
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 23 April 2021
Sec. Infectious Diseases: Pathogenesis and Therapy
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.615333
 Heloísa Antoniella Braz-de-Melo1
Heloísa Antoniella Braz-de-Melo1 Sara Socorro Faria1
Sara Socorro Faria1 Gabriel Pasquarelli-do-Nascimento1
Gabriel Pasquarelli-do-Nascimento1 Igor de Oliveira Santos1
Igor de Oliveira Santos1 Gary P. Kobinger2,3
Gary P. Kobinger2,3 Kelly Grace Magalhães1*
Kelly Grace Magalhães1*COVID-19 is spreading worldwide at disturbing rates, overwhelming global healthcare. Mounting death cases due to disease complications highlight the necessity of describing efficient drug therapy strategies for severe patients. COVID-19 severity associates with hypercoagulation and exacerbated inflammation, both influenced by ACE2 downregulation and cytokine storm occurrence. In this review, we discuss the applicability of the anticoagulant heparin and the anti-inflammatory corticosteroid dexamethasone for managing severe COVID-19 patients. The upregulated inflammation and blood clotting may be mitigated by administrating heparin and its derivatives. Heparin enhances the anticoagulant property of anti-thrombin (AT) and may be useful in conjunction with fibrinolytic drugs for severe COVID-19 patients. Besides, heparin can also modulate immune responses, alleviating TNF-α-mediated inflammation, impairing IL-6 production and secretion, and binding to complement proteins and leukotriene B4 (LTB4). Moreover, heparin may present anti-SARS-CoV-2 potential once it can impact viral infectivity and alter SARS-CoV-2 Spike protein architecture. Another feasible approach is the administration of the glucocorticoid dexamethasone. Although glucocorticoid's administration for viral infection managing is controversial, there is increasing evidence demonstrating that dexamethasone treatment is capable of drastically diminishing the death rate of patients presenting with Acute Respiratory Distress Syndrome (ARDS) that required invasive mechanical ventilation. Importantly, dexamethasone may be detrimental by impairing viral clearance and inducing hyperglycemia and sodium retention, hence possibly being deleterious for diabetics and hypertensive patients, two major COVID-19 risk groups. Therefore, while heparin's multitarget capacity shows to be strongly beneficial for severe COVID-19 patients, dexamethasone should be carefully administered taking into consideration underlying medical conditions and COVID-19 disease severity. Therefore, we suggest that the multitarget impact of heparin as an anti-viral, antithrombotic and anti-inflammatory drug in the early stage of the COVID-19 could significantly reduce the need for dexamethasone treatment in the initial phase of this disease. If the standard treatment of heparins fails on protecting against severe illness, dexamethasone must be applied as a potent anti-inflammatory shutting-down the uncontrolled and exacerbated inflammation.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiological agent of coronavirus disease (COVID-19), is rapidly spreading worldwide, being the cause of death of more than two million individuals globally in March of 2021 [World Health Organization (WHO), 2020]. Severe COVID-19 occurs due to complications, mostly affecting the lungs (1), heart (2), and kidney (3), impact more frequently the elderly and patients with certain comorbidities, like heart disease and hypertension, and usually correlates with intensive care unit (ICU) admission, mechanical ventilation requirement, and death (4). The elevated numbers of ill and deceased patients due to COVID-19 place it as a healthcare emergency and highlight the urgency of establishing effective drug therapy strategies for pandemics management.
Severe COVID-19 patients tend to show elevated circulating amounts of the proinflammatory mediators Interleukin-6 (IL-6), IL-8, IL-1β, and Monocyte Chemoattractant Protein-1 (MCP-1) (5). Even though plasma concentrations of IL-6, IL-8, and tumor necrosis factor-α (TNF-α) in severe COVID-19 patients may be lower than subjects affected by septic shock and similar to levels found in other patients in a critical state (6), the inflamed phenotype correlates with poor COVID-19 prognosis and is generated by cytokine storm, a condition characterized by macrophage activation syndrome, lymphopenia and organ mononuclear cell infiltration (7). Besides, SARS-CoV-2 infection leads to endothelial damage (8) and downregulation of its cell entry receptor ACE2 (9), further intensifying inflammatory responses and impacting coagulation processes. In this context, severely ill patients may benefit from anti-inflammatory therapies, including dexamethasone.
The hypercoagulable state characteristic of COVID-19 severity is illustrated by the consistently elevated D-dimer levels (10), disseminated intravascular coagulation (DIC), and subsequent consumption coagulopathy (11) displayed by affected individuals. These alterations explain the increased risk of developing life-threatening complications, such as pulmonary embolism (PE) (12) and myocardial infarction (13), by this group. Thus, administrating the anticoagulant drug heparin may also be useful for improving outcomes in severe cases.
Inflammatory and coagulation processes interplay intimately in many ways, including through Tissue Factor (TF), thrombin-dependent PAR activation, Toll-like Receptors (TLRs), and complement system (14). Elevated levels of COVID-19 proinflammatory markers, such as IL-6 and IL-8, also enhance the risk of thrombosis (15). High levels of IL-6 disrupt the procoagulant-anticoagulant balance by increasing the expression of clotting factors and diminishing the levels of thrombogenesis inhibitors (16). IL-8 also impacts hemostasis, significantly changing fibrin and thrombin amounts, and activating platelets (17). Once the intensity of inflammatory responses and clotting processes influence the prognosis of severe COVID-19 patients, investigating the effectiveness of anti-inflammatory and anticoagulant drugs may be crucial in decreasing morbidity and mortality caused by the COVID-19 pandemics. In this review, we discuss the use of the anticoagulant heparin and the anti-inflammatory dexamethasone as promising tools for the management of severe COVID−19 patients.
The emerging association between thromboembolic events and COVID-19 raised new insights about the physiopathology of the pandemic disease. Countless therapeutic targets were proposed through the knowledge of unbalanced procoagulant/anticoagulant factors that leads to the impairment of endogenous antithrombotic activity during SARS-CoV-2 infection (18–20). The great hypothesis was regarding the administration of anticoagulant drugs that could restore hemodynamic homeostasis and protect against the observed coagulopathy. Based on that, the study of frequently used anticoagulant drugs, especially heparin, increased worldwide and preliminary data suggest promising performance in clinical medicine.
Heparin is a polysaccharide originally isolated from mammalian animal tissue in 1916 (21). Thereafter the discovery, increasing research about the molecule chemical structure and mechanism of action brought new derivatives that improved the efficacy of antithrombotic activity and decreased side effects associated with the unfractionated heparin (UFH) form (22). The UFH was associated with an increased risk for thrombocytopenia and osteoporosis, causing a greater need for monitoring patients during the therapy (23, 24). Thereby, the development of low-molecular-weight heparin (LMWH) reached new perspectives and is currently the anticoagulant of choice for the treatment and prevention of coagulopathies.
Both UFH and LMWH have the ability of binding to anti-thrombin (AT) glycoprotein, enhancing AT inactivation of potent enzymes in the coagulation pathway, such as Factor Xa and Factor IIa (Thrombin). The heparin dependence of AT to prevent blood clot formation makes the drug an indirect antithrombotic agent and the absence of intrinsic fibrinolytic activity impairs thrombi breakdown when they are already shaped (25, 26). Besides, heparins also present an interesting immune-modulatory activity (27). One of the proposed mechanisms is the inhibition of different inflammatory responses mediated through necrosis factor-α (TNF-α), a potent pro-inflammatory cytokine (28, 29). As a result, the quick and efficient actions of heparin make the drug promising against inflammation, in addition to its anticoagulant properties. Thereby, different reports have been exploring this potential against SARS-CoV-2 infection, in which both inflammatory and coagulation disruption can lead to a complication during the disease progression.
