 Lili Ding
Lili Ding Juan Yang1
Juan Yang1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 27 April 2021
Sec. Pulmonary Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.616200
This article is part of the Research TopicOrgan Fibrosis: Pathogenesis, Biomarkers and Therapeutic TargetsView all 12 articles
Chronic inflammatory pulmonary diseases are characterized by recurrent and persistent inflammation of the airways, commonly associated with poor clinical outcomes. Although their etiologies vary tremendously, airway neutrophilia is a common feature of these diseases. Neutrophils, as vital regulators linking innate and adaptive immune systems, are a double-edged sword in the immune response of the lung involving mechanisms such as phagocytosis, degranulation, neutrophil extracellular trap formation, exosome secretion, release of cytokines and chemokines, and autophagy. Although neutrophils serve as strong defenders against extracellular pathogens, neutrophils and their components can trigger various cascades leading to inflammation and fibrogenesis. Here, we review current studies to elucidate the versatile roles of neutrophils in chronic pulmonary inflammatory diseases and describe the common pathogenesis of these diseases. This may provide new insights into therapeutic strategies for chronic lung diseases.
Pulmonary diseases are life-threatening conditions and an important cause of death worldwide. Chronic pulmonary diseases account for about 1 in 15 deaths in the United States, and mortality is increasing (1). According to current knowledge, chronic pulmonary diseases, including chronic obstructive pulmonary disease (COPD), asthma, cystic fibrosis (CF), and idiopathic pulmonary fibrosis (IPF), are characterized by chronic inflammation, repeated lung tissue injury and repair, and eventually pulmonary dysfunction. Although therapeutic strategies have been developed, effective therapy options are currently lacking for the middle and late stages of chronic pulmonary diseases (2). Fibrogenesis is a common phenomenon in the middle-to-late stages of these diseases, but the specific mechanisms are still unclear.
Generally, chronic inflammation may result from uncontrolled acute inflammation and may lead to fibrogenesis if repeated damage-repair cycles occur. Pulmonary fibrogenesis is a process of recurrent injury to the alveolar epithelium, followed by an uncontrolled proliferation of fibroblasts and overexpression of extracellular matrix (ECM) (3). Abnormal pneumocyte apoptosis, bronchiolar proliferation, and abnormal tissue remodeling are also involved in fibrogenesis (4). Fiber deposits in the alveolar interstitial matrix eventually leads to fibrosis. Neutrophils, a prominent subpopulation of immune cells in airways, have long been regarded as effector cells in the defense against extracellular pathogens during acute inflammation. However, neutrophils also participate in chronic inflammation in various diseases, such as rheumatoid arthritis (5, 6). Neutrophils play a modulating role in both innate and adaptive immune responses (7). On the one hand, neutrophils adhere to blood vessel walls, transmigrate to inflammatory sites following chemokine signals, and maintain cellular homeostasis by phagocytosis and degranulation or neutrophil extracellular trap (NET) formation (8). On the other hand, neutrophils and their effectors, including NETs, cytokines, exosomes, and autophagy, directly and indirectly activate fibroblasts, and promote ECM deposition. Most lung disorders, such as asthma, COPD, CF, and IPF, are inflammatory diseases that are accompanied by an increased number of neutrophils in the bronchoalveolar lavage fluid (BALF) or lung tissue (9–11). The long-term imbalance between pathogens and host defense contributes to the chronic inflammatory disease (12). High mobility group box protein 1 released by necrotic neutrophils induces inflammation and tissue remodeling by activating receptors for advanced glycation and toll-like receptor and CXC chemokine receptor (CXCR)4 signaling (13). As a result, chronic pulmonary interstitial inflammation, and fibrosis are induced and maintained.
In this review, we summarize how neutrophils restrict local inflammation and have a specific role in promoting chronic inflammation of the lung and pulmonary fibrosis. The review will reveal the various roles of neutrophils and explore common mechanisms of fibrogenesis in chronic lung diseases. This knowledge may inform treatment strategies for end-stage chronic lung diseases.
Neutrophils contribute to nearly 60% of all leukocytes in human body fluids (14). More neutrophils are found in the pulmonary capillaries than in the systemic circulation, which facilitates their rapid entry into the lung tissues in response to infection and inflammatory stimuli (15). Primarily, they are recruited in initial phases to exert pro-inflammatory effects. These cells are recruited within minutes after injury and involved in the removal of tissue debris. Neutrophils possess a series of effector mechanisms to play their roles in disease. Apart from phagocytosis, the recently discovered NET formation, exosomes, release of cytokines and chemokines, and autophagy all play a vital role in pulmonary diseases. These protective mechanisms favor pathogen elimination and minimization of collateral damage (16), and they have been observed in multiple respiratory diseases, such as CF and asthma (17, 18). Following phagocytosis, neutrophils activate apoptotic pathways to limit the inflammation in the lung tissue.
Although neutrophils exert powerful antibacterial and antiseptic properties, studies have found that the neutrophil/total cell ratio in lung tissue is positively correlated with the lung fibrosis degree (19). This indicates that neutrophils and/or their products may contribute to fibrogenesis (20). The earliest pathological change of pulmonary fibrosis is the injury of pulmonary endothelial cells, followed by chronic injury of lung epithelial cells and deposition of lung interstitial matrix. Neutrophils induce endothelial and epithelial cell death directly through NET formation (21) or release of invading substance, such as angiopoietin-2, which promotes vessel destabilization and facilitates neutrophil influx to lung tissue (22). Neutrophils recruited to inflammatory sites affect the function of parenchymal cells or via their mediators, such as exosomes or cytokines (23, 24). Neutrophils secrete toxic mediators, such as reactive oxygen species (ROS) and reactive nitrogen species, that lead directly to tissue injury, or even worse, induce another wave of chemokines that forms a positive feedback loop. Moreover, some pathogens possess properties to subvert the immune system, which causes neutrophils to respond more aggressively, leading to untoward tissue injury (25). For example, neutrophil drive the inflammation-induced tissue damage in pulmonary tuberculosis and as such, neutrophil counts are correlated with chronic inflammation in tuberculosis (26).
