
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 15 February 2021
Sec. Infectious Diseases – Surveillance, Prevention and Treatment
Volume 7 - 2020 | https://doi.org/10.3389/fmed.2020.616694
This article is part of the Research TopicSpotlight on the Relationship between Sepsis and Infection: from Mechanisms to TherapyView all 26 articles
 Brittany P. Fenner
Brittany P. Fenner D. B. Darden
D. B. Darden Lauren S. Kelly
Lauren S. Kelly Jaimar Rincon
Jaimar Rincon Scott C. Brakenridge
Scott C. Brakenridge Shawn D. LarsonFrederick A. Moore
Shawn D. LarsonFrederick A. Moore Philip A. EfronLyle L. Moldawer*
Philip A. EfronLyle L. Moldawer*Improved management of severe sepsis has been one of the major health care accomplishments of the last two decades. Due to enhanced recognition and improved management of severe sepsis, in-hospital mortality has been reduced by up to 40%. With that good news, a new syndrome has unfortunately replaced in-hospital multi-organ failure and death. This syndrome of chronic critical illness (CCI) includes sepsis patients who survive the early “cytokine or genomic storm,” but fail to fully recover, and progress into a persistent state of manageable organ injury requiring prolonged intensive care. These patients are commonly discharged to long-term care facilities where sepsis recidivism is high. As many as 33% of sepsis survivors develop CCI. CCI is the result, at least in part, of a maladaptive host response to chronic pattern-recognition receptor (PRR)-mediated processes. This maladaptive response results in dysregulated myelopoiesis, chronic inflammation, T-cell atrophy, T-cell exhaustion, and the expansion of suppressor cell functions. We have defined this panoply of host responses as a persistent inflammatory, immune suppressive and protein catabolic syndrome (PICS). Why is this important? We propose that PICS in survivors of critical illness is its own common, unique immunological endotype driven by the constant release of organ injury-associated, endogenous alarmins, and microbial products from secondary infections. While this syndrome can develop as a result of a diverse set of pathologies, it represents a shared outcome with a unique underlying pathobiological mechanism. Despite being a common outcome, there are no therapeutic interventions other than supportive therapies for this common disorder. Only through an improved understanding of the immunological endotype of PICS can rational therapeutic interventions be designed.
Sepsis afflicts over 1.7 million Americans annually, and accounts for over 250,000 deaths in the United States alone (1). More patients die from sepsis annually than from lung cancer, the number one cause of cancer-related deaths. Sepsis remains the most common cause of death in the intensive care unit (ICU), accounting for 1 in 3 hospital deaths (2). Sepsis is also the most expensive in-hospital diagnosis in the U.S. today (3). Even with these staggering numbers, the impact of sepsis on patients, their families, and the community is grossly underestimated as survivors experience multiple ongoing morbidities (4). Sepsis has an annual patient care cost approaching $23 billion (more than $55 million per day) (5), which again is likely an underestimate, as the chronic effects of sepsis months to years post-intensive care unit (ICU) discharge are unknown (6).
Sepsis induces a profound state of both acute and chronic immune dysregulation, which contributes to both the mortality and long-term morbidities (7). These long-lasting morbidities include frequent re-hospitalization within the year following sepsis diagnosis, with the most common admission diagnoses being pneumonia or urinary tract infection (8). Interestingly, there are no FDA-approved therapeutics for the immunologic treatment of sepsis despite over 150 clinical trials and successful pre-clinical testing (9). In-hospital management remains primarily supportive in nature. Post-hospitalization, Prescott and Angus in their 2018 JAMA review of enhancing recovery from sepsis had only three recommendations: (1) identify new problems and treat appropriately, (2) review and adjust long-term medications, and (3) evaluate for treatable conditions that result in rehospitalization (10). None of these approaches target the unique immunologic and physiologic consequences of critical illness, as our understanding of the acute sepsis survivor remains incomplete!
Sepsis is associated with an early/immediate “systemic inflammatory response” (11). Authors have used the terms “cytokine storm” and “genomic storm” for this early response (12, 13), but these are gross over-simplifications of an integrated innate and adaptive immune response. At its most fundamental, the host response we define as “sepsis” is due to the recognition and response of host protective immune systems to microbial pathogens and their products (termed pathogen-associated molecular patterns; PAMPS) or the consequential release of endogenous alarmins (danger-associated molecular patterns; DAMPs) (14, 15). Acutely, the response appears aimed at isolating microbial growth and limiting replication, although it is well-known that when the response is either exaggerated or becomes systemic, can be associated with microcirculatory defects, organ injury, and death (16, 17).
