 Minjie Wan
Minjie Wan Jiawen Han
Jiawen Han Lili Ding
Lili Ding Feng Hu
Feng Hu Pujun Gao
Pujun Gao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 01 April 2021
Sec. Pathology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.604894
This article is part of the Research TopicOrgan Fibrosis: Pathogenesis, Biomarkers and Therapeutic TargetsView all 12 articles
Liver fibrosis is a pathological process caused by persistent chronic injury of the liver. Kupffer cells, natural killer (NK) cells, NKT cells, and dendritic cells (DCs), which are in close contact with T and B cells, serve to bridge innate and adaptive immunity in the liver. Meanwhile, an imbalanced inflammatory response constitutes a challenge in liver disease. The dichotomous roles of novel immune cells, including T helper 17 (Th17), regulatory T cells (Tregs), mucosa-associated invariant T cells (MAIT), and innate lymphoid cells (ILCs) in liver fibrosis have gradually been revealed. These cells not only induce damage during liver fibrosis but also promote tissue repair. Hence, immune cells have unique, and often opposing, roles during the various stages of fibrosis. Due to this heterogeneity, the treatment, or reversal of fibrosis through the target of immune cells have attracted much attention. Moreover, activation of hepatic stellate cells (HSCs) constitutes the core of fibrosis. This activation is regulated by various immune mediators, including Th17, Th22, and Th9, MAIT, ILCs, and γδ T cells, as well as their related cytokines. Thus, liver fibrosis results from the complex interaction of these immune mediators, thereby complicating the ability to elucidate the mechanisms of action elicited by each cell type. Future developments in biotechnology will certainly aid in this feat to inform the design of novel therapeutic targets. Therefore, the aim of this review was to summarize the role of specific immune cells in liver fibrosis, as well as biomarkers and treatment methods related to these cells.
Liver fibrosis is a pathological process in which diffuse extracellular matrix (ECM) over precipitates in the liver due to abnormal hyperplasia of connective tissue caused by various pathogenic factors. The initiating event in liver fibrosis is the activation of hepatic stellate cells (HSCs), which promotes the production and accumulation of ECM (1). Liver fibrosis is a common pathological outcome of various chronic liver diseases (CLD), including chronic hepatitis B (CHB), chronic hepatitis C (CHC), non-alcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), autoimmune hepatitis (AIH), and primary biliary cirrhosis (PBC). The treatment and prognosis of chronic liver disease depends on the degree of liver fibrosis. However, currently no treatment has demonstrated the ability to reverse the progression of fibrosis in CLD. The aggravation of fibrosis may lead to cirrhosis, liver failure, or liver cancer, in which liver transplantation is performed as the last option (2). Therefore, early detection and inhibition of fibrosis progression is particularly important in the treatment of liver diseases.
The liver is an immune organ that plays a major role in innate and adaptive immunity. Its anatomical structure allows it to function as a filter for visceral blood, thus acting as the second line of defense for the intestinal immune system, preventing the entry of harmful substances from the intestinal tract, and their negative impact throughout the body (3). The high proportion of Kupffer cells (KCs), natural killer T (NKT) cells, γδ T cells, and dendritic cells (DCs), which are in close contact with antigen presenting cells, T cells, and B cells, serve to connect innate and adaptive immunity in the liver, while inducing immune tolerance, thereby avoiding immune responses from being mounted against foreign antigens that would otherwise cause tissue damage. These effects maintain the stability of hepatic microcirculation and tolerance to foreign antigens (4). Alternatively, inflammation generally precedes fibrosis, while immune cells are important factors in the regulation of fibrosis. Although immune cells can induce damage, they can also promote tissue repair in liver fibrosis (5). T cells and macrophages constitute the core of liver fibrosis pathogenesis with macrophage-derived transforming growth factor (TGF)-β1 known to be the strongest activator of HSCs (6). Recently, newly discovered immune cells, and their related cytokines, were shown to also participate in the process of liver fibrosis (Table 1). For instance, an imbalance in the ratio of regulatory T cells (Tregs)/T helper 17 cells (Th17) is characteristic of liver fibrosis progression. Indeed, some drugs function to restore the Tregs/Th17 balance, thereby alleviated liver fibrosis (7–11). Additionally, Th22, Th9, mucosa-associated invariant T (MAIT) cells, innate lymphoid cells (ILCs), γδ T cells, and their related cytokines, have been reported to regulate liver fibrosis (12–16).
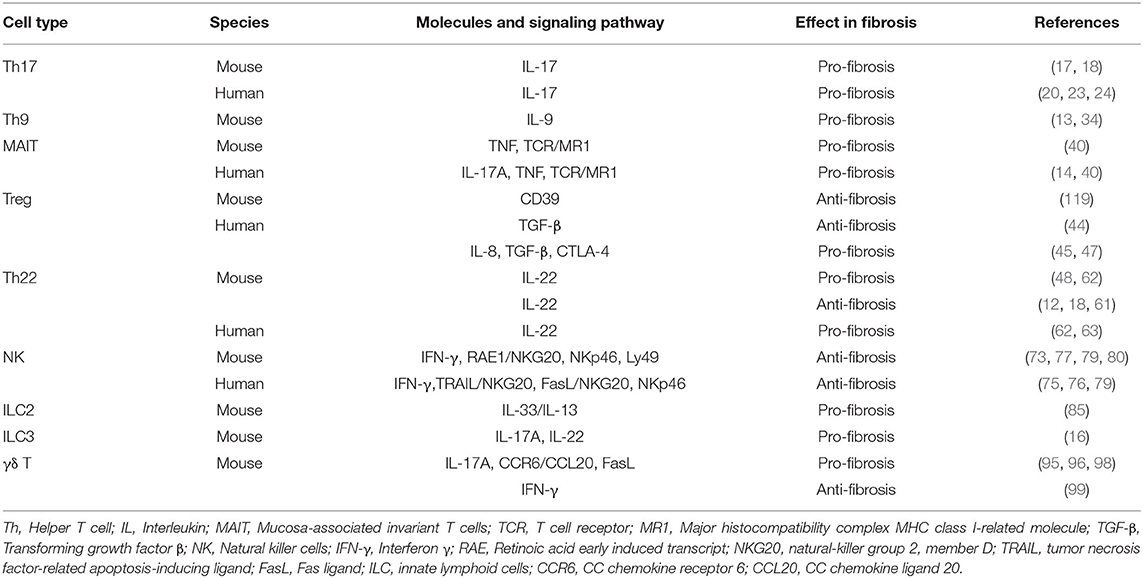
Table 1. The role of novel immune cells in liver fibrosis.
The aim of this review is to provide a summary of the current understanding regarding the roles of innate immune cells in liver fibrosis, and the recent diagnostic and treatment outcomes for liver fibrosis achieved through targeting newly discovered immune cells. First, the review deals with immune cells and their associated cytokines known to promote hepatic fibrosis. Second, the roles of immune cells and their related cytokines playing in anti-hepatic fibrosis are discussed. Third, the dichotomous roles of certain immune cell types in fibrosis is discussed. Finally, conclusions and future perspectives are provided.
Th17 cells are a subset of CD4+ T cells characterized by RORγt expression and interleukin (IL)-17, IL-22, and IL-23 production. In acute and chronic liver injury, the amount, and proportion, of Th17 cells in the liver and peripheral blood increases. These cells have clear fibrogenic properties (17–19), with high levels of intrahepatic Th17 and IL-17 commonly observed in liver fibrosis caused by various etiologies, such as HBV (20), HCV (21), cholestatic liver injury (22), autoimmune hepatitis (23), and NAFLD (24). In fact, within a bile duct ligation (BDL) murine model, knockout of IL-17A resulted in reduced liver damage and fibrosis, accompanied by decreased levels of tumor necrosis factor (TNF)-α, TGF-β, and type I collagen in the liver compared to wild-type (WT) mice (22). Moreover, in mice with liver fibrosis induced by carbon tetrachloride (CCl4), the concentration of collagen and TGF-β in the liver of WT mice was significantly higher than in the liver of IL-17RA deficient mice. Meanwhile, in vitro experiments confirmed that IL-17A activates HSCs to produce collagen through the ERK1/2 and p38 signaling pathways (17). Moreover, in animal models of liver fibrosis induced by CCl4 and BDL, serum or liver IL-17 expression was positively correlated with the degree of liver fibrosis, while blocking IL-17 signaling weakened liver fibrosis. Furthermore, it has been shown that IL-17A promotes the transformation of HSCs into myofibroblasts and the production of collagen through the STAT3 signaling pathway (18).
