 Peter Korsten
Peter Korsten Maximilian F. Konig
Maximilian F. Konig Björn Tampe
Björn Tampe Mehdi Mirsaeidi
Mehdi Mirsaeidi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
EDITORIAL article
Front. Med. , 20 January 2021
Sec. Pulmonary Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fmed.2020.644075
This article is part of the Research Topic Interstitial Lung Disease in the Context of Systemic Disease: Pathophysiology, Treatment and Outcomes View all 13 articles
Editorial on the Research Topic
Interstitial Lung Disease in the Context of Systemic Disease: Pathophysiology, Treatment and Outcomes
Interstitial lung disease (ILD) is an umbrella term for many different disease entities causing inflammation and fibrosis of the lung parenchyma. These can be broadly divided into five categories based on etiology (1): (1) ILD related to a distinct primary disease (e.g., sarcoidosis), (2) ILD related to environmental factors (e. g., hypersensitivity pneumonitis), (3) ILD induced by drugs or irradiation, (4) idiopathic interstitial pneumonias (e.g., idiopathic pulmonary fibrosis), and (5) ILD related to connective tissue diseases (CTD) (1). While all these entities require thorough and multidisciplinary assessment to ascertain a diagnosis, establish the need for diagnostic procedures, and recommend a patient-specific treatment plan, ILDs associated with systemic diseases are particularly challenging. In many cases, optimal treatment for involvement of other organ systems needs to be balanced with the choice of ILD-directed therapies.
For the highly heterogeneous group of patients who cannot be given a definite diagnosis of an autoimmune rheumatic disease but who demonstrate certain clinical, radiographic, and/or serological features suggestive of a CTD, the term interstitial pneumonia with autoimmune features (IPAF) has been coined (2), but its clinical value remains to be defined. Due to its complexity, management of ILDs associated with systemic disease requires multidisciplinary care, including pulmonologists, rheumatologists, and radiologists, often with critical input from other specialties, such as pathologists, dermatologists, or neurologists. In this Research Topic, we hope to present novel research and state-of-the-art reviews as relevant to the care of patients with ILD and systemic diseases.
The most frequently encountered ILDs in the setting of systemic diseases include parenchymal lung involvement with myositis (Myo-ILD), systemic sclerosis (SSc-ILD), and rheumatoid arthritis (RA-ILD). Less frequently reported are ILDs secondary to primary Sjögren's syndrome (pSS) or systemic lupus erythematosus (SLE). Figure 1 gives an overview of systemic diseases and frequently encountered autoantibodies that have been associated with the development of ILD in these diseases. For practical reasons, we find it helpful to distinguish these based on their predominant pattern on high-resolution computed tomography (HRCT), most frequently usual interstitial pneumonia (UIP) and non-specific interstitial pneumonia (NSIP). UIP changes tend to be less reversible and tend to confer a worse prognosis, whereas changes in NSIP (particularly in patients with non-fibrotic NSIP) may be more responsive to treatment. In different diseases, these lung disease patterns occur with varying frequency. In RA, UIP is often more frequently encountered than NSIP, although both may occur. Anti-cyclic citrullinated peptide (CCP) antibodies and rheumatoid factor (RF), present in 70–80% of patients with RA, are associated with RA-ILD. The same applies to SSc, where NSIP is common, but UIP also occurs. In pSS, pulmonary manifestations are relatively rare but have been reported to occur in about 16% of patients, and a consensus guideline on the diagnostic and therapeutic approach has recently been published (3). In pSS, the occurrence of a lymphocytic interstitial pneumonitis (LIP) is rare, but this radiologic pattern should prompt a search for other manifestations of pSS if ILD is the first presenting manifestation (4). ILD is common in myositis, especially in patients with antibodies directed against different aminoacyl-tRNA synthetases, a group collectively called the antisynthetase syndrome (ASyS), but also those with anti-Ro52 antibodies (5, 6). Anti-MDA5 antibodies are associated with clinically amyopathic dermatomyositis (CADM) which can present as a rapidly progressive ILD with high mortality despite aggressive immunosuppressive therapy (7). The following summarizes the papers publishes within this Research Topic.