The term “storm” is prescient, since the response, especially when the exposure is systemic, can produce devastating widespread host responses, including microcirculatory failure, profound vascular and organ injury, shock and death (18, 19). In the 1980's and early 1990's when many of the cytokines/mediators involved in this “storm” were originally identified and cloned, the complexity and breadth of the immediate host response to microbial products was not fully understood. Initial efforts to modify the host response to sepsis targeted this early/immediate response by interfering with individual microbial products and early cytokine appearance (20). Unfortunately, antibodies or immunoadhesins to endotoxin, TNFα, IL-1, IFNγ to name a very few, have all failed to improve outcomes to sepsis (21).
Although the reasons for the failure of these attempts to prevent this early “storm” are clearly multifactorial, including timing, redundancy of action, and heterogeneity of the patient population, there was also an over-assumption that treating sepsis would only require identifying the “silver bullet” responsible for the immediate organ damage (22). If there is anything that the failures of the past three decades have taught, it is that successful treatment of sepsis is, and will continue to be, an iterative process dependent upon both increasing our basic understanding of sepsis pathophysiology and translating this knowledge into improved clinical management.
What was learned was that one key to improving in-hospital survival was earlier sepsis recognition and initiation of treatment (23). Rapid diagnosis and initiation of sepsis treatment bundles have been a major hospital-systems' accomplishment (24). A second important key was the implementation of standardized best practices in the management of the sepsis patient (25). Almost two decades after implementation of the Surviving Sepsis Campaign, a 25% improvement in compliance with best-practice has resulted in a 9% absolute reduction in 28 day all-cause mortality (26). In the largest study to date, with over 1 million subjects, the state of New York-mandated early interventions significantly improved sepsis survival compared to states that did not mandate early intervention (27).
Earlier recognition and more consistent best-practice management have resulted in fewer patients dying early from the consequences of the “storm” (28, 29). This has resulted in more in-hospital survivors, and the appearance of late immunological complications of trauma and sepsis are now becoming the norm (30). As many as 33% of all sepsis survivors do not rapidly or fully recover but, instead, develop a new syndrome of “chronic critical illness” (CCI) (31–33). Chronic critical illness is represented by a persistent low grade inflammatory and chronic immunosuppressive phase associated with functional declines that can last from months to years following the acute event (Figure 1) (32).
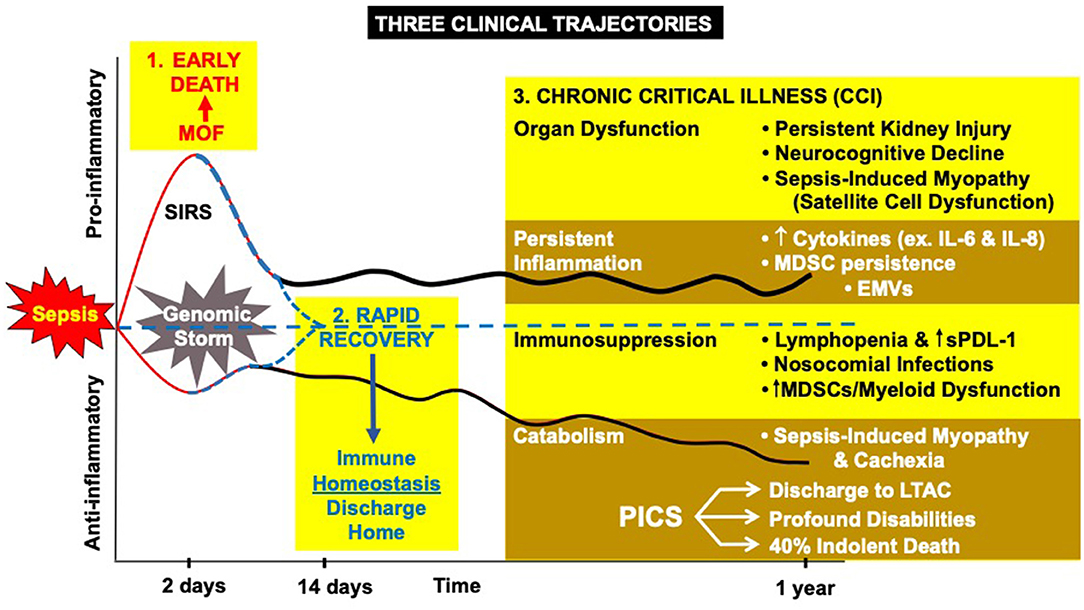
Figure 1. The host response to severe sepsis can have three different clinical trajectories: (1) early MOF leading to death, (2) rapid recovery, or (3) the new appearance of chronic critical illness (CCI) characterized by organ dysfunction, inflammation, immune suppression, and protein catabolism. MDSC, myeloid derived suppressor cells; EMVs, endothelial microvesicles; sPDL-1, soluble protein death ligand 1; LTAC, long-term acute care hospital.
