious viral diseases, including SARS-CoV-1 and severe influenza-related pathologies (125). Transfusion of anti-COVID-19 antibodies from recovered patients appears to be promising in severe patients. Recent studies showed that transfusion of convalescent plasma (CP) containing anti-COVID-19 neutralizing antibodies to COVID-19 critically ill patients, along with the conventional antiviral treatment, is associated with improvement in fever, inflammatory markers, lymphocyte count, viral clearance, and CT findings (126–128). In fact, Shen et al. conducted the first study describing the use of CP in COVID-19 patients. Indeed, 5 ARDS critically ill patients received CP from recovered healthy donors along with antiviral agents and methylprednisolone. All patients showed improvement of inflammatory markers, and the viral load became negative 12 days post transfusion (126). In another trial performed by Ye et al., 5 of 6 COVID-19 patients treated with CP demonstrated decreased pulmonary lesions based on their CT scan (129). Similar findings were reported by two other studies, revealing an overall improvement in clinical outcomes with no single death recorded during the treatment procedure (128, 130). Noteworthy, all the preliminary studies mentioned in Table 2 did not have control groups receiving CP alone, and the sample size of patients was generally limited in number. However, these studies established the safety and efficacy of anti-COVID-19 antibodies transfusion in critically ill patients. Therefore, CP was finally approved last August by the FDA as an investigational new drug for patients with life-threatening SARS-CoV-2 infection (144). On this basis, an FDA-initiated study on a cohort of 20,000 COVID-19 patients confirmed the safety of CP with low incidence of adverse events associated with transfusion (142). Recently, CP infusion was recommended during early stages of infection by Zeng et al. In this trial, 6 COVID-19 patients with respiratory failure received standard care along with CP treatment at a median of 21.5 days post-infection. Eventually, all patients tested negative for SARS-CoV-2 at 3 days post-infusion; however, 5 out of these 6 patients died, suggesting that CP is ineffective in reducing mortality in end-stage COVID-19 patients and should be initiated earlier (140). These findings were confirmed by Salazar et al., who reported a higher reduction in mortality rate in patients receiving CP transfusion within 44 hours of their hospitalization (143). These observations might be due to the late clinical deterioration observed in COVID-19 patients, related to hyper-immune attacks and inflammatory reactions, rather than a direct viral-effect since the peak of viral load is observed during the first week of infection (131). All clinical trials using CP for patients infected with SARS-CoV-1 or SARS-CoV-2 are presented in Table 2.
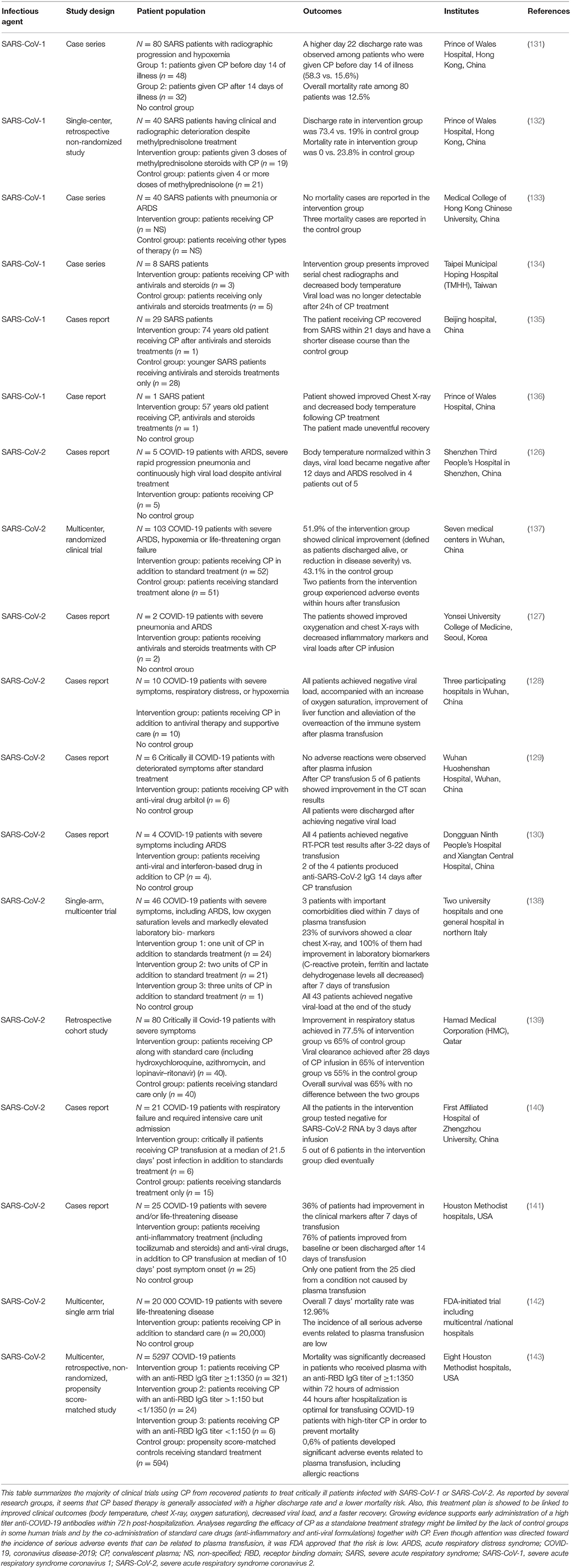
Table 2. The effect of convalescent plasma-based therapy on SARS and COVID-19 patients.
On the other hand, it is tempting to propose the use of Ang (1-7) agonists to overcome the harmful effects of SARS-CoV-2 infection in patients. Ang (1-7) has been reported to oppose the harmful effects of Ang II/AT1R axis by binding to MasR or to AT2R. In addition, it was shown to be cardiopulmonary protective through its anti-hypertensive, anti-thrombotic, anti-arrhythmic, and vasodilatory effects (145–147). Moreover, animal studies demonstrated that ARDS is associated with low Ang (1-7) levels and that Ang (1-7) upregulation reduces reactive oxygen species production and inhibits pulmonary fibrosis to control tissue damage. Besides, data showed that Ang (1-7)/MasR axis exhibits anti-inflammatory effects by inhibiting the NF-kB pathway and by reducing the production of pro-inflammatory cytokines such as TNF-α and IL-6 (53, 113, 148–151). In this context, Ang (1-7) oral formulation was also proved to attenuate the rupture of alveolar walls and behavioral changes in a mice model of elastase-induced emphysema (152).