Reports have shown that COVID-19 patients presented increased levels of procoagulation biomarkers and severe immune dysfunction that can lead to disseminated intravascular coagulation (DIC), increased risk for venous thromboembolism (VTE), and organ failure, which reflects in a high hospital mortality rate of 1:31 with a confidence interval (CI) of 95% (30–32). Using an animal model, the increased deposition of collagen, fibrin, and von Willebrand factor was linked to augmented thrombi formation and endothelialits establishment during COVID-19 (33). Moreover, the appearance of neurological alterations caused by cerebral venous thrombosis was also reported (34, 35). In this context, early anticoagulant treatment was applied and it was observed a decrease of D dimer levels, mechanical ventilation urgency, and a decrease of 24.2% in the mortality rate of patients presenting sepsis-induced coagulopathy (30, 32, 36, 37). The use of a high dose of UFH, such as 5,000 U, showed important protection against sepsis-induced VTE, a complication that presents a major impact on a patient's prognosis (32). Besides, ongoing clinical trials have been focusing on inhalational heparin treatment for hospitalized patients with or without mechanical ventilation urgency, especially due to several reports that addressed the protective role of nebulized UFH against intrapulmonary fibrin deposition and lung injury (38, 39). The use of this administration route can directly deliver the UFH to the lung microenvironment, being a potential mechanism to soften local hypercoagulation and hyperinflammation, preventing systemic harmful effects of anticoagulant treatment. The above-mentioned and the main in-progress clinical trials are summarized in Table 1.
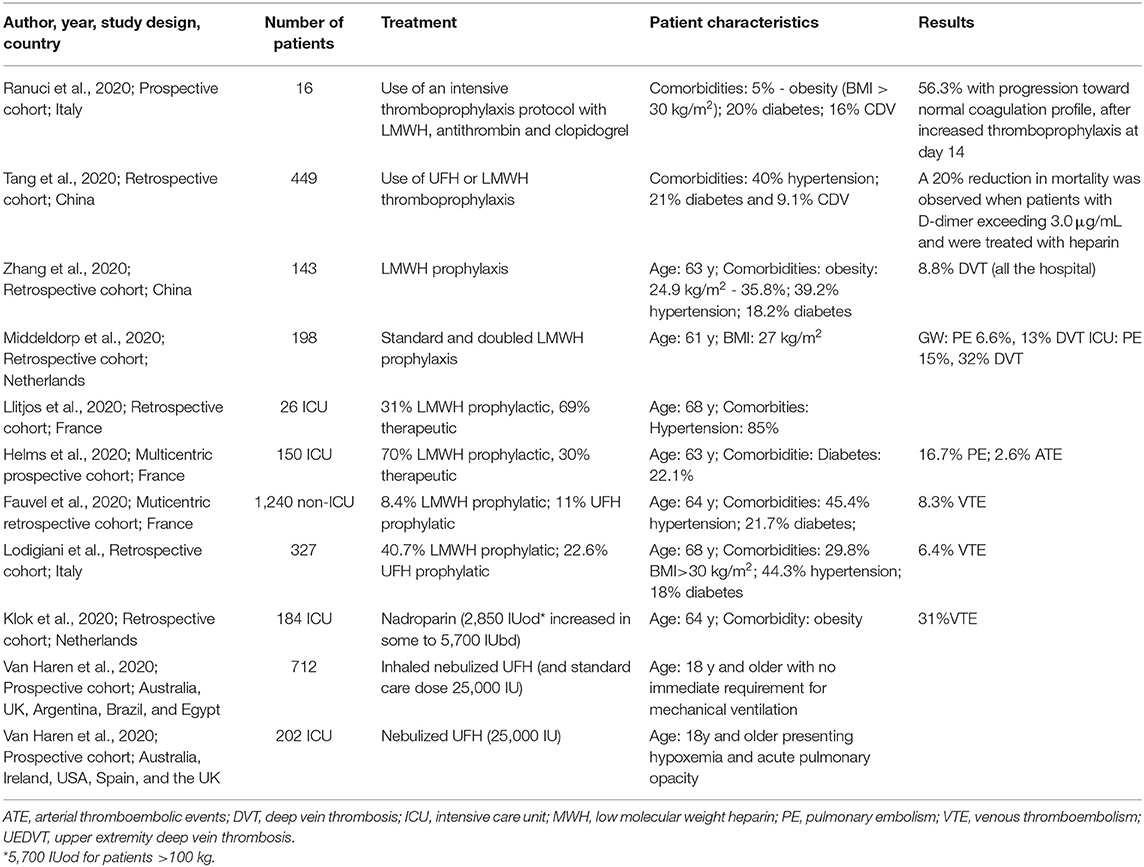
Table 1. Studies on heparin therapy for COVID-19.
It is important to emphasize the importance of indicating LMWH and UFH for both prophylaxis and early treatment, once these drugs are not able to promote fibrinolysis of pre-existing thrombi formed in the tissue. Aware of this fact, the combination of heparin and fibrinolytic drugs could be effective for the treatment of severe cases, once thrombolysis also presents clinical benefits against severe pulmonary embolism (40).
Beyond the expected effects of heparins on preventing clots formation through indirect inhibition of the coagulation pathway, other properties provided by these drugs can be the key for the positive data about their efficacy compared to single-target drugs. The exacerbated inflammation that is also found in severe COVID-19 cases is mediated especially through the uncontrolled production of pro-inflammatory cytokines such as interleukin-6 (IL-6), interleukin-8, monocyte chemoattractant protein-1 (MCP-1), and TNF-α in the cytokine release syndrome (41, 42). Research has been emphasizing how targeting TNF-α is important for a better prognosis against COVID-19-induced cytokine dysfunction, once its blockade can reduce both inflammatory and prothrombotic biomarkers (43). In this context, reports have shown that both LMWH and UFH soften TNF-α-induced inflammatory responses, such as the proper IL-6 and IL-8 production (28, 44, 45). This effect may occur through UFH inhibition of the nuclear transcription factor-κB (NF-κB) binding to the DNA, which is a crucial process for a broad range of cytokines signaling pathways (44). Besides, a non-anticoagulant fraction of enoxaparin was reported as a partial inhibitor of IL-6 release, indicating that the signalization range assigned to this cytokine may be mitigated in the presence of LMWH (46). Indeed, a retrospective review has demonstrated that treating COVID-19 cases with LMWH decreases IL-6 overproduction, thus being an important therapeutic choice to be considered, once inflammation and coagulopathies are closely related (37). Studies have also discussed that disturbances on the pro-inflammatory agents that compound the complement cascade may be modulated during COVID-19 illness (47, 48). It was demonstrated that the complement system plays a role in the SARS-CoV-2-induced endothelial damage in rhesus macaques (33). Thereby, it is important to highlight that UFH and LMWH also bind to complement proteins and reduce the classical cascade activity, thus being instigators of anti-inflammatory responses either through this pathway (49, 50).
Eicosanoid lipid mediators have been identified as important agents during the virus-induced inflammatory process of respiratory airways (51), as observed in Influenza (52) and acute severe respiratory syncytial virus bronchiolitis infection (53). Human bronchial epithelial cells and resident leukocytes of the lung are important sources of leukotriene B4 (LTB4), a potent pro-inflammatory mediator that is closely related to neutrophil activation (54). A recent review highlighted the association between SARS-CoV-2-induced endoplasmic reticulum stress to eicosanoid pathway activation, which could augment the proinflammatory storm linked to COVID-19 (55). In this context, previous reports have shown that heparin is capable of inhibiting LBT4 signalization, thus softening the inflammatory response assigned to this molecule (56). Besides, hyperventilation-induced bronchoconstriction was attenuated through inhibition of eicosanoids production by heparin in the animal model (57). Considering COVID-19-induced multi-organ damage through hyperinflammation, the countless immunomodulatory effects of heparin beyond anti-coagulant properties improve its potential against this pandemic disease.