However, Thomas et al. found that IL-17A does not directly promote HSC activation nor pro-fibrotic gene (COL1A1, TIMP-I, and ACTA2) expression, but rather requires TGF-β collaboration. Meanwhile, IL-17A upregulates and stabilizes TGF-β receptor II (TGF-βRII) expression on the surface of HSCs through the JNK signaling pathway and enhances SMAD2/3 phosphorylation to promote liver fibrosis (19). These differences may have been caused by differing experimental conditions. For instance, although both experiments conducted by Tan et al. (17) and Meng et al. (18), stimulated HSCs for 2–8 h, the latter study did not observe effects at this time point and thus, chose to further stimulate the cells for 48 h (19). Moreover, Meng et al. sought to exclude the effect of TGF-β in fetal bovine serum by performing the study under cell starvation conditions (19). Nevertheless, other studies have also confirmed that IL-17A does not induce the expression of fibrogenic genes, but rather promotes that of chemokines and pro-inflammatory factors in recruited macrophages, monocytes, and neutrophils (25, 26). Thus, IL-17 may recruit other cells to affect HSCs in complex hepatic fibrosis environments. Although advances have been made in identifying the underlying mechanism of Th17 cells and their cytokines in liver fibrosis, some challenges have arisen that require further clarification. For example, in HBV or HCV infected patients, the degree of liver fibrosis is significantly related to the virus replication rate in vivo. Moreover, Th17 cells and IL-17 promote viral clearance and have a certain antiviral role, similar to that of Th1 cells (27). However, both TH17 cells and IL-17 also aggravate inflammatory damage of the liver, leading to chronic HBV and HCV in patients. Due to the diverse functions of Th17 cells, determining how to exploit its anti-fibrotic effect while avoiding its pro-fibrosis potential, will serve to accelerate the clinical application of Th17 in the treatment of liver fibrosis.
Th9 cells are a newly distinguished CD4+ T cell subset characterized by the specific secretion of IL-9 and identified by PU.1 and IRF.4 (28, 29). IL-9 was originally mistaken as a type 2 cytokine until IL-4-induced differentiation of naïve CD4+ T cells was found to generate a group of IL-9+IL-10+Foxp3− T cells with no immunosuppressive capacity (30). IL-9 was further shown to be increased in the peripheral blood and liver of mice infected with Schistosoma japonicum, while its inhibition reduces procollagen-III (a fibrosis-related factor) expression in infected mice (31, 32). Consistently, intraperitoneal injection of anti-IL-9 antibody inhibits granulomatous inflammation in the liver and collagen deposition around the eggs of infected mice. Furthermore, direct stimulation of HSCs in vitro with IL-9 significantly increases the production of collagen and α-SMA (13). In addition, Th9 cells and IL-9 are increased in the blood of patients with HBV and HBV-related cirrhosis. This elevation is also present in the liver of mice with CCl4-induced liver fibrosis (33). Furthermore, Guo et al. demonstrated that CXCL10-induced IL-9 promotes liver fibrosis via the Raf/MEK/ERK signaling pathway in CCl4-induced mice (34). Hence, Th9 has clearly been shown to promote fibrosis. Consistent with studies in liver fibrotic diseases, IL-9 antibody treatment alleviates idiopathic pulmonary fibrosis and cystic fibrosis in mice (35, 36). However, Th9 cells have only been recently identified and investigated in the context of allergic reactions and parasitic infections. Therefore, the role and mechanism of Th9 cells in liver fibrosis require further analysis.
MAIT cells are a novel subset of innate-like T cells characterized by their invariant T cell receptor α-chain and their restrictive major histocompatibility complex related protein-1 (MR1), which are primarily distributed in the blood, liver, and intestinal mucosa (37). The innate functions of MAIT cells are similar to those of innate natural killer T cells (iNKT) and can be stimulated by IL-12 and IL-18 to secrete IFN-γ and granzyme (38). MAIT cells have antibacterial and immunological activities and present altered functions in chronic disease. The role of MAIT cells in liver fibrosis has been recognized due simply to their abundance in the liver, which accounts for ~30% of all CD3+ T cells present in the liver (39). In autoimmune liver disease, MAIT cells are significantly increased in the peripheral blood and liver; this increase is negatively correlated with the degree of liver fibrosis. In vitro studies further confirmed that IL-12 stimulates MAIT cells to produce large amounts of IL-17A. HSCs are activated by IL-17A and direct cell contact with MAIT cells, leading to HSC proliferation, pro-fibrosis, and pro-inflammatory gene expression (14). In animal models of alcoholic and non-alcoholic liver injury, MAIT cells promote the production of pro-inflammatory cytokines, such as IL-6 and IL-8 in mono-derived macrophages. Meanwhile, co-culture results demonstrate that MAIT cells promote fibroblast mitosis and pro-inflammatory properties through direct cell-cell contact (40). In addition, MR1−/− mice (MAIT-deficient) are resistant to liver fibrosis and have lower fibroblast density (40). Given the abovementioned results, MAIT cells play a crucial role in the process of liver fibrosis. However, the precise associated mechanism remains to be explored.
Tregs are a subset of immunosuppressive CD4+ T cells characterized by transcription factor forkhead box P3 (Foxp3) expression. The role of Tregs in liver fibrosis is complex and controversial. The number of circulating Tregs is positively correlated with the degree of liver fibrosis and serum HBV DNA load in HBV-infected patients (41). Meanwhile, Tregs inhibit HSC activation and proliferation, thereby ameliorating liver fibrosis (7, 8). However, given their immunosuppressive function, Tregs also act as a haven for hepatitis B viruses that are otherwise attacked by the immune system (41). Indeed, within HCV patients, hepatic CD4+Foxp3+ T cells are negatively correlated with liver fibrosis, whereas CD4+Foxp3+ Tregs in the blood of chronic HCV patients are less frequent than in healthy controls (42). Alternatively, Ward et al. observed no difference in the abundance of Foxp3+ cells between mild and severe fibrosis in portal tract areas from HCV patients (43). Moreover, it remains unclear whether hepatic Tregs directly control HSCs and immune cells in the liver, or whether Tregs in lymph nodes or the spleen suppress the activation and migration of effector cells before infiltrating into the liver. Notably, although TGF-β is a recognized pro-fibrotic factor, that produced by Tregs in HCV negatively correlates with liver inflammation and fibrosis, suggesting that TGF-β also has anti-fibrotic properties (44). The authors suggest that this dichotomy may be due to the numerous cytokines, including IL-10, that are produced by peripheral immune cells following TGF-β stimulation, thereby effectively balancing the fibrogenic effects of TGF-β produced by other cells in the liver. Additionally, IL-8+CD4+Foxp3+ T cells are abundant in the liver of HCV patients and are primarily distributed in the fibrosis and alpha-smooth muscle actin (α-SMA)+ region. Moreover, neutralization of IL-8 can block the activation of HSCs without affecting the immunosuppressive function of Tregs, suggesting that IL-8+ Tregs participate in the promotion of fibrosis (45). Hence different Treg subgroups appear to have opposing effects.
The mystery of Tregs in liver fibrosis is also reflected in their regulation of other immune cells. In the BDL model, Treg depletion promotes Th17 and CD8+ T cell infiltration in the fibrotic liver and increases the expression of inflammatory cytokines (IL-6, TNF-α, and IL-12p70) and chemokines (monocyte chemoattractant protein 1, macrophage inflammatory protein-1α, and regulated on activation, normal T-cell expressed and secreted chemokine), leading to the aggravation of fibrosis and suggesting that Tregs inhibit fibrosis by suppressing the formation of a pro-fibrotic niche by Th17 and CD8+ T cells (46). In contrast, Tregs are enriched in liver fibrosis tissues and protect HSCs from NK cell killing in HCV patients. Tregs inhibit NK cell killing of HSCs in two ways. The first involves inhibiting NK cells by direct contact with cytotoxic T lymphocyte associated antigen-4 (CTLA-4); whereas the second, involves the production of IL-8 and TGF-β to inhibit HSCs from producing major histocompatibility complex (MHC) class I and MHC class I chain related protein A or B (MIC-A/B), which are required for NK cell activation (47). Additionally, in severe liver fibrosis, the number of Tregs in the liver is higher than that in moderate fibrosis and is positively correlated with serum ALT levels, suggesting that Tregs may be recruited to control liver cell damage (48). In addition, the immunosuppressive regulatory effect of Tregs is conducive to the formation of chronic inflammation, which maintains liver fibrosis (49). Tregs also inhibit the secretion of matrix metalloproteinas (MMPs) by KCs through TGF-β, thereby limiting liver fibrosis regression. Meanwhile, depletion of Tregs with anti-CD25 antibodies accelerates fibrosis regression in CCl4-induced liver fibrosis mice (50). Therefore, the role of Tregs is not entirely opposed to that of Th17, and may depend on the cause of liver injury, the stage of fibrosis, and the interaction between different immune cells.
Recently, Tregs were identified in visceral adipose tissue (VAT) and are now widely accepted as associated with glucose metabolism and insulin resistance (51). In obese mice induced by a high-fat diet, metabolic syndrome and non-alcoholic steatohepatitis (NASH) occur accompanied by a decrease in the proportion of Tregs in VAT (52). VAT Tregs relieve insulin resistance and glucose metabolism disorders caused by a high-fat diet in mice (51). Conversely, consumption of VAT Tregs increases the expression of inflammatory cytokines, such as TNF-α, IL-6, and C-C chemokine ligand 5 (CCL5) and promotes insulin resistance in adipose tissues (53, 54). PPARγ, a transcription factor that regulates adipocyte differentiation, is specifically expressed in VAT Tregs (55). Disabling PPARγ on Tregs results in a decrease in VAT Tregs, while Tregs in the lymphoid organs are not affected (56). In contrast, exogenous injection of a PPAR agonist (pioglitazone) in high-fat diet mice increases the number of VAT Tregs, reduces local inflammation, and improves organic metabolism. Furthermore, mice with knocked out PPARγ expression in Tregs are less responsive to pioglitazone treatment, demonstrating that VAT Tregs constitute a key factor in the regulation of insulin sensitization (55). Considering that insulin resistance promotes the development from simple fatty liver to NASH and is a common risk factor for NAFLD (57, 58), it is reasonable to assume that VAT Tregs participate in the regulation of NASH development. Revealing the mechanism whereby VAT Tregs regulate NAFLD will, therefore, contribute to elucidating the role of VAT Tregs in liver fibrosis.