The cytoplasmic granules of neutrophils contain large amounts of lysozyme, alkaline phosphatase, antimicrobial proteins, defensin, and phagocytin. These substances eliminate foreign pathogens after their extracellular release by means of degranulation or after phagocytosis of the pathogen and fusion of the cytoplasmic granules to form the structure referred to as the phagolysosome (8). Phagolysosomes are formed from neutrophil granules (granule enzymes and ROS), lysosomes, and ingested bacteria (27). Pathogen killing is mediated by granule enzymes released from granules and ROS produced by mitochondria and NADPH oxidase (NOX) complexes. Mitochondria-generated ROS facilitate the innate immune function of neutrophils in a NOX-independent manner (28). By contrast, NOX is the only enzyme specifically used to produce ROS that participate in the regulation of infectious diseases and inflammation. However, NOX activity also leads to membrane depolarization of phagosomes to provide a proper environment for these cell organelles (29). The chemical elimination of pathogens prevents the further spread of infection and inflammation and protects the surrounding cells and tissues. Furthermore, proteinases and bactericidal proteins help in tissue repair and ECM reduction.
When phagocytosis is ineffective, degranulation occurs in response to stimuli. Neutrophils release granule contents in a degranulation manner. In this process, a series of enzymes, such as lysosomal enzymes and protease, are released. They help control bacterial challenge, but the ECM will be digested as a collateral side effect. Recurrent tissue damage and abnormal tissue repair contribute to lung tissue remodeling, degradation, and fibrogenesis. Neutrophil-derived matrix metalloproteinases (MMPs) digest the ECM, elastin, and collagen to allow for neutrophil transmigration from the bloodstream into tissues; but in some disease contexts, they weaken the host defense and contribute to tissue damage by degrading immune receptors and collectins (13). MMP2 and MMP9 are key orchestrators of emphysema (30, 31), indicating that MMPs are involved in pulmonary structural remodeling. In asthma, neutrophil phagocytosis is decreased (32), whereas, ROS generation is increased (33), which is highly correlated with tissue inflammation. In the COPD model, a reduction in MMP9 levels is correlated with disease alleviation (34). The dysregulated release of myeloperoxidase (MPO) and neutrophil elastase contributes to tissue damage and can exacerbate CF. Increased levels of MMP8/9 and neutrophil elastase are also detected in patients with CF (35). In the BALF and lung tissues from patients with IPF, high concentrations of neutrophil-derived MMP2 and MMP9 are detected, and MMP9 activity is positively correlated with the neutrophil count in the BALF (36). Collectively, the dysfunction of phagocytosis and degranulation favors ECM destruction, which is the basis for tissue over-repair, remodeling, and fibrogenesis.
NETs are a mechanism of activated neutrophils to control infection and inflammation by “trapping” pathogens extracellularly in a suicidal or vital manner (37). The progress of active NET release is known as NETosis. NETs can be produced in response to infectious or non-infectious stimuli. Following activation of NET cell death programs, suicidal NETs are released by dying neutrophils 2–4 h after their activation (38) and are composed of decondensed chromatin (including cathelicidin LL-37, α-defensin, and neutrophil elastase), dissolved nuclear membranes, and chromatin. NETosis can be accelerated by the presence of bacteria as a way to wall off the infection and to limit the pro-inflammatory cytokine secretion, which limits the inflammatory response (39). Various agents, including bacteria, fungi, protozoa, viruses, platelets, interferon (IFN)-α, autoantibody, nitric oxide donors, can induce NET formation (40, 41). Moreover, granular enzymes and ROS positively regulate NET formation (41). In vitro, lipopolysaccharides can also induce the generation of NETs (42). These results indicate that NETs play a critical role in pathogen clearance and tissue homeostasis.
Connective tissue growth factor (CTGF, CCN2) is a matricellular protein implicated in fibrosis and an important downstream mediator of TGF-β induced fibrosis (43). The expression of CTGF is elevated in IPF, and confined to proliferating type II alveolar cell and myofibroblasts (44). CTGF may play a role in promoting fibroblast proliferation and extracellular matrix production in pulmonary fibrosis (44). Studies show a closely relationship between NET and CTGF. Myofibroblast CTGF expression is enhanced by NETs inducer (45), which also promote lung fibroblasts activation and myofibroblast differentiation (45, 46). Fibrotic interstitial lung biopsies demonstrate a close proximity between NETs and alpha-smooth muscle actin (α-SMA)-expressing fibroblasts (45).
NETs are reported to participate in the pathogenesis of multiple lung diseases, including pneumonia, acute lung injury, asthma, COPD, and CF. Although NETs play an important role in pathogen elimination and host defense, other evidence reveals substantial damage to the lung epithelium and endothelium, either directly or indirectly (21). Much of the literature on NETs in pneumonia, acute lung injury, COPD, asthma, and CF has already been reviewed in detail, so we will summarize major findings here (47). Recent data suggest that NETs also play an important role in autoimmune and autoinflammatory pathologies (48, 49). NETs induce the activation of lung fibroblasts, stimulate their differentiation into the myofibroblast phenotype, and promote fibrotic activity via the expression of interleukin (IL)-17, a primary initiator of inflammation and fibrosis (45). In vitro, isolated NETs significantly increase the levels of cytokines and stimulate macrophages to secrete IL-1β, which promotes neutrophil and macrophage infiltration in airways and contributes to lung fibrosis (50). As a result, NETs amplify tissue inflammation along the NET-cytokine-cell axis. Peptidylarginine deiminase 4 (PAD4), an essential enzyme for NET formation (51), is elevated in the lungs of COPD patients (52). In addition, the concentrations of NETs and their components, including extracellular DNA, LL-37, and CXCL8/IL-8, are increased in patients with neutrophilic asthma and COPD, and these elevated levels are negatively correlated with lung function (53). In COPD patients, NET levels are also elevated in the sputum, and they are similarly negatively correlated with lung function. In mice, reduced NET levels following erythromycin administration can alleviate emphysema and decrease the numbers of Th1, Th17, and myeloid dendritic cells (54). Furthermore, NET components stimulate histamine and leukotriene release, which may lead to further inflammatory changes in asthma and COPD. Neutrophils of CF patients exhibit a prolonged lifespan, which promotes NET production and increased necrosis (55). Furthermore, the neutrophil count is increased in patients with CF, and the levels of NET components (extracellular DNA, neutrophil elastase, and MPO) are correlated with disease severity (56). In addition, increased levels of autoantibodies against NET components are correlated with decreased lung function in patients with CF (56, 57). Despite no currently proven direct connection between NETs and IPF, the relationship between NET and pulmonary fibrosis formation suggests a likely relationship between IPF and NETs. A recent study showed that pulmonary surfactant protein-D can inhibit NETosis in lipopolysaccharide-stimulated human neutrophils, providing the potential to eliminate negative NET effects (58).
