 Simona Pompili
Simona Pompili Giovanni Latella
Giovanni Latella Eugenio Gaudio3
Eugenio Gaudio3 Roberta Sferra
Roberta Sferra Antonella Vetuschi
Antonella Vetuschi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 16 April 2021
Sec. Gastroenterology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.610189
The intestinal extracellular matrix (ECM) represents a complex network of proteins that not only forms a support structure for resident cells but also interacts closely with them by modulating their phenotypes and functions. More than 300 molecules have been identified, each of them with unique biochemical properties and exclusive biological functions. ECM components not only provide a scaffold for the tissue but also afford tensile strength and limit overstretch of the organ. The ECM holds water, ensures suitable hydration of the tissue, and participates in a selective barrier to the external environment. ECM-to-cells interaction is crucial for morphogenesis and cell differentiation, proliferation, and apoptosis. The ECM is a dynamic and multifunctional structure. The ECM is constantly renewed and remodeled by coordinated action among ECM-producing cells, degrading enzymes, and their specific inhibitors. During this process, several growth factors are released in the ECM, and they, in turn, modulate the deposition of new ECM. In this review, we describe the main components and functions of intestinal ECM and we discuss their role in maintaining the structure and function of the intestinal barrier. Achieving complete knowledge of the ECM world is an important goal to understand the mechanisms leading to the onset and the progression of several intestinal diseases related to alterations in ECM remodeling.
The morphogenesis and homeostasis of the gastrointestinal tract are intimately linked to interactions between epithelial cells, arising from embryonic endoderm, and the stromal cells derived from mesenchyme. These cells contribute to the production and organization of the extracellular matrix (ECM). The extracellular matrix forms a complex network of proteins that not only acts as a support structure for resident cells but also interacts with them by modulating their phenotypes and functions. The resident cell, in turn, secretes numerous molecules that cooperate with the various components of the ECM, creating a specific local microenvironment.
The luminal surface of the intestinal mucosa (luminal side) is delimited by a monolayer of epithelial cells (enterocytes, enteroendocrine cells, goblet cells, and Paneth cells), which is an essential barrier against the external environment (1, 2). In this context, the ECM provides protection and mechanical support to cells conferring the elasticity and resilience to tensile forces to the organ.
The heterogeneous spatial and biochemical composition of the ECM modulates and synchronizes many cell functions and releases molecules such as integrins, cytokines, chemokines, and growth factors (3–5). The extracellular matrix and epithelial cells exchange biological and physical information to orchestrate organ function, showing that the ECM is not a static scaffolding, but rather a dynamic structure. Special ECM rearrangements build a specialized compartment in which unique cellular processes occur, such as the crypts and cradles of the intestinal stem cell pool. This niche nourishes the intestinal stem cells, which is pivotal for the self-renewal of the epithelium and tissue regeneration from injury (2, 6).
In this review, we give an overview of the intestinal ECM, describing its main components and their physiological functions.
In the scientific community, the ECM is considered the most complex structural organization of tissues in organisms. To date, the paradigm “no cells, no ECM” persists, but it was a long way to go to prove it, as the cells were discovered even thousands of years later than the ECM (7). Around the 1700's, we believed that tissues and organs were composed of different forms and arrangements of connective tissue fibers and arose spontaneously. For many years this “fibers theory” represented the most accepted explanation of the basis of life, and it took ~100 years for this view to change. In the early 1800's, Lorenz Oken formulated the hypothesis “omne vivum ex vivo,” which means that “life may originate exclusively from something already alive” (7). An important discovery milestone in this field was made by Rudolf Virchow that, in the 1850's, shocked the scientific community with his hypothesis “omnis cellula ex cellula,” asserting that there are no cells without other cells (7). The revolutionary assertion that “cells are life and make fibers” took about 50 years until being generally accepted, and only subsequently was attention shifted to the discovery of the relationship between cells and intercellular space composition.
At the end of the 19th century, through light microscopy and then by innovative chemical and physical methods, the identification of collagen and elastin fibers, as well as the observation of several macromolecules in the intercellular compartment, was possible, and the unique definition to denote the subcellular space became the “extracellular matrix.” In the following years, the discovery of even more effective analysis and instruments allowed us to constantly highlight new details of ECM components (Table 1) (7). The period of 1930–1973 yielded several important discoveries for the connective tissue characterization and mainly for collagen and elastin—two core components of ECM. Particularly, electron microscopy and X-ray diffraction, leading to other increasingly advanced techniques, made it possible to identify and quantify amino acid sequences of proteins forming collagen and elastin fibers. Finally, in the last 40 years, a breakthrough revealed the active role of the ECM in cellular regulation, and new research in these fields demonstrated that the ECM directly influences the functions of the resident cells. A fine-tuned crosstalk between epithelial cells, mesenchymal cells, and ECM components is an essential step for the regulation of several key processes, such as cell adhesion, proliferation, differentiation, and apoptosis. Furthermore, the ECM exerts not only structural support for the cells and a physical barrier against the external microenvironment but represents a reservoir of growth factors involved in the activation of molecular pathways that regulate cell behavior. In this context, the biological functions of ECM are constant (1–10).

Table 1. Timeline of our understanding of the extracellular matrix (ECM).
The ECM is composed of a complex and fine organized network of proteins and polysaccharide molecules, known as glycosaminoglycans (GAGs), and GAGs linked to protein forming proteoglycans (PGs). With ~300 different molecules interacting in the building of this amazing organization, the ECM represents an essential support structure for cells, tissues, and organs (4, 11). GAGs and PGs interact with several growth factors and ECM proteins and are involved in the regulation of cell proliferation. Their peculiar characteristics, such as buffering, hydration, and force-resistance confer to these molecules additional crucial functions. GAGs can interact also with water, acting as lubricants and supporting cell migration. They are involved in the organization of collagen deposition allowing the ECM to resist high compressive forces (4, 10–13).
The dynamic structure of the extracellular matrix includes two distinct entities, the basement membranes (BM) and the interstitial matrix (IM) that are intimately interconnected. The BM is located beneath the epithelial and endothelial cells, the IM in the lamina propria, submucosa, and in muscular and serosa layers.
In addition to the components of the basement membrane and the interstitial matrix, the transmembrane collagens and proteoglycans expressed by epithelial cells, including type XXIII collagen and syndecan-1, can also be identified (14, 15). Collagen XXIII appears to be involved in epithelial cell-to-cell contact and epithelial cell polarization. Syndecan-1 modulates epithelial cell adhesion, proliferation, and migration and stabilizes the tight junctions. The two compartments are schematized in Figure 1.
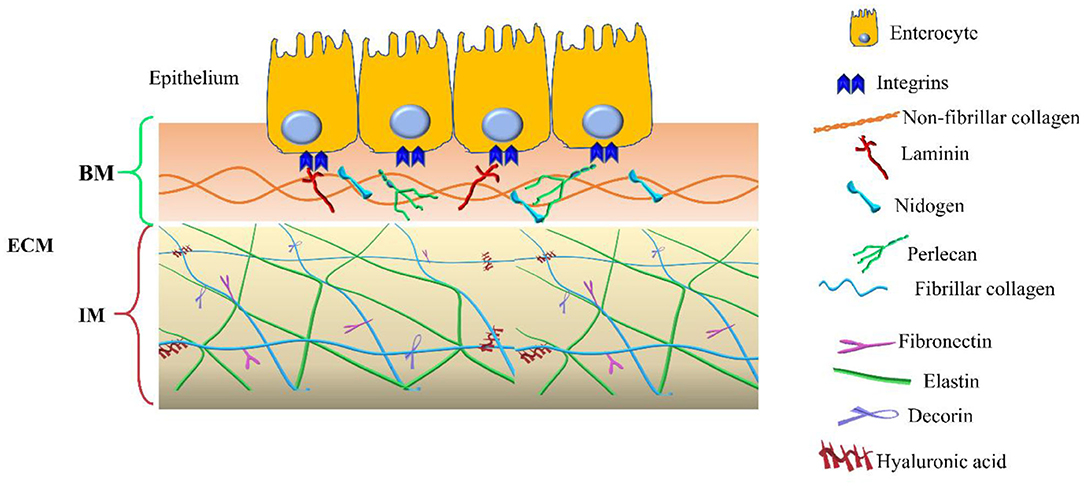
Figure 1. Extracellular matrix (ECM) compartments. Schematic representation of the main components of the two ECM compartments: basement membrane (BM) and interstitial matrix (IM). The legend indicates the identity of each ECM components.
The basement membrane is a specific 50–100 nm layer interposed between the epithelium and mesenchyme of lamina propria. The basement membrane represents a specialized form of ECM that controls cell organization and differentiation, interacting with cell surface receptors. The BM consists predominantly of collagen type IV, laminins, nidogens, and perlecan, also known as the basement membrane-specific heparan sulfate proteoglycan core protein (HSPG) or heparan sulfate proteoglycan 2 (HSPG2). Collagen and laminins can self-build more complex structures and represent the key components in BM stability, whereas nidogens and perlecan establish a complex link with laminins and collagen IV acting in the preservation of BM structural integrity (16, 17).
The BM components and their main functions are summarized in Table 2.
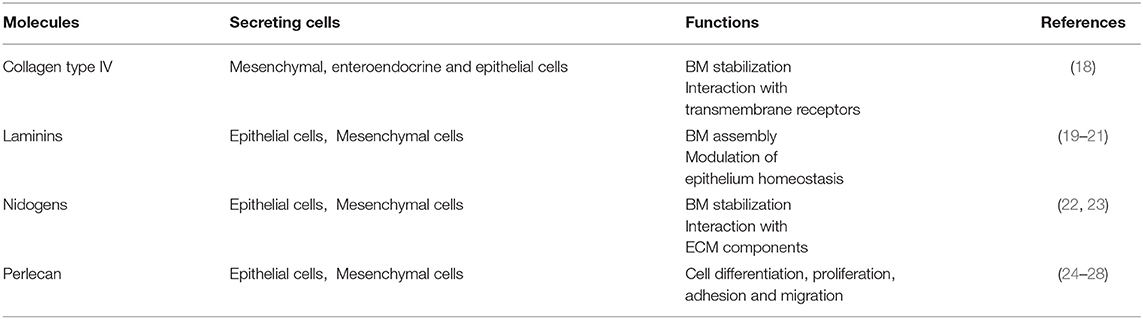
Table 2. The main components of the basement membrane and their functions.
Collagen type IV is the main component of BM and interacts with integrins, the transmembrane receptors that facilitate cell–ECM adhesion, acting as mechanical links between collagen matrices and the cell cytoskeleton (18, 22, 29–31). In the bowel, the main components of BM are α1, α2, α5, and α6 chains; however, at the mucosal surface, we can also find α3 and α4 chains (18). In physiological conditions, collagen IV is synthesized mainly by mesenchymal and enteroendocrine cells, although during BM restoration, epithelial cells can temporarily carry out this function (18). Type VI and type VIII collagens are also associated with BM. Type VI is expressed throughout the crypt-villus axis and mainly located in the interface between BM and IM and directly interacting with the type IV collagen and perlecan (18, 32). Type VIII collagen is mainly expressed by endothelial cells and seems to be involved in the homeostasis of these cells and angiogenesis. It is also partly expressed by smooth muscle cells and modulates their migration (18, 33).
The most abundant non-collagenous adhesive glycoproteins present in BM are laminins. These molecules can bind epithelial cells and are the basis for other BM proteins exerting a key role in the onset of BM assembly. Laminins are also able to bind collagens, particularly collagen IV and XVIII (22, 29). On the basolateral surface of the intestinal epithelial cells, each laminin isoforms exerts different functions: laminin α1β1γ1 can induce differentiation, whereas laminin α5β1γ1 and laminin α3β2γ2 induce adhesion and proliferation of the epithelial cells (19–21). Laminin α5β1γ1 is expressed in the upper crypt and in the base of the villus, laminin α3β2γ2 in the villus, and laminin α1β1γ1 is exclusively present in the intestinal crypt (20). Laminins are deposited by both epithelial and mesenchymal cells (19–21).
Laminins are binding sites for cellular integrins, which show a differential expression along with human small intestinal/colon tissue. In intestinal epithelial cells, the main laminin-binding integrins are α2β1, α3β1, α7β1, and α6β4 (32). In the basal domain of intestinal cells were found α2β1 (binding laminin α1β1γ1) and α3β1 (binding laminin α3β3γ2) integrins, particularly in the crypts and on the villus, respectively. Integrin α7β1 (binding laminins α1β1γ1 and α2β1γ1) is identified in the upper part of the crypt and in the lower region of the villus axis. Integrin α6β4 (binding laminins α1β1γ1, α2β1γ1, and α3β3γ2) was detected in equally distributed from the bottom of the crypt to the top of the villus (32). Integrins represent key regulators of cell-cell and cell-ECM interactions, thus influencing growth, differentiation, as well as wound healing, and development of fibrosis. In this context, an explicative example is provided by integrin avβ6. During the pathological condition, this molecule is overexpressed and it is able to locally switch latent TGF-β in activated TGF-β, finally fueling the TGF-β-mediated fibrotic process (32).
Collagen IV can create an interconnected network with the laminins forming two sheet like networks. These can interact with the nidogens, another important BM protein, also known as entactin (23). Epithelial and mesenchymal cells express nidogens, which not only acts to stabilize the BM, but also to enhance interactions with ECM components (i.e., perlecan, laminins, and fibulin) and to mediate signal transduction through integrins (Table 2) (1, 22, 23, 34).
Perlecan is present in the BM under physiological conditions, which is a large low-density heparan sulfate proteoglycan—a molecule with GAG chains but with an independent structural domain. Perlecan is crucial for tissue development, cell proliferation, differentiation, adhesion, and migration (35, 36). The protein core contains several binding sites for collagen IV, nidogens, and integrins other than for heparin. Perlecan can interact with important growth factors, especially with the vascular endothelial growth factor (VEGF). Perlecan is synthesized by epithelial cells mainly in the basolateral surface, enhancing intestinal regeneration through the modulation of Wnt/β catenin signaling (24–28).