Th22 cells constitute a newly discovered subset of CD4+ effector T cells that produce a high level of IL-22 rather than IL-17 or interferon (IFN)-γ. These cells are induced by IL-6 and TNF from naïve CD4+ T cells. The characteristic transcription factor of Th22 cells is the aryl hydrocarbon receptor (AHR) (59). Th22 cells participate in chronic inflammation, autoimmune diseases, and cancers. Moreover, the IL-22 receptor (IL-22R) is a heterodimer composed of IL-22R1 and IL-10R2. Among them, IL-22R1 is primarily expressed on epithelial cells located in the skin and the lumen of the digestive and respiratory tracts, thereby determining the primary locations where IL-22 exerts its effects (60). However, the role of Th22 cells and IL-22 in liver fibrosis remains controversial. In CCl4-induced liver fibrosis mice, the proportion of Th22 cells in the spleen is higher than that in WT mice, and is accompanied by increased IL-22 levels in the serum and liver, suggesting that the microenvironment of liver fibrosis is conducive to the differentiation and proliferation of Th22 cells (61). Researchers who hold the view that IL-22 has a pro-fibrotic effect have found that IL-22 relies on MAPK to promote TGF-β signaling in HSCs and induce HSCs to produce more α-SMA (48). In patients with hepatitis B cirrhosis, the infiltration of IL-22 positive cells in the liver is significantly higher than that in healthy individuals and is positively correlated with the stage of liver fibrosis. Furthermore, in HBV transgenic mice, increased IL-22 aggravates chronic liver inflammation and fibrosis by secreting CCL10 and CCL20 to recruit Th17 cells (62). Consistently, IL-22 and IL-22(+) cells are significantly increased in the peripheral blood of HCV patients, while the number of IL-22(+) cells in the liver is positively correlated with the liver fibrosis score. Further, IL-22(+) cells are primarily distributed within the fibrotic area. In vitro experiments have confirmed that IL-22 inhibits LX-2 (HSC line) cell apoptosis, while promoting their proliferation as well as the production of α-SMA and collagen (63).
However, several experiments have also confirmed the anti-fibrotic and protective effects of IL-22 in the liver. IL-22 resists fibrosis by inducing senescence of activated HSCs through SCOS3, p53, and STAT3 (12). Additionally, in vivo injection of IL-22 in BDL mice reduces collagen α1 (I) and α-SMA production to alleviate liver fibrosis (18). Moreover, administration of IL-22 inhibits HSC activation, reduces the production of pro-inflammatory factors (IL-1β, IL-6, and TNF-α), and ameliorates liver fibrosis in CCl4-induced liver fibrosis (61). In the liver of HFD-fed mice, CXCL1 is overexpressed and promotes steatosis-to-NASH progression by inducing neutrophil infiltration, oxidative stress, and stress kinase activation. However, IL-22 treatment blocks hepatic oxidative stress and its associated stress kinases via induction of metallothionein. Furthermore, although it does not target immune cells, IL-22 treatment attenuates the inflammatory functions of hepatocyte-derived, mitochondrial DNA-enriched extracellular vesicles, thereby suppressing liver inflammation in NASH (64). The functional differences of IL-22 may be related to the diversity of its sources. Indeed, various immune cells in the liver, such as Th1, Th17, Th22, γδ T, and NKT cells can produce IL-22 (65). However, none of the abovementioned studies has identified the specific cellular source of IL-22, the identification of which may provide targets for clinical therapeutic strategies. In addition, IL-22 and IL-17 are both type 3 cytokines, which can be produced simultaneously in chronic inflammatory diseases (48). In the absence of IL-17, Th22 has a protective effect against NASH. However, in the presence of IL-17, IL-22 recruits Th17 to aggravate liver fibrosis (66). Moreover, the pro-inflammatory and anti-inflammatory role of IL-22 has been shown to be regulated by IL-17 in airway inflammation (67). Thus, determining the source of IL-22 and the effect of other cytokines, such as IL-17, on IL-22 will help us better understand the role of IL-22 in liver fibrosis.
ILCs are a group of heterogeneous lymphocytes involved in innate immunity. They do not express the antigen-specific receptors of T or B cells and are largely distributed at mucosal barrier sites where they participate in immune surveillance and regulation (68). ILCs are divided into three groups: Group 1 (ILC1 and NK cells, dependent on T-bet and producing IFN-γ), Group 2 (ILC2, dependent on GATA3 and RORα and producing type 2 cytokines, such as IL-13 and IL-5), and Group 3 (ILC3 and lymphoid tissue-inducing cells, dependent on RORγt and producing IL-17 and IL-22). The functions of ILC1, ILC2, ILC3, and NK cells correspond to Th1, Th2, Th17, and CD8+ cytotoxic T cells, respectively (69). ILCs are distributed differently in different organs. The NKP44+ ILC3 type predominates in the gut, where it acts as a mucosal barrier by producing IL-22. However, the NKP44− ILC3 type predominates in the liver, and have the potential to differentiate into other ILCs. NKP44− ILC3 is the only type present in fetal liver, while other ILCs can be detected with prolonged pregnancy (70).
The role of ILC1 in liver fibrosis, however, is yet to be reported. Nabekura et al. demonstrate the protective role of ILC1s in a mouse model of CCl4-mediated moderate acute liver injury. CCl4-mediated acute liver injury results in ATP and IL-12 production from DCs that activates ILC1s to produce IFN-γ. This results in upregulation of Bcl-2 and Bcl-xL by hepatocytes leading to reduced cell death and liver damage (71). Furthermore, Wang et al. found that group 1 ILCs in adipose tissues aggravate adipose fibrosis and promote the development of diabetes (72). However, since NK cells and ILC1 were not studied separately, the role of ILC1 in fibrosis could not be clearly defined.
As for NK cells in liver fibrosis, activated NK cells kill HSCs by producing IFN-γ (73–75). In addition, NK cells induce apoptosis of HSCs by direct cell contact, which involves Fas ligand (FasL), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and natural killer group 2, member D (NKG2D) (76). Early activated HSCs produce large amounts of retinoic acid, leading to increased expression of RAE-1 (retinoic acid early inducible 1), a ligand that activates NK cell receptor NKG2D. RAE-1 and MICA synergically trigger NK cells to kill HSCs (77, 78). Additionally, Chamutal et al. reported that primary human and mouse HSCs express unknown ligands for human NKp46 and mouse NCR1 receptors, respectively, to mediate the killing of HSCs by NK cells (79). In addition to the activation-related receptors on NK cells, inhibitory receptors Ly-49 are also reportedly involved in NK cell killing of HSCs mediated by MHC I molecules (80, 81). Notably, NK cells preferentially help to eliminate senescent HSCs and contribute to the regression of liver fibrosis (78). Besides, other immune cells can also regulate the interaction between NK cells and HSCs. In immune stimulatory conditions, such as viral liver disease or Toll-like receptor stimulation, KCs and DCs promote NK cell activation (82–84). Meanwhile, Tregs can inhibit NK cell activation to protect HSCs (47). Although the molecular mechanism underlying the NK cell anti-liver fibrosis phenomena has been extensively studied, it remains unclear whether liver resident or non-resident NK cells limit fibrosis. Moreover, the details of the interaction between NK cells and HSCs muse be further revealed before NK cells can become an immune target for anti-fibrosis strategies.
It is widely accepted that ILC2 promotes liver fibrosis. ILC2 increases at the site of hepatic fibrosis and is positively correlated with the degree of hepatic fibrosis (15). In the CCl4-induced liver fibrosis model, collagen deposition is significantly reduced following ILC2 cell depletion (85). The fibrogenic effect of ILC2 is dependent on the IL-33/IL-13 signaling pathway. Meanwhile, during chronic liver injury, increased release of IL-33 leads to the accumulation and activation of ILC2 cells in the liver through ST2 receptors on the surface of ILC2. Activated ILC2 cells then produce IL-13, which in turn activates HSCs in an IL-4Rα- and STAT6-dependent manner to aggravate liver fibrosis (85). Meanwhile, liver fibrosis is alleviated in mice lacking ST2 or IL-13, however, transfusion of ILC2 restores fibrosis (85). In addition to IL-33, thymic stromal lymphopoietin (TSLP) and IL-25 are major cytokines that drive type 2 immunity. Moreover, the activation of ILCs by the above three cytokines has been reported to lead to fibrosis in the lung and skin (86–89). Consistent with the results in other organs, simultaneously blocking IL-33, IL-25, and TSLP can improve liver and lung fibrosis and reduce IL-13 production by ILC2 in mice injected with Schistosoma mansoni eggs. However, other studies have found that the proportion of CD4+IL-13+ T cells increases following infection with Schistosoma mansoni eggs, which is accompanied by decreased ILC2 activity, suggesting that adaptive immunity may gradually replace IL-33, IL-25, and TSLP-ILC2 to maintain liver fibrosis progression (86). Although the IL-33/IL13 axis clearly promotes liver fibrosis, the specific contribution of ILC2 and type 2 immune cells remains to be investigated.