These mechanisms underlying post-sepsis immunosuppression and inflammation are poorly understood, limiting our ability to prevent secondary infections and improve long-term outcomes in sepsis survivors. Chronic critical illness does not have a single consensus definition, like sepsis, but all define it as ongoing/persistent manageable organ dysfunction requiring hospitalization and increased resource utilization (e.g., ICU management) (31, 34). Based upon length-of-stay based mortality data from ICU patients at Shands UF Health, we set the duration of hospitalization at 2 weeks, and this definition has gained acceptance by others (35).
Our own studies have suggested that the severity of the initial acute phase, the age of the patient, the number of pre-existing comorbidities, and the influence of kidney injury, all impact the development of CCI (36). Sepsis survivors who develop CCI experience a higher frequency of secondary infections, have longer hospital stays, and poor disposition (37). Dramatically, 60% of CCI patients are readmitted in the first 6 months (38), usually for recurrent infections, and 40% of these patients are dead at 6 months (39). Not unexpectedly, 70% of deaths are preceded by a withdrawal of care (40, 41).
Chronic critical illness represents the clinical manifestation/endpoint of a complex immunological “dyscrasia” that results in increased susceptibility to secondary infections; poor functional, physical and cognitive outcomes (34, 38, 39). We have argued that the metabolic and immunologic underpinnings of this response is the immunological endotype we have termed the Persistent Inflammatory, immunosuppressive and protein Catabolic Syndrome (PICS) (32, 42, 43). As summarized in Table 1, sepsis survivors with CCI experience the very common PICS endotype reflected by persistently elevated inflammatory cytokines and DAMPs, immune suppression, and an increased number of opportunistic infections.
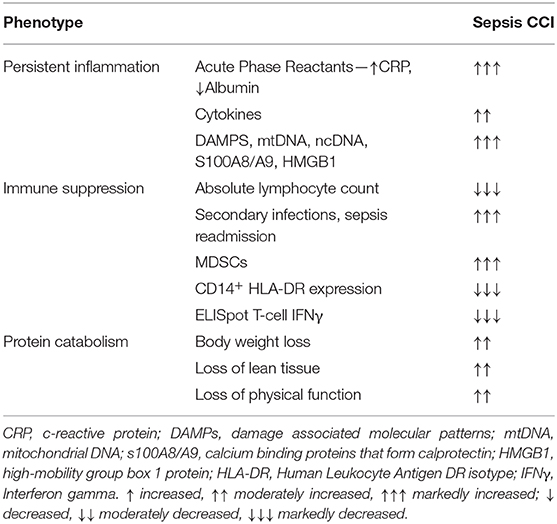
Table 1. CCI phenotype in severe trauma and sepsis survivors.
A “phenotype” is defined as a set of observable characteristics of an individual resulting from the interaction of its genotype with the environment. CCI can be classified as a phenotype because of its reproducible characteristics: ongoing manageable organ injury requiring at least 14 days of ICU care. In contrast, an endotype is defined as a subtype of a condition represented by a distinct functional or pathobiological mechanism (44). Importantly, endotypes differ from phenotypes because the former requires a common underlying mechanism. Endotypes, unlike phenotypes, can be associated with clusters of disease. PICS would classify as an endotype, since it may well be a common underlying mechanism that can explain not only sepsis and trauma CCI, but could also explain in part, cancer cachexia, and the chronic inflammation and lean tissue wasting associated with chronic obstructive pulmonary disease, cardiac cachexia and chronic renal disease.