Apart from these main constituents, the BM structure includes other fundamental molecules, such as the proteoglycan agrin, the glycoprotein fibulin, and the collagen-binding matricellular protein, also known as SPARC, Osteonectin (ON), or basement–membrane protein 40 (BM-40)—a molecule with anti-adhesion properties (23, 29, 34).
The interstitial matrix is located under the BM and acts as one of the major structural layers of the lamina propria and submucosa (18). The constituents of the IM cooperate in preserving the structural and functional integrity of this compartment of the intestinal wall (Figure 1).
The main IM components and their functions are summarized in Table 3.
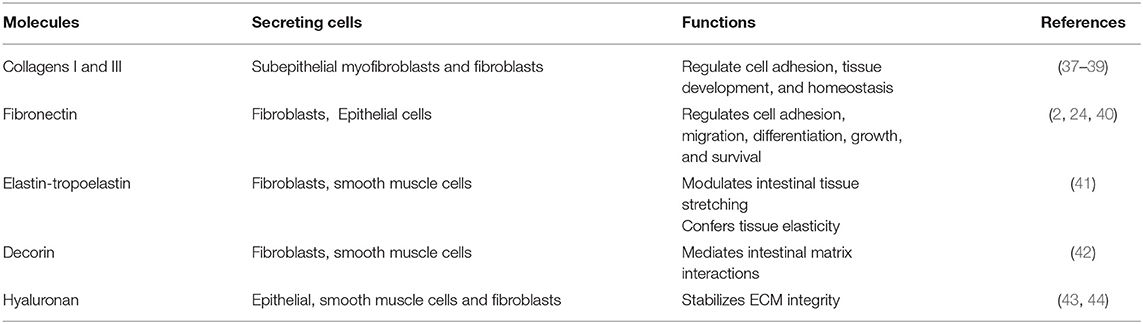
Table 3. Main components of the interstitial matrix.
Collagens represent important molecules involved in the regulation of cell adhesion and tissue homeostasis (22, 45). In the gut interstitial matrix, collagen types I and III are the most representative subtypes responsible for providing tensile strength to tissues (2, 37, 46, 47). These collagens (particularly collagen I) can interact with several proteins, such as proteolytic enzymes (Metalloproteinases: MMPs), surface receptors (i.e., integrins), and other ECM molecules like fibronectin, thrombospondin, SPARC, and proteoglycans (22). In the ECM of the IM, these two collagens are synthesized and released by subepithelial mesenchymal cells (37–39).
Type V collagen is directly involved in collagen fibril assembly by interacting with type I and III collagens and forms heterotypic fibrils (fibrils composed of type I, III, and V collagens) (18, 37, 47). Types XII, XVI, and XIX of the fibril-associated collagen with interrupted triple helices (FACIT) represent the main types present in the intestine IM. These are not involved in the building of collagen fibers, but connect collagen fibrils to other ECM molecules and promote the migration of intestinal myofibroblasts (18, 37, 48).
Fibronectin is a glycoprotein present as an insoluble form into the IM (2, 4, 22). It interacts with several ECM molecules such as collagens (including types II, III), heparin, tenascin-C, and cell surface receptors of the integrin superfamily. Fibronectin is involved in several cellular activities taking place in connection with the ECM, such as cell adhesion, growth, migration, differentiation, and survival (2, 11, 22, 49, 50). Fibronectin plays an important role in the homeostasis of the barrier function of the intestinal mucosa and is constantly exposed to luminal bacteria and toxins. During a mucosal injury, fibronectin participates in the restoration of epithelial integrity. Fibroblasts as well as epithelial cells are the main producers of the intestinal fibronectin (2, 24, 40).
Elastin is secreted as tropoelastin and represents another important component of the interstitial matrix that confers elasticity and resilience to intestinal tissue. It is responsible for the tissue's ability to recoil following repeated expansion and contraction stretching; however, its amazing elasticity is limited by the intimate association with collagen fibrils (51, 52). Tropoelastin is produced by fibroblasts, smooth muscle cells, and endothelial cells before it is processed to elastin by cleavage of its signal peptide (41).
Chondroitin sulfate proteoglycan decorin and the glycosaminoglycan hyaluronan are also present in the IM (18). These molecules can interact with several ECM proteins (i.e., collagens), cytokines (i.e., tumor necrosis factor-alpha, TNF-α), and growth factors such as transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF) (53–57). Decorin selectively interacts with different molecules: the isoforms of TGFβ, PDGF, vascular endothelial growth factor receptor-2 (VEGFR-2), epidermal growth factor receptor (EGFR), connective tissue growth factor (CTGF), thrombospondin, collagens, and fibronectin. Decorin is mainly expressed by fibroblasts, smooth muscle cells, and macrophages (42).
Hyaluronan (HA) is an abundant ECM component produced by epithelial cells, smooth muscle cells, and fibroblasts (43, 44). It exerts an important role in regulating the hydration of tissues, as well as affects cell adhesion, migration, and mitosis. It also acts as an anti-angiogenic and anti-inflammatory factor. Almost all of these effects are mediated by the hyaluronan receptor CD44, which is expressed by stromal and immune cells (58). Hyaluronan also acts in the homeostasis of intestinal stem cells (ISC) through the interaction with constituents of the extracellular matrix contributing to stabilizing its integrity. It is mainly expressed on the plasma membrane of ISCs and its structure contains binding sites for the Toll-like receptors activated in the response to commensal and pathogenic bacteria (59, 60). The hyaluronan level of polymerization indicates matrix integrity. In contrast, elevated hyaluronan' fragments were found to be associated with inflamed tissue in IBD inducing leukocyte infiltration into the intestine and innate immune activation (44, 61). Fragments of hyaluronan can promote wound healing, but also fibrosis, by inducing fibroblast proliferation and myofibroblast differentiation (59, 61).
The ECM does not represent a static structure, but a dynamic tissue component that constantly undergoes continuous remodeling (10, 11, 18, 22, 62). The homeostasis of healthy tissue is guaranteed by a continuous and balanced deposition, degradation, and modification of the ECM and an imbalance in this equilibrium can lead to pathological conditions (63–65). A large number of molecules and growth factors orchestrate this delicate process by regulating ECM amount, composition, and structure (10, 11, 18, 62). Intestinal fibrosis, characterized by an abnormal deposition of ECM, represents the main chronic complication of inflammatory bowel disease (IBD), chronic relapsing intestinal disorders including Crohn's disease, which can affect both the small and large intestine, and ulcerative colitis, which only affects the large intestine (5, 10).
The intestinal mucosa consists of three distinct portions: a single layer of epithelial cells, a connective tissue that keeps the epithelium (the lamina propria) in place, and a small layer of smooth muscle, called muscularis mucosae, which separates it from the underlying muscle layers. The various components of the ECM are produced by different types of intestinal cells. While the components of the basement membrane are produced by epithelial cells, those of the interstitial matrix are mainly produced by mesenchymal cells represented by fibroblasts, myofibroblasts, and smooth muscle cells. Endothelial cells, pericytes, and stellate cells also contribute to the release of ECM components. ECM-producing cells act synergistically and are under the control of numerous biological mediators. The ECM-producing cells and their main markers are summarized in Table 4.

Table 4. Intestinal ECM-producing cell types.
At least five cell types can be found in the intestinal mucosal epithelium: enterocytes, goblet cells, Paneth cells, enteroendocrine cells, and stem cells. They are found both in the intestinal glands and on the surface of the villi. The enterocytes are specialized in the absorption of water, electrolytes, and nutrients; the goblet cells secrete different types of mucins; the Paneth cells help to maintain mucosal immunity by secreting antimicrobial substances; the enteroendocrine cells produce various paracrine and endocrine hormones; the stem cells guarantee the physiological renewal of all of the above types of epithelial cells or when they are damaged. In chronic inflammatory bowel diseases, epithelial cells can undergo a well-known process of epithelial-to-mesenchymal phenotypic transformation, becoming one of the main sources of activated myofibroblasts, and thereby being directly involved in the processes of tissue repair and fibrogenesis (5, 66, 67).
Fibroblasts are a heterogeneous population of cells located in the interstitium of all normal tissues and organs where they are central in maintaining structural integrity, healing, and regeneration, by regulating matrix homeostasis. Fibroblasts are directly involved in the pathogenesis of intestinal fibrosis (69).
Myofibroblasts are highly contractile cells that exhibit a “hybrid” phenotype between fibroblasts and smooth muscle cells (SMCs) and, when activated, synthesize high levels of ECM, particularly collagen, glycosaminoglycans, tenascin-C, and fibronectin (5, 71, 78). Besides roles in tissue growth and differentiation, myofibroblasts are central to wound healing and fibrosis (5, 67). Two types of myofibroblasts are present in the intestinal mucosa in physiological conditions, the intestinal sub-epithelial myofibroblasts (SEMFs) and the interstitial cells of Cajal (ICC) (72, 79). Sub-epithelial myofibroblasts are located at the base of the intestinal crypts in the lamina propria, form a three-dimensional network, and are in connection with each other, but also maintain connections with epithelial cells. Myofibroblastic cells contain smooth muscle cytoskeletal markers (in particular α-smooth muscle actin: α-SMA) together with three filaments (vimentin, desmin, or myosin), with variable expression depending on tissue, species and environmental factors (72, 79). Activated myofibroblasts play the main role in the development of intestinal fibrosis (5, 67). Interstitial cells of Cajal are located in the submucosa and muscularis propria in association with the smooth muscle layer (80, 81). Interstitial cells of Cajal are pacemaker cells, which regulate intestinal smooth muscle motility.
Smooth muscle cells (SMCs) are one of the three interrelated cell phenotypes into which intestinal mesenchymal cells can differentiate (the other two being fibroblasts and myofibroblasts) (73). In chronic inflammation, SMCs can trans-differentiate into myofibroblasts, suggesting that a dynamic equilibrium thus exists between the myofibroblast and SMC phenotype (5, 67, 73).
Endothelial cells (ECs) are the major constituent of the microvasculature that line blood and lymphatic vessels. Normally, ECs provide an anti-adhesive and selectively permeable exchange barrier. Endothelial cells, by continual adjustments in structure and functions, coordinate vascular supply, immune cell migration, and regulation of the tissue environment. Inflammation induces changes in the endothelium of the intestinal vasculature in response to the cytokines, chemokines, and growth factors released by immune and non-immune cells, leading to decreased endothelial barrier function, adhesion molecule expression, leukocyte extravasation, and increased coagulation and angiogenesis (82). Intestinal inflammation, especially in chronic inflammatory bowel diseases, can induce endothelial cells to undergo endothelial-to-mesenchymal phenotypic transformation, becoming a further source of activated myofibroblasts, and thereby being directly involved in the processes of tissue repair and fibrogenesis (67, 75).
Pericytes are derived from undifferentiated mesenchymal cells and surround capillary and small blood vessel endothelial cells (76, 83). They reside at the interface between the endothelium and interstitium. Pericytes display an intermediate phenotype between vascular SMCs and fibroblasts. Pericytes control endothelial cell differentiation, endothelial signaling, angiogenesis, and ECM deposition (5, 67, 76). They represent a useful reserve of myofibroblasts during tissue repair and inflammation-associated fibrosis.
Stellate cells are mesenchymal cell precursors that contribute to retinoic acid metabolism, which impacts fibrosis and when activated, may differentiate into myofibroblasts (84, 85); however, limited information is available for intestinal stellate cells, although, in chronic inflammatory bowel diseases, we know that they differentiate into myofibroblasts faster than those from normal mucosa and proliferate faster, and produce collagen earlier and at higher levels (5, 67).
One of the main enzymes that can degrade ECM components is metzincins.
Metzincins represent a superfamily of zinc-dependent endopeptidases present in the ECM and are classified in: astacins, pappalysins, MMPs, serralysins, and adamalysins, including a-desintegrin and metalloproteinase (ADAMs) and a-desintegrin and metalloproteinase with thrombospondin motif (ADAMTS) (86–89) (Figure 2). From 1962, 23 MMPs, 21 ADAMs, and 19 secreted ADAMTS are identified in humans (87, 89). The main function of these proteases is the degradation of ECM proteins, but they also exert an important role in crucial physio-pathological processes such as enzymatic activities, protease inhibition, protein synthesis inhibition, cell proliferation, migration and apoptosis, inflammation, wound healing, fibrosis, angiogenesis, and carcinogenesis (86–110).
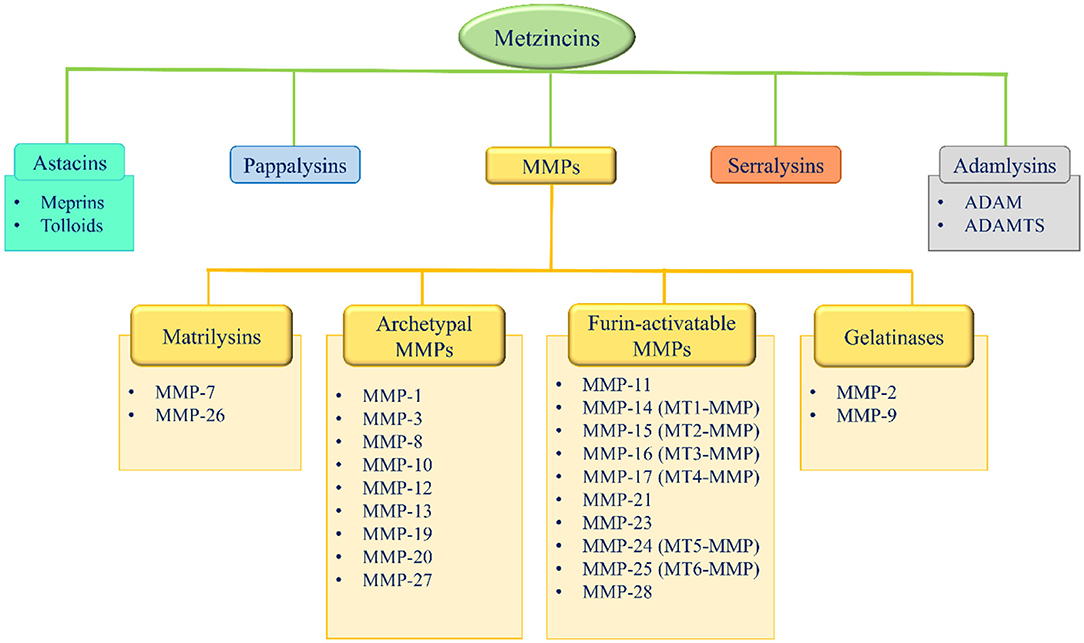
Figure 2. Metzincins superfamily members and classification of MMPs based on their domain arrangement. MMP, metalloproteinase; MT-MMP, membrane-type MMP; ADAM, a desintegrin and metalloproteinase; ADAMTS, a-desintegrin and metalloproteinase with thrombospondin motif.