The pathogenesis of SARS-CoV-2 infection is mediated through over-activation of the inflammatory response and an increased cytokine production (113, 148, 149). This could be related to the inhibition of the anti-inflammatory ACE2/Ang (1-7)/MasR axis by binding of the virus. Therefore, the severity of COVID-19 could be attenuated by Ang (1-7) administration, which may restore the anti-inflammatory response via MasR activation (153). Several Ang (1-7) agonists are available such as AVE-0991 (154), hydroxypropyl β-cyclodextrin (HPβCD)/Ang (1-7) (155, 156), cyclic angiotensin (1-7) (157), CGEN-856, and CGEN-857 (158). In animal models, these agonists exert their protective effects, such as vasodilation and improved cardiac remodeling, by binding to MasR, thus mimicking Ang (1-7) effects with high in vivo stability (158, 159). Unfortunately, not all Ang (1-7) agonists have been evaluated in human subjects; thus, the safety data is lacking for some of these drugs. Importantly, FDA has granted a pharmaceutical formulation of Ang (1-7), called TXA127, an orphan drug for the treatment of several conditions, including Duchenne muscular dystrophy and pulmonary arterial hypertension (160). Several clinical trials on TXA127 were announced since 2008, some of which were terminated or withdrawn for unknown reasons. For instance, phase I/II clinical trials conducted on cancer patients showed that Ang (1-7) drug is safe, well-tolerated, with no mortality rate and low-grade adverse events such as fatigue, injection site reaction, and flu-like symptoms (161, 162). Interestingly, two double-blind, placebo-control, randomized clinical trials are currently being conducted in Brazil (NCT04633772), Israel (NCT04605887), and New York (NCT04401423) on patients with severe COVID-19 cases. The purpose of these studies is to test the safety and efficacy of TXA127 and to determine whether Ang (1-7) infusions prevent respiratory failure, acute kidney injury, and multi-organ damage due to the management of inflammation. Considering the increasing spread and number of deaths due to COVID-19, it is extremely urgent to evaluate TXA127 and other Ang (1-7) agonists as possible treatments for seriously ill patients.
In the light of the vital role of Ang (1-7) in lung protection and the promising results of anti-COVID-19 antibodies based therapy, combination treatment of Ang (1-7) agonists along with anti-COVID-19 antibodies may be the ideal therapeutic intervention to alleviate COVID-19 severity (Figure 3).
This review summarizes the treatment strategies targeting ACE2 viral receptor, either directly or indirectly, in the context of COVID-19. Combination therapy using Ang (1-7) and CP in patients infected with SARS-CoV-2 appear to be the most promising alternative. Although the clinical potential of Ang (1-7) agonists has been evidenced in numerous human trials (161, 162); however, its use is limited due to the absence of studies validating its safety and efficacy in COVID-19 patients. In addition, a stable oral Ang (1-7) compound covering a broad range of patients is still lacking (163). Importantly, prolonged exposure to other immuno-suppressor drugs such as corticosteroids might increase the occurrence of secondary infections; thus, evaluating the risk of opportunistic infections in patients treated with Ang (1-7) appear to be of great value (164, 165). Furthermore, steroids possess salt retention activities that might increase the stress over the cardiovascular system.
On the other hand, CP therapy may encounter several challenges that should be taken into consideration despite its proven benefits. One such limitation is its availability; in fact, a shortage in the number of plasma donors could be seen in situations of rapid disease spread resulting in an increased number of infected patients compared to those that have recovered. This would be especially significant and serious for the rare blood group patients. In addition, potential risks can be directly associated with CP regimen. These risks include, among others, the transmission of harmful pathogens such as HIV, hepatitis B/C, and syphilis. Moreover, other transfusion-related events may occur following CP treatment, including allergic, anaphylactic, or hemolytic reactions, fever, and transfusion circulatory overload (166). Furthermore, transfusion can be associated with transfusion-related acute lung injury (167). Although this complication is not common; however, this possibility should not be ignored since COVID-19 patients are at high risk of developing pulmonary disease (168). Importantly, all studies on CP transfusion as a COVID-19 treatment showed that these severe adverse reactions are infrequent.
The presence of a confounding variable in most CP studies makes it difficult to prove the efficacy of CP transfusion as a stand-alone treatment because patients are mostly receiving a standard care treatment such as anti-viral and anti-inflammatory drugs along with anti-COVID-19 antibodies (128, 141). In addition, the safety and efficiency of CP in pregnant women and pediatric patients have not been evaluated yet. Noteworthy, several ongoing studies are covering both population groups (169). Finally, since the immune-competent population contributed to the generation of new viral strains in South Africa and the United Kingdom, new adaptations of the virus raise the concern about the possibility of escaping viral neutralization by convalescent antibodies. In this context, a very recent study reported that SARS-CoV-2 has the ability to generate new mutations in its viral spike, which is typically recognized by antibodies, thus facilitating the escape from neutralization (170).
Taken together, more clinical trials are warranted to prove the safety and efficacy as well as the synergistic therapeutic effects of this combination treatment procedure.
AN and KZ proposed the hypothesis and originated this work. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
α7-nAChR, α7 subtype of the nicotine acetylcholine receptor; ARDS, Acute respiratory distress syndrome; Ang I, angiotensin I; Ang (1-7), angiotensin (1-7); Ang (1-9), angiotensin (1-9); Ang II, angiotensin II; ACE, angiotensin converting enzyme; ACE2, angiotensin converting enzyme 2; ACEIs, angiotensin converting enzyme inhibitors; AT1R, angiotensin type 1 receptor; AT2R, angiotensin type 2 receptor; ARBs, angiotensin type 1 receptor blockers; Abs, antibodies; CV, cardiovascular; CVD, cardiovascular disease; CFR, case fatality rate; CCI, charlson comorbidity index; COPD, chronic obstructive pulmonary disease; CP, convalescent plasma; COVID-19, coronavirus disease-2019; CRP, C-reactive protein; DPP4, dipeptidyl peptidase 4; FP, fusion peptide; MasR, G-coupled receptor Mas; GPCR, G protein-coupled receptors; HR1, heptad repeat region 1; HR2, heptad repeat region 2; HT, hypertension; MERS-CoV, Middle East respiratory syndrome coronavirus; NS, non-specified; PCT, pro-calcitonin; RBD, receptor-binding domain; rhACE2, recombinant human ACE2; RAS, renin angiotensin system; RASIs, renin angiotensin system inhibitors; SARS-CoV-1, severe acute respiratory syndrome coronavirus; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; S protein, spike protein; S1, S protein subunit 1; S2, S protein subunit 2; TMPRSS2, transmembrane protease serine 2.
1. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet Lond Engl. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
2. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. (2020) 5:536–44. doi: 10.1038/s41564-020-0695-z
3. Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
4. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
5. Zhang W, Du R-H, Li B, Zheng X-S, Yang X-L, Hu B, et al. Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect. (2020) 9:386–9. doi: 10.1080/22221751.2020.1729071
6. van Doremalen N, Bushmaker T, Morris DH, Holbrook MG, Gamble A, Williamson BN, et al. Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1. N Engl J Med. (2020) 382:1564–7. doi: 10.1056/NEJMc2004973
7. Kolifarhood G, Aghaali M, Mozafar Saadati H, Taherpour N, Rahimi S, Izadi N, et al. Epidemiological and clinical aspects of COVID-19; a narrative review. Arch Acad Emerg Med. (2020) 8:e41.
8. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet Lond Engl. (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
9. Wong CK, Lam CWK, Wu AKL, Ip WK, Lee NLS, Chan IHS, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. (2004) 136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x
10. Chu KH, Tsang WK, Tang CS, Lam MF, Lai FM, To KF, et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. (2005) 67:698–705. doi: 10.1111/j.1523-1755.2005.67130.x
11. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. (2020) 382:1199–207. doi: 10.1056/NEJMoa2001316
12. Porcheddu R, Serra C, Kelvin D, Kelvin N, Rubino S. Similarity in Case Fatality Rates (CFR) of COVID-19/SARS-COV-2 in Italy and China. J Infect Dev Ctries. (2020) 14:125–8. doi: 10.3855/jidc.12600
13. Remuzzi A, Remuzzi G. COVID-19 and Italy: what next? Lancet Lond Engl. (2020) 395:1225–8. doi: 10.1016/S0140-6736(20)30627-9
14. Lu X, Zhang L, Du H, Zhang J, Li YY, Qu J, et al. SARS-CoV-2 infection in children. N Engl J Med. (2020) 382:1663–5. doi: 10.1056/NEJMc2005073
15. Guan W-J, Zhong N-S. Clinical characteristics of Covid-19 in China. Reply. N Engl J Med. (2020) 382:1861–2. doi: 10.1056/NEJMc2005203
16. Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
17. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. (2020) 5:562–9. doi: 10.1038/s41564-020-0688-y
18. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor Recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS Coronavirus. J Virol. (2020) 94:e00127-20. doi: 10.1128/JVI.00127-20
19. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. (2020) 367:1444–8. doi: 10.1126/science.abb2762
20. Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I, et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci USA. (2020) 117:7001–3. doi: 10.1073/pnas.2002589117
21. Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. (2015) 202:120–34. doi: 10.1016/j.virusres.2014.11.021
22. Wang P, Liu L, Iketani S, Luo Y, Guo Y, Wang M, et al. Increased resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7 to antibody neutralization. BioRxiv Prepr Serv Biol [Preprint]. (2021) doi: 10.1101/2021.01.25.428137
23. Santos JC, Passos GA. The high infectivity of SARS-CoV-2 B.1.1.7 is associated with increased interaction force between Spike-ACE2 caused by the viral N501Y mutation. Bioinformatics. [Preprint]. (2021) doi: 10.1101/2020.12.29.424708
24. Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–92.e6. doi: 10.1016/j.cell.2020.02.058
25. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. (2005) 436:112–6. doi: 10.1038/nature03712
26. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. (2005) 11:875–9. doi: 10.1038/nm1267
27. Wang K, Gheblawi M, Oudit GY. Angiotensin converting enzyme 2: a double-edged sword. Circulation. (2020) 142:426–8. doi: 10.1161/CIRCULATIONAHA.120.047049
28. Imai Y, Kuba K, Penninger JM. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. (2008) 93:543–8. doi: 10.1113/expphysiol.2007.040048
29. Kuba K, Imai Y, Penninger JM. Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol. (2006) 6:271–6. doi: 10.1016/j.coph.2006.03.001
30. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. (2000) 275:33238–43. doi: 10.1074/jbc.M002615200
31. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. (2005) 280:30113–9. doi: 10.1074/jbc.M505111200
32. Turner AJ, Hooper NM. Angiotensin-converting enzyme 2. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. Elsevier (2004). p. 349–51. doi: 10.1016/B978-0-12-079611-3.50092-6
33. Tikellis C, Thomas MC. Angiotensin-Converting Enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept. (2012) 2012:256294. doi: 10.1155/2012/256294
34. Unger T, Steckelings UM, Santos RS dos. The Protective Arm of the Renin Angiotensin: Functional Aspects and Therapeutic Implications. Academic Press (2015). p. 316.