Another approach that is being widely discussed is the antiviral potential provided by heparins. Previous to the COVID-19 pandemic, the antiviral activity of the current drug was observed in different experimental models and viruses, such as Human Immunodeficiency Virus (HIV) and Herpes Simplex Virus (HSV) (58, 59). This antiviral effect of heparins may be related to the direct competition for binding to the cell glycoprotein receptors or through softening harmful effects caused by the infection, as already demonstrated for Zika Virus (ZIKV) (60). Growing shreds of evidence suggest that heparin also presents an antiviral effect against SARS-CoV-2. A report has shown that the cellular invasion capability of the virus can be affected in the presence of heparins (61). In this study, it was noticed that heparan sulfate (HS) derivatives, such as UFH and LMWH, at feasible concentrations for clinical application, induces conformational alteration of the SARS-CoV-2 Spike protein which is a central molecule for the host's cell invasion. Another preprint demonstrated that UFH was also capable of affecting SARS-CoV-2 entry to the host cell, impacting its infectivity parameters (62). Moreover, an in vitro comparative analysis found that UFH presented a higher antiviral potential against SARS-CoV-2, suggesting that it could be more beneficial than LMWH (63, 64). It was also noticed an important role of cellular HS binding to SARS-CoV-2 that promotes S protein conformational change and binding to the ACE-2 receptor, suggesting that HS could be a coreceptor for the viral invasion (65). In this context, UFH was also reported as a blocking agent of this interaction between cellular HS molecule with SARS-CoV-2, reinforcing its antiviral activity.
Although antiviral evidence of both LMWH and UFH against SARS-CoV-2 are still preliminary, the other protective activities against COVID-19 complications are clear. The ideal therapeutic approach for treating such complex diseases is also using a multi-target molecule that restores different SARS-CoV-2-affected pathways. The anticoagulant, anti-inflammatory, and potential antiviral effects provided by heparins increase the perspectives of using these medications for creating a better prognosis for COVID-19 affected individuals (Figure 1). Considering the findings that the antiviral effect of UFH could be stronger than LMWH, the dose of choice must be decided carefully, once UFH presents a higher risk for bleeding than LMWH. Avoiding the increased risk, the most suitable dose for both UFH and LMWH is the standard care, which can be adjusted according to the body mass index (BMI) and kidney function (66). Besides, risk factors for bleeding must be considered before prescribing the anticoagulant drug as prophylaxis. Moreover, nebulized UFH is a great possibility to improve the performance of the current drug by directing the effect to the most required site and reducing risks for unwanted impact on systemic coagulation pathways.
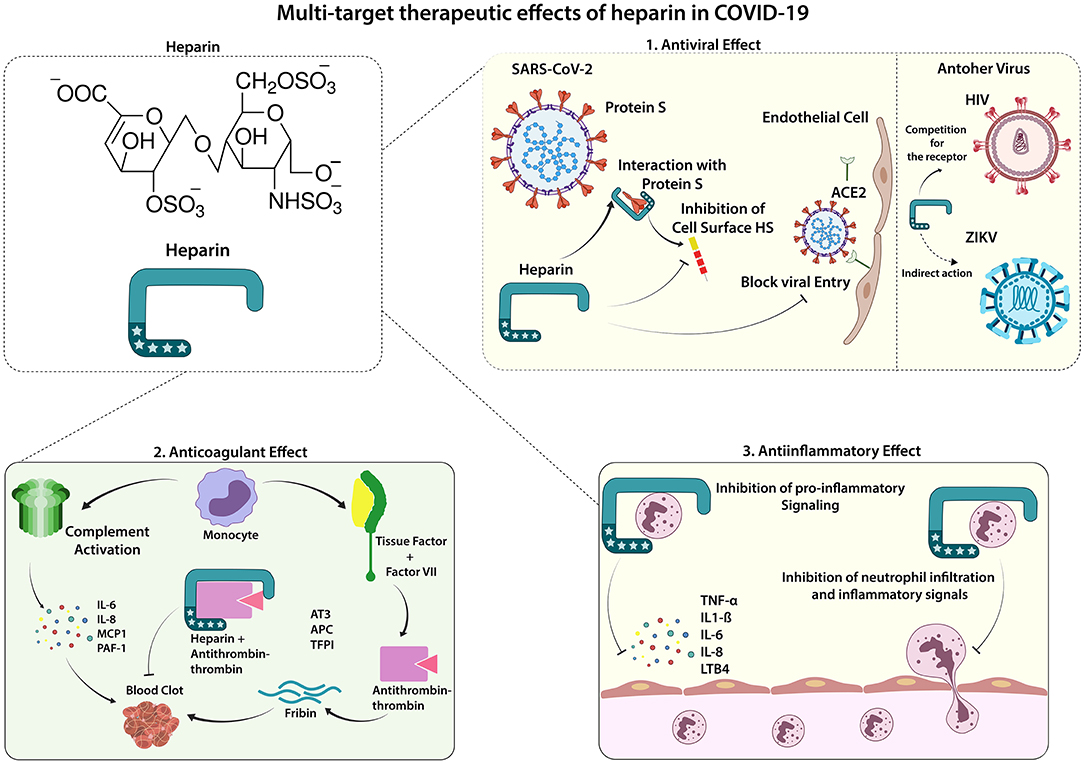
Figure 1. Therapeutic Effects of Heparin in COVID-19. Lung infection by SARS-CoV-2 can trigger a systemic inflammatory process, which is also associated with an increased occurrence of procoagulant factors found in severe cases of COVID-19. Low molecular weight Heparin (LMWH) shows effects in 3 main ways: 1. Antiviral effect: the entrance of SARS-CoV-2 in the endothelial and epithelial cells depends on its interaction with the cell surface heparan sulfate; thus, the binding of heparin to the viral spike protein can inhibit this interaction, decreasing viral cell invasion. Heparin has shown its antiviral effect on other viruses such as HIV (competing for the receptor) and ZIKV (indirect action, abrogating the viral-induced cytotoxic effect). 2. Anticoagulant effect: the uncontrolled blood clot formation can be controlled by the anticoagulant function inherent to heparin, mediated by the interaction of heparin and anti-thrombin-3 glycoprotein (AT3), potentiating the AT3 inactivation of thrombin, an essential factor for the formation of thrombi. 3. Anti-inflammatory effect: heparin has widely known anti-inflammatory effects, mainly canceling pro-inflammatory mediators, such as TNF-α, IL-6, and LTB4, which leads to decreased migration and activation of immune cells, preventing against the systemic inflammatory response. ACE2, Angiotensin-converting enzyme 2; APC, activated protein C; AT3, Antithrombin-III; CK, creatine kinase; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; HS, heparan sulfate; IL, interleukin; LDH, lactate dehydrogenase; PAI-1, Plasminogen activator inhibitor 1, TF, Tissue factor, TFPI, Tissue factor pathway inhibitor; TNF, Tumor necrosis factor; VII, Factor VII.
Based on available clinical data, around 20% of SARS-CoV-2-infected patients developed Acute Respiratory Distress Syndrome (ARDS), characterized by pulmonary pathological alterations as elevated dead space and decreased oxygenation (67–69). The massive release of proinflammatory cytokines and chemokines (5), coagulopathy and microvascular thrombosis (5, 10), hyaline membrane formation (68, 70), and intravascular DNA neutrophil extracellular traps (NETs) (71) are currently described as contributing factors for the diffuse alveolar damage commonly displayed in COVID-19 associated ARDS.
Consistently manifested by ARDS patients, pulmonary hyaline membrane formation impairs gas exchange, limits surfactant action, favors lung fibrosis, and induces lung microvascular thrombi (72, 73). Other factors that cope with thrombosis initiation and inflammation during ARDS onset are intravascular DNA neutrophil extracellular traps (NETs) and NETosis (71, 74). In addition to neutrophilia, the risk factors associated with the establishment of ARDS in COVID-19 patients were older age, organ dysfunction, and coagulopathies (75). The management of the cytokine and chemokine storm during COVID-19 represents a crucial and controversial point (76), considering that the use of systemic anti-inflammatory drugs can either inhibit the tissue damage or curb the cell-mediated immunity (77, 78).
Glucocorticoids (GCs) are steroid hormones derived from cholesterol metabolism. Both endogenous and synthetic forms of GCs share the same lipophilic chemical structure that allows the molecule to exert a broad range of endocrine effects in the organism (79). Currently, the synthetic GCs, such as dexamethasone, suffered alterations that significantly improved specificity, bioavailability, and potency, leading to a higher efficacy compared to the endogenous signaling pathway (80). The known immunosuppressive potential of dexamethasone made this anti-inflammatory molecule the first-line therapy for a great number of inflammatory diseases, such as autoimmune disorders and respiratory infections (81).