Meprins are members of astacins, existing in two isoforms (α and β), that play a key role in connective tissue homeostasis, especially in cell migration, differentiation, and proliferation (111, 112). These enzymes can cleave the N- and C-terminal pre-domains of procollagens I and III, an essential step for the correct assembly of collagen fibril (113, 114). Meprins are involved in collagen IV, nidogens, and fibronectin other than SPARC and fibulin cleavage (115–117). In the bowel, meprins are also crucial in the preservation of intestinal barrier functions. Particularly, meprin β is located on the apical side of the epithelial cells and prevents bacteria attachment and invasion; however, during pathological processes such as inflammation, the expression of fibrosis and cancer meprins increased (111, 118).
ADAMs are membrane-anchored metalloproteinases that exert proteolytic activity inhibiting the metalloproteinase domain. Likewise, they play a key role in cell–cell interactions by connecting heparan sulfate proteoglycans with integrin proteins (11, 119–122).
Beside ADAMs, other components of the adamalysins family are the ADAMTS. In these molecules, near to a disintegrin domain, there is a thrombospondin type 1-like repeats sequence that confers to ADAMTS the ability to bind ECM proteins (11, 123).
MMPs are produced in a precursor form (PRO-MMPs) and remain in a status of low activity. They are confined in specific areas, while their expression increases during processes of remodeling, repair, or in the presence of chronic inflammatory or neoplastic diseases in many organs, including the intestine (8, 11, 92–96). The presence of MMPs is generally low in normal or uninflamed tissues. The expression, secretion, activation, and activity of MMPs are tightly controlled (90). The activity of MMPs is regulated on several levels, including transcription, translation, secretion, activation by cleavage of the pro-domain, and inhibition by the endogenous TIMPs (61). MMPs play a crucial role in the physiological turnover of the ECM bowel.
A significant contribution to intestinal ECM remodeling is ascribable to MMPs, which can be classified mainly by two different criteria: based on their domain organization or their substrate preferences (86–89, 124–126).
According to their domain arrangement, MMPs are classified as matrilysins, archetypal MMPs, furin-activatable MMPs, and gelatinases (Figure 2) (86–89, 124–126).
According to their substrate specificity, MMPs are classified as collagenases (MMP-1, −8, −13, and −18, this latter is not expressed in mammals, but only in Xenopus), gelatinases (MMP-2 and 9), stromelysins (MMP-3, −10, and −11), metrilysins (MMP-7 and −26), membrane-type MMPs (MT-MMPs) (MT1-MMP, MT2-MMP, MT3-MMP, MT4-MMP, MT5-MMP, and MT6-MMP) and other MMPs (MMP-12, −19, −20, −21, −23, −27, and −28) (86–89, 113–115). Specific substrates have been identified for the majority of the MMPs (Table 5).

Table 5. Classification of MMPs ECM degrading enzymes and their main substrates.
Several ECM components undergo limited proteolysis generating bioactive fragments called matricryptins, which regulate many physiological and pathological processes through their binding to cell surface receptors. The major physiopathological processes regulated by matricryptins include enzymatic activities, protease inhibition, protein synthesis inhibition, cell proliferation, migration and apoptosis, inflammation, wound healing, fibrosis, angiogenesis, and carcinogenesis (91).
In addition to a crucial role in the ECM components degradation, MMPs have a vast range of extracellular, pericellular, and intracellular substrates (90). At the mucosal surface level, antibacterial molecules, such as the membrane-bound mucin-1 (MUC1) and defensins, can be modified by MMPs, leading to the alteration of host–bacterial interaction. Within the epithelial layer, MMPs can degrade intercellular junction molecules (e.g., cadherins, occludins, and claudins) and intracellular structural proteins (e.g., actins), leading to the alteration of the cell shapes and the barrier function. The degradation of ECM components (e.g., collagens) may release several chemotactic and angiogenic (e.g., endostatin) fragments. MMPs proteolytically activate or degrade a variety of non-matrix substrates, including chemokines, cytokines, adhesion molecules, growth factors, and survival molecules; therefore, MMPs are increasingly recognized as critical players in the intestinal inflammatory response, tissue repair, fibrogenesis, and carcinogenesis (90, 92–110).
The activity of the ECM degrading enzymes is balanced by specific inhibitors represented by the tissue inhibitors of metalloproteinases (TIMPs).
In the bowel, MMPs and TIMPs are mostly investigated in the mucosa but are also present in the submucosa and muscolaris propria. The healthy epithelium expresses a wide range of MMPs and TIMPs, but in the injured epithelium, MMP-7 and MMP-10 are more pronounced (90). TIMP-3 is mainly associated with a healthy intestine and is reduced in the inflamed intestine. Stromal cells such as fibroblasts also express many MMPs and TIMPs, but fibroblasts containing MMP-1, −3, −8, and −9 have been associated with inflamed intestine (90). Immune cells also contribute to MMP and TIMP expression in the mucosal and submucosal layers (90).
Metalloproteinases activity is fine regulated by activation of synthetic and endogenous (non-specific and specific) inhibitors (86–89, 124–126).
Synthetic inhibitors including hydroxamate-based inhibitors, non-hydroxamate-based inhibitors, catalytic domain inhibitors, allosteric and exosite inhibitors, and antibody-based inhibitors (124, 125).
Among the endogenous non-specific inhibitors there are α-macroglobulin, tissue factor pathway inhibitor (TFPI), membrane-bound β-amyloid precursor protein, C-terminal proteinases enhancer protein, reversion-inducing cysteine-rich protein with Kasal domain motifs (RECK), and GPI-anchored glycoprotein; however, the main regulators of MMPs other than the adamalysins activity, are the TIMPs (124, 125).
The tissue inhibitors of metalloproteinases family are composed of four members (TIMP-1, −2, −3, and −4) and an important parameter in the control of their function is their localization. While TIMP-1, −2, and −4 are present in soluble form, TIMP-3 is sequestered in the ECM through the interaction with HS and GAGs (11, 126–133). Although TIMPs globally inhibit all the MMPs, each TIMP showed a preferential substrate (124–133) (Table 6).
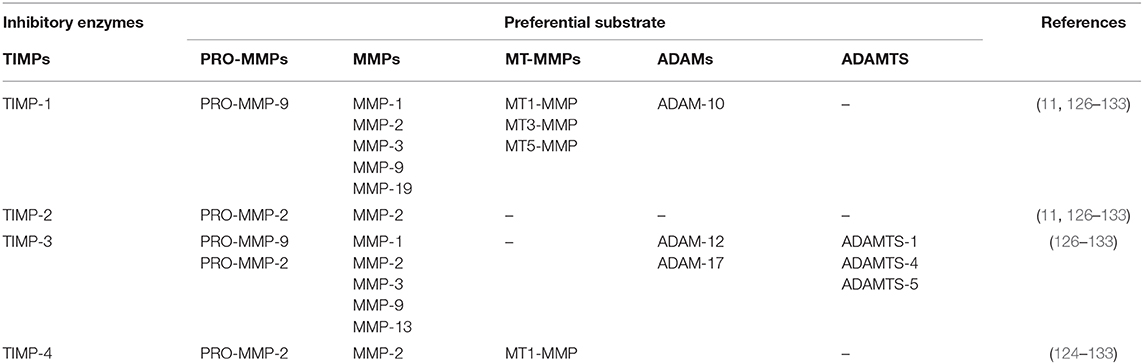
Table 6. MMPs inhibitors and their preferential substrates.
The extracellular matrix can act as a reservoir for growth factors released or activated upon MMPs mediated proteolysis, once released, these molecules affect cell recruitment and function, inducing ECM deposition and restoring its integrity. These growth factors are directly responsible for the maintenance of the tissue and repair of the intestinal epithelium (Figure 3).
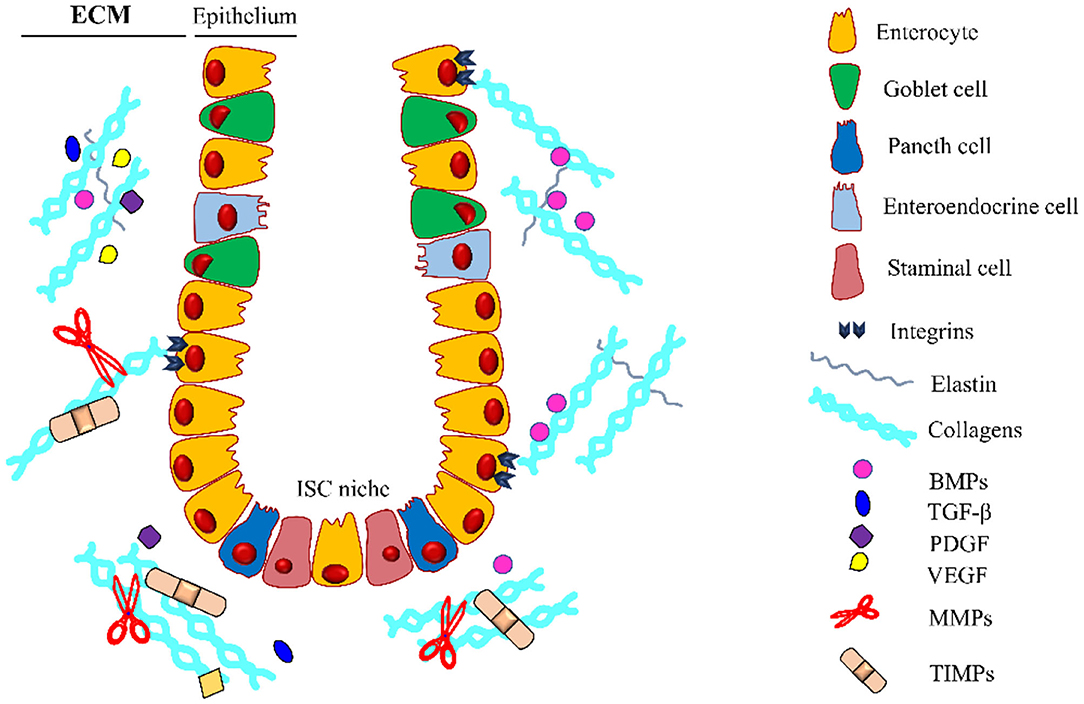
Figure 3. Intestinal crypt microenvironment. Schematic representation of an intestinal crypt with the cells and the molecules involved in its homeostasis. At the base of the invagination, there is the ISC niche, essential for supplying the staminal cell pool involved in the physiological self-renewal of the epithelium. Growth factors (GFs) are entrapped in the extracellular matrix (ECM), which is constantly remodeled by the coordinated activity of ECM-producing cells, metalloproteinases (MMPs), and tissue inhibitors of metalloproteinases (TIMPs). Along the crypt, there is a gradient for some GFs (i.e., BMPs) regulating the differentiation and proliferation of the intestinal stem cells (ISCs). BMPs, Bone Morphogenetic Proteins; TGF-β, Transforming Growth Factor-β; PDGF, Platelet-Derived Growth Factor; VEGF, Vascular Endothelial Growth Factor. The legend indicates the crypt components.
The main growth factors controlling ECM deposition are represented by TGF-β1, activins, CTGF, Fibroblast growth factor (FGF), PDGF, epidermal growth factor (EGF), insulin-like growth factor (IGF)-I and II, VEGF, bone morphogenetic proteins (BMPs), and hepatocyte growth factor (HGF).
Transforming growth factor-β is the crucial growth factor involved in the biological regulation of ECM protein synthesis. The main producers of TGF-β are represented by epithelial cells, immune cells, and fibroblasts, which are highly expressed in the lamina propria of a healthy bowel (134, 135). TGF-β exists in three different isoforms (TGF-β 1, 2, and 3) secreted in a latent state (LTGF-β) and often bound to a second protein, the latent TGF-β binding protein (LTBP). This complex facilitates the secretion and direction of ECM components, mainly fibronectins and fibrillins (134–143).
TGF-β interacts with the small mother against decapentaplegic (Smad) family proteins that induce its nuclear translocation, regulating its signaling, and the formation of transcriptionally active complexes (29, 144–149). During physiological intestinal ECM turnover, TGF-β plays a key role in the regulation of the expression of the collagen and the laminins proteins (22). Additionally, TGF-β1 through the interaction with Smad3 can induce procollagen I and III depositions by intestinal fibroblasts (143, 147). Besides Smads downstream pathways, TGF-β1 can also modulate, in a Smad/independent manner, other signal transduction pathways, such as ERK/cJUN/p38MAP kinases and the phosphoinositide-3 kinase (PI3-K) and its downstream target Akt, also known as protein kinase B (PKB), as well as members of the JAK and STAT protein family (143–147). It is not yet fully understood which of these transduction pathways mainly modulate the anti-inflammatory effect and which the pro-fibrotic effect of TGF- β (143–147); however, TGF-β exerts several crucial functions in the bowel. TGF-β plays an important role in the crosstalk between the host immune cells and the gut microbiota, both in the small intestine (regulating the complex microbiota including Lactobacillus sp., Streptococcus sp. Clostridium sp., and Escherichia coli) and in the colon (rich in Clostridium sp. and Bacteroides species). After an epithelial injury or an inflammation, intestinal epithelial cells increase the production of the TGF-β. In turn, the microbiota release molecules (butyrate, acetate, and propionate) to enhance the production of TGF-β by epithelial cells, regulating the immune response (150–154). Although TGF-β shows an anti-inflammatory effect during acute intestinal inflammation, in a chronic phase of the disease, it induces a pro-fibrotic effect. In this context, TGF-β is a key regulator of intestinal fibrosis (5, 63, 67).