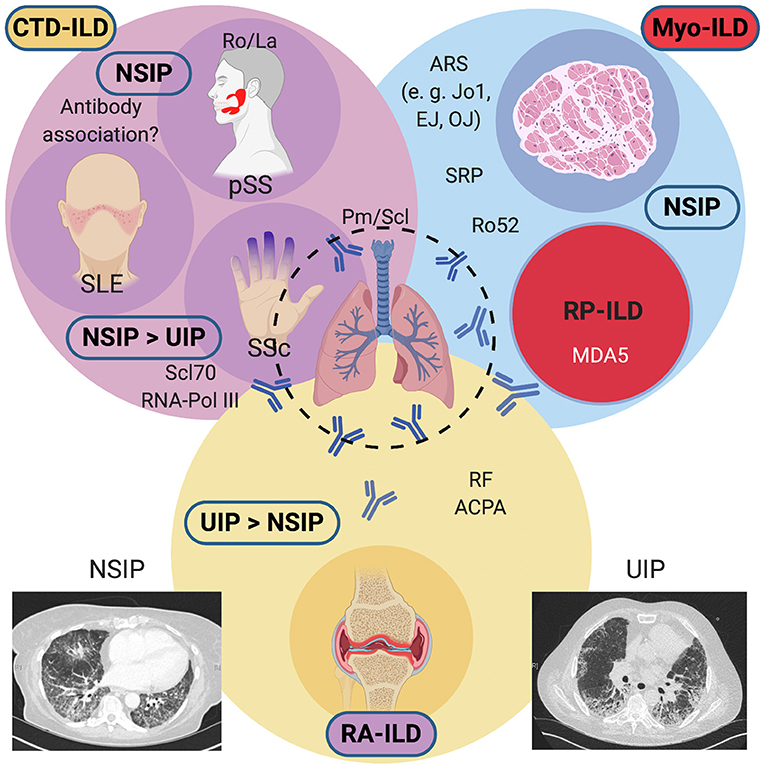
Figure 1. Overview of systemic diseases causing interstitial lung diseases (ILDs) and their associated antibodies. Conditions associated with a predominant NSIP pattern on HRCTs include Myositis-associated ILD (antisynthetase syndrome, anti-RO52 positive overlap myositis, or RP-ILD), Systemic Sclerosis, primary Sjögren's syndrome, and rarely, Systemic Lupus Erythematosus. In SSc, a UIP pattern may also be commonly found. RA-ILD frequently shows a UIP pattern and confers a guarded prognosis. An NSIP pattern may also be encountered on HRCTs. ACPA, anti-citrullinated peptide antibodies; ARS, aminoacyl-tRNA synthetase antibodies; CTD, connective tissue disease; HRCT, high-resolution computed tomography; MDA5; melanocyte-differentiation antigen 5; Myo, myositis; NSIP, non-specific interstitial pneumonia; Pm/Scl, polymyositis-scleroderma; pSS; primary Sjögren's syndrome; RA, rheumatoid arthritis; RF, rheumatoid factor; RNA Pol III, RNA polymerase III; RP, rapidly progressive; Scl70, topoisomerase I; SLE, systemic lupus erythematosus, Sm, Smith; SRP, signal recognition particle; SSc, systemic sclerosis; U1-snRNP, U1 small nuclear ribonucleoprotein; UIP, usual interstitial pneumonia. Created with BioRender.com.
In the first paper, Furini et al. systematically investigated the evidence for multidisciplinary conferences (MDC) to assess ILDs. They found that most MDC evaluated patients with history taking and clinical assessment, HRCT, pulmonary function tests (PFTs), lung biopsy (in most MDCs), and serological testing. Less consensus existed on the use of additional tests, such as nailfold video capillaroscopy (NVC) and 6-min walking distance. Only seven studies evaluated the rheumatologist's role in MDCs, but the authors suggest that MDCs include serological testing and rheumatology expertise to help classify CTD-ILD and IPAF with more certainty. The second paper by Tirelli et al. reported results from a retrospective cohort study from Pavia, Italy. In their cohort, the authors found that 15% of all patients were diagnosed with CTD-ILD, 33% were classified as IPAF; the remainder had no underlying systemic disease. They found the application of a standardized screening questionnaire, laboratory testing, and the inclusion of NVC useful for detecting these entities. Karampitsakos et al. summarized the current use and ongoing clinical trials of biological therapies in sarcoidosis and idiopathic pulmonary fibrosis, which emphasizes a potentially useful role of rheumatologists in the management of ILDs given their extensive expertise in the use of these drugs and management of complications.
One paper specifically reported findings in SLE, pSS, and SSc patients. Patients with SLE and pSS tend to have less ILD. Therefore, it is even more critical to identify individuals at risk and potential overlap syndromes among patients with these relatively common autoimmune rheumatic diseases. Amarnani et al. reviewed the pulmonary manifestations of SLE, including ILDs. Interestingly, the development of an ILD in SLE has not been associated with anti-dsDNA antibodies but rather anti-Ro or anti-U1-snRNP antibodies, which are also associated with mixed connective tissue disease (MCTD). It is, therefore, unclear whether these patients instead represent an overlap population. Sogkas et al. reported their single-center experience from a large cohort of pSS patients with ILD. At their center, 13% of patients were eventually diagnosed with ILD. Of note, almost two thirds (61%) were diagnosed with ILD at presentation and, surprisingly, UIP was the most commonly encountered pattern on HRCT (43%). Lastly, Mirsaeidi et al. reviewed the current treatment options for SSc-ILD, an emerging and rapidly changing topic with the recently approved anti-fibrotic nintedanib, and several ongoing clinical trials.