35. Simões e Silva AC, Pinheiro S, Pereira R, Ferreira A, Santos R. The therapeutic potential of Angiotensin-(1-7) as a novel renin- angiotensin system mediator. Mini-Rev Med Chem. (2006) 6:603–609. doi: 10.2174/138955706776876203
36. Turner AJ, Tipnis SR, Guy JL, Rice G, Hooper NM. ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ACE inhibitors. Can J Physiol Pharmacol. (2002) 80:346–53. doi: 10.1139/y02-021
37. Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. (2004) 383:45–51. doi: 10.1042/BJ20040634
38. Ferrario CM, Ahmad S, Nagata S, Simington SW, Varagic J, Kon N, et al. An evolving story of angiotensin-II-forming pathways in rodents and humans. Clin Sci. (2014) 126:461–9. doi: 10.1042/CS20130400
39. Santos RAS, Simoes e Silva AC, Maric C, Silva DMR, Machado RP, de Buhr I, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA. (2003) 100:8258–63. doi: 10.1073/pnas.1432869100
40. Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, Sexton PM, et al. G-Protein–coupled receptor mas is a physiological antagonist of the Angiotensin II Type 1 receptor. Circulation. (2005) 111:1806–13. doi: 10.1161/01.CIR.0000160867.23556.7D
41. Nehme A, Zibara K. Efficiency and specificity of RAAS inhibitors in cardiovascular diseases: how to achieve better end-organ protection? Hypertens Res. (2017) 40:903–9. doi: 10.1038/hr.2017.65
42. Wehbe Z, Hammoud S, Soudani N, Zaraket H, El-Yazbi A, Eid AH. Molecular insights into SARS COV-2 interaction with cardiovascular disease: role of RAAS and MAPK signaling. Front Pharmacol. (2020) 11:836. doi: 10.3389/fphar.2020.00836
43. Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. (2006) 86:747–803. doi: 10.1152/physrev.00036.2005
44. Magalhães GS, Rodrigues-Machado MG, Motta-Santos D, Silva AR, Caliari MV, Prata LO, et al. Angiotensin-(1-7) attenuates airway remodelling and hyperresponsiveness in a model of chronic allergic lung inflammation. Br J Pharmacol. (2015) 172:2330–42. doi: 10.1111/bph.13057
45. Nehme A, Cerutti C, Dhaouadi N, Gustin MP, Courand P-Y, Zibara K, et al. Atlas of tissue renin-angiotensin-aldosterone system in human: a transcriptomic meta-analysis. Sci Rep. (2015) 5:10035. doi: 10.1038/srep10035
46. Imai Y, Kuba K, Penninger JM. Angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Cell Mol Life Sci CMLS. (2007) 64:2006–12. doi: 10.1007/s00018-007-6228-6
47. Li Y, Zeng Z, Cao Y, Liu Y, Ping F, Liang M, et al. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci Rep. (2016) 6:27911. doi: 10.1038/srep27911
48. Magalhaes GS, Barroso LC, Reis AC, Rodrigues-Machado MG, Gregório JF, Motta-Santos D, et al. Angiotensin-(1-7) promotes resolution of eosinophilic inflammation in an experimental model of asthma. Front Immunol. (2018) 9:58. doi: 10.3389/fimmu.2018.00058
49. Magalhães GS, Rodrigues-Machado MG, Motta-Santos D, Alenina N, Bader M, Santos RA, et al. Chronic allergic pulmonary inflammation is aggravated in angiotensin-(1-7) Mas receptor knockout mice. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L1141–8. doi: 10.1152/ajplung.00029.2016
50. Specks U, Martin WJ, Rohrbach MS. Bronchoalveolar lavage fluid angiotensin-converting enzyme in interstitial lung diseases. Am Rev Respir Dis. (1990) 141:117–23. doi: 10.1164/ajrccm/141.1.117
51. Fourrier F, Chopin C, Wallaert B, Mazurier C, Mangalaboyi J, Durocher A. Compared evolution of plasma fibronectin and angiotensin-converting enzyme levels in septic ARDS. Chest. (1985) 87:191–5. doi: 10.1378/chest.87.2.191
52. Idell S, Kueppers F, Lippmann M, Rosen H, Niederman M, Fein A. Angiotensin converting enzyme in bronchoalveolar lavage in ARDS. Chest. (1987) 91:52–6. doi: 10.1378/chest.91.1.52
53. Chen Q, Yang Y, Huang Y, Pan C, Liu L, Qiu H. Angiotensin-(1-7) attenuates lung fibrosis by way of Mas receptor in acute lung injury. J Surg Res. (2013) 185:740–7. doi: 10.1016/j.jss.2013.06.052
54. Marshall RP, Gohlke P, Chambers RC, Howell DC, Bottoms SE, Unger T, et al. Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2004) 286:L156–64. doi: 10.1152/ajplung.00313.2002
55. Jia HP, Look DC, Shi L, Hickey M, Pewe L, Netland J, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. (2005) 79:14614–21. doi: 10.1128/JVI.79.23.14614-14621.2005
56. Rivellese F, Prediletto E. ACE2 at the centre of COVID-19 from paucisymptomatic infections to severe pneumonia. Autoimmun Rev. (2020) 19:102536. doi: 10.1016/j.autrev.2020.102536
57. Wu Y. Compensation of ACE2 function for possible clinical management of 2019-nCoV-Induced acute lung injury. Virol Sin. (2020) 35:256–8. doi: 10.1007/s12250-020-00205-6
58. Hirano T, Murakami M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity. (2020) 52:731–3. doi: 10.1016/j.immuni.2020.04.003
59. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. (2020) 367:1260–3. doi: 10.1126/science.abb2507
60. Rossi GP, Sanga V, Barton M. Potential harmful effects of discontinuing ACE-inhibitors and ARBs in COVID-19 patients. eLife. (2020) 9:e57278. doi: 10.7554/eLife.57278
61. Ortega JT, Serrano ML, Pujol FH, Rangel HR. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: an in silico analysis. EXCLI J. (2020) 19:410–7. doi: 10.17179/excli2020-1167
62. Chen J, Wang R, Wang M, Wei G-W. Mutations strengthened SARS-CoV-2 infectivity. J Mol Biol. (2020) 432:5212–26. doi: 10.1016/j.jmb.2020.07.009
63. Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. (2020) 182:812–27.e19. doi: 10.1016/j.cell.2020.06.043
64. Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, et al. Spike mutation D614G alters SARS-CoV-2 fitness and neutralization susceptibility. BioRxiv Prepr Serv Biol [Preprint]. (2020) doi: 10.1101/2020.09.01.278689