Dexamethasone mechanisms of action are characterized by exclusive binding to the classical cytosolic GC receptor (cGCR), which provides the majority of anti-inflammatory effects (82). The drug can reduce the inflammatory process by controlling the transcription of many pro-inflammatory genes that encode cytokines, cell adhesion molecules, and receptors related to inflammation (83). Dexamethasone is associated with decreased capillaries permeability, in addition to reduced neutrophil and lymphocyte migration into the inflammatory sites (84–86). An important advantage of the current drug is its extended half-life in the organism, which may decrease the time required for therapy compared to alternative GCs (74). The dose of choice is based on the desired effect, once low doses are associated with anti-inflammatory modulation and higher doses are associated with the immunosuppressive activity (87).
As previously discussed, endogenous GCs are central for metabolic homeostasis and systemic inflammatory events during tissue repair and pathogens elimination (88). Due to this fact, long-term exogenous intake of synthetic GCs can provide adverse effects, such as extreme shut down of inflammatory responses, leading to a higher susceptibility to secondary infections establishment (87, 89). Besides, GCs play a role in glucose metabolism through alterations of insulin signaling, leading ta reduced uptake and increased concentration of plasmatic glucose levels (90). This intervention in the metabolism gives rise to insulin resistance (IR) conditions caused by supra-physiologic or prolonged synthetic GCs medication, which can evolve to type-2 diabetes (T2D) and other metabolic disorders (91, 92). For this reason, synthetic GCs medication needs to be handled carefully, avoiding complications rather than therapeutic effects.
At first, it was not clear whether patients diagnosed with COVID-19 could take advantage of the use of dexamethasone, especially taking into account the above-mentioned points (93). Reports have shown that dexamethasone medication against influenza-induced pneumonia, SARS-CoV-1, and Middle East respiratory syndrome coronavirus (MERS-CoV) presented harmful effects that could negatively impact the disease prognosis (94–97). However, specifically in patients who develop ARDS during COVID-19, dexamethasone treatment showed to be effective in down-regulating systemic and pulmonary inflammation, restoring tissue homeostasis by accelerating resolution of diffuse alveolar damage, leading to protection against extrapulmonary organ dysfunction (98, 99). Besides, experimental and clinical studies have demonstrated that GR-α expression in myeloid cells of bronchoalveolar lavage is associated with significant protection against severe COVID-19 symptoms, especially through reduction of NETosis and lung neutrophilic inflammation (100). Of note, when lung damage has already occurred, the case fatality rate of COVID-19 is high (101). If dexamethasone could alleviate the clinical progression at this stage, then the therapy may decrease the cases of severe illness and therefore lower the case fatality rate of COVID-19 (102). A comparative scenario of COVID-19 progression in the presence and absence of dexamethasone was summarized (Figure 2).
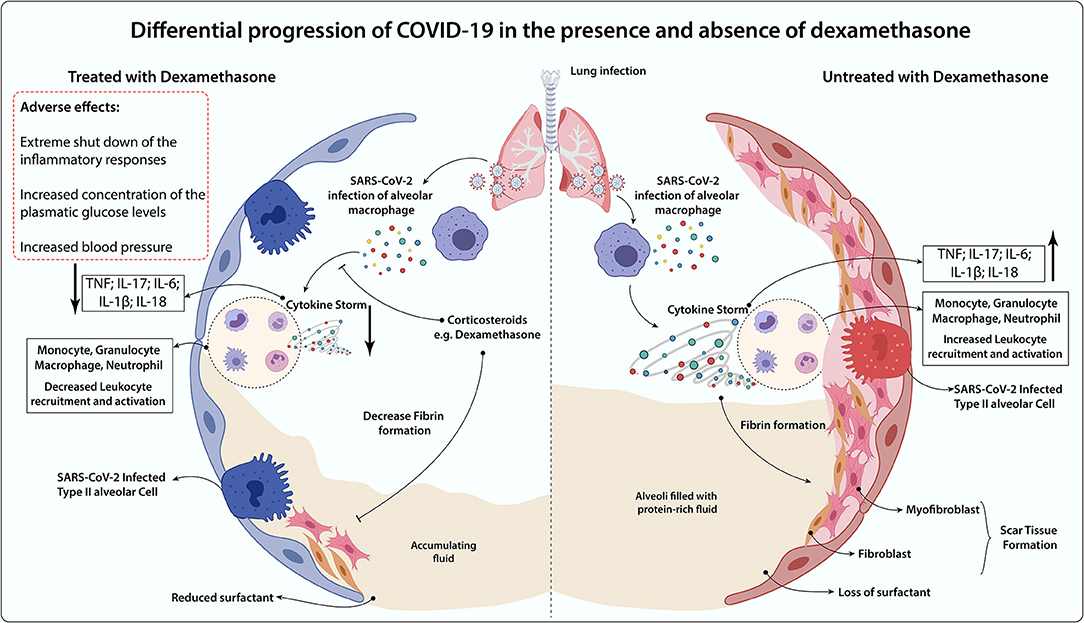
Figure 2. Differential progression of COVID-19 in the presence and absence of dexamethasone. In the absence of an anti-inflammatory treatment, COVID-19-induced lung dysfunction is triggered by hyperactivation of alveolar macrophages infected by SARS-CoV-2 and immune cells (Monocytes, Granulocytes, Macrophages, Neutrophils) recruitment to the lung surroundings, which leads to the massive secretion of inflammatory mediators (TNF, IL-17, IL-6, IL-1b, IL-18) in the cytokine storm release. The persistence of the inflammatory process gives rise to the increased fibroblasts and myofibroblast invasion throughout the scar tissue formation, loss of natural pulmonary surfactant, and increased alveolar fluid. Dexamethasone administration may benefit through the impairment of cytokine storm occurrence and leukocyte lung infiltration, which decreases tissue fibrosis and alveolar fluid accumulation. However, this glucocorticoid is associated with immunosuppression, augmented blood pressure, and glycemia as major side effects. Thereby, it may be detrimental for some groups, including non-severe COVID-19 cases, diabetics, and hypertensive subjects.
Faced with the complex scenario of severely ill patients with COVID-19, diverse protocols employing complementary treatments (94) have been developed, some of them using GCs for the treatment of hospitalized patients with COVID-19 (103). Members of the WHO and the Chinese Thoracic Society have conflicting opinions regarding the use of corticosteroids in COVID-19 (104). According to the WHO guidelines, dexamethasone should only be used under clinical trial conditions (105). Russell and colleagues concluded that there is no reason to expect that individuals with COVID-19 will benefit from corticosteroid treatment, based on the increased mortality and risk of secondary infection in influenza, impaired clearance of SARS-CoV, and MERS-CoV (93). However, the high potential of dexamethasone for cytokine storm mitigation brought proposals to overcome the impairment of viral clearance and increased risk of developing secondary infections, and the main in-progress clinical trials are summarized in Table 2.
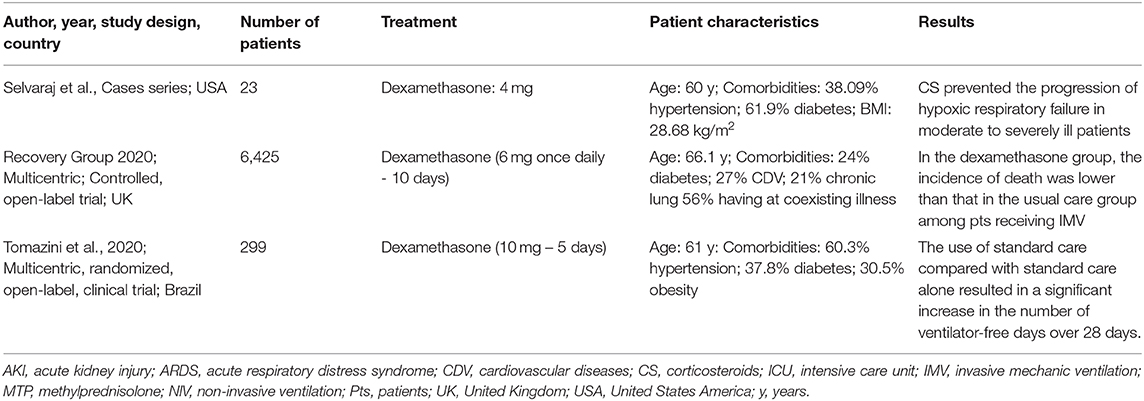
Table 2. Studies on dexamethasone for COVID-19.