The role of ILC3 in fibrosis has only recently been discovered. In HBV patients, ILC3 is increased in peripheral blood and is positively correlated with the degree of fibrosis and inflammation. Moreover, co-culturing ILC3 with LX-2 cells demonstrated that ILC3 cells activate HSCs by producing IL-17A and IL-22. Furthermore, transferring ILC3 from normal mice to CCl4-induced Rag-1−/− mice leads to HSC activation, ECM accumulation, and aggravation of hepatic fibrosis (16). Additionally, RORγt+ ILCs exert a partial protective role in the hepatic immune response induced by CCl4 (90). However, the study does not distinguish between ILC3 and lymphoid tissue-inducing cells (LTi).
LTi cells are essential for peripheral lymphoid organ and tissue development (91). These cells secrete IL-17 and IL-22 in groups, both during embryo development and after birth. Activated LTi cells also produce a large number of cytokines and chemokines during induction of peripheral lymphoid organ/tissue formation, leading to lymphocyte and DC cell aggregation (92). Therefore, LTi cells have pro-inflammatory properties and are likely to participate in the inflammatory processes associated with most diseases, including liver fibrosis. However, the specific molecular mechanisms remain to be investigated.
γδ T cells make up 3–5% of total lymphocytes and 15–25% of T cells in the liver (93). These cells represent a double-edged sword in liver fibrosis. Wang et al. demonstrate that macrophages increase the number of IL-17A-producing γδT cells through the HMGB1-TLR4-IL-23 signaling pathway, recruit neutrophils to infiltrate the liver, and aggravate liver inflammation (94). Furthermore, in mice infected with Schistosoma japonicum, Vγ2 γδ T cells recruit neutrophils to granuloma and the liver by producing IL-17A, thereby aggravating liver fibrosis (95). However, γδ T cells do not only interact with immune cells to affect the fibrosis process but are also regulated by HSCs. In CCl4-induced acute liver injury and early stage liver fibrosis, exosomes released by hepatocytes bind to TLR3 and activate HSCs to produce IL-17A, which promotes the production of IL-17A by hepatic γδ T cells to aggravate liver fibrosis (96). However, it has also been argued that exosomes directly promote the production of IL-17 by γδ T cells (97). In addition to promoting fibrosis, γδ T cells have also been found to ameliorate liver inflammation and fibrosis. In two chronic liver injury mouse models (CCl4 and methionine-choline-deficient diet), γδ T cells are recruited to the liver through the activation of the CCR6/CCL20 signaling pathway and directly promote HSC apoptosis in a FasL-dependent manner to limit liver fibrosis (98). Liu et al. found that γδ T cells (particularly IFN-γ-producing subsets) protect the liver from fibrosis by killing activated HSCs directly or indirectly by enhancing NK cell-mediated cytotoxicity (99). Collectively, inducing the cytotoxicity of γδ T cells against HSCs can display an anti-fibrosis role, while promoting the production of IL-17 by γδ T cells aggravates fibrosis.
The liver is exposed to gut-derived bacterial metabolites and their products through the portal vein (100). Normally, the liver maintains a delicate balance between inflammatory and regulatory immune responses. However, when gut microbiota becomes altered, microbial stimuli affect the function of immune cells in the liver and ultimately lead to the development of liver disease (101). Liver inflammation reshapes intestinal microbiota through an unknown mechanism, leading to increased Lactobacillus (especially L. johnsonii) abundance in the gut. During the recovery stage of acute liver injury induced by concanavalin A, Lactobacillus was found to activate intestinal ILC3 cells to produce IL-22, which repairs the intestinal mucosal barrier and blocks further metastasis of gut microbiota to the liver. Moreover, IL-22 can induce the production of IL-10 and TGF-β by recruiting regulatory DC cells to the liver to maintain immune tolerance (102). Additionally, Hendrikx et al. report that the gut microbiota regulate ILC3 cells to reduce progression of ALD. In chronic-binge ethanol feeding mice, intestinal microbiota derived AHR ligand indole-3-acetic acid are reduced, resulting in decreased IL-22 production by ILC3s. IL-22 can also regulate the expression of intestinal REG3G, which protects mice against ethanol-induced liver disease by reducing bacterial translocation. In fact, supplementation with Lactobacillus to produce IL-22 effectively reduces liver damage and bacterial translocation to the liver (103). Hence, considering that systemic injections of IL-22 increase the risk of hepatocellular carcinoma in patients with CLD (104–106), altering the gut microbiota to regulate the immune cells that produce IL-22 may offer a more viable option for liver injury therapeutic interventions.
In addition to ILC3, MAIT cells are also influenced by gut microbiota in ALD. Fecal extracts from patients with ALD have reduced blood MAIT cells that are hyperactivated and exhibit defective antibacterial cytokine/cytotoxic responses (37). Moreover, in an intrahepatic cholangitis model, gut L gasseri are enriched and translocate to the liver, where they amplify IL-17+ γδ T cells to promote liver fibrosis and inflammation (107). Microbial-derived lipids are presented to γδ T cell receptors through CD1d on hepatocytes, which activates γδ T cells to express IL-17A, thereby aggravating NAFLD. Notably, this is unique to hepatic γδ T cells, and cannot be applied to circulating γδ T cells (108). Overall, these studies suggest that gut microbiota helps to shape the liver immune response. Although evidence does not directly suggest that an altered gut microbiota affects the role of novel immune cells in liver fibrosis, intestinal microbiota represents the core of the gut-liver axis that has been shown to drive many liver diseases to different stages. Therefore, it has important value and far-reaching significance for research in this field.
Inflammation is present in all the stages of liver fibrosis, cirrhosis, and hepatocellular carcinoma. Meanwhile, persistent activation of inflammatory responses contributes to the expansion of liver fibrosis (109). Degeneration and necrosis of hepatic parenchyma cells caused by various factors leads to the release of the inflammasome, which recruits and activates inflammatory cells. Activated inflammatory cells (particularly KCs) secrete TGF-β1, TNF-α, PDGF, and other factors, which promote the transformation of HSCs, or other fibrogenic cells, into myofibroblasts. Myofibroblasts continue to secrete and deposit extracellular matrix, and ultimately form hepatic fibrosis (110). Intrinsic cells and immune cells in the liver and HSCs together establish a complex regulatory system. These novel immune cells and their related cytokines do not only directly regulate HSCs and fibrogenesis, but also indirectly affect liver fibrosis by influencing the inflammatory microenvironment. For instance, IL-17 stimulates STAT3-mediated human endothelial cell activation and production of GRO-α, GM-CSF and IL-8, which regulate neutrophil recruitment to the liver (111). IL-17 can also induce HepG2 to produce IL-6 through activation of MAPK. Consequently, IL-6 stimulates Th17 cells and forms a positive feedback loop in AIH (23). Meanwhile, IL-17 recruits other inflammatory cells and promotes the synthesis of pro-inflammatory cytokines to exacerbate the inflammatory process, which triggers and maintains the differentiation of profibrogenic cells into myofibroblasts to amplify fibrosis. In addition to IL-17, TNF signaling controls NLRP3 inflammasome activation in myeloid derived cells to initiate liver inflammation, via recruitment of neutrophils and pro-inflammatory macrophages, leading to subsequent activation of fibrogenic pathways (112). Furthermore, the inhibitory regulation of Tregs favors the formation of chronic inflammation and contributes to the persistence of liver fibrosis. Specifically, Tregs suppress NK cells, M1 KCs, and CD8+ T cells to maintain chronic liver inflammation and fibrosis (49). Hence, application of drugs capable of regulating the liver immune microenvironment while inducing the related cells to support the reversal of liver fibrosis may represent a new strategy for treating liver fibrosis.
The novel immune cells discussed in this review are important players in the pathogenesis of liver fibrosis. Hence, regulating their functions may represent a therapeutic strategy for the treatment of liver fibrosis. Currently, studies have shown that abrogating Th17/IL-17 signaling alleviates liver fibrosis. For instance, in Schistosoma japonicum-infected mice, a selective RhoA-Rho-associated kinase (ROCK) inhibitor (fasudil) limited liver fibrosis by inhibiting Th17 differentiation and IL-17 production, and upregulating Tregs (113). Moreover, abrogating inducible co-stimulator (ICOS) signaling reportedly inhibits Th17 cells, and their related cytokines, thereby reducing granulomatous inflammation and liver fibrosis around the eggs in a Schistosoma japonicum infection model (114). Mesenchymal stem cells have also been reported to restrict liver fibrosis by inhibiting Th17 cells (115, 116). In addition, miR-29a/miR-652, 1, 25(OH)2D3, as well as certain drugs, such as rapamycin and tofacitinib, have been shown to attenuate liver fibrosis by regulating Th17 cells (10, 11, 117, 118). Meanwhile, low dose IL-2 specifically expands and activates Treg cell populations thereby controlling autoimmune diseases and inflammation. Additionally, IL-2 and IL-2 immune complexes promote the expression of CD39 on hepatic Tregs, which inhibits the proliferation of CD8+ T cells and reduces the expression of osteopontin and TNF-α to diminish biliary fibrosis in murine sclerosing cholangitis (119). Cumulatively, these results provides a theoretical basis for the treatment of fibrosing cholangiopathies with low dose IL-2. In addition to limiting liver fibrosis by targeting the novel immune cells, as described above, they may also be applied for disease prediction. In fact, γδ T cells gene signature can predict the overall and recurrence free survival of patients with HCC. Tumor microenvironments recruit γδ T cells from peripheral or peritumor regions into tumors to elicit anti-tumor effects (120). Chronic liver inflammation and fibrosis are necessary processes in the development of HCC. Therefore, if we can effectively monitor these novel immune cells, and their related molecules, as non-invasive diagnostic markers, it will be of great benefit to patients with chronic liver disease.