The immunological dyscrasia that defines PICS is multifactorial. Primary mechanisms leading to sepsis-induced impairment of adaptive immune system include: (i) apoptosis-induced T-cell depletion, (ii) T-cell exhaustion due to upregulation of inhibitory receptors or downregulation of essential co-stimulatory receptors, (iii) decreased bone marrow lymphopoiesis, and (iv) myeloid-based T-cell suppression, and myeloid cell dysregulation (45–47).
“Emergency myelopoiesis” is defined as inflammation-induced hematopoiesis, which is critical for the immediate management of tissue injury and controlling infection (48–50). In contrast to adaptive immune cells, such as T cells and B cells, that proliferate in response to their specific antigens, innate myeloid populations are continuously replenished from hematopoietic stem cells (HSCs) and progenitors in bone marrow (BM) and extramedullary (42, 51). However, the molecular mechanism of emergency myelopoiesis during infection remains incompletely understood. HSCs and hematopoietic progenitors can directly sense the presence of pathogens or endogenous alarmins via pattern recognition receptors (PRRs) such as Toll-like or NOD-like receptors (TLRs, NLRs), and they can also respond to pro-inflammatory cytokines such as interferon (IFN)-α, IFN-γ, interleukin (IL)-1, tumor necrosis factor (TNF)-α, and granulocyte colony-stimulating factor (G-CSF) (42). IFN-α and IFN-γ have pleiotropic effects on many cell types, including HSCs and hematopoietic progenitors (52). Importantly, these cytokines, along with IL-27, have been demonstrated to induce an expansion of HSCs and myeloid progenitors, leading to the production of differentiated PMNs, macrophages and dendritic cells at the cost of both lymphopoiesis and erythropoiesis (49, 50).
Dmitry Gabrilovich has argued that the long-term host response to cancer, chronic infection, and sepsis (Figure 2B) results in what he has termed “pathological activation of neutrophils and monocytes” (53). Weak activation signals that occur in sepsis survivors during CCI, such as endogenous alarmins released from low grade organ injury, secondary colonization and infection from invasive ICU procedures, immobility, and delirium, all result in the mild but consistent elevated production of inflammatory cytokines and signals that drive persistent “emergency myelopoiesis.” What Gabrilovich has argued (54), and we have experimentally demonstrated in both trauma and sepsis (55), is that long-duration, low-grade inflammation drives pathologic myeloid cell activation and T-cell exhaustion, leading to both persistent inflammation and immune suppression (PICS).
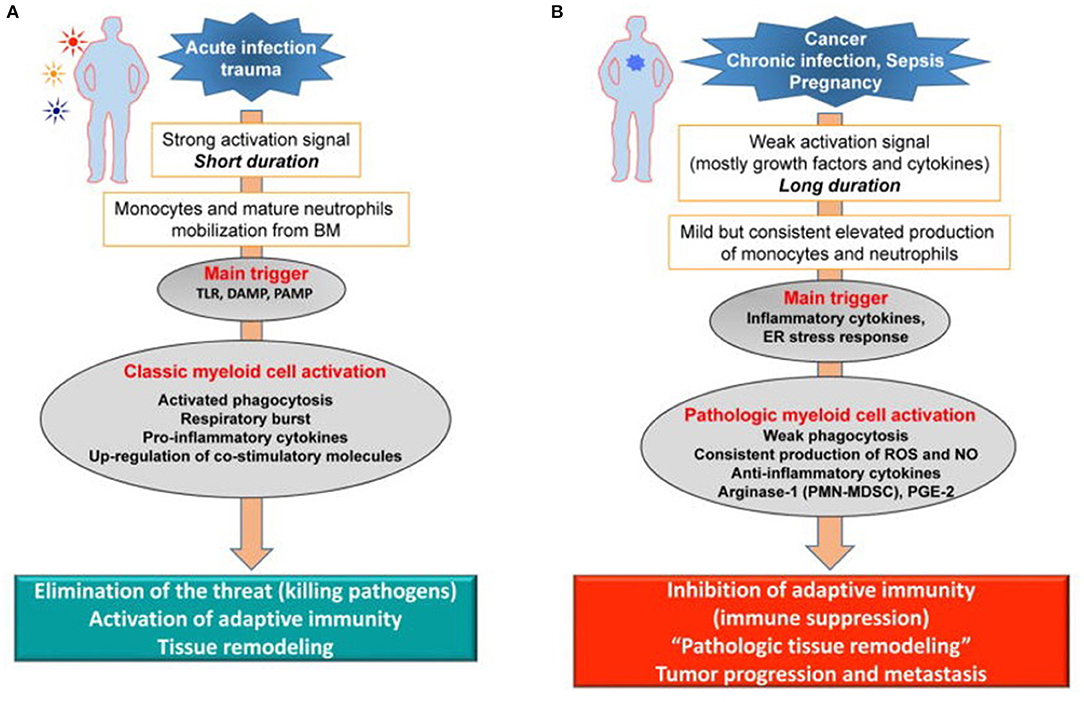
Figure 2. Pathologic activation of neutrophils and monocytes. (A) In the presence of strong activation signals coming from pathogens in the form of toll-like receptors ligands (TLRL), damage associated molecular pattern (DAMP), pathogen-associated molecular patterns (PAMP) molecules monocytes and neutrophils are mobilized from the BM. This response results in classic myeloid cell activation. (B) In the presence of weak activation signal mediated mostly by growth factors and cytokines, myeloid cells undergo modest but continuous expansion. Pro-inflammatory cytokines and ER stress responses contribute to pathologic myeloid cells activation that manifests in weak phagocytic activity, increased production of reactive oxygen species (ROS), nitric oxide (NO), arginase 1 (not expressed in human monocytes and M-MDSC) and prostaglandin-E2 (PGE2). This results in immune suppression. Endoplasmic reticulum = ER. Modified from Veglia et al. (53).