The preliminary results of a large randomized, controlled, open-label trial conducted in the United Kingdom are in favor of dexamethasone use. In this trial, Dexamethasone arm constituted 2,104 patients receiving 6 mg dexamethasone (oral or intravenous) once daily for up to 10 days and 4,321 patients receiving standard care. Dexamethasone reduced mortality by 35% in patients receiving invasive mechanical ventilation. Besides, the prospective meta-analysis from the WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group recommended the independent use of corticosteroids in patients with COVID-19. An important correlation between the administration of systemic GCs and reduced mortality was found among critically ill patients with COVID-19 (106). Selvaraj et al. reported that short-term use of dexamethasone by hospitalized patients with COVID-19 was well tolerated and increased the patients' prognosis (107).
Although dexamethasone promising performance for severe COVID-19, there are still concerns regarding this anti-inflammatory drug prescription for indiscriminately cases. Until now, the available data suggests a beneficial role of dexamethasone treatment on hospitalized patients, especially for those who received intensive oxygen therapy (108, 109). A possible explanation can be based on the association between hyperinflammation and the development of pulmonary damage, increasing urgency for ICU. The excessive inflammation needs to be handled by a strong anti-inflammatory treatment, which could be accessed by dexamethasone therapy. In the absence of an unbalanced inflammatory process as in the early stage of the disease, dexamethasone treatment might disturb the development of the host's natural immunity and abrogate anti-viral response, which could lead to a delayed viral clearance.
Besides the previously discussed impairment of viral clearance when applied in the early stage of the disease (95), the increasing risk of secondary infections associated with high doses of systemic GCs was noticed (87, 89). A report regarding Strongyloides hyperinfection, a neglected nematode disease, brought the current concern about dexamethasone application without concomitant vermifuge use (110).
Besides that, GCs modulation of glucose metabolism leads to intensive care on diabetic patients during the treatment (92). As discussed worldwide, diabetes is a major risk factor for severe COVID-19 and dexamethasone treatment in these individuals could be associated with the development of hyperglycemic condition (111–113). The increased concentration of glucose levels in the bloodstream could be associated with a poor prognosis of the disease, considering the recent discovery about SARS-CoV-2 dependency of glucose for viral replication in vitro (114). With increasing glucose concentration, the viral load increased concomitantly, favoring the infection and the severity of the disease (114). Besides, hypertensive patients, who are also at risk for COVID-19 severity, should be at constant monitoring during dexamethasone treatment, once GCs-induced hypertension is frequently observed (115). High dexamethasone doses are associated with increased sodium retention, which leads to the elevation of blood pressure, in addition to chemical alteration of peripheral nerves homeostasis (87, 116, 117). This effect could lead to complications rather than COVID-19 treatment, increasing the associated mortality risks of the affected individuals.
Severe cases of COVID-19 are marked by intense inflammation and the presence of thrombotic events. Exacerbated inflammation that mediates the characteristic cytokine storm observed in the severe COVID-19, and the blood clots in the lungs that can compromise oxygenation, lead to worsening clinical outcomes of SARS-CoV-2 infection. Several recent reports have demonstrated a beneficial effect of the use of heparin/low molecular weight heparin and corticosteroids, such as dexamethasone, on mortality in COVID-19.
The advantageous and successful effect of heparin underlying treatment of COVID-19 patients could be explained not only by its anticoagulant properties but also due to its non-anticoagulant mechanisms, which include anti-viral and anti-inflammatory actions such as (I) decrease of SARS-CoV-2 host cell entry, (II) inhibition of pro-inflammatory cytokines and chemokines, (III) inhibition of vascular permeability and leukocyte migration.
Despite the controversial role of corticosteroids in treating severe infectious diseases, several clinical studies have provided increasing evidence that dexamethasone could function widely as an available treatment for the most severely ill patients with COVID-19. However, many clinically important questions remain open, and determination of optimal initiation period, dosing, and duration of the dexamethasone treatment might be considered to avoid serious adverse effects during COVID-19 management.
While UFH or LMWH are indicated as prophylactic agents for the initial phase of the disease, which could impair clots formation in addition to abrogate viral cell entry, dexamethasone must be prescribed only for severe cases, when the disease reaches a highly inflammatory state. Taking this into account, we suggest that the multitarget impact of heparin as an anti-viral, antithrombotic and anti-inflammatory drug in the early stage of the COVID-19 could significantly reduce the need for dexamethasone treatment in the initial phase of this disease. If the standard treatment of heparins fails on protecting against severe illness, dexamethasone must be applied as a potent anti-inflammatory shutting-down the uncontrolled and exacerbated inflammation.
Overall, the association of anti-coagulant heparin and the corticosteroid dexamethasone could be a very effective and promising therapeutic tool in avoiding COVID-19 complications when used for severely ill patients.
GP-d-N, HB-d-M, SF, GK, and KM wrote different sections of the manuscript. KM revised, wrote, and prepared the manuscript. IS prepared the figures. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. (2020) 46:1089–98. doi: 10.1007/s00134-020-06062-x
2. Driggin E, Madhavan M V., Bikdeli B, Chuich T, Laracy J, Biondi-Zoccai G, et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. J Am Coll Cardiol. (2020) 75:2352–71. doi: 10.1016/j.jacc.2020.03.031
3. Batlle D, Soler MJ, Sparks MA, Hiremath S, South AM, Welling PA, et al. Acute kidney injury in COVID-19: emerging evidence of a distinct pathophysiology. J Am Soc Nephrol. (2020) 31:1380–3. doi: 10.1681/ASN.2020040419
4. Espinosa OA, Zanetti A dos S, Antunes EF, Longhi FG, Matos TA de, Battaglini PF. Prevalence of comorbidities in patients and mortality cases affected by SARS-CoV2: a systematic review and meta-analysis. Rev Inst Med Trop Sào Paulo. (2020) 62:159–66. doi: 10.1590/s1678-9946202062043
5. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
6. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. (2012) 76:16–32. doi: 10.1128/MMBR.05015-11
7. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
8. Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol. (2020) 20:389–91. doi: 10.1038/s41577-020-0343-0