65. Zhang L, Jackson CB, Mou H, Ojha A, Rangarajan ES, Izard T, et al. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. BioRxiv Prepr Serv Biol [Preprint]. (2020) doi: 10.1101/2020.06.12.148726
66. Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. Epidemiology MedRxiv [Preprint]. (2020) doi: 10.1101/2020.12.21.20248640
67. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
68. Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Krämer-Kühl A, et al. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol. (2013) 87:5502–11. doi: 10.1128/JVI.00128-13
69. Glowacka I, Bertram S, Müller MA, Allen P, Soilleux E, Pfefferle S, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. (2011) 85:4122–34. doi: 10.1128/JVI.02232-10
70. Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol. (2019) 93:e01815-18. doi: 10.1128/JVI.01815-18
71. Muruato AE, Fontes-Garfias CR, Ren P, Garcia-Blanco MA, Menachery VD, Xie X, et al. A high-throughput neutralizing antibody assay for COVID-19 diagnosis and vaccine evaluation. Nat Commun. (2020) 11:4059. doi: 10.1038/s41467-020-17892-0
72. Li G, He X, Zhang L, Ran Q, Wang J, Xiong A, et al. Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J Autoimmun. (2020) 112:102463. doi: 10.1016/j.jaut.2020.102463
73. Shi Y, Yu X, Zhao H, Wang H, Zhao R, Sheng J. Host susceptibility to severe COVID-19 and establishment of a host risk score: findings of 487 cases outside Wuhan. Crit Care Lond Engl. (2020) 24:108. doi: 10.1186/s13054-020-2833-7
74. Singh AK, Gupta R, Misra A. Comorbidities in COVID-19: outcomes in hypertensive cohort and controversies with renin angiotensin system blockers. Diabetes Metab Syndr. (2020) 14:283–7. doi: 10.1016/j.dsx.2020.03.016
75. Lippi G, Wong J, Henry BM. Hypertension in patients with coronavirus disease 2019 (COVID-19): a pooled analysis. Pol Arch Intern Med. (2020) 130:304–9. doi: 10.20452/pamw.15272
76. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. (2020) 14:185–92. doi: 10.1007/s11684-020-0754-0
77. Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. (2020) 26:681–7. doi: 10.1038/s41591-020-0868-6
78. Cai G, Bossé Y, Xiao F, Kheradmand F, Amos CI. Tobacco smoking increases the lung gene expression of ACE2, the Receptor of SARS-CoV-2. Am J Respir Crit Care Med. (2020) 201:1557–9. doi: 10.1164/rccm.202003-0693LE
79. Kashyap VK, Dhasmana A, Massey A, Kotnala S, Zafar N, Jaggi M, et al. Smoking and COVID-19: adding fuel to the flame. Int J Mol Sci. (2020) 21:6581. doi: 10.3390/ijms21186581
80. Russo P, Bonassi S, Giacconi R, Malavolta M, Tomino C, Maggi F. COVID-19 and smoking: is nicotine the hidden link? Eur Respir J. (2020) 55:2001116. doi: 10.1183/13993003.01116-2020
81. Gülsen A, Yigitbas BA, Uslu B, Drömann D, Kilinc O. The effect of smoking on COVID-19 symptom severity: systematic review and meta-analysis. Pulm Med. (2020) 2020:7590207. doi: 10.1101/2020.08.15.20102699
82. Lawrence H, Hunter A, Murray R, Lim WS, McKeever T. Cigarette smoking and the occurrence of influenza - systematic review. J Infect. (2019) 79:401–6. doi: 10.1016/j.jinf.2019.08.014
83. Duffney PF, Embong AK, McGuire CC, Thatcher TH, Phipps RP, Sime PJ. Cigarette smoke increases susceptibility to infection in lung epithelial cells by upregulating caveolin-dependent endocytosis. PLoS ONE. (2020) 15:e0232102. doi: 10.1371/journal.pone.0232102
84. Duffney PF, McCarthy CE, Nogales A, Thatcher TH, Martinez-Sobrido L, Phipps RP, et al. Cigarette smoke dampens antiviral signaling in small airway epithelial cells by disrupting TLR3 cleavage. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L505–13. doi: 10.1152/ajplung.00406.2017
85. Staudt MR, Salit J, Kaner RJ, Hollmann C, Crystal RG. Altered lung biology of healthy never smokers following acute inhalation of E-cigarettes. Respir Res. (2018) 19:78. doi: 10.1186/s12931-018-0778-z
86. Shastri MD, Shukla SD, Chong WC, Kc R, Dua K, Patel RP, et al. Smoking and COVID-19: what we know so far. Respir Med. (2020) 176:106237. doi: 10.1016/j.rmed.2020.106237
87. Leung JM, Yang CX, Tam A, Shaipanich T, Hackett T-L, Singhera GK, et al. ACE-2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID-19. Eur Respir J. (2020) 55:2000688. doi: 10.1101/2020.03.18.20038455
88. Tomchaney M, Contoli M, Mayo J, Baraldo S, Shuaizhi L, Cabel CR, et al. Paradoxical effects of cigarette smoke and COPD on SARS-CoV2 infection and disease. BioRxiv Prepr Serv Biol [Preprint]. (2020) doi: 10.1101/2020.12.07.413252
89. Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. (2004) 43:970–6. doi: 10.1161/01.HYP.0000124667.34652.1a
90. Wang X, Ye Y, Gong H, Wu J, Yuan J, Wang S, et al. The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang(1-7)-Mas axes in pressure overload-induced cardiac remodeling in male mice. J Mol Cell Cardiol. (2016) 97:180–90. doi: 10.1016/j.yjmcc.2016.05.012
91. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. (2005) 111:2605–10. doi: 10.1161/CIRCULATIONAHA.104.510461
92. Kow CS, Zaidi STR, Hasan SS. Cardiovascular disease and use of renin-angiotensin system inhibitors in COVID-19. Am J Cardiovasc Drugs Drugs Devices Interv. (2020) 20:217–21. doi: 10.1007/s40256-020-00406-0
93. Burrell LM, Risvanis J, Kubota E, Dean RG, MacDonald PS, Lu S, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. (2005) 26:369–75; discussion 322–324. doi: 10.1093/eurheartj/ehi114
94. Burchill LJ, Velkoska E, Dean RG, Griggs K, Patel SK, Burrell LM. Combination renin-angiotensin system blockade and angiotensin-converting enzyme 2 in experimental myocardial infarction: implications for future therapeutic directions. Clin Sci Lond Engl. (2012) 123:649–58. doi: 10.1042/CS20120162
95. Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med. (2020) 382:1653–9. doi: 10.1056/NEJMsr2005760
96. Furuhashi M, Moniwa N, Mita T, Fuseya T, Ishimura S, Ohno K, et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am J Hypertens. (2015) 28:15–21. doi: 10.1093/ajh/hpu086
97. Danser AHJ, Epstein M, Batlle D. Renin-angiotensin system blockers and the COVID-19 pandemic: at present there is no evidence to abandon renin-angiotensin system blockers. Hypertension. (2020) 75:1382–5. doi: 10.1161/HYPERTENSIONAHA.120.15082
98. Groß S, Jahn C, Cushman S, Bär C, Thum T. SARS-CoV-2 receptor ACE2-dependent implications on the cardiovascular system: from basic science to clinical implications. J Mol Cell Cardiol. (2020) 144:47–53. doi: 10.1016/j.yjmcc.2020.04.031
99. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. (2020) 46:586–90. doi: 10.1007/s00134-020-05985-9
100. Abassi ZA, Skorecki K, Heyman SN, Kinaneh S, Armaly Z. Covid-19 infection and mortality: a physiologist's perspective enlightening clinical features and plausible interventional strategies. Am J Physiol Lung Cell Mol Physiol. (2020) 318:L1020–22. doi: 10.1152/ajplung.00097.2020
101. Lei C, Qian K, Li T, Zhang S, Fu W, Ding M, et al. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat Commun. (2020) 11:2070. doi: 10.1038/s41467-020-16048-4
102. Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble Human ACE2. Cell. (2020) 181:905–13.e7. doi: 10.1016/j.cell.2020.04.004
103. Dimitrov DS. The secret life of ACE2 as a receptor for the SARS Virus. Cell. (2003) 115:652–3. doi: 10.1016/S0092-8674(03)00976-0
104. Alhenc-Gelas F, Drueke TB. Blockade of SARS-CoV-2 infection by recombinant soluble ACE2. Kidney Int. (2020) 97:1091–3. doi: 10.1016/j.kint.2020.04.009
105. Ciaglia E, Vecchione C, Puca AA. COVID-19 infection and circulating ACE2 levels: protective role in women and Children. Front Pediatr. (2020) 8:206. doi: 10.3389/fped.2020.00206
106. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. (2020) 63:364–74. doi: 10.1007/s11427-020-1643-8
107. Mentz RJ, Bakris GL, Waeber B, McMurray JJV, Gheorghiade M, Ruilope LM, et al. The past, present and future of renin-angiotensin aldosterone system inhibition. Int J Cardiol. (2013) 167:1677–87. doi: 10.1016/j.ijcard.2012.10.007
108. Saavedra JM. Angiotensin receptor blockers and COVID-19. Pharmacol Res. (2020) 156:104832. doi: 10.1016/j.phrs.2020.104832
109. Fedson DS. Treating the host response to emerging virus diseases: lessons learned from sepsis, pneumonia, influenza and Ebola. Ann Transl Med. (2016) 4:421. doi: 10.21037/atm.2016.11.03
110. Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. (2020) 8:e21. doi: 10.1016/S2213-2600(20)30116-8
111. Jakovac H. COVID-19: is the ACE2 just a foe? Am J Physiol-Lung Cell Mol Physiol. (2020) 318:L1025–6. doi: 10.1152/ajplung.00119.2020
112. Jia H. Pulmonary Angiotensin-Converting Enzyme 2 (ACE2) and inflammatory lung disease. Shock Augusta Ga. (2016) 46:239–48. doi: 10.1097/SHK.0000000000000633
113. Wösten-van Asperen RM, Lutter R, Specht PA, Moll GN, van Woensel JB, van der Loos CM, et al. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J Pathol. (2011) 225:618–27. doi: 10.1002/path.2987
114. Tan WSD, Liao W, Zhou S, Mei D, Wong W-SF. Targeting the renin-angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr Opin Pharmacol. (2018) 40:9–17. doi: 10.1016/j.coph.2017.12.002
115. Feng Y, Ling Y, Bai T, Xie Y, Huang J, Li J, Xiong W, et al. COVID-19 with different severities: a multicenter study of clinical features. Am J Respir Crit Care Med. (2020) 201:1380–8. doi: 10.1164/rccm.202002-0445OC
116. Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, et al. Association of inpatient use of angiotensin converting enzyme inhibitors and angiotensin ii receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res. (2020) 126:1671–81. doi: 10.1161/CIRCRESAHA.120.317134
117. Tedeschi S, Giannella M, Bartoletti M, Trapani F, Tadolini M, Borghi C, et al. Clinical impact of renin-angiotensin system inhibitors on in-hospital mortality of patients with hypertension hospitalized for COVID-19. Clin Infect Dis. (2020) 71:899–901. doi: 10.1093/cid/ciaa492
118. Meng J, Xiao G, Zhang J, He X, Ou M, Bi J, et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect. (2020) 9:757–60. doi: 10.1080/22221751.2020.1746200
119. Yang G, Tan Z, Zhou L, Yang M, Peng L, Liu J, et al. Effects of ARBs and ACEIs on virus infection, inflammatory status and clinical outcomes in COVID-19 patients with hypertension: a single center retrospective study. Hypertension. (2020) 76:51–8. doi: 10.1161/HYPERTENSIONAHA.120.15143
120. Mehta N, Kalra A, Nowacki AS, Anjewierden S, Han Z, Bhat P, et al. Association of use of angiotensin-converting enzyme inhibitors and Angiotensin II receptor blockers with testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:1020–6. doi: 10.1001/jamacardio.2020.1855
121. Li J, Wang X, Chen J, Zhang H, Deng A. Association of renin-angiotensin system inhibitors with severity or risk of death in patients with hypertension hospitalized for coronavirus disease 2019 (COVID-19) infection in Wuhan, China. JAMA Cardiol. (2020) 5:825–30. doi: 10.1001/jamacardio.2020.1624
122. Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Renin–Angiotensin–aldosterone system blockers and the risk of Covid-19. N Engl J Med. (2020) 382:2431–40. doi: 10.1056/nejmoa2006923
123. Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB, et al. Renin–Angiotensin–aldosterone system inhibitors and risk of Covid-19. N Engl J Med. (2020) 382:2441–8. doi: 10.1056/nejmoa2008975
124. Kaddoura M, AlIbrahim M, Hijazi G, Soudani N, Audi A, Alkalamouni H, et al. COVID-19 therapeutic options under investigation. Front Pharmacol. (2020) 11:1196. doi: 10.3389/fphar.2020.01196