During unresolved inflammation, such as following microbial infection, ongoing tissue injury, and other chronic conditions; the nature of signals affecting T-cells and myeloid cells differs from that seen during the early or immediate “genomic or cytokine storm” (56). Reduced bone marrow and thymic generation of new T-cells and increased expression of immunosuppressive receptors favors exhaustion and apoptosis of T-cell populations resulting in lymphopoiesis. The expansion of myeloid-derived suppressor cells (MDSCs) in sepsis is a complex and gradual phenomenon governed by multiple factors. Gabrilovich has argued that accumulation of MDSC requires two groups of signals: the first leading to expansion of immature myeloid cells and the second, pathological activation as MDSCs. The first group of signals is driven as a direct host response to the microbial pathogen and includes: GM-CSF, G-CSF, M-CSF, SCF, VEGF, and polyunsaturated fatty acids (PUFAs) (53, 57). Transcriptional factors/regulators including STAT3, STAT5, IRF8, C/EBPβ, and NOTCH play a major role in this process (58). Other factors involved in this process include adenosine receptors A2b, NLRP3, and alarmins S100A8 and A9 (59). Importantly, the second group of signals, resulting in the pathological activation of MDSCs, does not require an infectious process and can be provided by inflammatory cytokines and endogenous alarmins alone, which include interferon (IFN)-γ, IL-1β, IL-4, IL-6, IL-13, IL-27, TNF-α, and the TLR ligand, HMGB1 (59).
First and foremost, these stimuli drive the expansion of bone marrow and extramedullary myelopoiesis. Neutrophils and monocytes generated under these conditions display a variant phenotype and morphology. They are characterized by relatively weak phagocytic activity, increased levels of reactive oxygen species (ROS) and nitric oxide (NO) production, and high expression of arginase 1, PGE2, and a number of anti-inflammatory cytokines (60, 61). Most of these features are absent in classically activated neutrophils and monocytes, which is why Gabrilovich has characterized this activation as “pathologic” (54). This state of activation leads not to the elimination of the threat or activation of host protective immunity, but to the inhibition of adaptive and innate immunity. Cells in this pathologic state of activation can be identified functionally, biochemically, and, to some extent, phenotypically, and are now termed MDSCs. The longer the myeloid compartment is exposed to the effects of factors described above, the more potent the pathologic activation of these MDSCs. Therefore, at any given moment, there is a heterogeneous population of cells in tissues represented by classically activated neutrophils, monocytes, and pathologically activated MDSCs (Figure 2). In the early stages of sepsis, bona-fide immune suppressive MDSCs are rarely detected (62, 63). However, there are cells with some biochemical and genomic characteristics of MDSCs, which probably represent an intrinsic part of MDSC development (53, 62).