9. Imai Y, Kuba K, Penninger JM. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. (2008) 93:543–8. doi: 10.1113/expphysiol.2007.040048
10. Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med. (2020) 382:e38. doi: 10.1056/NEJMc2007575
11. Li X, Ma X. The role of heparin in sepsis: much more than just an anticoagulant. Br J Haematol. (2017) 179:389–98. doi: 10.1111/bjh.14885
12. Bompard F, Monnier H, Saab I, Tordjman M, Abdoul H, Fournier L, et al. Pulmonary embolism in patients with COVID-19 pneumonia. Eur Respir J. (2020) 56:2001365. doi: 10.1183/13993003.01365-2020
13. Bonow RO, Fonarow GC, O'Gara PT, Yancy CW. Association of coronavirus disease 2019 (COVID-19) with myocardial injury and mortality. JAMA Cardiol. (2020) 323:1061–9. doi: 10.1001/jamacardio.2020.1105
14. Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res. (2016) 118:1392–408. doi: 10.1161/CIRCRESAHA.116.306853
15. Magro G. COVID-19: review on latest available drugs and therapies against SARS-CoV-2. Coagulation and inflammation cross-talking. Virus Res. (2020) 286:198070. doi: 10.1016/j.virusres.2020.198070
16. Kerr R, Stirling D, Ludlam CA. Interleukin 6 and haemostasis. Br J Haematol. (2001) 115:3–12. doi: 10.1046/j.1365-2141.2001.03061.x
17. Regnault V, De Maistre E, Carteaux JP, Gruel Y, Nguyen P, Tardy B, et al. Platelet activation induced by human antibodies to interleukin-8. Blood. (2003) 101:1419–21. doi: 10.1182/blood-2002-02-0620
18. Filisbino MM, Baldi BG. COVID-19 pneumonia : a risk factor for pulmonary thromboembolism? J Bras Pneumol. (2020) 46:3–4. doi: 10.36416/1806-3756/e20200168
19. Wang T, Chen R, Liu C, Liang W, Guan W, Tang R, et al. Attention should be paid to venous thromboembolism prophylaxis in the management of COVID-19. Lancet Haematol. (2020) 7:e362–3. doi: 10.1016/S2352-3026(20)30109-5
20. Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir. (2019) 2019:2019–20. doi: 10.1016/S2213-2600(20)30216-2
22. Hoppensteadt D, Walenga JM, Fareed J, Bick RL. Heparin, low-molecular-weight heparins, and heparin pentasaccharide basic and clinical differentiation. Hematol Oncol Clin North Am. (2003) 17:313–41. doi: 10.1016/S0889-8588(02)00091-6
23. Sancar Eke SN. Heparin-induced thrombocytopenia. J Emerg Trauma Shock. (2011) 4:97–102. doi: 10.4103/0974-2700.76843
24. Signorelli SS, Scuto S, Marino E, Giusti M, Xourafa A, Gaudio A. Anticoagulants and osteoporosis. Int J Mol Sci. (2019) 20:5275–87. doi: 10.3390/ijms20215275
25. Oduah EI, Linhardt RJ, Sharfstein ST. Heparin: past, present, and future. Pharmaceuticals. (2016) 9:1–12. doi: 10.3390/ph9030038
26. Alquwaizani M, Buckley L, Adams C, Fanikos J. Anticoagulants: a review of the pharmacology, dosing, and complications. Curr Emerg Hosp Med Rep. (2013) 1:83–97. doi: 10.1007/s40138-013-0014-6
27. Ludwig R. Therapeutic use of heparin beyond anticoagulation. Curr Drug Discov Technol. (2009) 6:281–9. doi: 10.2174/157016309789869001
28. Salas A, Sans M, Soriano A, Piqué JM, Panés J, Reverter JC, et al. Heparin attenuates TNF-α induced inflammatory response through a CD11b dependent mechanism. Gut. (2000) 47:88–96. doi: 10.1136/gut.47.1.88
29. Wan MX, Zhang XW, Törkvist L, Thorlacius H. Low molecular weight heparin inhibits tumor necrosis factor α-induced leukocyte rolling. Inflamm Res. (2001) 50:581–4. doi: 10.1007/PL00000237
30. Kollias A, Kyriakoulis KG, Dimakakos E, Poulakou G, Stergiou GS, Syrigos K. Thromboembolic risk and anticoagulant therapy in COVID-19 patients: emerging evidence and call for action. Br J Haematol. (2020) 189:846–7. doi: 10.1111/bjh.16727
31. Klok FA, Kruip MJHA, Meer NJM, Van Der Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
32. Negri EM, Piloto BM, Morinaga LK, Jardim CV, Lamy SA, Ferreira MA, et al. Heparin therapy improving hypoxia in COVID-19 patients - a case series. Front Physiol. (2020) 11:573044. doi: 10.3389/fphys.2020.573044
33. Aid M, Busman-Sahay K, Vidal SJ, Maliga Z, Bondoc S, Starke C, et al. Vascular disease and thrombosis in SARS-CoV-2-infected rhesus macaques. Cell. (2020) 183:1–13. doi: 10.1016/j.cell.2020.10.005
34. Goldberg MF, Goldberg MF, Cerejo R, Tayal AH. Cerebrovascular disease in COVID-19. Am J Neuroradiol. (2020) 41:1170–2. doi: 10.3174/AJNR.A6588
35. Reddy ST, Garg T, Shah C, Nascimento FA, Imran R, Kan P, et al. Cerebrovascular disease in patients with COVID-19: a review of the literature and case series. Case Rep Neurol. (2020) 77030:199–209. doi: 10.1159/000508958
36. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. (2020) 18:1094–9. doi: 10.1111/jth.14817
37. Shi C, Wang C, Wang H, Yang C, Cai FEI, Zeng F, et al. The potential of low molecular weight heparin to mitigate cytokine storm in severe covid-19 patients: a retrospective clinical study. Clin Transl Sci. (2020) 13:1087–95. doi: 10.1101/2020.03.28.20046144
38. Van Haren FMP, Richardson A, Yoon H-J, Artigas A, Laffey JG, Dixon B, et al. INHALEd nebulised unfractionated HEParin for the treatment of hospitalised patients with COVID-19 (INHALE-HEP): protocol and statistical analysis plan for an investigator-initiated international meta-trial of randomised studies. Br J Clin Pharmacol. (2020) 39:471. doi: 10.1111/bcp.14714
39. Glas GJ, Van Der Sluijs KF, Schultz MJ, Hofstra JJH, Van Der Poll T, Levi M. Bronchoalveolar hemostasis in lung injury and acute respiratory distress syndrome. J Thromb Haemost. (2013) 11:17–25. doi: 10.1111/jth.12047
40. Agnelli G, Becattini C, Kirschstein T. Thrombolysis vs heparin in the treatment of pulmonary embolism: a clinical outcome-based meta-analysis. Arch Intern Med. (2002) 162:2537–41. doi: 10.1001/archinte.162.22.2537
41. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
42. Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun. (2020) 111:2–9. doi: 10.1016/j.jaut.2020.102452
43. Robinson PC, Richards D, Tanner HL, Feldmann M. Accumulating evidence suggests anti-TNF therapy needs to be given trial priority in COVID-19 treatment. Lancet Rheumatol. (2020) 2:e653–5. doi: 10.1016/S2665-9913(20)30309-X
44. Spratte J, Meyer zu schwabedissen H, Endlich N, Zygmunt M, Fluhr H. Heparin inhibits TNF-α signaling in human endometrial stromal cells by interaction with NF-kB. Mol Hum Reprod. (2013) 19:227–36. doi: 10.1093/molehr/gas060
45. Zhang N, Zhan Y, Shi P, Ye Y, Li J, Zhao L, et al. LMWH inhibits TNF-α and IL-6 in placental villous explants and its effects are attenuated by TLR-4/NF-κB p65 blocking in JEG-3 cells. Int J Clin Exp Med. (2018) 11:674–83. doi: 10.1111/j.1365-2141.2006.05959.x
46. Shastri MD, Stewart N, Horne J, Peterson GM, Gueven N, Sohal SS, et al. In-vitro suppression of IL-6 and IL-8 release from human pulmonary epithelial cells by non-anticoagulant fraction of enoxaparin. PLoS ONE. (2015) 10:1–23. doi: 10.1371/journal.pone.0126763
47. Risitano AM, Mastellos DC, Huber-Lang M, Yancopoulou D, Garlanda C, Ciceri F, et al. Complement as a target in COVID-19? Nat Rev Immunol. (2020) 20:343–4. doi: 10.1038/s41577-020-0320-7
48. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
49. Oberkersch R, Attorresi AI, Calabrese GC. Low-molecular-weight heparin inhibition in classical complement activaton pathway during pregnancy. Thromb Res. (2010) 125:e240–5. doi: 10.1016/j.thromres.2009.11.030