The cellular and molecular mechanisms of liver fibrosis are currently under intense investigation. Although the reversibility of liver fibrosis provides an effective early opportunity for treatment, no ideal anti-fibrotic drug is currently available for clinical practice. The activation of HSCs constitutes the core of fibrosis and is regulated by various immune mediators. Recently, novel immune cells have been discovered whose role in liver fibrosis has also been gradually recognized (Figure 1). Additionally, the proportion of regulatory B cells (Bregs) in peripheral blood has been positively correlated with the stage of liver fibrosis in HBV patients. Bregs inhibit effector T cells, however, enhance the function of Tregs to regulate immune tolerance in HBV-infected patients (121). In addition to regulating fibrosis by acting on HSCs, other cells also affect fibrosis controlled by the novel immune subsets and cytokines. For instance, in collaboration with TNF-α, IL-17 promotes HepG2 cells to produce more periostin, which induces fibroblasts to synthesize additional type I collagen and aggravate liver fibrosis (122). IL-17A induces intrahepatic biliary epithelial cells to undergo epithelial to mesenchymal transition, during which cells obtain fibroblast-related characteristics to promote fibrosis in PBC (123). Due to the complex microenvironment of liver fibrosis, the role of one single cell type cannot be discussed while ignoring others. For example, changes in Tregs and Th17 tend to occur simultaneously and are accompanied by Th1/Th2 shifts in the initial stages of liver fibrosis. In addition, each type of immune cell produces many different cytokines, which leads to the diversity of immune cell function. Fortunately, the application of single-cell RNA sequencing technology (scRNA-seq) in liver disease enables us to identify some subsets of cells that are historically difficult to isolate (124). In fact, scRNA-seq has identified a specific subset of macrophages (TREM2+CD9+MNDA+ scar-associated macrophages) in human fibrotic liver that is primarily distributed in scarring regions. This subset promotes the production of collagen and proliferation of HSCs (125). Thus, these technologies allow us to capture information about key cell populations and discover new therapeutic targets. The development of biotechnology will facilitate the identification of new cell populations involved in liver fibrosis as well as to elucidate the mechanisms underlying liver fibrosis.
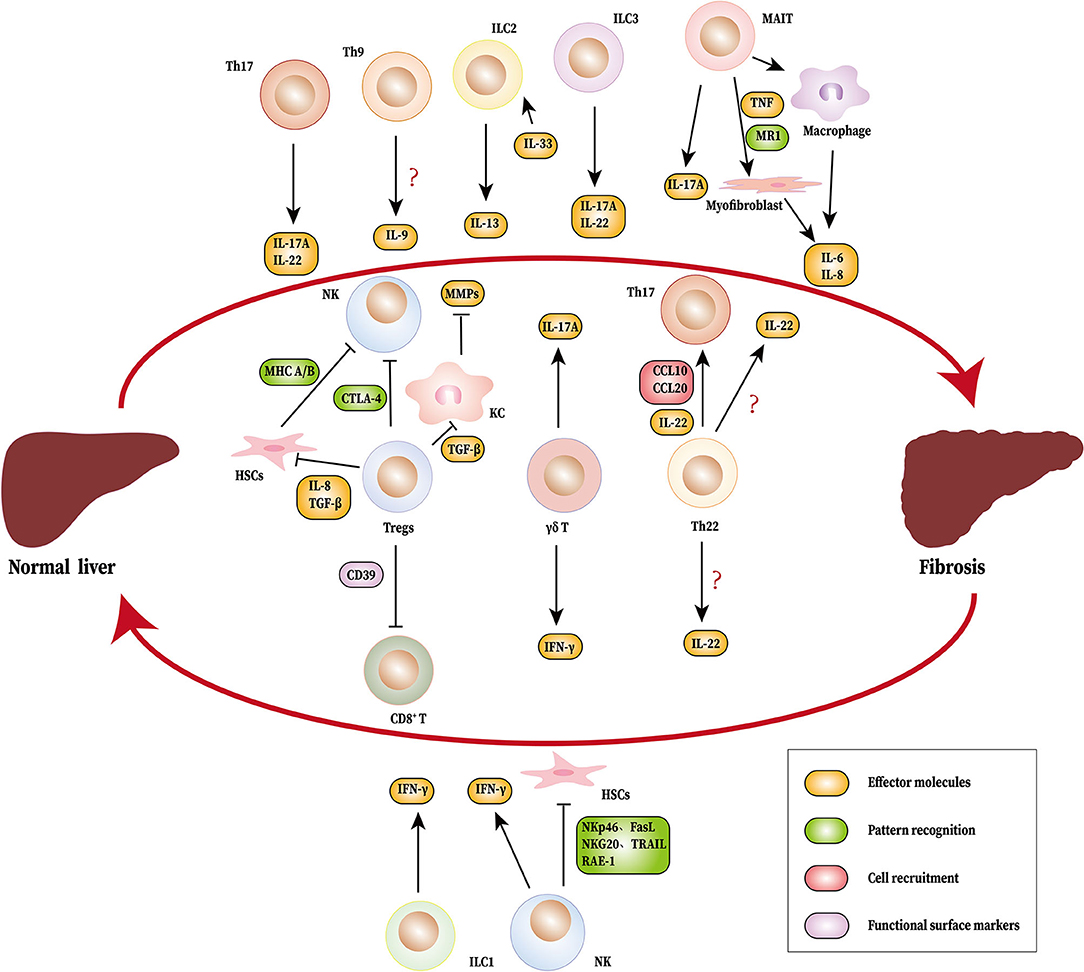
Figure 1. Novel immune cells and related cytokines regulate liver fibrosis. During the process of liver fibrosis, Th17 (IL-17A and IL-22), MAIT cells (IL-17A), ILC2 (IL-13), ILC3 (IL-17A, IL-22), and γδ T cells (IL-17A) promote fibrosis. MAIT cells can induce myofibroblasts and macrophages to produce IL-6 and IL-8. Tregs inhibit NK cells directly or indirectly by HSCs. Tregs also suppress KC to produce MMPs by TGF-β. Th22 cells recruits Th17 to the liver through IL-22, CCL6, and CCL20. NK cells, ILC1 and γδ T cells produce IFN-γ, which limits fibrosis. NK cells also suppress liver fibrosis by inhibiting HSCs. The source of IL-9 and IL-22 have not been identified. HSCs, hepatic stellate cells; Th, T helper cells; MAIT cells, mucosa-associated invariant T cells; ILC, innate lymphoid cell; NK cells, natural killer cells; KC, Kupffer cells; IL, interleukin; IFN-γ, Interferon γ; TGF-β, Transforming growth factor β; MMPs, matrix metalloproteinas; RAE-1, Retinoic acid early induced transcript 1; NKG20, natural killer group 2, member D; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; FasL, Fas ligand; CCL, CC chemokine ligand; TNF, tumor necrosis factor; CTLA-4, cytotoxic T lymphocyte antigen 4; MHC A/B, major histocompatibility complex A/B.
MW wrote the manuscript. JH and LD collected related literature. FH drew the figures. PG revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the grant from the National Natural Science Foundation of China (81671592), the Science and Technology Department of Jilin Province (20190201140JC), the National Natural Science Foundation of Jilin Province (2018SCZWSZX-003, JLSCZD2019-008), the National Science and Technology Major Project (2017ZX10202202,2018ZX10302206, and 2018ZX10723203).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Thanks are due to Dr. Huanfa Yi for critical comments and invaluable advice on this manuscript, and to the central laboratory for its support. We would like to thank Editage (www.editage.cn) for English language editing.