Exosomes are extracellular vesicles <150 nm in size that are released into the plasma by budding and carry proteins, lipids, and nucleic acids. Exosomes are secreted by a wide range of cells in response to different stimuli and play a role in information transmission (59). As an intercellular communicator, exosomes can reach distant cells quickly and affect their behavior via interaction with surface markers. Exosomes acquiring the CD63/CD66b phenotype are from neutrophils, with which they can be traced extracellularly. Studies have shown that exosomes are released into the lungs, and they are related to the pathophysiology of lung diseases (60). Functionally, exosomes eliminate pro-inflammatory chromosomal DNA fragments to maintain cellular homeostasis (61). Functional inhibition of exosomes triggers the innate immune response to generate more ROS by accumulating nuclear DNA.
Exosomes from activated neutrophils express higher neutrophil enzyme than quiescent exosomes, which indicate that exosomes show higher destructive effect in lung (62). Neutrophil-derived exosomes can be immediately internalized by airway smooth muscle cells, thereby altering their proliferative properties and contributing to airway remodeling (17). This is associated with excessive neutrophil-driven inflammation and local elevation in neutrophil elastase levels. Exosomes from neutrophils enhance the proliferation of airway smooth muscle cells to promote airway remodeling in patients with asthma, and they play an important role in the progression of asthma (17). In COPD, exosomes containing neutrophil elastase, which is resistant to alpha1-antitrypsin, destroy the tissue architecture via integrin Mac-1 and neutrophil elastase (62). Although studies concerning neutrophil-derived exosomes and lung fibrotic disease have not been identified, such exosomes potentially contribute to the pulmonary architectural distortion in chronic lung diseases. In turn, exosomes from lung fibroblasts activate DNA damage response and epithelial cell senescence (63). In systemic sclerosis, a systemic fibrotic disease, neutrophil-derived exosomes inhibit the proliferation, and migration of human dermal microvascular endothelial cells via S100A8/A9 in neutrophils, this result indicates that exosomes from neutrophils prevent lung remodeling (64). In mice with bleomycin-induced pulmonary fibrosis, exosomes containing microRNA-22 can ameliorate pulmonary fibrosis by suppressing transforming growth factor (TGF)-β1-induced expression of α-smooth muscle actin (65). This contrary effect of exosomes in fibric diseases is mainly caused by the different functional components contained in the released exosomes.
Autophagy is the lysosome-dependent process of cell organelle self-degradation that is mediated under various cellular stress conditions to maintain cellular homeostasis of cellular protein synthesis, degradation, and recycling (66). Autophagy is crucial for cellular function and can be seen in both physiological and pathological processes in most cells, including neutrophils. In recent studies, autophagy has shown a complicated and important moderator function in pulmonary diseases (67). In chronic lung diseases, autophagy helps to maintain the cell cycle of lung fibroblasts and reduce the production of ECM.
In patients with asthma, autophagy levels of peripheral blood cells in the sputum and peripheral blood eosinophils are increased (68). Similarly, autophagy levels in airway tissues are also increased in a murine asthma model. Inhibiting autophagy reduces airway responsiveness, eosinophilia, and inflammation in this murine asthma model (69). Smoke-exposed neutrophils present increased autophagy and lose the ability to ingest the respiratory pathogen Staphylococcus aureus (70). Moreover, cigarette smoke exposure induces autophagic cell death in neutrophils, leading to the development of emphysema (71). These results indicate that the upregulation of autophagy in neutrophils contributes to persistent inflammation and the development of COPD.
However, studies concerning the autophagy of neutrophils in CF and IPF are lacking. The level of autophagic Beclin-1 is decreased in fibroblasts in fibrotic autoimmune diseases, suggesting an inhibited autophagy status (72). Autophagy stimulated by rapamycin (an mTOR inhibitor promoting autophagy) causes the reduced expression of fibronectin and α-smooth muscle actin in fibroblasts in vitro and exerts an anti-fibrotic effect in the bleomycin model in vivo (73). By contrast, inhibiting autophagy leads to an increase in extracellular collagen production and fibroblast-myofibroblast differentiation in pulmonary fibrosis (74, 75). In IPF, insufficient autophagy promotes fibrogenesis by stimulating fibroblast proliferation (76) and accelerates cellular senescence and myofibroblast differentiation of lung fibroblasts, suggesting the pro-fibrotic functions of insufficient autophagy in IPF (77, 78). Macroautophagy and mitophagy are decreased in lung epithelial cells and lung fibroblasts of IPF patients (54). Mitophagy, a crucial process in cellular energy homeostasis, modulates macrophage apoptosis and stabilizes macrophages to release TGF-β1, thereby stimulating local fibroblast activation. Aging mitochondria and impaired autophagy/mitophagy are involved in the pathogenesis of IPF (75, 79). This result caters to the fact that aging is a major risk factor for IPF (80). Blocking mitophagy in alveolar macrophages protects against bleomycin-induced fibrosis in mice (81). These results indicate an intimate connection between autophagy and persistent inflammation, as well as fibrosis, but more studies are needed to verify the specific function of neutrophil autophagy in these diseases.
Some drugs show to be highly associated with autophagy. For example, hydroxychloroquine blocks autophagy by impairing the autophagosome-lysosome fusion and the degradation of the autophagosome contents (82). Metformin, an insulin-sensitizing drug, stimulates AMP-activated protein kinase, which enhances autophagy by ULK1 (a key regulating protein of autophagy) activation (83). Moreover, autophagy positively regulates NET formation (84, 85), proposing that inhibition of autophagy using pharmacological inhibitors may hinder the release of NETs. Consequently, modulation of autophagy emerges as a promising therapeutic option for the treatment of NET-driven disorders.