50. Haining Yu, Eva M. Munoz, R. Erik Edens RJL. Kinetic studies on the interactions of heparin and complement proteins using surface plasmon resonance. Biochim Biophys Acta Mol Cell Biol Lipids. (2014) 23:1–7. doi: 10.1016/j.bbagen.2005.08.003
51. McCarthy MK, Weinberg JB. Eicosanoids and respiratory viral infection: coordinators of inflammation and potential therapeutic targets. Mediators Inflamm. (2012) 2012:1–13. doi: 10.1155/2012/236345
52. Lee SMY, Cheung CY, Nicholls JM, Hui KPY, Leung CYH, Uiprasertkul M, et al. Hyperinduction of cyclooxygenase-2-mediated proinflammatory cascade: a mechanism for the pathogenesis of avian influenza H5N1 infection. J Infect Dis. (2008) 198:525–35. doi: 10.1086/590499
53. Sznajer Y, Westcott JY, Wenzel SE, Mazer B, Tucci M, Joseph Toledano B. Airway eicosanoids in acute severe respiratory syncytial virus bronchiolitis. J Pediatr. (2004) 145:115–8. doi: 10.1016/j.jpeds.2004.03.049
54. Jame AJ, Lackie PM, Cazaly AM, Sayers I, Penrose JF, Holgate ST, et al. Human bronchial epithelial cells express an active and inducible biosynthetic pathway for leukotrienes B4 and C4. Clin Exp Allergy. (2007) 37:880–92. doi: 10.1111/j.1365-2222.2007.02733.x
55. Hammock BD, Wang W, Gilligan MM, Panigrahy D. Eicosanoids: the overlooked storm in coronavirus disease 2019 (COVID-19)? Am J Pathol. (2020) 190:1782–8. doi: 10.1016/j.ajpath.2020.06.010
56. Lappegård KT, Riesenfeld J, Brekke OL, Bergseth G, Lambris JD, Mollnes TE. Differential effect of heparin coating and complement inhibition on artificial surface-induced eicosanoid production. Ann Thorac Surg. (2005) 79:917–23. doi: 10.1016/j.athoracsur.2004.08.015
57. Suzuki R, Freed AN. Heparin inhibits eicosanoid metabolism and hyperventilation-induced bronchoconstriction in dogs. Am J Respir Crit Care Med. (2000) 161:1850–4. doi: 10.1164/ajrccm.161.6.9907013
58. Baba M, Pauwels R, Balzarini J, Desmyter J, De Clercq E. Antiviral activity of heparin and dextran sulfate against human immunodeficiency virus (HIV) in vitro. Ann N Y Acad Sci. (1989) 556:419–21. doi: 10.1111/j.1749-6632.1989.tb22523.x
59. Nahmias AJ, Kibrick S. Inhibitory effect of heparin on herpes simplex virus. J Bacteriol. (1964) 87:1060–6. doi: 10.1128/JB.87.5.1060-1066.1964
60. Ghezzi S, Cooper L, Rubio A, Pagani I, Capobianchi MR, Ippolito G, et al. Heparin prevents Zika virus induced-cytopathic effects in human neural progenitor cells. Antiviral Res. (2017) 140:13–7. doi: 10.1016/j.antiviral.2016.12.023
61. Mycroft-West CJ, Su D, Pagani I, Rudd TR, Elli S, Guimond SE, et al. Heparin inhibits cellular invasion by SARS-CoV-2: structural dependence of the interaction of the surface protein (spike) S1 receptor binding domain with heparin. Thromb Haemost. (2020) 12:1700–15. doi: 10.1055/s-0040-1721319
62. Partridge LJ, Green LR, Monk PN. Unfractionated heparin potently inhibits the binding of SARS-CoV-2 spike protein to a human cell line. bioRxiv. (2020) 2020.05.21:1–31. doi: 10.1101/2020.05.21.107870
63. Tree JA, Turnbull JE, Buttigieg KR, Elmore MJ, Coombes N, Hogwood J, et al. Unfractionated heparin inhibits live wild-type SARSCoV-2 cell infectivity at therapeutically relevant concentrations. Br J Pharmacol. (2020) 178:626–35. doi: 10.1111/bph.15304
64. Kwon PS, Oh H, Kwon SJ, Jin W, Zhang F, Fraser K, et al. Sulfated polysaccharides effectively inhibit SARS-CoV-2 in vitro. Cell Discov. (2020) 6:4–7. doi: 10.1038/s41421-020-00192-8
65. Clausen TM, Sandoval DR, Spliid C, Pihl J, Perrett HR, Painter CD, et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. (2020) 7:1043–57. doi: 10.1016/j.cell.2020.09.033
66. Orsi FA, De Paula E V., Santos F, de O, Teruchkin MM, Campêlo DHC, Mello TT, et al. Guidance on diagnosis, prevention and treatment of thromboembolic complications in COVID-19: a position paper of the Brazilian Society of thrombosis and hemostasis and the thrombosis and hemostasis committee of the brazilian association of hematology, hemotherapy and cellular therapy. Hematol Transfus Cell Ther. (2020) 42:300–8. doi: 10.1016/j.htct.2020.06.001
67. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
68. Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med. (2020) 173:268–77. doi: 10.7326/M20-2003
69. Rubin JE, Crowe SE. Annals of internal medicine®. Ann Intern Med. (2020) 172:ITC1–14. doi: 10.7326/AITC202001070
70. Adachi T, Chong JM, Nakajima N, Sano M, Yamazaki J, Miyamoto I, et al. Clinicopathologic and immunohistochemical findings from autopsy of patient with COVID-19, Japan. Emerg Infect Dis. (2020) 26:2157–61. doi: 10.3201/eid2609.201353
71. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. (2020) 217:1–7. doi: 10.1084/jem.20200652
72. Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med. (2003) 31:213–20. doi: 10.1097/01.CCM.0000057846.21303.AB
73. Günther A, Ruppert C, Schmidt R, Markart P, Grimminger F, Walmrath D, et al. Surfactant alteration and replacement in acute respiratory distress syndrome. Respir Res. (2001) 2:353–64. doi: 10.1186/rr86
74. Shefrin AE, Goldman RD. Use of dexamethasone and prednisone in acute asthma exacerbations in pediatric patients. Can Fam Physician. (2009) 55:704–6.
75. Mangalmurti NS, Reilly JP, Cines DB, Meyer NJ, Hunter CA, Vaughan AE. COVID-19-associated acute respiratory distress syndrome clarified: a vascular endotype? Am J Respir Crit Care Med. (2020) 202:750–3. doi: 10.1164/rccm.202006-2598LE
76. Favalli EG, Ingegnoli F, De Lucia O, Cincinelli G, Cimaz R, Caporali R. COVID-19 infection and rheumatoid arthritis: Faraway, so close! Autoimmun Rev. (2020) 19:102523. doi: 10.1016/j.autrev.2020.102523
77. Mahallawi WH, Khabour OF, Zhang Q, Makhdoum HM, Suliman BA. Cytokine MERS-CoV infection in humans is associated with a pro-in fl ammatory Th1 and Th17 cytokine profile. Cytokine. (2018) 104:8–13. doi: 10.1016/j.cyto.2018.01.025
78. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X. COVID-19 infection: the perspectives on immune responses. Cell Death Differ. (2020) 27:1451–4. doi: 10.1038/s41418-020-0530-3
79. Williams DM. Clinical pharmacology of corticosteroids. Respir Care. (2018) 63:655–70. doi: 10.4187/respcare.06314
80. Buchwald P, Bodor N. Soft glucocorticoid design: structural elements and physicochemical parameters determining receptor-binding affinity. Pharmazie. 59:396–404.
81. Janahi IA, Rehman A, Baloch NU-A. “Corticosteroids and their use in respiratory disorders,” In: Corticosteroids, Vol. 55, (IntechOpen) (2017). p. 2–55. doi: 10.5772/intechopen.72147
82. Levin ER, Hammes SR. Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat Rev Mol Cell Biol. (2016) 17:783–97. doi: 10.1038/nrm.2016.122
83. Ronchetti S, Migliorati G, Bruscoli S, Riccardi C. Defining the role of glucocorticoids in inflammation. Clin Sci. (2018) 132:1529–43. doi: 10.1042/CS20171505
84. Almawi WY, Melemedjian OK. Negative regulation of nuclear factor-κB activation and function by glucocorticoids. J Mol Endocrinol. (2002) 28:69–78. doi: 10.1677/jme.0.0280069
85. Ristimäki A, Narko K, Hla T. Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem J. (1996) 318:325–31. doi: 10.1042/bj3180325
86. Johnson DB, Lopez MJ, Kelley B. Dexamethasone. Treasure Island, FL: StatPearls Publishing (2020). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK482130/ (Accessed September 26, 2020).