Other members of TGF-β superfamily are the BMPs and activins. BMPs play an important role during homeostasis by controlling cellular differentiation, proliferation, and apoptosis (155–157). In the intestine, the ECM regulates the position, the timing, and the intensity of the BMPs activity (158). The BMPs are sequestered in ECM, mainly interacting with fibrillin and collagen IV (159). BMPs promote epithelial differentiation in the crypts and inhibit the expansion of the stem cells pool (160, 161). A peculiar gradient of BMPs is reported in the intestine, degrading from the villus toward the base of the crypt (161). Endogenous BMP antagonists exist, such as Gremlin1, Gremlin2, and Chordin, secreted by the intestinal subepithelial myofibroblasts and smooth muscle cells (162). Furthermore, BMP2 and 7 are inhibited by angiopoietin-like-protein 2 expressed by subepithelial myofibroblasts to maintain ISC homeostasis (163, 164). Activins represent an important player in the regulation of the intestinal epithelial cell functions. Activin A and its receptors modulate epithelial cells migration and proliferation exerting a positive role during the intestinal inflammation and wound healing processes (164). Activins activate Smad transcription factors and the MAP kinase signaling pathways (165).
The connective tissue growth factor is a downstream mediator of TGF-β. It is co-expressed with TGF-β and stimulates cell proliferation and ECM synthesis. Its expression is controlled by TGF-β in a Smad-dependent manner. In IBD, the activation of the TGF-β pathway induces an increased expression of CTGF that leads to abnormal local deposition of ECM components and intestinal fibrosis. In addition to TGF-β, other modulators of CTGF expression include VEGF, TNF-α, and ROS (166, 167).
Fibroblast growth factor (FGF) is intimately associated with ECM, especially with HSPG present in the BM (29). The interaction with its receptor (FGFR), expressed on fibroblasts and epithelial cells of the intestinal crypt, can regulate the synthesis of specific ECM components such as collagens, laminins, and fibronectin (22, 168–170).
Another molecule associated with ECM proteins is represented by PDGF. This factor is mainly expressed by the epithelial cells, but also endothelial cells, fibroblasts, and smooth muscle cells it is a positive regulator of stromal cell proliferation (171). Plated-derived growth factor-AA and its receptor (PDGFR), are crucial for the proper structure of the intestinal mucosa. Knockout mice for PDGF-AA or PGFR-A showed a reduction in the enterocyte turnover, and the subepithelial mesenchymal cluster aggregation, as well as a decrease in villus formation (172). The activity of PDGF and its diffusion in the tissue interstitium is regulated by the binding with ECM proteins, decorin, HS, and SPARC, that sequester PDGF in extracellular space in an inactive form and inhibit its action (36, 140, 173).
Similarly, the VEGF is secreted by the enterocytes, endothelium, and muscularis layer and remains spatially confined through HS interaction. Once ECM is remodeled, VEGF is released and controls cell proliferation, migration, and differentiation, and angiogenesis (174, 175).
Insulin-like growth factor-I and II and their respective receptors are expressed in the intestine and interact principally with fibroblasts, epithelial, and endothelial cells and regulate collagen deposition. IGF-1 enhances myofibroblast migration and increases intestinal SMC and myofibroblasts (36, 168–178). Epidermal growth factor is the prototype member of a family comprising different peptides with a similar primary structure that binds to a family of EGF receptors. It regulates transcription, translation, cell architecture, and cell proliferation. EGF can be isolated from the intestine and its receptors are located on monocytes and myofibroblasts. EGF stimulates fibroblast proliferation and ECM production and regulates human colonic fibroblast and myofibroblast migration (179–181).
Hepatocyte growth factor (HGF) and BMP-7 are natural inhibitors of the TGF-β/Smad pathway (182, 183). HGF exerts several biological activities on myofibroblasts, including the inhibition of growth and ECM deposition and the increase of MMP expression (184, 185). BMP-7 downregulates α-SMA and phosphorylated Smad2/3 (183).
ECM organization and functions are under the control of metabolism and metabolic regulators (186–188). The main cellular metabolic sensor, AMP-activated protein kinase (AMPK) joins the cell energetic status with the cell-to-ECM adhesion and the ECM deposition (186). In fact, AMPK has been reported to inhibiting the expression of the integrin-binding proteins, tensin 1 and tensin 3 in several systems. Moreover, AMPK is able to phosphorylate proteins of cell-to-cell adhesion, such as claudin 1, claudin 4, and cingulin, enhancing the epithelial barrier and cell polarity (186–188). Activation of AMPK induces the suppression of the TGF-β1 signaling and prevents abnormal ECM remodeling with excessive collagen synthesis deposition and tissue fibrosis (189). Another important metabolic mediator involved in ECM function is the yes-associated protein 1/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) pathway (190, 191). YAP/TAZ signaling integrates the energetic status to adhesion on ECM and activation of YAP facilitates the cell contact to a stiff ECM. Interestingly, YAP is also reported to be involved in tissue regeneration and ISC activation. Upon injuring conditions, ECM releases factors binding the integrin β1 and activating focal adhesion kinase (FAK)/Src pathway. Finally, YAP/TAZ is triggered and leads to stem cell activation and tissue repair (190–192). Furthermore, key regulators of lipids, and carbohydrates metabolisms, such as PPAR-γ, mTOR, and the adipokine leptin, are able to highly modulate ECM functions (134, 149, 193, 194). Indeed, these molecules influence ECM behavior acting on ECM-producing cells. Particularly, PPAR-γ leads to a decrease of ECM proteins deposition exerting an anti-fibrotic effect, while mTOR and leptin activate myofibroblasts resulting in abnormal deposition of ECM, and the onset of the fibrotic process (134, 149, 193, 194).
The intestinal mucosa is constantly subject to mechanical stimuli that can activate molecular signals on the epithelial cells and leads to alterations of the epithelium biology. Mainly two forms of mechanical forces act on the intestinal wall: contractile activities (i.e., peristalsis) and shear stress (due to the presence of food, gas, and fluids) (192, 195, 196). These forces heavily impact bowel morphogenesis. An interesting study conducted on rat's small intestine revealed that ECM fibers are organized as two interwoven arrays running diagonally around the crypt wall: one set clockwise and the other one set counterclockwise and oriented at a range of angle ± 30–50° (192, 195–197). This spatial arrangement provides mucosa with the right flexibility to fit stretch forces (192, 195–197). The ECM conveys this mechanical information to cells through an integrin network, activates focal adhesion kinase (FAK) and the extracellular signal-regulated kinase (ERK) signaling, finally leading to cell proliferation. Shear stress and mechanical stretch were also reported to induce cyclo-oxygenase-2 and other pro-inflammatory mediators [interleukin (IL)-6, IL-8, and monocyte chemoattractant protein 1] (198).
Furthermore, a high impact on ECM deposition is provided by changes in the tensile forces and elasticity of the intestinal wall. Indeed, a reduction of these parameters leads to alterations of ECM physical properties, such as ECM stiffness. Tissue stiffness is established by ECM composition and contraction, as well as by ECM-producing cells (fibroblast, myofibroblasts, and smooth muscle cells) contractility, under the control of the master regulator TGF-β (63).
The ECM can undergo peculiar compartmentalization, creating specialized tridimensional invagination in both the small and large intestines, known as the crypt, the hub for stem cell pool, allowing fast turnover (4–5 days) of the epithelium (185, 199–201). At the base of the crypt reside the intestinal stem cells (ISCs), a heterogeneous group of cells nourished by a surrounding cell niche. This niche, known as the intestinal stem cells niche, acts in physiological epithelial tissue regeneration, self-renewal, and differentiation other than in the repair of intestinal mucosal epithelium after injury (199–201). Several studies highlighted that ISCs are regulated by many signaling pathways, mainly by Wnt, bone morphogenic proteins (BMPs), and Notch signaling, involved both in physiologic and pathologic conditions (202–205). Particularly, in ISCs, Wnt is involved in the regulation of intestinal epithelial renewal in physiological conditions, BMPs acts in the intestinal development and the epithelial homeostasis, and Notch ensures that the crypt compartment remains in an undifferentiated and proliferative state during gut development (206–210). The ISC niche can be described as a combination of physical and cellular niches (2). The physical niche includes the complex network of ECM proteins (i.e., fibronectins and collagens) that provides a crucial scaffold to maintain the correct architecture of the intestine (1, 211, 212) (Figure 3). The cellular niche is the stromal microenvironment composed of all resident cells dipped in the ECM. These cells included myofibroblasts, fibroblasts, smooth muscle cells, endothelial cells, and pericytes that cooperated to secrete several growth factors and matrix components that are crucial for maintaining the homeostasis of ISCs by regulating their proliferation and differentiation (73, 200, 213).
The intestinal ECM is a complex structure consisting of a mixture of proteins, the glycosaminoglycan hyaluronan and proteoglycans that interact to make a scaffold for the resident cells. The core matrisome of the ECM consists of more than 300 proteins, each of them with unique biochemical properties conferring exclusive biological functions (5, 10). These proteins give rise to distinct ECM compartments: the basement membrane (BM) and the interstitial matrix (IM). The structural proteins (collagens) in ECM provides a scaffold for the tissue and mainly endures the tensile strength, limiting the stretching of the organ. Glycoproteins (laminins, elastins, fibronectin, and nidogens) are involved in the ECM assembly and cell-to-ECM interaction by binding cell receptors as integrins. The PGs (perlecan and decorin) and the GAG (hyaluronan) retain water in the tissue, conferring hydration, and entrap growth factors that influence cell behavior. The ECM contributes to establishing a selective barrier to the external environment (5, 10). The extracellular matrix is a dynamic structure frequently remodeled by the orchestrated activity of degrading enzymes and their inhibitors (10, 11, 90, 92). During this remodeling, ECM releases growth factors affecting and regulating resident cell function and finally ECM deposition. This cell-to-ECM crosstalk is pivotal for the physiological homeostasis of the intestine, but the plethora of mechanisms controlling this reciprocal communication are not yet fully known. In this context, the extracellular vesicles (EVs), which are complex phospholipidic structures (20 up to 1,000 nm) released by prokaryotic and eukaryotic cells in the biological fluids, appear to play an important role. During their biogenesis, EVs acquire the molecular legacy of the donor cells and can shuttle biological information far away from the cells (214–219). For this reason, EVs are recognized as a new means of intercellular and ECM-cell communication, cooperating in the physiological and pathological regulation in many organs (214, 220–223). In this context, intestinal epithelial cells (IECs) produce EVs that can regulate the TGF-β pathway, which is pivotal for intestinal homeostasis (224, 225). EVs released by IECs contain TGF-β that can bind its receptor on target cells, but also carry the integrin αvβ6 that induces the activation of the LTGF-β in ECM, releasing the active factor. Metalloproteinases have also been described to be shuttled by EVs exerting their activity, such as MMP-2, −3, −9, −13, and −14 as well as TIMPs (TIMP-2) (225–228).
All that we have described and shown in this ECM overview is only a piece of a bigger and more complex puzzle. The intestinal homeostasis is correctly prompted by a multifaced interaction between different systems. One of the main interlocutors is undoubtedly the intestinal microbiota, the large mass of symbiotic commensal microorganisms presents in the gut lumen. The presence of microorganisms strongly regulates the health and physiology of the intestine, since commensal bacteria, especially those attached to the mucous surface, contribute together with the intestinal epithelium to the integrity of the important intestinal barrier function. The intestinal microbiota continuously interacts with the local immune system, creating a mutually beneficial balance between them. Interestingly, this interaction between the microbiota-gut-immune system is partially modulated by EVs (219). Gut microbiota physiologically produces numerous metabolites, such as short-chain fatty acids and Krebs cycle anions which have significant effects on the homeostasis of intestinal epithelial cells, but also of ECM-producing cells and immune cells. Microbiota produce several proteases that can alter the barrier functions of the intestinal mucosa.
Acute intestinal damage activates the acute inflammatory response which, in turn, initiates the well-known mucosal healing processes mediated by the ECM remodeling. Unfortunately, the factors and/or mechanisms that lead to an abnormal ECM turnover in the course of chronic intestinal inflammation, which is also responsible for complications that can develop over time, such as intestinal fibrosis and intestinal cancer, are not yet known. It is now established that disturbance of the fine-tuned regulation of ECM remodeling is the cornerstone of several gut diseases, including chronic inflammation, fibrosis, and cancer (64, 67, 229, 230). Although huge steps in the knowledge of the ECM have been made over the last decade, further investigations are needed. Achieving complete knowledge of the various components of the ECM and understanding the multitudes of interactions and functions carried out by the intestinal ECM structure represents an important objective to unravel the mechanisms leading to the onset and the progression of several intestinal diseases related to alteration ECM remodeling, such as inflammatory bowel disease and its complications (intestinal fibrosis and cancer).