Neutrophils secrete a vast number of cytokines and chemokines, such as pro-inflammatory cytokines (IL-1, IL-6, IL-17, and IL-18), anti-inflammatory cytokines (IL-10, TGF-β1, and TGF-β2), chemokines (CXCL1–11, CCL2–4, and CCL17–22), and immunoregulatory cytokines IFN-γ, IL-12, and IL-23), all of which act directly or indirectly on other immune cells, contributing to tissue damage or repair (8, 29, 86). As a key connector of the innate and adaptive immune system, neutrophils interact with other immune cells, including T cells, B cells, natural killer cells, macrophages, dendritic cells, and mesenchymal stem cells (7). Particularly, IFN-γ, TGF-β, IL-6, IL-17A, and IL-17F are functionally important in lung diseases.
IFN-γ shows anti-fibrotic effects by inhibiting the collagen formation in fibroblasts in vitro. It also decreases the gene expression of pro-fibrogenic factors, including TGF-β1 and CTGF (87). Clinically, INF-γ is used to treat lung fibrosis in IPF, despite its effectiveness being quite controversial (88).
TGF-β is a strong pro-fibrotic factor in the lung (89). Three major mammalian isoforms of TGF-β have been identified: TGF-β1, TGF-β2, and TGF-β3. TGF-β1 and TGF-β2 are secreted by neutrophils. TGF-β induces the proliferation of fibroblasts and macrophages via platelet-derived growth factor expression and stimulates fibroblasts to generate superabundant ECM. Besides, activated macrophages also express a vast number of pro-inflammatory and fibrogenic cytokines, such as tumor necrosis factor-α, IL-1β, and IL-13, triggering persistent inflammation downstream and chronic lung fibrosis (89). Pulmonary TGF-β1 levels are increased in a model of experimental lung fibrosis, and TGF-β1 overexpression induces persistent pulmonary fibrosis via the SMAD3 signaling pathway (90). Smad3 gene knockout protects mice from TGF-β1- and bleomycin-induced pulmonary fibrosis (91).
In patients with IPF or acute exacerbation of IPF, IL-6 levels in the BALF and serum are significantly higher compared to healthy controls (92–94), indicating a close connection between this cytokine and fibrosis pathogenesis. In IPF, IL-6 enhances the proliferation of lung fibroblasts via SHP-2/ERK/MAPK signaling (95). In addition, IL-6 promotes in IPF the resistance of lung fibroblasts to Fas-induced apoptosis by overexpression of the anti-apoptotic protein BCL-2 (96).
In human lung fibroblasts, IL-17A stimulates their proliferation, generation of ECM, and differentiation into the myofibroblast phenotype via the NF-κB pathway. IL-17 also promotes fibrosis via NETosis (97). IL-17 expresses in NETs and promotes the fibrotic activity of lung fibroblasts (45). Study shows that IL-17-producing cells and NETosis are synchronous increased in psoriatic lesions. The expression of IL-17 is increased in presence of NETs in vitro (97). Neutralization of IL-17A can ameliorate bleomycin-induced lung fibrosis in mice (98). These results indicate a pro-fibrotic role for IL-17A in human lung tissue remodeling through direct effects on lung fibroblasts (99). Superabundant neutrophils also lead to tissue damage, and pulmonary fibrosis is significantly alleviated when neutrophils are depleted (100). Collectively, neutrophils, as a major source of inflammatory factors, play vital pro- or anti-fibrotic roles in the lung. This dual effect is mainly caused by differences in environmental conditions of the distinct pulmonary diseases. Changing disease-related factors may reverse the disease progression and promote recovery.
Chronic inflammatory lung diseases are a group of neutrophil-related disorders with poor prognosis in middle-to-late stages. To sum up the common features of these diseases, first, neutrophilia can be detected in the lung tissue or BALF. Second, protective factors and pro-inflammatory factors coexist, but the balance is disturbed under disease conditions. Third, specific neutrophil functions are altered, such as enhanced ROS production, aberrant NET formation, increased autophagy, and abnormal secretion of cytokines. In contrast to the traditional view on these short-lived cells, research corroborates the hypothesis that neutrophils and their products contribute to inflammation removal, but also chronic inflammation and fibrosis of lung tissue (Figure 1). The shift in the balance toward tissue destruction may result in persistent inflammation and fibrogenesis. These mechanisms also explain the acute exacerbation of some chronic lung diseases after experiencing infection. Avoiding infection is an important preventive measure to control pulmonary fibrosis.