125. Mair-Jenkins J, Saavedra-Campos M, Baillie JK, Cleary P, Khaw F-M, Lim WS, et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. J Infect Dis. (2015) 211:80–90. doi: 10.1093/infdis/jiu396
126. Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J, et al. Treatment of 5 Critically Ill patients with COVID-19 with convalescent plasma. JAMA. (2020) 323:1582–9. doi: 10.1001/jama.2020.4783
127. Ahn JY, Sohn Y, Lee SH, Cho Y, Hyun JH, Baek YJ, et al. Use of convalescent plasma therapy in two COVID-19 patients with acute respiratory distress syndrome in Korea. J Korean Med Sci. (2020) 35:e149. doi: 10.3346/jkms.2020.35.e149
128. Duan K, Liu B, Li C, Zhang H, Yu T, Qu J, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci USA. (2020) 117:9490–6. doi: 10.1073/pnas.2004168117
129. Ye M, Fu D, Ren Y, Wang F, Wang D, Zhang F, et al. Treatment with convalescent plasma for COVID-19 patients in Wuhan, China. J Med Virol. (2020) 92:1890–901. doi: 10.1002/jmv.25882
130. Zhang B, Liu S, Tan T, Huang W, Dong Y, Chen L, et al. Treatment with convalescent plasma for Critically Ill patients with severe acute respiratory syndrome Coronavirus 2 infection. Chest. (2020) 158:e9–e13. doi: 10.1016/j.chest.2020.03.039
131. Cheng Y, Wong R, Soo YOY, Wong WS, Lee CK, Ng MHL, et al. Use of convalescent plasma therapy in SARS patients in Hong Kong. Eur J Clin Microbiol Infect Dis. (2005) 24:44–6. doi: 10.1007/s10096-004-1271-9
132. Soo YOY, Cheng Y, Wong R, Hui DS, Lee CK, Tsang KK, Ng MH, et al. Retrospective comparison of convalescent plasma with continuing high-dose methylprednisolone treatment in SARS patients. Clin Microbiol Infect. (2004) 10:676–8. doi: 10.1111/j.1469-0691.2004.00956.x
133. Nie QH, Luo XD, Hui WL. Advances in clinical diagnosis and treatment of severe acute respiratory syndrome. World J Gastroenterol. (2003) 9:1139–43. doi: 10.3748/wjg.v9.i6.1139
134. Yeh KM, Chiueh TS, Siu LK, Lin JC, Chan PK, Peng MY, et al. Experience of using convalescent plasma for severe acute respiratory syndrome among healthcare workers in a Taiwan hospital. J Antimicrob Chemother. (2005) 56:919–22. doi: 10.1093/jac/dki346
135. Zhou X, Zhao M, Wang FS, Jiang TJ, Li YG, Nie WM, et al. Epidemiologic features, clinical diagnosis and therapy of first cluster of patients with severe acute respiratory syndrome in Beijing area. Zhonghua yi xue za zhi. (2003) 83:1018–22.
136. Wong VW, Dai D, Wu AK, Sung JJ. Treatment of severe acute respiratory syndrome with convalescent plasma. Hong Kong Med J. (2003) 9:199–201.
137. Li L, Zhang W, Hu Y, Tong X, Zheng S, Yang J, et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. JAMA. (2020) 324:460–70. doi: 10.1001/jama.2020.10044
138. Perotti C, Baldanti F, Bruno R, Del Fante C, Seminari E, Casari S, et al. Mortality reduction in 46 severe Covid-19 patients treated with hyperimmune plasma. a proof of concept single arm multicenter trial. Haematologica. (2020) 105:2834–40. doi: 10.3324/haematol.2020.261784
139. Omrani AS, Zaqout A, Baiou A, Daghfal J, Elkum N, Alattar RA, et al. Convalescent plasma for the treatment of patients with severe coronavirus disease 2019: a preliminary report. J Med Virol. (2020) 93:1678–86. doi: 10.1002/jmv.26537
140. Zeng Q-L, Yu Z-J, Gou J-J, Li G-M, Ma S-H, Zhang G-F, et al. Effect of convalescent plasma therapy on viral shedding and survival in patients with coronavirus disease 2019. J Infect Dis. (2020) 222:38–43. doi: 10.1093/infdis/jiaa228
141. Salazar E, Perez KK, Ashraf M, Chen J, Castillo B, Christensen PA, et al. Treatment of Coronavirus Disease 2019 (COVID-19) patients with convalescent plasma. Am J Pathol. (2020) 190:1680–90. doi: 10.1016/j.ajpath.2020.08.001
142. Joyner MJ, Bruno KA, Klassen SA, Kunze KL, Johnson PW, Lesser ER, et al. Safety update. Mayo Clin Proc. (2020) 95:1888–97. doi: 10.1016/j.mayocp.2020.06.028
143. Salazar E, Christensen PA, Graviss EA, Nguyen DT, Castillo B, Chen J, et al. Significantly decreased mortality in a large cohort of coronavirus disease 2019 (COVID-19) patients transfused early with convalescent plasma containing high-titer anti-severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) spike protein IgG. Am J Pathol. (2021) 191:90–107. doi: 10.1016/j.ajpath.2020.10.008
144. Recommendations for Investigational COVID-19 Convalescent Plasma/FDA.7. Available online at: https://www.fda.gov/vaccines-blood-biologics/investigational-new-drug-ind-or-device-exemption-ide-process-cber/recommendations-investigational-covid-19-convalescent-plasma
145. Nehme A, Zouein FA, Zayeri ZD, Zibara K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J Cardiovasc Dev Dis. (2019) 6:14. doi: 10.3390/jcdd6020014
146. Santos RA. Angiotensin-(1-7). Hypertension. (2014) 63:1138–47. doi: 10.1161/HYPERTENSIONAHA.113.01274
147. Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Díez-Freire C, Dooies A, et al. The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. (2010) 182:1065–72. doi: 10.1164/rccm.200912-1840OC
148. Rodrigues Prestes TR, Rocha NP, Miranda AS, Teixeira AL, Simoes-e-Silva AC. The anti-inflammatory potential of ACE2/Angiotensin-(1-7)/Mas receptor axis: evidence from basic and clinical research. Curr Drug Targets. (2017) 18:1301–13. doi: 10.2174/1389450117666160727142401
149. Chakraborty C, Sharma AR, Sharma G, Bhattacharya M, Lee SS. SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): diagnostic and proposed therapeutic options. Eur Rev Med Pharmacol Sci. (2020) 24:4016–26. doi: 10.26355/eurrev_202004_20871