87. Yasir M, Sonthalia S. Corticosteroid Adverse Effects. 1st ed. StatPearls Publishing (2019). Available at: http://www.ncbi.nlm.nih.gov/pubmed/30285357
88. Cruz-Topete D, Cidlowski JA. One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation. (2014) 22:20–32. doi: 10.1159/000362724
89. Gensler LS. Glucocorticoids: complications to anticipate and prevent. Neurohospitalist. (2013) 3:92–7. doi: 10.1177/1941874412458678
90. Kuo T, Harris CA, Wang J, States U, States U, Francisco S, et al. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol Cell Endocrinol. (2016) 380:79–88. doi: 10.1016/j.mce.2013.03.003
91. Rice JB, White AG, Scarpati LM, Wan G, Nelson WW. Long-term systemic corticosteroid exposure : a systematic literature review. Clin Ther. (2017) 39:2216–29. doi: 10.1016/j.clinthera.2017.09.011
92. Willms A, Schumm-Draeger PM, Siegmund T. Glucocorticoids and diabetes. Diabetologe. (2016) 12:127–38. doi: 10.1007/s11428-016-0070-0
93. Russell CD, Millar JE, Baillie JK. Comment clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet. (2020) 6736:2019–21. doi: 10.1016/S0140-6736(20)30317-2
94. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. J Am Med Assoc. (2020) 2019:1824–36. doi: 10.1001/jama.2020.6019
95. Hui DS. Systemic corticosteroid therapy may delay viral clearance in patients with middle east respiratory syndrome coronavirus infection. Am J Respir Crit Care Med. (2018) 197:700–1. doi: 10.1164/rccm.201712-2371ED
96. Ni YN, Chen G, Sun J, Liang BM, Liang ZA. The effect of corticosteroids on mortality of patients with influenza pneumonia: a systematic review and meta-analysis. Crit Care. (2019) 23:1–9. doi: 10.1186/s13054-019-2395-8
97. Jung K, Alekseev KP, Zhang X, Cheon D-S, Vlasova AN, Saif LJ. Altered pathogenesis of porcine respiratory coronavirus in pigs due to immunosuppressive effects of dexamethasone: implications for corticosteroid use in treatment of severe acute respiratory syndrome coronavirus. J Virol. (2007) 81:13681–93. doi: 10.1128/JVI.01702-07
98. Meduri GU, Annane D. Activation and regulation of systemic inflammation in ARDS rationale for prolonged glucocorticoid therapy. Chest. (2009) 136:1631–43. doi: 10.1378/chest.08-2408
99. Villar J, Ferrando C, Martínez D, Ambrós A, Muñoz T, Soler JA, et al. Dexamethasone treatment for the acute respiratory distress syndrome: a multicentre, randomised controlled trial. Lancet Respir Med. (2020) 8:267–76. doi: 10.1016/S2213-2600(19)30417-5
100. Park JH, Lee HK. Re-analysis of single cell transcriptome reveals that the NR3C1-CXCL8-neutrophil axis determines the severity of COVID-19. Front Immunol. (2020) 11:1–9. doi: 10.3389/fimmu.2020.02145
101. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. (2020) 2600:1–7. doi: 10.1016/S2213-2600(20)30079-5
102. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
103. Russell B, Moss C, Rigg A, Van Hemelrijck M. COVID-19 and treatment with NSAIDs and corticosteroids: should we be limiting their use in the clinical setting? Ecancermedicalscience. (2020) 14:1–3. doi: 10.3332/ecancer.2020.1023
104. Shang L, Zhao J, Hu Y, Du R, Cao B. On the use of corticosteroids for 2019-nCoV pneumonia. Lancet. (2020) 395:683–4. doi: 10.1016/S0140-6736(20)30361-5
105. Jin Y, Cai L, Cheng Z, Cheng H, Deng T, Fan Y, et al. A rapid advice guideline for the diagnosis and treatment of 2019 novel coronavirus (2019-nCoV) infected pneumonia (standard version). Mil Med Res. (2020) 7:1–23. doi: 10.1186/s40779-020-0233-6
106. Sterne JAC, Murthy S, Diaz J V., Slutsky AS, Villar J, Angus DC, et al. Association between administration of systemic corticosteroids and mortality among critically ill patients with COVID-19: a meta-analysis. J Am Med Assoc. (2020) 324:1330–41. doi: 10.1001/jama.2020.17023
107. Selvaraj V, Dapaah-afriyie K, Finn A, Flanigan TP. Short-term dexamethasone in Sars-CoV-2 patients. R I Med J. (2020). 103:39–43.
108. RECOVERY Collaborative Group, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in Hospitalized Patients with Covid-19. N Engl J Med. (2021) 384:693–704. doi: 10.1056/nejmoa2021436
109. Tomazini BM, Maia IS, Cavalcanti AB, Berwanger O, Rosa RG, Veiga VC, et al. Effect of dexamethasone on days alive and ventilator-free in patients with moderate or severe acute respiratory distress syndrome and COVID-19: the CoDEX randomized clinical trial. J Am Med Assoc. (2020) 324:1307–16. doi: 10.1001/jama.2020.17021
110. Stauffer WM, Alpern J, Walker PF. COVID-19 and dexamethasone: a potential strategy to avoid steroid-related strongyloides hyperinfection. J Am Med Assoc. (2020) 55455:5–6. doi: 10.1001/jama.2020.13170
111. Deng F, Gao D, Ma X, Guo Y, Wang R, Jiang W, et al. Corticosteroids in diabetes patients infected with COVID-19. Ir J Med Sci. (2021) 190:29–31. doi: 10.1007/s11845-020-02287-3
112. O'Connell RS, Clinger BN, Donahue EE, Celi FS, Golladay GJ. Dexamethasone and postoperative hyperglycemia in diabetics undergoing elective hip or knee arthroplasty: a case control study in 238 patients. Patient Saf Surg. (2018) 12:1–8. doi: 10.1186/s13037-018-0178-9
113. Lukins MB, Manninen PH. Hyperglycemia in patients administered dexamethasone for craniotomy. Anesth Analg. (2005) 100:1129–33. doi: 10.1213/01.ANE.0000146943.45445.55
114. Codo AC, Davanzo GG, Monteiro L de B, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab. (2020) 32:437–46. doi: 10.1016/j.cmet.2020.07.007
115. Goodwin JE, Geller DS. Glucocorticoid-induced hypertension. Pediatr Nephrol. (2012) 27:1059–66. doi: 10.1007/s00467-011-1928-4
116. Ong SLH, Zhang Y, Sutton M, Whitworth JA. Hemodynamics of dexamethasone-induced hypertension in the rat. Hypertens Res. (2009) 32:889–94. doi: 10.1038/hr.2009.118
Keywords: COVID-19, heparin, dexamethasone, anticoagulant, corticosteroid
Citation: Braz-de-Melo HA, Faria SS, Pasquarelli-do-Nascimento G, Santos IO, Kobinger GP and Magalhães KG (2021) The Use of the Anticoagulant Heparin and Corticosteroid Dexamethasone as Prominent Treatments for COVID-19. Front. Med. 8:615333. doi: 10.3389/fmed.2021.615333
Received: 08 October 2020; Accepted: 26 March 2021;
Published: 23 April 2021.
Edited by:
Ana Afonso, University of São Paulo, BrazilReviewed by:
Frank M. P. Van Haren, Australian National University, AustraliaCopyright © 2021 Braz-de-Melo, Faria, Pasquarelli-do-Nascimento, Santos, Kobinger and Magalhães. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelly Grace Magalhães, a2VsbHltYWdhbGhhZXNAdW5iLmJy; orcid.org/0000-0002-7435-5272
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.