1. Shang L, Hosseini M, Liu X, Kisseleva T, Brenner DA. Human hepatic stellate cell isolation and characterization. J Gastroenterol. (2018) 53:6–17. doi: 10.1007/s00535-017-1404-4
2. Manka P, Zeller A, Syn W-K. Fibrosis in Chronic liver disease: an update on diagnostic and treatment modalities. Drugs. (2019) 79:903–27. doi: 10.1007/s40265-019-01126-9
3. Kubes P, Jenne C. Immune responses in the liver. Annu Rev Immunol. (2018) 36:247–77. doi: 10.1146/annurev-immunol-051116-052415
4. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
5. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. (2014) 14:181–94. doi: 10.1038/nri3623
6. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. (2017) 127:55–64. doi: 10.1172/JCI88881
7. Sun X-F, Gu L, Deng W-S, Xu Q. Impaired balance of T helper 17/T regulatory cells in carbon tetrachloride-induced liver fibrosis in mice. World J Gastroenterol. (2014) 20:2062–70. doi: 10.3748/wjg.v20.i8.2062
8. Yu X, Guo R, Ming D, Su M, Lin C, Deng Y, et al. Ratios of regulatory T cells/T-helper 17 cells and transforming growth factor-β1/interleukin-17 to be associated with the development of hepatitis B virus-associated liver cirrhosis. J Gastroenterol Hepatol. (2014) 29:1065–72. doi: 10.1111/jgh.12459
9. Li J, Qiu S-J, She W-M, Wang F-P, Gao H, Li L, et al. Significance of the balance between regulatory T (Treg) and T helper 17 (Th17) cells during hepatitis B virus related liver fibrosis. PLoS ONE. (2012) 7:e39307. doi: 10.1371/journal.pone.0039307
10. Gu L, Deng W-S, Sun X-F, Zhou H, Xu Q. Rapamycin ameliorates CCl4-induced liver fibrosis in mice through reciprocal regulation of the Th17/Treg cell balance. Mol Med Rep. (2016) 14:1153–61. doi: 10.3892/mmr.2016.5392
11. Wang H, Feng X, Han P, Lei Y, Xia Y, Tian D, et al. The JAK inhibitor tofacitinib ameliorates immune-mediated liver injury in mice. Mol Med Rep. (2019) 20:4883–92. doi: 10.3892/mmr.2019.10750
12. Kong X, Feng D, Wang H, Hong F, Bertola A, Wang F-S, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. (2012) 56:1150–9. doi: 10.1002/hep.25744
13. Zhan T, Ma H, Jiang S, Zhong Z, Wang X, Li C, et al. Interleukin-9 blockage reduces early hepatic granuloma formation and fibrosis during Schistosoma japonicum infection in mice. Immunology. (2019) 158:296–303. doi: 10.1111/imm.13111
14. Böttcher K, Rombouts K, Saffioti F, Roccarina D, Rosselli M, Hall A, et al. MAIT cells are chronically activated in patients with autoimmune liver disease and promote profibrogenic hepatic stellate cell activation. Hepatology. (2018) 68:172–86. doi: 10.1002/hep.29782
15. Gonzalez-Polo V, Pucci-Molineris M, Cervera V, Gambaro S, Yantorno SE, Descalzi V, et al. Group 2 innate lymphoid cells exhibit progressively higher levels of activation during worsening of liver fibrosis. Ann Hepatol. (2019) 18:366–72. doi: 10.1016/j.aohep.2018.12.001
16. Wang S, Li J, Wu S, Cheng L, Shen Y, Ma W, et al. Type 3 innate lymphoid cell: a new player in liver fibrosis progression. Clin Sci. (2018) 132:2565–82. doi: 10.1042/CS20180482
17. Tan Z, Qian X, Jiang R, Liu Q, Wang Y, Chen C, et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol. (2013) 191:1835–44. doi: 10.4049/jimmunol.1203013
18. Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. (2012) 143:765–76.e3. doi: 10.1053/j.gastro.2012.05.049
19. Fabre T, Kared H, Friedman SL, Shoukry NH. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol. (2014) 193:3925–33. doi: 10.4049/jimmunol.1400861
20. Sun HQ, Zhang JY, Zhang H, Zou ZS, Wang FS, Jia JH. Increased Th17 cells contribute to disease progression in patients with HBV-associated liver cirrhosis. J Viral Hepat. (2012) 19:396–403. doi: 10.1111/j.1365-2893.2011.01561.x
21. Chang Q, Wang Y-K, Zhao Q, Wang C-Z, Hu Y-Z, Wu B-Y. Th17 cells are increased with severity of liver inflammation in patients with chronic hepatitis C. J Gastroenterol Hepatol. (2012) 27:273–8. doi: 10.1111/j.1440-1746.2011.06782.x
22. Hara M, Kono H, Furuya S, Hirayama K, Tsuchiya M, Fujii H. Interleukin-17A plays a pivotal role in cholestatic liver fibrosis in mice. J Surg Res. (2013) 183:574–82. doi: 10.1016/j.jss.2013.03.025
23. Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS ONE. (2011) 6:e18909. doi: 10.1371/journal.pone.0018909
24. Rau M, Schilling A-K, Meertens J, Hering I, Weiss J, Jurowich C, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol. (2016) 196:97–105. doi: 10.4049/jimmunol.1501175
25. Yan S, Wang L, Liu N, Wang Y, Chu Y. Critical role of interleukin-17/interleukin-17 receptor axis in mediating Con A-induced hepatitis. Immunol Cell Biol. (2012) 90:421–8. doi: 10.1038/icb.2011.59
26. Guillot A, Hamdaoui N, Bizy A, Zoltani K, Souktani R, Zafrani E-S, et al. Cannabinoid receptor 2 counteracts interleukin-17-induced immune and fibrogenic responses in mouse liver. Hepatology. (2014) 59:296–306. doi: 10.1002/hep.26598
27. Wu W, Li J, Chen F, Zhu H, Peng G, Chen Z. Circulating Th17 cells frequency is associated with the disease progression in HBV infected patients. J Gastroenterol Hepatol. (2010) 25:750–7. doi: 10.1111/j.1440-1746.2009.06154.x
28. Kaplan MH. Th9 cells: differentiation and disease. Immunol Rev. (2013) 252:104–15. doi: 10.1111/imr.12028
29. Zhao P, Xiao X, Ghobrial RM, Li XC. IL-9 and Th9 cells: progress and challenges. Int Immunol. (2013) 25:547–51. doi: 10.1093/intimm/dxt039
30. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. (2008) 9:1347–55. doi: 10.1038/ni.1677
31. Li L, Xie H, Wang M, Qu J, Cha H, Yang Q, et al. Characteristics of IL-9 induced by Schistosoma japonicum infection in C57BL/6 mouse liver. Sci Rep. (2017) 7:2343. doi: 10.1038/s41598-017-02422-8
32. Barreto AVMS, Lacerda GANd, Figueiredo ALdC, Diniz GTN, Gomes ECS, Domingues ALC, et al. Evaluation of serum levels of IL-9 and IL-17 in human Schistosoma mansoni infection and their relationship with periportal fibrosis. Immunobiology. (2016) 221:1351–4. doi: 10.1016/j.imbio.2016.07.014
33. Qin S-y, Lu D-h, Guo X-y, Luo W, Hu B-l, Huang X-l, et al. A deleterious role for Th9/IL-9 in hepatic fibrogenesis. Sci Rep. (2016) 6:18694. doi: 10.1038/srep18694
34. Guo X, Cen Y, Wang J, Jiang H. CXCL10-induced IL-9 promotes liver fibrosis via Raf/MEK/ERK signaling pathway. Biomed Pharmacother. (2018) 105:282–9. doi: 10.1016/j.biopha.2018.05.128
35. Sugimoto N, Suzukawa M, Nagase H, Koizumi Y, Ro S, Kobayashi K, et al. IL-9 blockade suppresses silica-induced lung inflammation and fibrosis in mice. Am J Respir Cell Mol Biol. (2019) 60:232–43. doi: 10.1165/rcmb.2017-0287OC
36. Moretti S, Renga G, Oikonomou V, Galosi C, Pariano M, Iannitti RG, et al. A mast cell-ILC2-Th9 pathway promotes lung inflammation in cystic fibrosis. Nat Commun. (2017) 8:14017. doi: 10.1038/ncomms14017
37. Riva A, Patel V, Kurioka A, Jeffery HC, Wright G, Tarff S, et al. Mucosa-associated invariant T cells link intestinal immunity with antibacterial immune defects in alcoholic liver disease. Gut. (2018) 67:918–30. doi: 10.1136/gutjnl-2017-314458
38. Ussher JE, Klenerman P, Willberg CB. Mucosal-associated invariant T-cells: new players in anti-bacterial immunity. Front Immunol. (2014) 5:450. doi: 10.3389/fimmu.2014.00450
39. Dusseaux M, Martin E, Serriari N, Péguillet I, Premel V, Louis D, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood. (2011) 117:1250–9. doi: 10.1182/blood-2010-08-303339
40. Hegde P, Weiss E, Paradis V, Wan J, Mabire M, Sukriti S, et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat Commun. (2018) 9:2146. doi: 10.1038/s41467-018-04450-y
41. Xu D, Fu J, Jin L, Zhang H, Zhou C, Zou Z, et al. Circulating and liver resident CD4+CD25+ regulatory T cells actively influence the antiviral immune response and disease progression in patients with hepatitis B. J Immunol. (2006) 177:739–47. doi: 10.4049/jimmunol.177.1.739
42. Claassen MAA, de Knegt RJ, Tilanus HW, Janssen HLA, Boonstra A. Abundant numbers of regulatory T cells localize to the liver of chronic hepatitis C infected patients and limit the extent of fibrosis. J Hepatol. (2010) 52:315–21. doi: 10.1016/j.jhep.2009.12.013
43. Ward SM, Fox BC, Brown PJ, Worthington J, Fox SB, Chapman RW, et al. Quantification and localisation of FOXP3+ T lymphocytes and relation to hepatic inflammation during chronic HCV infection. J Hepatol. (2007) 47:316–24. doi: 10.1016/j.jhep.2007.03.023