The relevance of complete knowledge of the ECM is underlying also by its increasing use in regenerative medicine, an innovative area based on the use of tissue engineering, stem cell biology, immunology, and bioengineering, to restore damaged tissues (231–240). Biomaterials used for this purpose may be distinct into synthetic and natural materials (231–233). The latter can be obtained or using the entire ECM or single components derived from ECM degradation such as collagen, laminin, glycosaminoglycans, and fibronectin (233). Among them, collagens and HA are the most widely used for biological folders (231). In the gastrointestinal tract, biological scaffolds represent a great promise as a new substrate to promote tissue remodeling in situ and replace damaged areas occurring for example during neoplasia and IBD and induce partial restoration and function of the organ. In vivo studies revealed that tubular porcine small intestine submucosa (SIS) bio-scaffold implantation can regenerate mucosa and smooth muscle in a damaged tissue after ileostomy (235). Few months after implantation, the neo-intestine presented restoration of the mucosa, smooth muscle, and serosa layers. Similarly, other authors reported that SIS bioscaffolds induced intestinal regeneration in an animal model of celiotomy (236). Following implantation, epithelium barrier renewal and regeneration of mucosa and submucosa layers were observed. ECM-derived scaffolds have proved to be effective also in the large intestine tract. Indeed, SIS biomaterials modulate the immune response in a rat model of colitis, reducing the local secretion of pro-inflammatory cytokines and enhancing the recovery of the colonic mucosa (237). Those mentioned are only a few examples of ECM bioscaffolds applications, but well-showed the importance of this innovative frontier, which could represent a promising therapeutic approach in many unresolved diseases (238, 239).
SP and GL performed the literature review, wrote the manuscript, and prepared the illustrations. RS and AV reviewed the manuscript and suggested important research points. EG reviewed the manuscript and provided critical comments. All authors have read and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Simon-Assmann P, Kedinger M, De Arcangelis A, Rousseau A, Simo P. Extracellular matrix components in intestinal development. Experientia. (1995) 51:883–900. doi: 10.1007/BF01921739
2. Meran L, Baulies A, Li VSW. Intestinal stem cell niche: the extracellular matrix and cellular components. Stem Cells Int. (2017) 2017:7970385. doi: 10.1155/2017/7970385
3. Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci. (2009) 122:159–63. doi: 10.1242/jcs.018093
4. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at glance. J. of Cell Science. (2010) 123:4195–4200. doi: 10.1242/jcs.023820
5. Speca S, Giusti I, Rieder F, Latella G. Cellular and molecular mechanisms of intestinal fibrosis. World J Gastroenterol. (2012) 18:3635–61. doi: 10.3748/wjg.v18.i28.3635
6. Pastula A, Marcinkiewicz J. Cellular interactions in the intestinal stem cell niche. Arch Immunol Terap Experi. (2019) 67:19–26. doi: 10.1007/s00005-018-0524-8
7. Piez KA. History of extracellular matrix: a personal view. Matrix Biol. (1997) 16:85–92. doi: 10.1016/S0945-053X(97)90037-8
8. Hynes RO. Extracellular matrix: not just pretty fibrils. Science. (2009) 326: 1216–9. doi: 10.1126/science.1176009
9. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Prospect Biol. (2011) 3:a005058. doi: 10.1101/cshperspect.a005058
10. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. (2014) 15:786–801. doi: 10.1038/nrm3904
11. Yue B. Biology of the extracellular matrix: an overview. J Glaucoma. (2014) 23:S20–3. doi: 10.1097/IJG.0000000000000108
12. Murch SH, MacDonald TT, Walker-Smith JA, Lionetti P, Levin M, Klein J, et al. Disruption of sulphated glycosaminoglycans in intestinal inflammation. Lancet. (1993) 341:711–4. doi: 10.1016/0140-6736(93)90485-Y
13. Hodde JP, Badylak S, Brightman AO, Voytik-Harbin SL. Glycosaminoglycan content of small intestinal submucosa: a bioscaffold for tissue replacement. Tissue Eng. (1996) 2:209–17. doi: 10.1089/ten.1996.2.209
14. Koch M, Veit G, Stricker S, Bhatt P, Kutsch S, Zhou P, et al. Expression of type XXIII collagen mRNA and protein. J Biol Chem. (2006) 281:21546–57. doi: 10.1074/jbc.M604131200
15. Wang Z, Li R, Tan J, Peng L, Wang P, Liu J, et al. Syndecan-1 Acts in synergy with tight junction through Stat3 signaling to maintain intestinal mucosal barrier and prevent bacterial translocation. Inflamm Bowel Dis. (2015) 21:1894–907. doi: 10.1097/MIB.0000000000000421
16. Miner JH, Li C, Mudd JL, Go G, Sutherland AE. Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation. Development. (2004) 131:2247–56. doi: 10.1242/dev.01112
17. Poschl E, Schlotzer-Schreharland U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. (2004) 131:1619–28. doi: 10.1242/dev.01037
18. Mortensen JH, Lindholm M, Langholm LL, Kjeldsen J, Bay-Jensen AC, Karsdal MA, et al. The intestinal tissue homeostasis - the role of extracellular matrix remodeling in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. (2019) 13:977–93. doi: 10.1080/17474124.2019.1673729
19. Teller IC, Beaulieu JF. Interactions between laminin and epithelial cells in intestinal health and disease. Expert Rev Mol Med. (2001) 3:1–18. doi: 10.1017/S1462399401003623
20. Turck N, Gross I, Gendry P, Stutzmann J, Freund JN, Kedinger M, et al. Laminin isoforms: biological roles and effects on the intracellular distribution of nuclear proteins in intestinal epithelial cells. Exp Cell Res. (2005) 303:494–503. doi: 10.1016/j.yexcr.2004.10.025
21. Kedinger M, Lefebvre O, Duluc I, Freund JN, Simon-Assmann P. Cellular and molecular partners involved in gut morphogenesis and differentiation. Phil Trans R Soc Lond B. (1998) 353:847–56. doi: 10.1098/rstb.1998.0249
22. Akalu A, Brooks PC. Matrix, Extracellular and Interstitial. Encyclop Mol Cell Biol Mol Med. (2005) 8:45–79. doi: 10.1002/3527600906.mcb.200400091
23. Chung AE, Dong LJ, Wu C, Durkin ME. Biological functions of entactin. Kidney Int. (1993) 43:13–9. doi: 10.1038/ki.1993.4
24. Simon-Assmann P, Kedinger M, Haffen K. Immunocytochemical localization of extracellular-matrix proteins in relation to rat intestinal morphogenesis. Different Res Biol Diversity. (1986) 32:59–66. doi: 10.1111/j.1432-0436.1986.tb00556.x
25. Oshiro M, Ono K, Suzuki Y, Ota H, Katsuyama T, Mori N. Immunohistochemical localization of heparan sulfate proteoglycan in human gastrointestinal tract. Histochem Cell Biol. (2001) 115:373–80. doi: 10.1007/s004180100271
26. Iozzo R. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. (2005) 6:646–56. doi: 10.1038/nrm1702
27. Yamamoto S, Nakase H, Matsuura M, Uza N, Yamaguchi Y, Mizoguchi E, et al. Heparan sulfate on intestinal epithelial cells plays a critical role in intestinal crypt homeostasis via Wnt/beta-catenin signaling. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G241–9. doi: 10.1152/ajpgi.00480.2012
28. Alkim C, Alkim H, Koksal AR, Boga S, Sen I. Angiogenesis in inflammatory bowel disease. Int J Inflam. (2015) 2015:970890. doi: 10.1155/2015/970890
29. LeBlau VS, MacDonald B, Kalluri R. Structure and function of basement membranes. Exp Biol Med. (2007) 232:1121–9. doi: 10.3181/0703-MR-72
30. Zeltz C, Orgel J, Gullberg D. Molecular composition and function of integrin-based collagen glues-introducing COLIBRIs. Biochim Biophys Acta. (2014) 1840:2533–48. doi: 10.1016/j.bbagen.2013.12.022
31. Zeltz C, Gullberg D. The integrin-collagen connection-a glue for tissue repair? J Cell Sci. (2016) 129:653–64. doi: 10.1242/jcs.180992
32. Kuo HJ, Maslen L, Keene DR, Glanville RW. Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. J Biol Chem. (1997) 272:26522–9. doi: 10.1074/jbc.272.42.26522
33. Kittelberger R, Davis PF, Flynn DW, Greenhill NS. Distribution of type VIII collagen in tissues: an immunohistochemical study. Connect Tissue Res. (1990) 24:303–18. doi: 10.3109/03008209009152157
34. Erickson A, Couchman JR. Still more complexity in mammalian basement membranes. J Histochem Cytochem. (2000) 48:1291–306. doi: 10.1177/002215540004801001
35. Zhang X, Reagan MR, Kaplan DL. Electrospun silk biomaterial scaffolds for regenerative medicine. Adv Drug Deliv Rev. (2009) 61:988–1006. doi: 10.1016/j.addr.2009.07.005
36. Kjellen L, Lindahl U. Proteoglycans: structures and interactions. Annu Rev Biochem. (1991) 60:443–75. doi: 10.1146/annurev.bi.60.070191.002303
37. Gelse K, Pöschl E, Aigner T. Collagens - structure, function, and biosynthesis. Adv Drug Deliv Rev. (2003) 55:1531–46. doi: 10.1016/j.addr.2003.08.002
38. De Wever O, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. (2008) 123:2229–38. doi: 10.1002/ijc.23925
39. Hulmes DJS. Collagen diversity, synthesis and assembly. In: Fratzl P, editor. Collagen Structure and Mechanisms. Boston, MA: Springer (2008). p. 15–47.
40. Quaroni K, Isselbacher J, Ruoslahti E. Fibronectin synthesis by epithelial crypt cells of rat small intestine. Proc Natl Acad Sci USA. (1978) 75:5548–52. doi: 10.1073/pnas.75.11.5548
41. Kristensen JH, Karsda MA. Biochemistry of Collagens, Laminins and Elastin. Structure, Function and Biomarkers. Amsterdam (2016).
42. Zhang W, Ge Y, Cheng Q, Zhang Q, Fang L, Zheng J. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget. (2018) 9:5480–91. doi: 10.18632/oncotarget.23869
43. de la Motte CA, Hascall VC, Drazba J, Bandyopadhyay S, Strong SA. Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyI:C:inter-α-trypsin inhibitor is crucial to structure and function. Am J Pathol. (2003) 163:21–133. doi: 10.1016/S0002-9440(10)63636-X
44. Kessler S, Rho H, West G, Fiocchi C, Drazba J, de la Motte C. Hyaluronan (HA) deposition precedes and promotes leukocyte recruitment in intestinal inflammation. Clin Transl Sci. (2008) 1:57–61. doi: 10.1111/j.1752-8062.2008.00025.x
45. Rozario T, DeSimone DW. The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol. (2010) 341:126–40. doi: 10.1016/j.ydbio.2009.10.026
46. Haffen K, Lacroix B, Kedinger M, Simon-Assmann PM. Inductive properties of fibroblastic cell cultures derived from rat intestinal mucosa on epithelial differentiation. Differentiation. (1983) 23:226–33. doi: 10.1111/j.1432-0436.1982.tb01287.x
47. Graham MF, Diegelmann RF, Elson CO, Lindblad WJ, Gotschalk N, Gay S, et al. Collagen content and types in the intestinal strictures of Crohn's disease. Gastroenterol. (1988) 94:257–65. doi: 10.1016/0016-5085(88)90411-8
48. Ricard-Blum S, Ruggiero F. The collagen superfamily: from the extracellular matrix to the cell membrane. Pathol Biol. (2005) 53:430–42. doi: 10.1016/j.patbio.2004.12.024
49. Trebaul A, Chan EK, Midwood KS. Regulation of fibroblast migration by tenascin-C. Biochem Soc Trans. (2007) 35:695–7. doi: 10.1042/BST0350695
50. Tucker RP, Chiquet-Ehrismann R. Evidence for the evolution of tenascin and fibronectin early in the chordate lineage. Int J Biochem. Cell Biol. (2009) 41:424–34. doi: 10.1016/j.biocel.2008.08.003
51. Wise SG, Weiss AS. Tropoelastin. Int J Biochem Cell Biol. (2009) 41:494–7. doi: 10.1016/j.biocel.2008.03.017
52. Mecham RO. Overview of extracellular matrix. Curr Protoc Cell Biol. (2012) 57:10. doi: 10.1002/0471143030.cb1001s57
53. Imai K, Hiramatsu A, Fukushima D, Pierschbacher MD, Okada Y. Degradation of decorin by matrix metalloproteinases: identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem J. (1997) 322:809–14. doi: 10.1042/bj3220809
54. Schönherr E, Lugering N, Stoll R, Domschke W, Kresse H. Differences in decorin and biglycan expression in patients with gastric ulcer healing. Scand J Gastroenterol. (1997) 32:785–90. doi: 10.3109/00365529708996535
55. Reinboth B, Hanssen E, Cleary EG, Gibson MA. Molecular interactions of biglycan and decorin with elastic fiber components: biglycan forms a ternary complex with tropoelastin and microfibril-associated glycoprotein 1. J Biol Chem. (2002) 277:3950–7. doi: 10.1074/jbc.M109540200
56. Nastase MV, Young MF, Schaefer L. Biglycan: a multivalent proteoglycan providing structure and signals. J Histochem Cytochem. (2012) 60:963–75. doi: 10.1369/0022155412456380