The majority of papers addressed Myo-ILD. The review by Hervier and Uzunhan represents a timely and current overview of the diagnostic and therapeutic approach to Myo-ILD. This is expanded by original data from Asian cohorts on rapidly-progressive ILD (Li et al.), acute exacerbations of ILD in Myo-ILD (Liang et al.), and the understudied role of plasma exchange in the treatment of refractory Myo-ILD (Ning et al.). Finally, Korsten et al. report their findings on the efficacy of immunosuppression in ASyS-ILD. They specifically report equal usefulness of rituximab (RTX) compared to other conventional immunosuppressive drugs, especially in patients with more frequent clinical myositis and progressive or relapsing pulmonary involvement.
The recent observation of MUC5B promoter variants as a risk factor for RA-ILD, similar to patients with idiopathic pulmonary fibrosis, has generated significant interest in understanding the potential role of antifibrotic strategies in the treatment of this subset of ILD (8). Emerging data from cohort studies examining different immunosuppressive drugs [e.g., abatacept or RTX; (9, 10)] is of similar interest. Fragoulis et al. provided an overview of the difficult topic of RA-ILD, which is an area of active investigation. The authors summarize data on methotrexate-induced pneumonitis and pulmonary fibrosis associated with RA.
PK wrote the first draft and created the figure. BT, MK, and MM edited and reviewed the draft. All authors approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the reviewers and additional external editors for their time and thoroughness in assessing the submitted papers.
1. Wijsenbeek M, Cottin V. Spectrum of Fibrotic Lung Diseases. N Engl J Med. (2020) 383:958–68. doi: 10.1056/NEJMra2005230
2. Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. (2015) 46:976–87. doi: 10.1183/13993003.00150-2015
3. Lee AS, Scofield RH, Hammitt KM, Gupta N, Thomas DE, Moua T, et al. Consensus guidelines for evaluation and management of pulmonary disease in Sjögren's. Chest. (2020). doi: 10.1016/j.chest.2020.10.011
4. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren's syndrome. Eur Respir Rev. (2016) 25:110–23. doi: 10.1183/16000617.0011-2016
5. Ferreira JP, Almeida I, Marinho A, Cerveira C, Vasconcelos C. Anti-ro52 antibodies and interstitial lung disease in connective tissue diseases excluding scleroderma. ISRN Rheumatol. (2012) 2012:415272. doi: 10.5402/2012/415272
6. Xing X, Li A, Li C. Anti-Ro52 antibody is an independent risk factor for interstitial lung disease in dermatomyositis. Respir Med. (2020) 172:106134. doi: 10.1016/j.rmed.2020.106134
7. Chino H, Sekine A, Baba T, Kitamura H, Iwasawa T, Okudela K, et al. Interstitial lung disease with anti-melanoma differentiation-associated protein 5 antibody: rapidly progressive perilobular opacity. Intern Med. (2019) 58:2605–13. doi: 10.2169/internalmedicine.2328-18
8. Juge P-A, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med. (2018) 379:2209–19. doi: 10.1056/NEJMoa1801562
9. Vadillo C, Nieto MA, Romero-Bueno F, Leon L, Sanchez-Pernaute O, Rodriguez-Nieto MJ, et al. Efficacy of rituximab in slowing down progression of rheumatoid arthritis-related interstitial lung disease: data from the NEREA Registry. Rheumatology. (2020) 59:2099–108. doi: 10.1093/rheumatology/kez673
Keywords: interstitial lung disease, autoimmune diseases, rheumatoid arthritis, systemic sclerosis (scleroderma), myositis
Citation: Korsten P, Konig MF, Tampe B and Mirsaeidi M (2021) Editorial: Interstitial Lung Disease in the Context of Systemic Disease: Pathophysiology, Treatment and Outcomes. Front. Med. 7:644075. doi: 10.3389/fmed.2020.644075
Received: 19 December 2020; Accepted: 24 December 2020;
Published: 20 January 2021.
Edited and reviewed by: Laurent Pierre Nicod, University of Lausanne, Switzerland
Copyright © 2021 Korsten, Konig, Tampe and Mirsaeidi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Korsten, cGV0ZXIua29yc3RlbkBtZWQudW5pLWdvZXR0aW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.