44. Li S, Vriend LEM, Nasser IA, Popov Y, Afdhal NH, Koziel MJ, et al. Hepatitis C virus-specific T-cell-derived transforming growth factor beta is associated with slow hepatic fibrogenesis. Hepatology. (2012) 56:2094–105. doi: 10.1002/hep.25951
45. Langhans B, Krämer B, Louis M, Nischalke HD, Hüneburg R, Staratschek-Jox A, et al. Intrahepatic IL-8 producing Foxp3?CD4? regulatory T cells and fibrogenesis in chronic hepatitis C. J Hepatol. (2013) 59:229–35. doi: 10.1016/j.jhep.2013.04.011
46. Roh YS, Park S, Lim CW, Kim B. Depletion of Foxp3+ regulatory T cells promotes profibrogenic milieu of cholestasis-induced liver injury. Dig Dis Sci. (2015) 60:2009–18. doi: 10.1007/s10620-014-3438-2
47. Langhans B, Alwan AW, Krämer B, Glässner A, Lutz P, Strassburg CP, et al. Regulatory CD4+ T cells modulate the interaction between NK cells and hepatic stellate cells by acting on either cell type. J Hepatol. (2015) 62:398–404. doi: 10.1016/j.jhep.2014.08.038
48. Fabre T, Molina MF, Soucy G, Goulet J-P, Willems B, Villeneuve J-P, et al. Type 3 cytokines IL-17A and IL-22 drive TGF-β-dependent liver fibrosis. Sci Immunol. (2018) 3:eaar7754. doi: 10.1126/sciimmunol.aar7754
49. Zhang X, Lou J, Bai L, Chen Y, Zheng S, Duan Z. Immune regulation of intrahepatic regulatory T cells in fibrotic livers of mice. Med Sci Monit. (2017) 23:1009–16. doi: 10.12659/MSM.899725
50. Zhang X, Feng M, Liu X, Bai L, Kong M, Chen Y, et al. Persistence of cirrhosis is maintained by intrahepatic regulatory T cells that inhibit fibrosis resolution by regulating the balance of tissue inhibitors of metalloproteinases and matrix metalloproteinases. Transl Res. (2016) 169:67–79.e1-2. doi: 10.1016/j.trsl.2015.10.008
51. Zhu F, Wang A, Li Y, Liang R, Li D, Li B. Adipose tissue-resident regulatory T cells. Adv Exp Med Biol. (2017) 1011:153–62. doi: 10.1007/978-94-024-1170-6_4
52. Van Herck MA, Vonghia L, Kwanten WJ, Julé Y, Vanwolleghem T, Ebo DG, et al. Diet reversal and immune modulation show key role for liver and adipose tissue T cells in murine nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. (2020) 10:467–90. doi: 10.1016/j.jcmgh.2020.04.010
53. Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes. (2011) 60:2954–62. doi: 10.2337/db11-0358
54. Panduro M, Benoist C, Mathis D. Tissue tregs. Annu Rev Immunol. (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
55. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. (2012) 486:549–53. doi: 10.1038/nature11132
56. Cipolletta D. Adipose tissue-resident regulatory T cells: phenotypic specialization, functions and therapeutic potential. Immunology. (2014) 142:517–25. doi: 10.1111/imm.12262
57. Kitade H, Chen G, Ni Y, Ota T. Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients. (2017) 9:387. doi: 10.3390/nu9040387
58. Brunt EM, Wong VWS, Nobili V, Day CP, Sookoian S, Maher JJ, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. (2015) 1:15080. doi: 10.1038/nrdp.2015.80
59. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. (2009) 10:857–63. doi: 10.1038/ni.1767
60. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov. (2014) 13:21–38. doi: 10.1038/nrd4176
61. Lu D-H, Guo X-Y, Qin S-Y, Luo W, Huang X-L, Chen M, et al. Interleukin-22 ameliorates liver fibrogenesis by attenuating hepatic stellate cell activation and downregulating the levels of inflammatory cytokines. World J Gastroenterol. (2015) 21:1531–45. doi: 10.3748/wjg.v21.i5.1531
62. Zhao J, Zhang Z, Luan Y, Zou Z, Sun Y, Li Y, et al. Pathological functions of interleukin-22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology. (2014) 59:1331–42. doi: 10.1002/hep.26916
63. Wu L-Y, Liu S, Liu Y, Guo C, Li H, Li W, et al. Up-regulation of interleukin-22 mediates liver fibrosis via activating hepatic stellate cells in patients with hepatitis C. Clin Immunol. (2015) 158:77–87. doi: 10.1016/j.clim.2015.03.003
64. Hwang S, He Y, Xiang X, Seo W, Kim S-J, Ma J, et al. Interleukin-22 ameliorates neutrophil-driven nonalcoholic steatohepatitis through multiple targets. Hepatology. (2020) 72:412–29. doi: 10.1002/hep.31031
65. Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev. (2013) 252:116–32. doi: 10.1111/imr.12027
66. Rolla S, Alchera E, Imarisio C, Bardina V, Valente G, Cappello P, et al. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci. (2016) 130:193–203. doi: 10.1042/CS20150405
67. Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. (2010) 207:1293–305. doi: 10.1084/jem.20092054
68. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
69. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
70. Forkel M, Berglin L, Kekäläinen E, Carlsson A, Svedin E, Michaëlsson J, et al. Composition and functionality of the intrahepatic innate lymphoid cell-compartment in human nonfibrotic and fibrotic livers. Eur J Immunol. (2017) 47:1280–94. doi: 10.1002/eji.201646890
71. Nabekura T, Riggan L, Hildreth AD, O'Sullivan TE, Shibuya A. Type 1 innate lymphoid cells protect mice from acute liver injury via interferon-γ secretion for upregulating bcl-xl expression in hepatocytes. Immunity. (2020) 52:96–108.e9. doi: 10.1016/j.immuni.2019.11.004
72. Wang H, Shen L, Sun X, Liu F, Feng W, Jiang C, et al. Adipose group 1 innate lymphoid cells promote adipose tissue fibrosis and diabetes in obesity. Nat Commun. (2019) 10:3254. doi: 10.1038/s41467-019-11270-1
73. Jeong W-I, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology. (2008) 134:248–58. doi: 10.1053/j.gastro.2007.09.034
74. Jeong W-I, Park O, Suh Y-G, Byun J-S, Park S-Y, Choi E, et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology. (2011) 53:1342–51. doi: 10.1002/hep.24190
75. Krämer B, Körner C, Kebschull M, Glässner A, Eisenhardt M, Nischalke H-D, et al. Natural killer p46High expression defines a natural killer cell subset that is potentially involved in control of hepatitis C virus replication and modulation of liver fibrosis. Hepatology. (2012) 56:1201–13. doi: 10.1002/hep.25804
76. Glässner A, Eisenhardt M, Krämer B, Körner C, Coenen M, Sauerbruch T, et al. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Invest. (2012) 92:967–77. doi: 10.1038/labinvest.2012.54
77. Radaeva S, Wang L, Radaev S, Jeong W-I, Park O, Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. (2007) 293:G809–16. doi: 10.1152/ajpgi.00212.2007
78. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. (2008) 134:657–67. doi: 10.1016/j.cell.2008.09.015
79. Gur C, Doron S, Kfir-Erenfeld S, Horwitz E, Abu-Tair L, Safadi R, et al. NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut. (2012) 61:885–93. doi: 10.1136/gutjnl-2011-301400
80. Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, et al. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. (2006) 45:60–71. doi: 10.1016/j.jhep.2005.12.025
81. Muhanna N, Abu Tair L, Doron S, Amer J, Azzeh M, Mahamid M, et al. Amelioration of hepatic fibrosis by NK cell activation. Gut. (2011) 60:90–8. doi: 10.1136/gut.2010.211136
82. Tu Z, Bozorgzadeh A, Pierce RH, Kurtis J, Crispe IN, Orloff MS. TLR-dependent cross talk between human Kupffer cells and NK cells. J Exp Med. (2008) 205:233–44. doi: 10.1084/jem.20072195
83. Okazaki A, Hiraga N, Imamura M, Hayes CN, Tsuge M, Takahashi S, et al. Severe necroinflammatory reaction caused by natural killer cell-mediated Fas/Fas ligand interaction and dendritic cells in human hepatocyte chimeric mouse. Hepatology. (2012) 56:555–66. doi: 10.1002/hep.25651
84. Zhou Z, Yu X, Zhang J, Tian Z, Zhang C. TLR7/8 agonists promote NK-DC cross-talk to enhance NK cell anti-tumor effects in hepatocellular carcinoma. Cancer Lett. (2015) 369:298–306. doi: 10.1016/j.canlet.2015.09.017
85. McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. (2013) 39:357–71. doi: 10.1016/j.immuni.2013.07.018
86. Vannella KM, Ramalingam TR, Borthwick LA, Barron L, Hart KM, Thompson RW, et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci Transl Med. (2016) 8:337ra65. doi: 10.1126/scitranslmed.aaf1938
87. Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci USA. (2014) 111:367–72. doi: 10.1073/pnas.1315854111
88. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. (2013) 210:2939–50. doi: 10.1084/jem.20130351
89. Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci USA. (2013) 110:13921–6. doi: 10.1073/pnas.1307321110
90. Matsumoto A, Kanai T, Mikami Y, Chu P-S, Nakamoto N, Ebinuma H, et al. IL-22-producing RORγt-dependent innate lymphoid cells play a novel protective role in murine acute hepatitis. PLoS ONE. (2013) 8:e62853. doi: 10.1371/journal.pone.0062853
91. Castellanos JG, Longman RS. The balance of power: innate lymphoid cells in tissue inflammation and repair. J Clin Invest. (2019) 129:2640–50. doi: 10.1172/JCI124617
92. Lane PJL, Gaspal FM, McConnell FM, Kim MY, Anderson G, Withers DR. Lymphoid tissue inducer cells: innate cells critical for CD4+ T cell memory responses? Ann N Y Acad Sci. (2012) 1247:1–15. doi: 10.1111/j.1749-6632.2011.06284.x
93. Gao B, Jeong W-I, Tian Z. Liver: An organ with predominant innate immunity. Hepatology. (2008) 47:729–36. doi: 10.1002/hep.22034
94. Wang X, Sun R, Wei H, Tian Z. High-mobility group box 1 (HMGB1)-Toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: Interaction of γδ T cells with macrophages. Hepatology. (2013) 57:373–84. doi: 10.1002/hep.25982
95. Zheng L, Hu Y, Wang Y, Huang X, Xu Y, Shen Y, et al. Recruitment of neutrophils mediated by Vγ2 γδ T cells deteriorates liver fibrosis induced by Schistosoma japonicum infection in C57BL/6 mice. Infect Immun. (2017) 85:e01020-16. doi: 10.1128/IAI.01020-16
96. Seo W, Eun HS, Kim SY, Yi H-S, Lee Y-S, Park S-H, et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by γδ T cells in liver fibrosis. Hepatology. (2016) 64:616–31. doi: 10.1002/hep.28644
97. Ni M, Gu J, Rao J, Zhang Y, Ding Z, Wang X, et al. Comment on exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by γδ T cells in liver fibrosis. Hepatology. (2016) 64:2271–2. doi: 10.1002/hep.28729
98. Hammerich L, Bangen JM, Govaere O, Zimmermann HW, Gassler N, Huss S, et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology. (2014) 59:630–42. doi: 10.1002/hep.26697
99. Liu M, Hu Y, Yuan Y, Tian Z, Zhang C. γδT cells suppress liver fibrosis via strong cytolysis and enhanced NK cell-mediated cytotoxicity against hepatic stellate cells. Front Immunol. (2019) 10:477. doi: 10.3389/fimmu.2019.00477
100. Balmer ML, Slack E, de Gottardi A, Lawson MAE, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med. (2014) 6:237ra66. doi: 10.1126/scitranslmed.3008618
101. Tiegs G, Lohse AW. Immune tolerance: what is unique about the liver. J Autoimmun. (2010) 34:1–6. doi: 10.1016/j.jaut.2009.08.008
102. Nakamoto N, Amiya T, Aoki R, Taniki N, Koda Y, Miyamoto K, et al. Commensal lactobacillus controls immune tolerance during acute liver injury in mice. Cell Rep. (2017) 21:1215–26. doi: 10.1016/j.celrep.2017.10.022
103. Hendrikx T, Duan Y, Wang Y, Oh J-H, Alexander LM, Huang W, et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut. (2019) 68:1504–15. doi: 10.1136/gutjnl-2018-317232
104. Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y, et al. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. (2011) 54:900–9. doi: 10.1002/hep.24486
105. Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S, et al. In vivo consequences of liver-specific interleukin-22 expression in mice: Implications for human liver disease progression. Hepatology. (2011) 54:252–61. doi: 10.1002/hep.24339
106. Waidmann O, Kronenberger B, Scheiermann P, Köberle V, Mühl H, Piiper A. Interleukin-22 serum levels are a negative prognostic indicator in patients with hepatocellular carcinoma. Hepatology. (2014) 59:1207. doi: 10.1002/hep.26528
107. Tedesco D, Thapa M, Chin CY, Ge Y, Gong M, Li J, et al. Alterations in intestinal microbiota lead to production of interleukin 17 by intrahepatic γδ T-cell receptor-positive cells and pathogenesis of cholestatic liver disease. Gastroenterology. (2018) 154:2178–93. doi: 10.1053/j.gastro.2018.02.019
108. Li F, Hao X, Chen Y, Bai L, Gao X, Lian Z, et al. The microbiota maintain homeostasis of liver-resident γδT-17 cells in a lipid antigen/CD1d-dependent manner. Nat Commun. (2017) 7:13839. doi: 10.1038/ncomms13839
109. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. (2015) 61:1066–79. doi: 10.1002/hep.27332
110. Parola M, Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. (2019) 65:37–55. doi: 10.1016/j.mam.2018.09.002
111. Yuan S, Zhang S, Zhuang Y, Zhang H, Bai J, Hou Q. Interleukin-17 stimulates STAT3-mediated endothelial cell activation for neutrophil recruitment. Cell Physiol Biochem. (2015) 36:2340–56. doi: 10.1159/000430197
112. Wree A, McGeough MD, Inzaugarat ME, Eguchi A, Schuster S, Johnson CD, et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology. (2018) 67:736–49. doi: 10.1002/hep.29523
113. Zhou W, Yang Y, Mei C, Dong P, Mu S, Wu H, et al. Inhibition of rho-kinase downregulates Th17 cells and ameliorates hepatic fibrosis by infection. Cells. (2019) 8:1262. doi: 10.3390/cells8101262
114. Wang B, Liang S, Wang Y, Zhu X-Q, Gong W, Zhang H-Q, et al. Th17 down-regulation is involved in reduced progression of schistosomiasis fibrosis in ICOSL KO mice. PLoS Negl Trop Dis. (2015) 9:e0003434. doi: 10.1371/journal.pntd.0003434
115. Milosavljevic N, Gazdic M, Simovic Markovic B, Arsenijevic A, Nurkovic J, Dolicanin Z, et al. Mesenchymal stem cells attenuate liver fibrosis by suppressing Th17 cells - an experimental study. Transpl Int. (2018) 31:102–15. doi: 10.1111/tri.13023
116. Huang B, Cheng X, Wang H, Huang W, la Ga Hu Z, Wang D, et al. Mesenchymal stem cells and their secreted molecules predominantly ameliorate fulminant hepatic failure and chronic liver fibrosis in mice respectively. J Transl Med. (2016) 14:45. doi: 10.1186/s12967-016-0792-1
117. Xuan J, Guo S-L, Huang A, Xu H-B, Shao M, Yang Y, et al. MiR-29a and miR-652 attenuate liver fibrosis by inhibiting the differentiation of CD4+ T cells. Cell Struct Funct. (2017) 42:95–103. doi: 10.1247/csf.17005
118. Gu L, Xu Q, Cao H. 1,25(OH)2D3 protects liver fibrosis through decreasing the generation of TH17 cells. Med Sci Monit. (2017) 23:2049–58. doi: 10.12659/MSM.904271
119. Taylor AE, Carey AN, Kudira R, Lages CS, Shi T, Lam S, et al. Interleukin 2 promotes hepatic regulatory T cell responses and protects from biliary fibrosis in murine sclerosing cholangitis. Hepatology. (2018) 68:1905–21. doi: 10.1002/hep.30061
120. Zhao N, Dang H, Ma L, Martin SP, Forgues M, Ylaya K, et al. Intratumoral γδ T-cell infiltrates, CCL4/5 protein expression and survival in patients with hepatocellular carcinoma. Hepatology. (2020) 73:1045–60. doi: 10.1002/hep.31412
121. Liu Y, Cheng L-S, Wu S-d, Wang S-Q, Li L, She W-M, et al. IL-10-producing regulatory B-cells suppressed effector T-cells but enhanced regulatory T-cells in chronic HBV infection. Clin Sci. (2016) 130:907–19. doi: 10.1042/CS20160069
122. Amara S, Lopez K, Banan B, Brown S-K, Whalen M, Myles E, et al. Synergistic effect of pro-inflammatory TNFα and IL-17 in periostin mediated collagen deposition: potential role in liver fibrosis. Mol Immunol. (2015) 64:26–35. doi: 10.1016/j.molimm.2014.10.021
123. Huang Q, Chu S, Yin X, Yu X, Kang C, Li X, et al. Interleukin-17A-induced epithelial-mesenchymal transition of human intrahepatic biliary epithelial cells: implications for primary biliary cirrhosis. Tohoku J Exp Med. (2016) 240:269–75. doi: 10.1620/tjem.240.269
124. Ramachandran P, Matchett KP, Dobie R, Wilson-Kanamori JR, Henderson NC. Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat Rev Gastroenterol Hepatol. (2020) 17:457–72. doi: 10.1038/s41575-020-0304-x
Keywords: liver fibrosis, T helper cells, mucosa-associated invariant T cells, innate lymphoid cells, regulatory T cells, hepatic stellate cells
Citation: Wan M, Han J, Ding L, Hu F and Gao P (2021) Novel Immune Subsets and Related Cytokines: Emerging Players in the Progression of Liver Fibrosis. Front. Med. 8:604894. doi: 10.3389/fmed.2021.604894
Received: 10 September 2020; Accepted: 05 March 2021;
Published: 01 April 2021.
Edited by:
Peter Olinga, University of Groningen, NetherlandsReviewed by:
Christoph Siegfried Niki Klose, Charité – Universitätsmedizin Berlin, GermanyCopyright © 2021 Wan, Han, Ding, Hu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pujun Gao, Z3BqQGpsdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.