57. Karsdal MA, Nielsen MJ, Sand JM, Henriksen K, Genovese F, Bay-Jensen AC, et al. Extracellular matrix remodeling: the common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev Technol. (2013) 11:70–92. doi: 10.1089/adt.2012.474
58. Sammarco G, Shalaby M, Elangovan S, Petti L, Roda G, Restelli S, et al. Hyaluronan accelerates intestinal mucosal healing through interaction with TSG-6. Cells. (2019) 11:70–92. doi: 10.3390/cells8091074
59. de la Motte CA. Hyaluronan in intestinal homeostasis and inflammation: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. (2011) 301:G945–9. doi: 10.1152/ajpgi.00063.2011
60. Riehl TE, Foster L, Stenson WF. Hyaluronic acid is radioprotective in the intestine through a TLR4 and COX-2-mediated mechanism. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G309–16. doi: 10.1152/ajpgi.00248.2011
61. Shimshoni E, Yablecovitch D, Baram L, Dotan I, Sagi I. ECM remodelling in IBD: innocent bystander or partner in crime? The emerging role of extracellular molecular events in sustaining intestinal inflammation. Gut. (2015) 64:367–72. doi: 10.1136/gutjnl-2014-308048
62. Lee SE, Massie I, Meran L, Li VSW. Extracellular matrix remodeling in intestinal homeostasis and disease. Adv Stem Cells Their Niches. (2018) 2:99–140. doi: 10.1016/bs.asn.2018.01.001
63. Latella G, Rieder F. Intestinal fibrosis: ready to be reversed. Curr Opin Gastroenterol. (2017) 33:239–45. doi: 10.1097/MOG.0000000000000363
64. Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci. (2020) 6:160. doi: 10.3389/fmolb.2019.00160
65. Pompili S, Sferra R, Gaudio E, Viscido A, Frieri G, Vetuschi A, et al. Can Nrf2 Modulate the development of intestinal fibrosis and cancer in inflammatory bowel disease? Int J Mol Sci. (2019) 20:4016. doi: 10.3390/ijms20164061
66. Flier SN, Tanjore H, Kokkotou EG, Sugimoto H, Zeisberg M, Kalluri R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J Biol Chem. (2010) 285:20202–12. doi: 10.1074/jbc.M110.102012
67. Lawrance IC, Rogler G, Bamias G, Breynaert C, Florholmen J, Pellino G, et al. Cellular and molecular mediators of intestinal fibrosis. J Crohns Colitis. (2017) 11:1491–503. doi: 10.1016/j.crohns.2014.09.008
68. Gracz AD, Fuller MK, Wang F, Li L, Stelzner M, Dunn JCY, et al. CD24 and CD44 mark human intestinal epithelial cell populations with characteristics of active and facultative stem cells. Stem Cells. (2013) 31:2024–30. doi: 10.1002/stem.1391
69. McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. (2007) 39:666–71. doi: 10.1016/j.biocel.2006.11.005
70. Kisselbach L, Merges M, Bossie A, Boyd A. CD90 expression on human primary cells and elimination of contaminating fibroblasts from cell cultures. Cytotechnology. (2009) 59:31–44. doi: 10.1007/s10616-009-9190-3
71. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. (2012) 18:1028–40. doi: 10.1038/nm.2807
72. Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. (1999) 277:C1–9. doi: 10.1152/ajpcell.1999.277.1.C1
73. Rieder F, Fiocchi C. Intestinal fibrosis in IBD–a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. (2009) 6:228–35. doi: 10.1038/nrgastro.2009.31
74. Wedel T, Van Yes GJJM, Waltregny D, Glènisson W, Castronovo V, Vanderwinden JM, et al. Novel smooth muscle markers reveal abnormalities of the intestinal musculature in severe colorectal motility disorders neurogastroenterol. Motil. (2006) 18:526–38. doi: 10.1111/j.1365-2982.2006.00781.x
75. Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C. Inflammation induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. (2011) 179:2660–73. doi: 10.1016/j.ajpath.2011.07.042
76. Allt G, Lawrenson JG. Pericytes: cell biology and pathology. Cells Tissues Organs. (2001) 169:1–11. doi: 10.1159/000047855
77. Shang L, Hosseini M, Liu X, Kisseleva T, Brenner DA. Human hepatic stellate cell isolation and characterization. J Gastroenterol. (2018) 53:6–17. doi: 10.1007/s00535-017-1404-4
78. Mifflin RC, Pinchuk IV, Saada JI, Powell DW. Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G684–96. doi: 10.1152/ajpgi.00474.2010
79. Powell DW, Pinchuk IV, Saada JI, Chen X, Mifflin RC. Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol. (2011) 73:213–37. doi: 10.1146/annurev.physiol.70.113006.100646
80. Sanders KM, Ordög T, Ward SM. Physiology and pathophysiology of the interstitial cells of cajal: from bench to bedside. IV. Genetic and animal models of gi motility disorders caused by loss of interstitial cells of cajal. Am J Physiol Gastrointest Liver Physiol. (2002) 282:G747–56. doi: 10.1152/ajpgi.00362.2001
81. Vetuschi A, Sferra R, Latella G, D'Angelo A, Catitti V, Zanninelli G, et al. Smad3-null mice lack interstitial cells of cajal in the colonic wall. Eur J Clin Invest. (2006) 36:41–8. doi: 10.1111/j.1365-2362.2006.01593.x
82. Cromer WE, Mathis JM, Granger DN, Chaitanya GV, Alexander JS. Role of the endothelium in inflammatory bowel diseases. World J Gastroenterol. (2011) 17:578–93. doi: 10.3748/wjg.v17.i5.578
83. Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. (2003) 314:15–23. doi: 10.1007/s00441-003-0745-x
84. Nagy NE, Holven KB, Roos N, Senoo H, Kojima N, Norum R, et al. Storage of vitamin A in extrahepatic stellate cells in normal rats. J Lipid Res. (1997) 38:645–58. doi: 10.1016/S0022-2275(20)37232-1
85. Rieder F, Fiocchi C. Intestinal fibrosis in inflammatory bowel disease: progress in basic and clinical science. Curr Opin Gastroenterol. (2008) 24:462–8. doi: 10.1097/MOG.0b013e3282ff8b36
86. Kappor C, Vaidya S, Wadhwan V, Kaur HG, Pathak A. Seesaw of matrix metalloproteinases (MMPs). J Cancer Res Therapeutics. (2016) 12:28–35. doi: 10.4103/0973-1482.157337
87. Ågren MS, auf dem Keller U. Matrix metalloproteinases: how much can they do? Int J Mol Sci. (2020) 21:2678. doi: 10.3390/ijms21082678
88. Laronha H, Caldeira J. Structure and function of human matrix metalloproteinases. Cells. (2020) 9:1076. doi: 10.3390/cells9051076
89. Li K, Tay RF, Yung Yiu CK. The past, present and future perspectives of matrix metalloproteinase inhibitors. Pharmacol Therap. (2020) 207:107465. doi: 10.1016/j.pharmthera.2019.107465
90. de Bruyn M, Vandooren J, Ugarte-Berzal E, Arijs I, Vermeire S, Opdenakker G. The molecular biology of matrix metalloproteinases and tissue inhibitors of metalloproteinases in inflammatory bowel diseases. Crit Rev Biochem Mol Biol. (2016) 51:295–358. doi: 10.1080/10409238.2016.1199535
91. Ricard-Blum S, Vallet SD. Proteases decode the extracellular matrix cryptome. Biochimie. (2016) 122:300–13. doi: 10.1016/j.biochi.2015.09.016
92. Nagase H, Woessner JF. Matrix metalloproteinases. J Biolo Chem. (1999) 274:21491–4. doi: 10.1074/jbc.274.31.21491
93. Dollery CM, Libby P. Atherosclerosis and proteinase activation. Cardiovasc Res. (2006) 69:625–35. doi: 10.1016/j.cardiores.2005.11.003
94. Cappariello A, Paone R, Maurizi A, Capulli M, Rucci N, Muraca M, et al. Biotechnological approach for systemic delivery of membrane receptor activator of NF-κB ligand (RANKL) active domain into the circulation. Biomaterials. (2015) 46:58–69. doi: 10.1016/j.biomaterials.2014.12.033
95. Vetuschi A, D'Alfonso A, Sferra R, Zanelli D, Pompili S, Patacchiola F, et al. Changes in muscularis propria of anterior vaginal wall in women with pelvic organ prolapse. Eur J Histochem. (2016) 60:2604. doi: 10.4081/ejh.2016.2604
96. Garcia-Fernandez N, Jacobs-Cachá C, Mora-Gutiérrez JM, Vergara A, Orbe J, Soler J, et al. Matrix metalloproteinases in diabetic kidney disease. J Clin Med. (2020) 9:472. doi: 10.3390/jcm9020472
97. Löffek S, Schilling O, Franzke CW. Biological role of matrix metalloproteinases: a critical balance. Eur Respir J. (2011) 38:191–208. doi: 10.1183/09031936.00146510
98. Corbel M, Boichot E, Lagente V. Role of gelatinases MMP-2 and MMP-9 in tissue remodeling following acute lung injury. Braz J Med Biol Res. (2000) 33:749–54. doi: 10.1590/S0100-879X2000000700004
99. Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. (2011) 41:271–90. doi: 10.1007/s00726-010-0689-x
100. Manon-Jensen T, Itoh Y, Couchman JR. Proteoglycans in health and disease: the multiple roles of syndecan shedding. FEBS J. (2010) 277:3876–89. doi: 10.1111/j.1742-4658.2010.07798.x
101. Itoh Y. Membrane-type matrix metalloproteinases: their functions and regulations. Matrix Biol. (2015) 44–6:207–23. doi: 10.1016/j.matbio.2015.03.004
102. Shitomi Y, Thøgersen IB, Ito N, Leitinger B, Enghild JJ, Itoh Y. ADAM10 controls collagen signaling and cell migration on collagen by shedding the ectodomain of discoidin domain receptor 1 (DDR1). Mol Biol Cell. (2015) 15: 659–73. doi: 10.1091/mbc.E14-10-1463
103. de Bruyn M, Breynaert C, Arijs I, De Hertogh G, Geboes K, Thijs G, et al. Inhibition of gelatinase B/MMP-9 does not attenuate colitis in murine models of inflammatory bowel disease. Nat Commun. (2017) 8:15384. doi: 10.1038/ncomms15384
104. Fingleton B. Matrix metalloproteinases as regulators of inflammatory processes. Mol Cell Res. (2017) 1864:2036–42. doi: 10.1016/j.bbamcr.2017.05.010
105. Itoh Y. Metalloproteinases in rheumatoid arthritis: potential therapeutic targets to improve current therapies. Prog Mol Biol Transl Sci. (2017) 148:327–38. doi: 10.1016/bs.pmbts.2017.03.002
106. Majkowska I, Shitomi Y, Ito N, Gray NS, Itoh Y. Discoidin domain receptor 2 mediates collagen-induced activation of membrane-type 1 matrix metalloproteinase in human fibroblasts. J Biol Chem. (2017) 292:6633–43. doi: 10.1074/jbc.M116.770057
107. Coufal S, Galanova N, Bajer L, Gajdarova Z, Schierova D, Jiraskova Z, et al. Inflammatory bowel disease types differ in markers of inflammation, gut barrier and in specific anti-bacterial response. Cells. (2019) 8:719. doi: 10.3390/cells8070719
108. Cuffaro D, Nuti E, Gifford V, Ito N, Camodeca C, Tuccinardi T, et al. Design, synthesis and biological evaluation of bifunctional inhibitors of membrane type 1 matrix metalloproteinase (MT1-MMP). Bioorg Med Chem. (2019) 27:196–207. doi: 10.1016/j.bmc.2018.11.041
109. Gifford V, Itoh Y. MT1-MMP-dependent cell migration: proteolytic and non-proteolytic mechanisms. Biochem Soc Trans. (2019) 47:811–26. doi: 10.1042/BST20180363
110. Esteban S, Clemente C, Koziol A, Gonzalo P, Rius C, Martínez F, et al. Endothelial MT1-MMP targeting limits intussusceptive angiogenesis and colitis via TSP1/nitric oxide axis. EMBO Mol Med. (2020) 12:e10862. doi: 10.15252/emmm.201910862
111. Broder C, Becker-Pauly C. The metalloproteases meprin α and meprin β: unique enzymes in inflammation, neurodegeneration, cancer and fibrosis. Biochem J. (2013) 450:253–64. doi: 10.1042/BJ20121751
112. Prox J, Arnold P, Becker-Pauly C. Meprin α and meprin β: procollagen proteinases in health and disease. Matrix Biol. (2015) 44–6:7–13. doi: 10.1016/j.matbio.2015.01.010
113. Ishmael FT, Shier VK, Ishmael SS, Bond JS. Intersubunit and domain interactions of the meprin B metalloproteinase. J Biol Chem. (2005) 280:13895–901. doi: 10.1074/jbc.M414218200
114. Kronenberg D, Bruns BC, Moali C, Vadon-LeGoff S, Sterchi EE, Traupe H, et al. Processing of procollagen III by meprins: new players in extracellular matrix assembly? J Invest Dermatol. (2010) 130:2727–35. doi: 10.1038/jid.2010.202
115. Kohler D, Kruse M, Stocker W, Sterchi EE. Heterologously overexpressed, affnity-purified human meprin alpha is functionally active and cleaves components of the basement membrane in vitro. FEBS Lett. (2000) 465:2–7. doi: 10.1016/S0014-5793(99)01712-3
116. Yan Q, Weaver M, Perdue N, Sage EH. Matricellular protein SPARC is translocated to the nuclei of immortalized murine lens epithelial cells. J Cell Physiol. (2005) 203:286–94. doi: 10.1002/jcp.20226
117. Sterchi EE, Stocker W, Bond JS. Meprins, membrane-bound and secreted astacin metalloproteinases. Mol Aspects Med. (2008) 29:309–28. doi: 10.1016/j.mam.2008.08.002
118. Arnold P, Otteb A, Becker-Pauly C. Meprin metalloproteases: molecular regulation and function in inflammation and fibrosis. BBA-Mol Res. (2017) 1864:2096–104. doi: 10.1016/j.bbamcr.2017.05.011