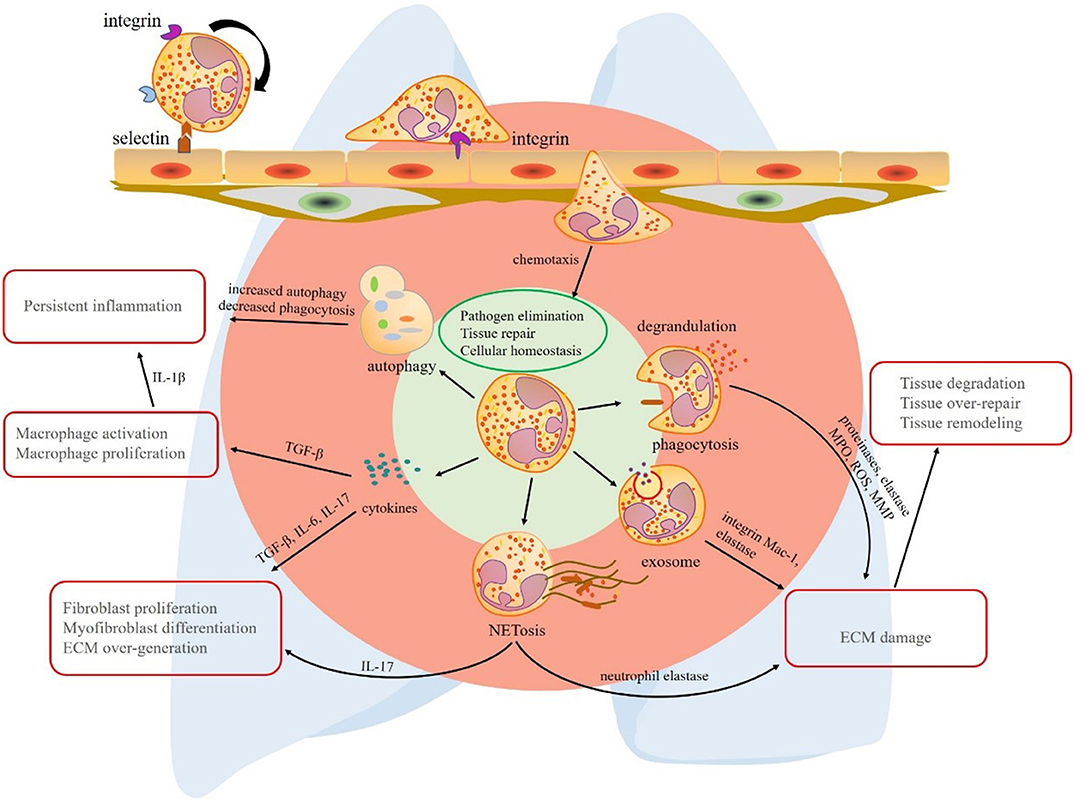
Figure 1. The multiple effects of neutrophils in lung fibrosis. Circulating neutrophils roll, adhere, crawl, and transmigrate to chemokines to lung tissue. In the lung tissue, neutrophils play their roles via phagocytosis, degranulation, neutrophil extracellular trap (NET) formation, exosome secretion, release of cytokines, and autophagy redox balance. The primary common features of these mechanisms are pathogen elimination, tissue repair, and cellular homeostasis, respectively. However, the mechanisms that promote the progression of chronic inflammatory lung disease are quite different. Phagocytosis, degrandulation, NET formation, and exosomes contribute to ECM injury and tissue damage-repair-remodeling. In addition to ECM damage, NETs play a role in activating immune responses and release pro-fibrotic factor IL-17. Although neutrophil autophagy effect is limited, increased proportion of autophagy in neutrophil deduces the ability of eliminating inflammation. A vast of pro-fibrotic cytokines released by neutrophils contribute to lung fibrosis formation, respectively. Transforming growth factor (TGF)-β promotes persistent inflammation by macrophage activation and proliferation. interleukin (IL)-17 derived from neutrophils or NETs induces fibroblast proliferation and myofibroblast differentiation. As a result, extracellular matrix (ECM) is over-expressed in interstitial tissue of lung. TGF-β, transforming growth factor-β; IL-17, interleukin-17; NET, neutrophil extracellular trap; ECM, extracellular matrix; MPO, myeloperoxidase; ROS, reactive oxygen species; MMP, matrix metalloproteinase.
Given the role of neutrophils in fibrosis, strategies focusing on neutrophil components may be effective, such as reducing neutrophil numbers in the airway, decreasing protease and ROS generation, decorating NETs, regulating autophagy, reducing the expression or activity of TGF-β protein, and providing exogenously exosomes containing microRNA. DNase and NET-associated elastase that affect the formation of NET may be helpful. Compound, such as hydroxychloroquine, which can reduce NETs via blocking autophagy is also considerable. Moreover, based on the tightly correlation of NETs and lung function, NET is expected to be a biomarker for evaluating criteria of lung function and fibrosis. However, the effectiveness of these strategies is limited by their off-target effects. More studies are needed to explore the precise role of neutrophils in lung fibrotic diseases, which will provide better evidence for the treatment of these diseases.
LD wrote this review. JY and CZ consulted relevant literature. XZ drew the figures. PG revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was sponsored by the National Science and Technology Major Project (2017ZX10202202, 2018ZX10302206, 2018ZX10723203), National Key Research Plan Precision Medicine Research Key Project (2017YFC0908103), the National Natural Science Foundation of Jilin Province (2018SCZWSZX-003, JLSCZD2019-008), Program for JLU Science and Technology Innovative Research Team (2017TD-08), and the Fundamental Research Funds for the Central Universities.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We appreciate the critical comments of Dr. Huanfa Yi from Central Laboratory, The First Hospital of Jilin University.
1. Belchamber KBR, Donnelly LE. Targeting defective pulmonary innate immunity - a new therapeutic option? Pharmacol Ther. (2020) 209:107500. doi: 10.1016/j.pharmthera.2020.107500
2. Takeda Y, Tsujino K, Kijima T, Kumanogoh A. Efficacy and safety of pirfenidone for idiopathic pulmonary fibrosis. Patient Prefer Adherence. (2014) 8:361–70. doi: 10.2147/PPA.S37233
3. Poletti V, Ravaglia C, Buccioli M, Tantalocco P, Piciucchi S, Dubini A, et al. Idiopathic pulmonary fibrosis: diagnosis and prognostic evaluation. Respiration. (2013) 86:5–12. doi: 10.1159/000353580
4. Chilosi M, Doglioni C, Murer B, Poletti V. Epithelial stem cell exhaustion in the pathogenesis of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. (2010) 27:7–18.
5. Edilova M, Akram A, Abdul-Sater AA. Innate immunity drives pathogenesis of rheumatoid arthritis. Biomed J. (2020). doi: 10.1016/j.bj.2020.06.010. [Epub ahead of print].