150. Gwathmey TM, Pendergrass KD, Reid SD, Rose JC, Diz DI, Chappell MC. Angiotensin-(1-7)-Angiotensin-Converting Enzyme 2 attenuates reactive oxygen species formation to Angiotensin II within the cell nucleus. Hypertension. (2010) 55:166–71. doi: 10.1161/HYPERTENSIONAHA.109.141622
151. Klein N, Gembardt F, Supé S, Kaestle SM, Nickles H, Erfinanda L, et al. Angiotensin-(1-7) protects from experimental acute lung injury. Crit Care Med. (2013) 41:e334–43. doi: 10.1097/CCM.0b013e31828a6688
152. Bastos AC, Magalhães GS, Gregório JF, Matos NA, Motta-Santos D, Bezerra FS, et al. Oral formulation angiotensin-(1-7) therapy attenuates pulmonary and systemic damage in mice with emphysema induced by elastase. Immunobiology. (2020) 225:151893. doi: 10.1016/j.imbio.2019.12.002
153. Magalhaes GS, Rodrigues-Machado M da G, Motta-Santos D, Campagnole-Santos MJ, Santos RAS. Activation of Ang-(1-7)/Mas receptor is a possible strategy to treat coronavirus (SARS-CoV-2) infection. Front Physiol. (2020) 11:730. doi: 10.3389/fphys.2020.00730
154. Carvalho MBL, Duarte FV, Faria-Silva R, Fauler B, da Mata Machado LT, de Paula RD, et al. Evidence for Mas-mediated bradykinin potentiation by the angiotensin-(1-7) nonpeptide mimic AVE 0991 in normotensive rats. Hypertension. (2007) 50:762–7. doi: 10.1161/HYPERTENSIONAHA.107.094987
155. Zhang F, Liu J, Li S-F, Song J-X, Ren J-Y, Chen H. Angiotensin-(1-7): new perspectives in atherosclerosis treatment. J Geriatr Cardiol JGC. (2015) 12:676–82. doi: 10.11909/j.issn.1671-5411.2015.06.014
156. Marques FD, Ferreira AJ, Sinisterra RDM, Jacoby BA, Sousa FB, Caliari MV, et al. An oral formulation of angiotensin-(1-7) produces cardioprotective effects in infarcted and isoproterenol-treated rats. Hypertension. (2011) 57:477–83. doi: 10.1161/HYPERTENSIONAHA.110.167346
157. Durik M, van Veghel R, Kuipers A, Rink R, Haas Jimoh Akanbi M, Moll G, et al. The effect of the thioether-bridged, stabilized Angiotensin-(1-7) analogue cyclic ang-(1-7) on cardiac remodeling and endothelial function in rats with myocardial infarction. Int J Hypertens. (2012) 2012:536426. doi: 10.1155/2012/536426
158. Savergnini SQ, Beiman M, Lautner RQ, de Paula-Carvalho V, Allahdadi K, Pessoa DC, et al. Vascular relaxation, antihypertensive effect, and cardioprotection of a novel peptide agonist of the MAS receptor. Hypertension. (2010) 56:112–20. doi: 10.1161/HYPERTENSIONAHA.110.152942
159. Povlsen AL, Grimm D, Wehland M, Infanger M, Krüger M. The vasoactive mas receptor in essential hypertension. J Clin Med. (2020) 9:267. doi: 10.3390/jcm9010267
160. National Center for Advancing Translational Sciences. ANGIOTENSIN 1-7. Available online at: https://drugs.ncats.io/substance/IJ3FUK8MOF (accessed March 04, 2021).
161. Savage PD, Lovato J, Brosnihan KB, Miller AA, Petty WJ. Phase II Trial of Angiotensin-(1-7) for the treatment of patients with metastatic sarcoma. Sarcoma. (2016) 2016:4592768. doi: 10.1155/2016/4592768
162. Rodgers KE, Oliver J, diZerega GS. Phase I/II dose escalation study of angiotensin 1-7 [A(1-7)] administered before and after chemotherapy in patients with newly diagnosed breast cancer. Cancer Chemother Pharmacol. (2006) 57:559–68. doi: 10.1007/s00280-005-0078-4
163. Machado-Silva A, Passos-Silva D, Santos RA, Sinisterra RD. Therapeutic uses for Angiotensin-(1-7). Expert Opin Ther Pat. (2016) 26:669–78. doi: 10.1080/13543776.2016.1179283
164. van Paassen J, Vos JS, Hoekstra EM, Neumann KMI, Boot PC, Arbous SM. Corticosteroid use in COVID-19 patients: a systematic review and meta-analysis on clinical outcomes. Crit Care Lond Engl. (2020) 24:696. doi: 10.1186/s13054-020-03400-9
165. Use of Angiotensin-(1-7) in COVID-19 (NCT04633772). Available online at: https://clinicaltrials.gov/ct2/show/NCT04633772 (accessed March 04, 2021).
166. Piyush R, Rajarshi K, Khan R, Ray S. Convalescent plasma therapy: a promising coronavirus disease 2019 treatment strategy. Open Biol. (2020) 10:200174. doi: 10.1098/rsob.200174
167. Mora-Rillo M, Arsuaga M, Ramírez-Olivencia G, de la Calle F, Borobia AM, Sánchez-Seco P, et al. Acute respiratory distress syndrome after convalescent plasma use: treatment of a patient with Ebola virus disease contracted in Madrid, Spain. Lancet Respir Med. (2015) 3:554–62. doi: 10.1016/S2213-2600(15)00180-0
168. Gajic O, Rana R, Winters JL, Yilmaz M, Mendez JL, Rickman OB, et al. Transfusion-related acute lung injury in the critically ill: prospective nested case-control study. Am J Respir Crit Care Med. (2007) 176:886–91. doi: 10.1164/rccm.200702-271OC
169. COVID-19 Treatment Guidelines. Available online at: https://www.covid19treatmentguidelines.nih.gov/immune-based-therapy/blood-derived-products/convalescent-plasma/ (accessed March 04, 2021).
Keywords: ACE2, SARS-CoV-2, COVID-19, lung pathology, cardiovascular pathology, convalescent plasma (CP), Angiotensin 1-7 (Ang1-7), combination therapy
Citation: Issa H, Eid AH, Berry B, Takhviji V, Khosravi A, Mantash S, Nehme R, Hallal R, Karaki H, Dhayni K, Faour WH, Kobeissy F, Nehme A and Zibara K (2021) Combination of Angiotensin (1-7) Agonists and Convalescent Plasma as a New Strategy to Overcome Angiotensin Converting Enzyme 2 (ACE2) Inhibition for the Treatment of COVID-19. Front. Med. 8:620990. doi: 10.3389/fmed.2021.620990
Received: 26 October 2020; Accepted: 22 February 2021;
Published: 18 March 2021.
Edited by:
Reza Lashgari, Institute for Research in Fundamental Sciences, IranReviewed by:
Dongdong Li, Sichuan University, ChinaCopyright © 2021 Issa, Eid, Berry, Takhviji, Khosravi, Mantash, Nehme, Hallal, Karaki, Dhayni, Faour, Kobeissy, Nehme and Zibara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazem Zibara, a3ppYmFyYUB1bC5lZHUubGI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.