119. Przemyslaw L, Boguslaw HA, Elzbieta S, Malgorzata SM. ADAM and ADAMTS family proteins and their role in the colorectal cancer etiopathogenesis. BMB Rep. (2013) 46:139–50. doi: 10.5483/BMBRep.2013.46.3.176
120. Kang TB, Zhao YG, Pei DQ, Sucic JF, Sang QXA. Intracellular activation of human adamalysin 19/disintegrin and metalloproteinase 19 by furin occurs via one of the two consecutive recognition sites. J Biol Chem. (2002) 277:25583–91. doi: 10.1074/jbc.M203532200
121. White JM. ADAMs: modulators of cell–cell and cell–matrix interactions. Curr Opin Cell Biol. (2003) 15:598–606. doi: 10.1016/j.ceb.2003.08.001
122. Arribas J, Bech-Serra JJ, Santiago-Josefat B. ADAMs, cell migration and cancer. Cancer Metastasis Rev. (2006) 25:57–68. doi: 10.1007/s10555-006-7889-6
123. Kuno K, Matsushima K. ADAMTS-1 protein anchors at the extracellular matrix through the thrombospondin type I motifs and its spacing region. J Biol Chem. (1998) 273:13912–7. doi: 10.1074/jbc.273.22.13912
124. Fontani F, Domazetovic V, Marcucci T, Vincenzini MT, Iantomasi T. MMPs, ADAMs and their natural inhibitors in inflammatory bowel disease: involvement of oxidative stress. J Clin Gastroenterol Treat. (2017) 3:039. doi: 10.23937/2469-584X/1510039
125. Larohna H, Carpinteiro I, Portugal J, Azul A, Polido M, Petrova KT, et al. Challenges in matrix metalloproteinases inhibition. Biomolecules. (2020) 10:717. doi: 10.3390/biom10050717
126. Raeeszadeh-Sarmazdeh M, Do LD, Hritz BG. Metalloproteinases and their inhibitors: potential for the development of new therapeutics. Cells. (2020) 9:1313. doi: 10.3390/cells9051313
127. Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. (2006) 69:562–73. doi: 10.1016/j.cardiores.2005.12.002
128. Wojtowicz-Praga SM, Dickson RB, Hawkins MJ. Matrix metalloproteinase inhibitors. Investig New Drugs. (1997) 15:61–75. doi: 10.1023/A:1005722729132
129. Yu WH, Yu S, Meng Q, Brew K, Woessner JF. TIMP-3 binds to sulfated glycosaminoglycans of the extracellular matrix. J Biol Chem. (2000) 275:31226–32. doi: 10.1074/jbc.M000907200
130. Mochizuki S, Shimoda M, Shiomi T, Fujii Y, Okada Y. ADAM28 is activated by MMP-7 (matrilysin-1) and cleaves insulin-like growth factor binding protein-3. Biochem Biophys Res Commun. (2004) 315:79–84. doi: 10.1016/j.bbrc.2004.01.022
131. Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. (2010) 1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003
132. Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. (2000) 1477:267–83. doi: 10.1016/S0167-4838(99)00279-4
133. Baker A, Edwards D, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. (2002) 115:3719–27. doi: 10.1242/jcs.00063
134. Latella G, Rogler G, Bamias G, Breynaert C, Florholmen J, Pellino G, et al. Results of the 4th scientific workshop of the ECCO (I): pathophysiology of intestinal fibrosis in IBD. J Crohns Colitis. (2014) 8:1147–65. doi: 10.1016/j.crohns.2014.03.008
135. Latella G, Di Gregorio J, Flati V, Rieder F, Lawrance IC. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol. (2015) 50:53–65. doi: 10.3109/00365521.2014.968863
136. Babyatsky MW, Rossiter G, Podolsky DK. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterol. (1996) 110:975–84. doi: 10.1053/gast.1996.v110.pm8613031
137. Di Sabatino A, Pickard KM, Rampton D, Kruidenier L, Rovedatti L, Leakey NA, et al. Blockade of transforming growth factor beta upregulates T-box transcription factor T-bet, and increases T helper cell type 1 cytokine and matrix metalloproteinase-3 production in the human gut mucosa. Gut. (2008) 57:605–12. doi: 10.1136/gut.2007.130922
138. Massam-Wu T, Chiu M, Choudhury R, Choudhury SS, Baldwin AK, McGovern A, et al. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF beta. J Cell Sci. (2010) 123:3006–18. doi: 10.1242/jcs.073437
139. Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Courousse T, Sakai LY, et al. Specificity of latent TGF-beta binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J Cell Physiol. (2012) 227:3828–36. doi: 10.1002/jcp.24094
140. Jenkins G. The role of proteases in transforming growth factor beta activation. Int J Biochem Cell Biol. (2008) 40:1068–78. doi: 10.1016/j.biocel.2007.11.026
141. Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur J Cell Biol. (2008) 87:601–15. doi: 10.1016/j.ejcb.2008.01.012
142. Taipale J, Miyazono K, Heldin CH, Keski-Oja J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J Cell Biol. (1994) 124:171–81. doi: 10.1083/jcb.124.1.171
143. Latella G. Redox imbalance in intestinal fibrosis: beware of the TGFβ-1, ROS, and Nrf2 connection. Digestive Dis Sci. (2018) 63:312–20. doi: 10.1007/s10620-017-4887-1
144. Latella G, Vetuschi A, Sferra R, Catitti V, D'Angelo A, Zanninelli G, et al. Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int. (2009) 29:997–1009. doi: 10.1111/j.1478-3231.2009.02011.x
145. Sferra R, Pompili S, Festuccia C, Marampon F, Gravina GL, Ventura L, et al. The possible prognostic role of histone deacetylase and transforming growth factor β/Smad signaling in high grade gliomas treated by radio-chemotherapy: a preliminary immunohistochemical study. Eur J Histochem. (2017) 61:2732. doi: 10.4081/ejh.2017.2732
146. Luna J, Masamunt MC, Llach J, Delgado S, Sans M. Palm oil tocotrienol rich fraction reduces extracellular matrix production by inhibiting transforming growth factor-beta 1 in human intestinal fibroblasts. Clin Nutr. (2011) 30:858–64. doi: 10.1016/j.clnu.2011.07.001
147. Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. (2001) 276:17058–62. doi: 10.1074/jbc.M100754200
148. Roberts AB, Russo A, Felici A, Flanders KC. Smad3: a key player in pathogenetic mechanisms dependent on TGF-beta. Ann N Y Acad Sci. (2003) 995:1–10. doi: 10.1111/j.1749-6632.2003.tb03205.x
149. Latella G, Vetuschi A, Sferra R, Speca S, Gaudio E. Localization of ανβ6 integrin-TGF-β1/Smad3, mTOR and PPARγ in experimental colorectal fibrosis. Eur J Histochem. (2013) 57:e40. doi: 10.4081/ejh.2013.e40
150. Tsukada S, Westwick JK, Ikejima K, Sato N, Rippe RA. SMAD and p38 MAPK signaling pathways independently regulate alpha1(I) collagen gene expression in unstimulated and transforming growth factor-beta-stimulated hepatic stellate cells. J Biol Chem. (2005) 280:10055–64. doi: 10.1074/jbc.M409381200
151. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science. (2011) 331:337–41. doi: 10.1126/science.1198469
152. Atarashi K, Tanoue K, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of clostridia strains from the human microbiota. Nature. (2013) 500:232–6. doi: 10.1038/nature12331
153. Brown EM, Sadarangani M, Finlay BB. The role of the immune system in governing host-microbe interactions in the intestine. Nat Immunol. (2013) 14:660–7. doi: 10.1038/ni.2611
154. Bauchè D, Marie JC. Transforming growth factor β: a master regulator of the gut microbiota and immune cell interactions. Clin Transl Immunol. (2017) 6:e136. doi: 10.1038/cti.2017.9
155. Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. (2003) 113:685–700. doi: 10.1016/S0092-8674(03)00432-X
156. Wu MY, Hill CS. Tgf-β superfamily signaling in embryonic development and homeostasis. Dev Cell. (2009) 16:329–43. doi: 10.1016/j.devcel.2009.02.012
157. Cappariello A, Ponzetti M, Rucci N. The “soft” side of the bone: unveiling its endocrine functions. Horm Mol Biol Clin Investig. (2016) 28:5–20. doi: 10.1515/hmbci-2016-0009
158. Umulis D, O'Connor MB, Blair SS. The extracellular regulation of bone morphogenetic protein signaling. Development. (2009) 136:3715–28. doi: 10.1242/dev.031534
159. Massagué J, Chen YG. Controlling TGF-β signaling. Genes Dev. (2000) 14:627–44. doi: 10.1101/gad.14.6.627
160. He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat Gene. (2004) 36:1117–21. doi: 10.1038/ng1430
161. Qi Z, Li Y, Zhao B, Xu C, Liu Y, Li H, et al. BMP restricts stemness of intestinal Lgr5+ stem cells by directly suppressing their signature genes. Nat Commun. (2017) 8:13824. doi: 10.1038/ncomms13824
162. Pinchuk IV, Mifflin RC, Saada JI, Powell DW. Intestinal mesenchymal cells. Curr Gastroenterol Rep. (2010) 12:310–8. doi: 10.1007/s11894-010-0135-y
163. Kosinski C, Li VSW, Chan ASY, Zhang J, Ho C, Tsui WY, et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci USA. (2007) 104:15418–23. doi: 10.1073/pnas.0707210104
164. Horiguchi H, Endo M, Kawane K, Kadomatsu T, Terada K, Morinaga J, et al. ANGPTL2 expression in the intestinal stem cell niche controls epithelial regeneration and homeostasis. EMBO J. (2017) 36:409–24. doi: 10.15252/embj.201695690
165. Werner S, Alzheimer C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. (2006) 17:157–71. doi: 10.1016/j.cytogfr.2006.01.001
166. Grotendorst GR. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. (1997) 8:171–9. doi: 10.1016/S1359-6101(97)00010-5
167. Blom IE, Goldschmeding R, Leask A. Gene regulation of connective tissue growth factor: new targets for antifibrotic therapy? Matrix Biol. (2002) 21:473–82. doi: 10.1016/S0945-053X(02)00055-0
168. Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier FG, et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem. (1996) 281:15292–7. doi: 10.1074/jbc.271.25.15292
169. Visco V, Belleudi F, Marchese C, Leone L, Aimati L, Cardinali G, et al. Differential response to keratinocyte growth factor receptor and epidermal growth factor receptor ligands of proliferating and differentiating intestinal epithelial cells. J Cell Physiol. (2004) 200:31–44. doi: 10.1002/jcp.10385
170. Matsuda Y, Ueda J, Ishiwata T. Fibroblast growth factor receptor 2: expression, roles, and potential as a novel molecular target for colorectal cancer. Pathol Res Int. (2012) 2012:574768. doi: 10.1155/2012/574768
171. Sukhotnik I, Mogilner JG, Pollak Y, Blumenfeld S, Bejar J, Coran AG. PDGF-α stimulates intestinal epithelial cell turnover after massive small bowel resection in a rat. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1274–81. doi: 10.1152/ajpgi.00532.2011
172. Raines EW, Lane TF, Iruela-Arispe ML, Ross R, Sage EH. The extracellular glycoprotein SPARC interacts with platelet-derived growth factor (PDGF)-AB and -BB and inhibits the binding of PDGF to its receptors. Proc Natl Acad Sci USA. (1992) 89:1281–5. doi: 10.1073/pnas.89.4.1281
173. Arita Y, Kihara S, Ouchi N, Maeda K, Kuriyama H, Okamoto Y, et al. Adipocyte-derived plasma protein adiponectin acts as a platelet-derived growth factor-BB-binding protein and regulates growth factor-induced common postreceptor signal in vascular smooth muscle cell. Circulation. (2002) 105:2893–8. doi: 10.1161/01.CIR.0000018622.84402.FF
174. Ruhrberg C, Gerhardt H, Golding M, Watson R, Ioannidou S, Fujisawa H, et al. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. (2002) 16:2684–98. doi: 10.1101/gad.242002
175. Frysz-Naglak D, Fryc B, Klimacka-Nawrot E, Mazurek U, Suchecka V, Kajor M, et al. Expression, localization and systemic concentration of vascular endothelial growth factor (VEGF) and its receptors in patients with ulcerative colitis. Int Immunopharmacol. (2011) 11:220–5. doi: 10.1016/j.intimp.2010.11.023
176. Simmons JG, Pucilowska JB, O Keku T, Lund PK. IGF-I and TGF-beta1 have distinct effects on phenotype and proliferation of intestinal fibroblasts. Am J Physiol Gastrointest Liver Physiol. (2002) 283:G809–18. doi: 10.1152/ajpgi.00057.2002
177. Leeb SN, Vogl D, Falk W, Schölmerich J, Rogler G, Gelbmann CM. Regulation of migration of human colonic myofibroblasts. Growth Factors. (2002) 20:81–91. doi: 10.1080/08977190290031941
178. Simmons JG, Ling Y, Wilkins H, Fuller CR, D'Ercole AJ, Fagin J, et al. Cell-specific effects of insulin receptor substrate-1 deficiency on normal and IGF-I-mediated colon growth. Am J Physiol Gastrointest Liver Physiol. (2007) 293:G995–1003. doi: 10.1152/ajpgi.00537.2006
179. Hoffmann P, Zeeh JM, Lakshmanan J, Wu S, Procaccino F, Reinshagen M, et al. Increased expression of transforming growth factor alpha precursors in acute experimental colitis in rats. Gut. (1997) 41:195–202. doi: 10.1136/gut.41.2.195
180. Earp HS, Calvo BF, Sartor CI. The EGF receptor family–multiple roles in proliferation, differentiation, and neoplasia with an emphasis on HER4. Trans Am Clin Climatol Assoc. (2003) 114:315–33.