6. Cabrini G, Rimessi A, Borgatti M, Lampronti I, Finotti A, Pinton P, et al. Role of cystic fibrosis bronchial epithelium in neutrophil chemotaxis. Front Immunol. (2020) 11:1438. doi: 10.3389/fimmu.2020.01438
7. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
8. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
9. Richter AG, Perkins GD, Chavda A, Sapey E, Harper L, Thickett DR. Neutrophil chemotaxis in granulomatosis with polyangiitis (Wegener's) and idiopathic pulmonary fibrosis. Eur Respir J. (2011) 38:1081–8. doi: 10.1183/09031936.00161910
10. Nathan S, Brown A, Mogulkoc N, Soares F, Collins A, Cheng J, et al. The association between white blood cell count and outcomes in patients with idiopathic pulmonary fibrosis. Respir Med. (2020) 170:106068. doi: 10.1016/j.rmed.2020.106068
11. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2016) 138:16–27. doi: 10.1016/j.jaci.2016.05.011
12. Yang F, Feng C, Zhang X, Lu J, Zhao Y. The diverse biological functions of neutrophils, beyond the defense against infections. Inflammation. (2017) 40:311–23. doi: 10.1007/s10753-016-0458-4
13. Christoffersson G, Vågesjö E, Vandooren J, Lidén M, Massena S, Reinert R, et al. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood. (2012) 120:4653–62. doi: 10.1182/blood-2012-04-421040
14. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. (2016) 273:11–28. doi: 10.1111/imr.12440
15. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
16. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
17. Vargas A, Roux-Dalvai F, Droit A, Lavoie JP. Neutrophil-derived exosomes: a new mechanism contributing to airway smooth muscle remodeling. Am J Respir Cell Mol Biol. (2016) 55:450–61. doi: 10.1165/rcmb.2016-0033OC
18. Pham DL, Ban GY, Kim SH, Shin YS, Ye YM, Chwae YJ, et al. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clin Exp Allergy. (2017) 47:57–70. doi: 10.1111/cea.12859
19. Pardo A, Barrios R, Gaxiola M, Segura-Valdez L, Carrillo G, Estrada A, et al. Increase of lung neutrophils in hypersensitivity pneumonitis is associated with lung fibrosis. Am J Respir Crit Care Med. (2000) 161:7. doi: 10.1164/ajrccm.161.5.9907065
20. Nordin J, Lee Y, Vader P, Mäger I, Johansson H, Heusermann W, et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine. (2015) 11:879–83. doi: 10.1016/j.nano.2015.01.003
21. Saffarzadeh M, Juenemann C, Queisser M, Lochnit G, Barreto G, Galuska S, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
22. Lomas-Neira J, Venet F, Chung C, Thakkar R, Heffernan D, Ayala AJAjorc, et al. Neutrophil-endothelial interactions mediate angiopoietin-2-associated pulmonary endothelial cell dysfunction in indirect acute lung injury in mice. Am J Respir Cell Mol Biol. (2014) 50:193–200. doi: 10.1165/rcmb.2013-0148OC
23. Rani S, O'Brien K, Kelleher F, Corcoran C, Germano S, Radomski M, et al. Isolation of exosomes for subsequent mRNA, MicroRNA, and protein profiling. Methods Mol Biol. (2011) 784:181–95. doi: 10.1007/978-1-61779-289-2_13
24. Baranyai T, Herczeg K, Onódi Z, Voszka I, Módos K, Marton N, et al. Isolation of exosomes from blood plasma: qualitative and quantitative comparison of ultracentrifugation and size exclusion chromatography methods. PLoS ONE. (2015) 10:e0145686. doi: 10.1371/journal.pone.0145686
25. Bhattacharya M, Berends ETM, Chan R, Schwab E, Roy S, Sen CK, et al. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc Natl Acad Sci U S A. (2018) 115:7416–21. doi: 10.1073/pnas.1721949115
26. Cruz A, Fraga A, Fountain J, Rangel-Moreno J, Torrado E, Saraiva M, et al. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med. (2010) 207:1609–16. doi: 10.1084/jem.20100265
27. Jasper AE, McIver WJ, Sapey E, Walton GM. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research. (2019) 8:557. doi: 10.12688/f1000research.18411.1
28. Ravindran M, Khan MA, Palaniyar N. Neutrophil extracellular trap formation: physiology, pathology, and pharmacology. Biomolecules. (2019) 9. doi: 10.3390/biom9080365
29. Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. (2013) 210:1283–99. doi: 10.1084/jem.20122220
30. Finlay G, O'Driscoll L, Russell K, D'Arcy E, Masterson J, FitzGerald M, et al. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med. (1997) 156:240–7. doi: 10.1164/ajrccm.156.1.9612018
31. Russell R, Culpitt S, DeMatos C, Donnelly L, Smith M, Wiggins J, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2002) 26:602–9. doi: 10.1165/ajrcmb.26.5.4685
32. Alexis N, Eldridge M, Peden D. Effect of inhaled endotoxin on airway and circulating inflammatory cell phagocytosis and CD11b expression in atopic asthmatic subjects. J Allergy Clin Immunol. (2003) 112:353–61. doi: 10.1067/mai.2003.1651
33. Qu J, Li Y, Zhong W, Gao P, Hu C. Recent developments in the role of reactive oxygen species in allergic asthma. J Thorac Dis. (2017) 9:E32–43. doi: 10.21037/jtd.2017.01.05
34. Lee J, Lee D, Kim E, Choe K, Oh Y, Shim T, et al. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med. (2005) 172:987–93. doi: 10.1164/rccm.200501-041OC
35. Forrester DL, Barr HL, Fogarty A, Knox AJPP. sTREM-1 is elevated in cystic fibrosis and correlates with proteases. Pediatr Pulmonol. (2017) 52:467–71. doi: 10.1002/ppul.23650
36. Suga M, Iyonaga K, Okamoto T, Gushima Y, Miyakawa H, Akaike T, et al. Characteristic elevation of matrix metalloproteinase activity in idiopathic interstitial pneumonias. Am J Respir Crit Care Med. (2000) 162:1949–56. doi: 10.1164/ajrccm.162.5.9906096
37. Yipp B, Kubes P. NETosis: how vital is it? Blood. (2013) 122:2784–94. doi: 10.1182/blood-2013-04-457671
38. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:4. doi: 10.1126/science.1092385
39. Keitelman I, Sabbione F, Shiromizu C, Giai C, Fuentes F, Rosso D, et al. Short-term fever-range hyperthermia accelerates NETosis and reduces pro-inflammatory cytokine secretion by human neutrophils. Front Immunol. (2019) 10:2374. doi: 10.3389/fimmu.2019.02374
40. Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog. (2015) 11:e1004651. doi: 10.1371/journal.ppat.1004651
41. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2017) 18:134–47. doi: 10.1038/nri.2017.105
42. Claushuis T, van der Donk L, Luitse A, van Veen H, van der Wel N, van Vught L, et al. Klebsiella pneumoniae-Role of Peptidylarginine Deiminase 4 in Neutrophil Extracellular Trap Formation and Host Defense during Induced Pneumonia-Derived Sepsis. J Immunol. (2018) 201:1241–52. doi: 10.4049/jimmunol.1800314
43. Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. (2008) 19:133–44. doi: 10.1016/j.cytogfr.2008.01.002
44. Pan LH, Yamauchi K, Uzuki M, Nakanishi T, Takigawa M, Inoue H, et al. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Cytokine Growth Factor Rev. (2001) 17:1220–7. doi: 10.1183/09031936.01.00074101
45. Chrysanthopoulou A, Mitroulis I, Apostolidou E, Arelaki S, Mikroulis D, Konstantinidis T, et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J Pathol. (2014) 233:294–307. doi: 10.1002/path.4359
46. Zhang S, Jia X, Zhang Q, Zhang L, Yang J, Hu C, et al. Neutrophil extracellular traps activate lung fibroblast to induce polymyositis-related interstitial lung diseases via TLR9-miR-7-Smad2 pathway. J Cell Mol Med. (2020) 24:1658–69. doi: 10.1111/jcmm.14858
47. Twaddell SH, Baines KJ, Grainge C, Gibson PG. The emerging role of neutrophil extracellular traps in respiratory disease. Chest. (2019) 156:774–82. doi: 10.1016/j.chest.2019.06.012
48. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross W, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. (2009) 15:623–5. doi: 10.1038/nm.1959
49. Fuchs T, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
50. Chen X, Li Y, Qin L, He R, Hu C. Neutrophil extracellular trapping network promotes the pathogenesis of neutrophil-associated asthma through macrophages. Immunol Invest. (2020) 18:1–18. doi: 10.1080/08820139.2020.1778720
51. Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol. (2012) 3:307. doi: 10.3389/fimmu.2012.00307
52. Lugli EB, Correia R, Fischer R, Lundberg K, Bracke KR, Montgomery AB, et al. Expression of citrulline and homocitrulline residues in the lungs of non-smokers and smokers: implications for autoimmunity in rheumatoid arthritis. Arthritis Res Ther. (2015) 17:1–8. doi: 10.1186/s13075-015-0520-x
53. Wright T, Gibson P, Simpson J, McDonald V, Wood L, Baines KJR. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology. (2016) 21:467–75. doi: 10.1111/resp.12730
54. Zhang H, Qiu S, Tang Q, Zhou X, Zhang J, He Z, et al. Erythromycin suppresses neutrophil extracellular traps in smoking-related chronic pulmonary inflammation. Cell Death Dis. (2019) 10:678. doi: 10.1038/s41419-019-1909-2
55. Gray R, Hardisty G, Regan K, Smith M, Robb C, Duffin R, et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax. (2018) 73:134–44. doi: 10.1136/thoraxjnl-2017-210134
56. Rada B. Neutrophil extracellular trap release driven by bacterial motility: relevance to cystic fibrosis lung disease. Commun Integr Biol. (2017) 10:e1296610. doi: 10.1080/19420889.2017.1296610
57. Skopelja S, Hamilton B, Jones J, Yang M, Mamula M, Ashare A, et al. The role for neutrophil extracellular traps in cystic fibrosis autoimmunity. JCI Insight. (2016) 1:e88912. doi: 10.1172/jci.insight.88912
58. Arroyo R, Khan MA, Echaide M, Pérez-Gil J, Palaniyar N. SP-D attenuates LPS-induced formation of human neutrophil extracellular traps (NETs), protecting pulmonary surfactant inactivation by NETs. Commun Biol. (2019) 2:470. doi: 10.1038/s42003-019-0662-5
59. Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. (2017) 27:172–88. doi: 10.1016/j.tcb.2016.11.003
60. Qazi K, Torregrosa Paredes P, Dahlberg B, Grunewald J, Eklund A, Gabrielsson SJT. Proinflammatory exosomes in bronchoalveolar lavage fluid of patients with sarcoidosis. Thorax. (2010) 65:1016–24. doi: 10.1136/thx.2009.132027
61. Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. (2017) 8:15287. doi: 10.1038/ncomms15287
62. Genschmer KR, Russell DW, Lal C, Szul T, Bratcher PE, Noerager BD, et al. Activated PMN exosomes: pathogenic entities causing matrix destruction and disease in the lung. Cell. (2019) 176:113–26.e15. doi: 10.1016/j.cell.2018.12.002
63. Kadota T, Yoshioka Y, Fujita Y, Araya J, Minagawa S, Hara H, et al. Extracellular vesicles from fibroblasts induce epithelial cell senescence in pulmonary fibrosis. Am J Respir Cell Mol Biol. (2020) 63:623–36. doi: 10.1165/rcmb.2020-0002OC
64. Li L, Zuo X, Xiao Y, Liu D, Luo H, Zhu HJB, et al. Neutrophil-derived exosome from systemic sclerosis inhibits the proliferation and migration of endothelial cells. Biochem Biophys Res Commun. (2020) 526:334–40. doi: 10.1016/j.bbrc.2020.03.088
65. Kuse N, Kamio K, Azuma A, Matsuda K, Inomata M, Usuki J, et al. Exosome-derived microRNA-22 ameliorates pulmonary fibrosis by regulating fibroblast-to-myofibroblast differentiation in vitro and in vivo. J Nippon Med Sch. (2020) 87:118–28. doi: 10.1272/jnms.JNMS.2020_87-302
66. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
67. Xu S, Liu C, Ji H. Concise review: therapeutic potential of the mesenchymal stem cell derived secretome and extracellular vesicles for radiation-induced lung injury: progress and hypotheses. Stem Cells Transl Med. (2019) 8:344–54. doi: 10.1002/sctm.18-0038
68. Ban G, Pham D, Trinh T, Lee S, Suh D, Yang E, et al. Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: a new therapeutic target. Clin Exp Allergy. (2016) 46:48–59. doi: 10.1111/cea.12585
69. Cho I, Choi Y, Gong J, Shin D, Kang M, Kang Y. Astragalin inhibits autophagy-associated airway epithelial fibrosis. Respir Res. (2015) 16:51. doi: 10.1186/s12931-015-0211-9
70. Guzik K, Skret J, Smagur J, Bzowska M, Gajkowska B, Scott D, et al. Cigarette smoke-exposed neutrophils die unconventionally but are rapidly phagocytosed by macrophages. Cell Death Dis. (2011) 2:e131. doi: 10.1038/cddis.2011.13
71. Lv X, Liu S, Li K, Cui B, Liu C, Hu Z. Cigarette smoke promotes COPD by activating platelet-activating factor receptor and inducing neutrophil autophagic death in mice. Oncotarget. (2017) 8:74720–35. doi: 10.18632/oncotarget.20353
72. Del Principe D, Vona R, Giordani L, Straface E, Giammarioli A. Defective autophagy in fibroblasts may contribute to fibrogenesis in autoimmune processes. Curr Pharm Des. (2011) 17:3878–87. doi: 10.2174/138161211798357791
73. Patel A, Lin L, Geyer A, Haspel J, An C, Cao J, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE. (2012) 7:e41394. doi: 10.1371/journal.pone.0041394
74. Mi S, Li Z, Yang H, Liu H, Wang J, Ma Y, et al. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol. (2011) 187:3003–14. doi: 10.4049/jimmunol.1004081
75. Sosulski M, Gongora R, Danchuk S, Dong C, Luo F, Sanchez C. Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFβ1. Aging Cell. (2015) 14:774–83. doi: 10.1111/acel.12357
76. Wang K, Chen Y, Zhang P, Lin P, Xie N, Wu M. Protective features of autophagy in pulmonary infection and inflammatory diseases. Cells. (2019) 8:123. doi: 10.3390/cells8020123
77. Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L56–69. doi: 10.1152/ajplung.00213.2012
78. Araya J, Hara H, Kuwano K. Autophagy in the pathogenesis of pulmonary disease. Intern Med. (2013) 52:2295–303. doi: 10.2169/internalmedicine.52.1118
79. Nho R, Hergert P. IPF fibroblasts are desensitized to type I collagen matrix-induced cell death by suppressing low autophagy via aberrant Akt/mTOR kinases. PLoS ONE. (2014) 9:e94616. doi: 10.1371/journal.pone.0094616
80. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. (2017) 3:17074. doi: 10.1038/nrdp.2017.74
81. Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity. (2016) 44:582–96. doi: 10.1016/j.immuni.2016.01.001
82. Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. (2011) 17:654. doi: 10.1158/1078-0432.CCR-10-2634
83. Egan D, Shackelford D, Mihaylova M, Gelino S, Kohnz R, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. (2011) 331:456–61. doi: 10.1126/science.1196371
84. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. (2011) 18:581–8. doi: 10.1038/cdd.2011.1
85. Park S, Shrestha S, Youn Y, Kim J, Kim S, Kim H, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respir Crit Care Med. (2017) 196:577–89. doi: 10.1164/rccm.201603-0596OC
86. Scapini P, Cassatella M. Social networking of human neutrophils within the immune system. Blood. (2014) 124:710–9. doi: 10.1182/blood-2014-03-453217
87. Ulloa L, Doody J, Massagué J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. (1999) 397:710–3. doi: 10.1038/17826
88. Muntasir MA, Samman YS, Wali SO, Hamad MMA. Treatment of idiopathic pulmonary fibrosis Is there anything new?. Respirology. (2005) 10:5. doi: 10.1111/j.1440-1843.2005.00712.x
89. Fernandez IE, Eickelberg O. The impact of TGF-β on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc. (2012) 9:111–6. doi: 10.1513/pats.201203-023AW
90. Westergren-Thorsson G, Hernnäs J, Sarnstrand B, Oldberg A, Heinegard D, Malmström A. Altered expression of small proteoglycans, collagen, and transforming growth Factor-fl1 in developing bleomycin-induced pulmonary fibrosis in rats. J Clin Invest. (1993) 92:632–7. doi: 10.1172/JCI116631
91. Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, et al. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol. (2004) 173:2099–108. doi: 10.4049/jimmunol.173.3.2099
92. Antoniou K, Tzouvelekis A, Alexandrakis M, Sfiridaki K, Tsiligianni I, Rachiotis G, et al. Different angiogenic activity in pulmonary sarcoidosis and idiopathic pulmonary fibrosis. Chest. (2006) 130:982–8. doi: 10.1378/chest.130.4.982
93. Takizawa H, Satoh M, Okazaki H, Matsuzaki G, Suzuki N, Ishii A, et al. Increased IL-6 and IL-8 in bronchoalveolar lavage fluids (BALF) from patients with sarcoidosis: correlation with the clinical parameters. Clin Exp Immunol. (1997) 107:175–81. doi: 10.1046/j.1365-2249.1997.d01-905.x
94. Weng D, Chen XQ, Qiu H, Zhang Y, Li QH, Zhao MM, et al. The role of infection in acute exacerbation of idiopathic pulmonary fibrosis. Mediators Inflamm. (2019) 2019:5160694. doi: 10.1155/2019/5160694
95. Knight D, Ernst M, Anderson G, Moodley Y, Mutsaers S. The role of gp130/IL-6 cytokines in the development of pulmonary fibrosis: critical determinants of disease susceptibility and progression? Pharmacol Ther. (2003) 99:327–38. doi: 10.1016/S0163-7258(03)00095-0
96. Moodley Y, Scaffidi A, Misso N, Keerthisingam C, McAnulty R, Laurent G, et al. Fibroblasts isolated from normal lungs and those with idiopathic pulmonary fibrosis differ in interleukin-6/gp130-mediated cell signaling and proliferation. Am J Pathol. (2003) 163:345–54. doi: 10.1016/S0002-9440(10)63658-9
97. Lambert S, Hambro C, Johnston A, Stuart P, Tsoi L, Nair R, et al. Neutrophil extracellular traps induce human Th17 cells: effect of psoriasis-associated TRAF3IP2 genotype. J Invest Dermatol. (2019) 139:1245–53. doi: 10.1016/j.jid.2018.11.021
98. Cipolla E, Fisher A, Gu H, Mickler E, Agarwal M, Wilke C, et al. IL-17A deficiency mitigates bleomycin-induced complement activation during lung fibrosis. FASEB J. (2017) 31:5543–56. doi: 10.1096/fj.201700289R
99. Zhang J, Wang D, Wang L, Wang S, Roden AC, Zhao H, et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L487–97. doi: 10.1152/ajplung.00301.2018
Keywords: neutrophils, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, fibrosis, fibrogenesis
Citation: Ding L, Yang J, Zhang C, Zhang X and Gao P (2021) Neutrophils Modulate Fibrogenesis in Chronic Pulmonary Diseases. Front. Med. 8:616200. doi: 10.3389/fmed.2021.616200
Received: 11 October 2020; Accepted: 19 February 2021;
Published: 27 April 2021.
Edited by:
Peter Olinga, University of Groningen, NetherlandsReviewed by:
Gunnar N. Hillerdal, Karolinska University Hospital, SwedenCopyright © 2021 Ding, Yang, Zhang, Zhang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pujun Gao, Z3BqQGpsdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.