181. Kong Q, Majeska RJ, Vazquez M. Migration of connective tissue-derived cells is mediated by ultra-low concentration gradient fields of EGF. Exp Cell Res. (2011) 317:1491–502. doi: 10.1016/j.yexcr.2011.04.003
182. Panganiban RAM, Day RM. Hepatocyte growth factor in lung repair and pulmonary fibrosis. Acta Pharmacol Sin. (2011) 32:12–20. doi: 10.1038/aps.2010.90
183. Weiskirchen R, Meurer SK, Gressner OA, Herrmann J, Borkham-Kamphorst E, Gressner AM. BMP-7 as antagonist of organ fibrosis. Front Biosci. (2009) 14:4992–5012. doi: 10.2741/3583
184. Mizuno S, Matsumoto K, Nakamura T. HGF as a renotrophic and anti-fibrotic regulator in chronic renal disease. Front Biosci. (2008) 13:7072–86. doi: 10.2741/3211
185. Chakraborty S, Chopra P, Hak A, Dastidar SG, Ray A. Hepatocyte growth factor is an attractive target for the treatment of pulmonary fibrosis. Expert Opin Investig Drugs. (2013) 22:499–515. doi: 10.1517/13543784.2013.778972
186. Georgiadou M, Lilja J, Jacquemet G, Guzmán C, Rafaeva M, Alibert C, et al. AMPK negatively regulates tensin-dependent integrin activity. J Cell Biol. (2017) 216:1107–21. doi: 10.1083/jcb.201609066
187. Georgiadou M, Ivaska J. Tensins: bridging AMP-activated protein kinase with integrin activation. Trends Cell Biol. (2017) 10:703–11. doi: 10.1016/j.tcb.2017.06.004
188. Zhu MJ, Sun X, Du M. AMPK in regulation of apical junctions and barrier function of intestinal epithelium. Tissue Barriers. (2018) 6:1–13. doi: 10.1080/21688370.2018.1487249
189. Luo T, Nocon A, Fry J, Sherban A, Rui X, Jiang B, et al. AMPK activation by metformin suppresses abnormal extracellular matrix remodeling in adipose tissue and ameliorates insulin resistance in obesity. Diabetes. (2016) 65:2295–310. doi: 10.2337/db15-1122
190. Chen R, Guan KL. Colonic epithelium rejuvenation through YAP/TAZ. EMBO J. (2018) 37:164–6. doi: 10.15252/embj.201798618
191. Yui S, Azzolin L, Maimets M, Pedersen MT, Fordham RP, Hansen SL, et al. YAP/TAZ-dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell. (2018) 22:35–49.e7. doi: 10.1016/j.stem.2017.11.001
192. Romani P, Valcarcel-Jimenez L, Frezza C, Dupont S. Crosstalk between mechanotransduction and metabolism. Nat Rev Mol Cell Biol. (2021) 22:22–38. doi: 10.1038/s41580-020-00306-w
193. Di Gregorio J, Sferra R, Speca S, Vetuschi A, Dubuquoy C, Desremaux P, et al. Role of glycogen synthase kinase-3β and PPAR-γ on epithelial–to-mesenchymal transition in DSS- induced colorectal fibrosis. PLoS ONE. (2017) 12:e0171093. doi: 10.1371/journal.pone.0171093
194. Speca S, Rousseaux C, Dubuquoy C, Rieder F, Vetuschi A, Sferra R, et al. Novel PPARγ modulator GED-0507-34 levo ameliorates inflammation-driven intestinal fibrosis. Inflamm Bowel Dis. (2016) 22:279–92. doi: 10.1097/MIB.0000000000000618
195. Basson MD. Paradigms for mechanical signal transduction in the intestinal epithelium. Digestion. (2003) 68:217–225. doi: 10.1159/000076385
196. Onfroy-Roy L, Hamel D, Foncy J, Malaquin L, Ferrand A. Extracellular matrix mechanical properties and regulation of the intestinal stem cells: when mechanics control fate. Cells. (2020) 9:2629. doi: 10.3390/cells9122629
197. Komuro T. The lattice arrangement of the collagen fibres in the submucosa of the rat small intestine: scanning electron microscopy. Cell Tissue Res. (1988) 251:117–21. doi: 10.1007/BF00215455
198. Shi XZ. Mechanical regulation of gene expression in gut smooth muscle cells. Front Physiol. (2017) 8:1000. doi: 10.3389/fphys.2017.01000
199. Yen TH, Wright NA. The gastrointestinal tract stem cell niche. Stem Cell Rev. (2006) 2:203–12. doi: 10.1007/s12015-006-0048-1
200. Humphries A, Wright NA. Colonic crypt organization and tumorigenesis. Nat Rev Cancer. (2008) 8:415–2. doi: 10.1038/nrc2392
201. Beumer J, Clevers H. Regulation and plasticity of intestinal stem cells during homeostasis and regeneration. Company Biol. (2016) 143:3639–49. doi: 10.1242/dev.133132
202. Haramis AP, Begthel H, Van de Born M, van Es J, Jonkheer S, Offerhaus GJA, et al. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science. (2004) 303:1684–6. doi: 10.1126/science.1093587
203. Batts LE, Polk DB, Dubois RN, Kulessa H. Bmp signaling is required for intestinal growth and morphogenesis. Dev Dyn. (2006) 235:1563–70. doi: 10.1002/dvdy.20741
204. Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet. (2006) 7:349–59. doi: 10.1038/nrg1840
205. Capulli M, Hristova D, Valbret Z, Carys K, Arjan R, Maurizi A, et al. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. BJC. (2019) 121:157–71. doi: 10.1038/s41416-019-0501-y
206. Moore KA, Lemischka IR. Stem cells and their niches. Science. (2006) 311:1880–5. doi: 10.1126/science.1110542
207. Scoville DH, Sato T, He XC, Li L. Current view: intestinal stem cells and signaling. Gastroenterology. (2008) 134:849–64. doi: 10.1053/j.gastro.2008.01.079
208. Van der Flier LG, Clevers H. Stem cells, self-renewal and differentiation in the intestinal epithelium. Annu Rev Physiol. (2009) 71:241–60. doi: 10.1146/annurev.physiol.010908.163145
209. Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. (2003) 17:1709–13. doi: 10.1101/gad.267103
210. Gjorevski N, Ordóñez-Morán P. Intestinal stem cell niche insights gathered from both in vivo and novel in vitro models. Stem Cells Int. (2017) 2017:8387297. doi: 10.1155/2017/8387297
211. Kolachala VL, Bajaj R, Wang L, Yan Y, Ritzenthaler JD, Gewirtz AT, et al. Epithelial-derived fibronectin expression, signaling, and function in intestinal inflammation. J Biol Chem. (2007) 282:32965–73. doi: 10.1074/jbc.M704388200
212. Groulx JF, Gagné D, Benoit YD, Martel D, Basora N, Beaulieu F. Collagen VI is a basement membrane component that regulates epithelial cell-fibronectin interactions. Matrix Biol. (2011) 30:195–206. doi: 10.1016/j.matbio.2011.03.002
213. Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, et al. Paneth cells constitute the niche for Lgr5 stem cells inintestinal crypts. Nature. (2011) 469:415–8. doi: 10.1038/nature09637
214. van Niel G, Raposo G, Candalh C, Boussac M, Hershberg R, Cerf-Bensussan N, et al. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterol. (2001) 121:337–49. doi: 10.1053/gast.2001.26263
215. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. (2013) 200:373–83. doi: 10.1083/jcb.201211138
216. Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. (2016) 164:1226–32. doi: 10.1016/j.cell.2016.01.043
217. Cappariello A, Loftus A, Muraca M, Maurizi A, Rucci N, Teti A. Osteoblast-derived extracellular vesicles are biological tools for the delivery of active molecules to bone. J Bone Miner Res. (2018) 33:517–33. doi: 10.1002/jbmr.3332
218. Pieters BCH, Cappariello A, van den Bosch MHJ, van Lent P, Teti A, van de Loo AJ, et al. Macrophage-derived extracellular vesicles as carriers of alarmins and their potential involvement in bone homeostasis. Front Immunol. (2019) 10:1901. doi: 10.3389/fimmu.2019.01901
219. Chang X, Wang SL, Zhao SB, Shi YH, Pan P, Gu L, et al. Extracellular vesicles with possible roles in gut intestinal tract homeostasis and IBD. Media Inflamm. (2020) 2020:1945832. doi: 10.1155/2020/1945832
220. Mitsuhashi S, Feldbrügge L, Csizmadia E, Mitsuhashi M, Robson SC, Moss C. Luminal Extracellular Vesicles (EVs) in inflammatory bowel disease (IBD) exhibit pro-inflammatory effects on epithelial cells and macrophages. Inflamm Bowel Dis. (2016) 22:1587–95. doi: 10.1097/MIB.0000000000000840
221. Cappariello A, Rucci N. Tumour-Derived Extracellular Vesicles (EVs): a dangerous “message in a bottle” for bone. Int J Mol Sci. (2019) 20:4805. doi: 10.3390/ijms20194805
222. Loftus A, Cappariello A, George C, Ucci A, Shefferd K, Green A, et al. Extracellular vesicles from osteotropic breast cancer cells affect bone resident cells. J Bone Miner Res. (2020) 35:396–412. doi: 10.1002/jbmr.3891
223. Macia L, Nanan R, Hosseini-Beheshti E, Grau GE. Host- and microbiota-derived extracellular vesicles, immune function, and disease development. Int J Mol Sci. (2020) 21:107. doi: 10.3390/ijms21010107
224. Xiao C, Chun-Hua S, Bai-Sui F, Fong-Li L, Ping L, Zheng PY, et al. Intestinal epithelial cell-derived integrin αβ6 plays an important role in the induction of regulatory T cells and inhibits an antigenspecific Th2 response. J Leukocyte Biol. (2011) 90:751–9. doi: 10.1189/jlb.1210696
225. Liang L, Shen Y, Guoetal D. EpCAM-dependent extracellular vesicles from intestinal epithelial cells maintain intestinal tract immune balance. Nat Commun. (2016) 7:13045. doi: 10.1038/ncomms13045
226. Dean DD, Schwartz Z, Muniz OE, Gomez R, Swain LD, Howell DS, et al. Matrix vesicles are enriched in metalloproteinases that degrade proteoglycans. Calcif Tissue Int. (1992) 50:342–9. doi: 10.1007/BF00301632
227. Hakulinen J, Sankkila L, Sugiyama N, Lehti K, Keski-Oja J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J Cell Biochem. (2008) 105:1211–8. doi: 10.1002/jcb.21923
228. Han KY, Dugas-Ford J, Seiki M, Chang JH, Azar DT. Evidence for the Involvement of MMP14 in MMP2 processing and recruitment in exosomes of corneal fibroblasts. Invest Ophthalmol Vis Sci. (2015) 56:5323–9. doi: 10.1167/iovs.14-14417
229. Crotti S, Piccoli M, Rizzolio F, Giordano A, Nitti D, Agostini, et al. Extracellular matrix and colorectal cancer: how surrounding microenvironment affects cancer cell behavior? J Cell Physiol. (2017) 232:967–75. doi: 10.1002/jcp.25658
230. Vetuschi A, Pompili S, Gaudio E, Latella G, Sferra R. PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur Rev Med Pharmacol Sci. (2018) 22:8839–48. doi: 10.26355/eurrev_201812_16652
231. Hussey GS, Keane TJ, Badylak SF. The extracellular matrix of the gastrointestinal tract: a regenerative medicine platform. Nat Rev Gastroenterol Hepatol. (2017) 14:540–52. doi: 10.1038/nrgastro.2017.76
232. Hussey GS, Dziki JL, Badylak SF. Extracellular matrix-based materials for regenerative medicine. Nat Rev Mater. (2018) 3:159–73. doi: 10.1038/s41578-018-0023-x
233. Hussey GS, Cramer MC, Badylak SF. Extracellular matrix bioscaffolds for building gastrointestinal tissue. Cell Mol Gastroenterol Hepatol. (2017) 5:1–13. doi: 10.1016/j.jcmgh.2017.09.004
234. Chen MK, Badylak SF. Small bowel tissue engineering using small intestinal submucosa as a scaffold. J Surg Res. (2001) 99:352–8. doi: 10.1006/jsre.2001.6199
235. Wang ZQ, Watanabe Y, Toki A. Experimental assessment of small intestinal submucosa as a small bowel graft in a rat model. J Pediatr Surg. (2003) 38:1596–601. doi: 10.1016/S0022-3468(03)00567-0
236. Ansaloni L, Bonasoni P, Cambrini P, Catena F, De Cataldis A, Gagliardi S, et al. Experimental evaluation of surgisis as scaffold for neointestine regeneration in a rat model. Transplant Proc. (2006) 38:1844–8. doi: 10.1016/j.transproceed.2006.05.004
237. Demirbilek S, Kanmaz T, Ozardali I, Edali MN, Yucesan S. Using porcine small intestinal submucosa in intestinal regeneration. Pediatr Surg Int. (2003) 19:588–92. doi: 10.1007/s00383-003-1025-2
238. Keane TJ, Dziki J, Sobieski E, Smoulder A, Castleton A, Turner N, et al. Restoring mucosal barrier function and modifying macrophage phenotype with an extracellular matrix hydrogel: potential therapy for ulcerative colitis. J Crohns Colitis. (2017) 11:360–8. doi: 10.1093/ecco-jcc/jjw149
239. Engin AB, Nikitovic D, Neagu M, Henrich-Noack P, Docea AO, Shtilman MI, et al. Mechanistic understanding of nanoparticles' interactions with extracellular matrix: the cell and immune system. Part Fibre Toxicol. (2017) 14:22. doi: 10.1186/s12989-017-0199-z
Keywords: bowel, intestinal wall, extracellular matrix, basement membrane, interstitial matrix, MMPs, TIMPs
Citation: Pompili S, Latella G, Gaudio E, Sferra R and Vetuschi A (2021) The Charming World of the Extracellular Matrix: A Dynamic and Protective Network of the Intestinal Wall. Front. Med. 8:610189. doi: 10.3389/fmed.2021.610189
Received: 25 September 2020; Accepted: 22 March 2021;
Published: 16 April 2021.
Edited by:
Pedro M. Baptista, Health Research Institute of Aragon (IIS Aragon), SpainReviewed by:
Xiaofei Sun, University of California, San Francisco, United StatesCopyright © 2021 Pompili, Latella, Gaudio, Sferra and Vetuschi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberta Sferra, cm9iZXJ0YS5zZmVycmFAdW5pdmFxLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.