The evidence that expansion of immunosuppressive MDSCs is a constant response to prolonged sepsis is incontrovertible. As early as 2007, we demonstrated that by 7 days post-sepsis, up to 95% of bone marrow cells are of myeloid lineage, mostly immature and functional MDSCs (64). These cells also overwhelm secondary lymph tissues, such as the spleen, lymph nodes, and reticuloendothelial tissues (such as the lung and liver) (65). Importantly, we have demonstrated that rapid, sustained presence of MDSCs, and their quantitative levels are strong predictors of nosocomial infections and poor discharge outcomes in sepsis patients (7). These findings were confirmed by Uhel, who established that sepsis survivors with expanded MDSC populations had a higher rate of reinfection and hospital readmission (66).
Additionally, McCall et al. demonstrated that these cells evolve functionally over time, becoming more immunosuppressive (32). With regard to MDSC suppressor activity, Hollen et al. have observed considerable time-dependent differences in MDSC suppression of T-cell proliferation. Much to our surprise, but consistent with Gabrilovich's overarching hypothesis (Figure 2), pathological activation of MDSCs in humans did not occur immediately after sepsis, but required 7–14 days to develop fully. Regardless of whether MDSCs came from sepsis survivors who developed CCI or rapidly recovered, PBMC-derived CD11b+CD33+HLA-DRdim MDSCs obtained prior to day 7 were not immunosuppressive, while MDSCs obtained at or after day 14 (all CCI patients) suppressed both autologous CD4+ and CD8+ T-cell proliferation to antiCD3/CD28 (Figure 3). Also, septic MDSCs from day 14 (late), but not from day 4 (early), potently suppressed stimulated T-cell production of IL-2 and, to a lesser extent, IFNγ.
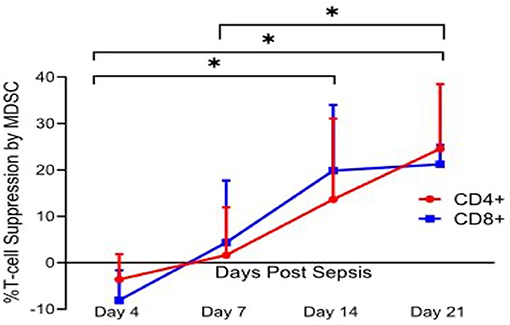
Figure 3. Percent T lymphocyte suppression by MDSCs. Immature myeloid cells with the surface markers CD33+CD11b+HLA-DRdim were isolated on days 4, 7, 14, and 21 after sepsis, and from healthy control subjects. Autologous T lymphocytes were stimulated with soluble anti-CD3/28 and seeded in a co-culture with MDSCs in a 1:1 ratio. T cells were labeled with CellTrace Violet to detect proliferation, and a proliferation index (PI) was calculated for both CD4+ and CD8+ T cells using flow cytometric analysis. Percent suppression was calculated as the ratio of PI from stimulated T cells in the presence of MDSCs and the PI of stimulated T cells in culture medium alone. Percent suppression for both CD4+ and CD8+ T cells was significantly different between day 4 vs. 14 (p = 0.0402 and 0.0012), day 4 vs. 21 (p = 0.0225 and <0.0001), and day 7 vs. 21 (p = 0.037 and 0.045). There was no significance noted of percent suppression of CD4+ and CD8+ T cells between days 7 and 14 (p = 0.17 and 0.08). This T cell suppression was not seen in age-matched healthy control subjects. Modified from Hollen et al. (62). *indicates statistically significant intervals (p < 0.05).
More interestingly, most of the MDSCs in septic CCI patients were granulocytic with a gene expression profile reflective of a highly inflammatory and immunosuppressive transcriptome (67). Analysis of individual gene transcripts from bulk cell-sorted human CD11b+CD33+HLA-DRdim MDSCs was consistent with suppressed HLA gene expression and up-regulated inflammatory gene expression (Figure 4). Canonical Pathway and Causal Network Analysis supported these pathway alterations and a pattern of simultaneous low-grade inflammation with immunosuppression.
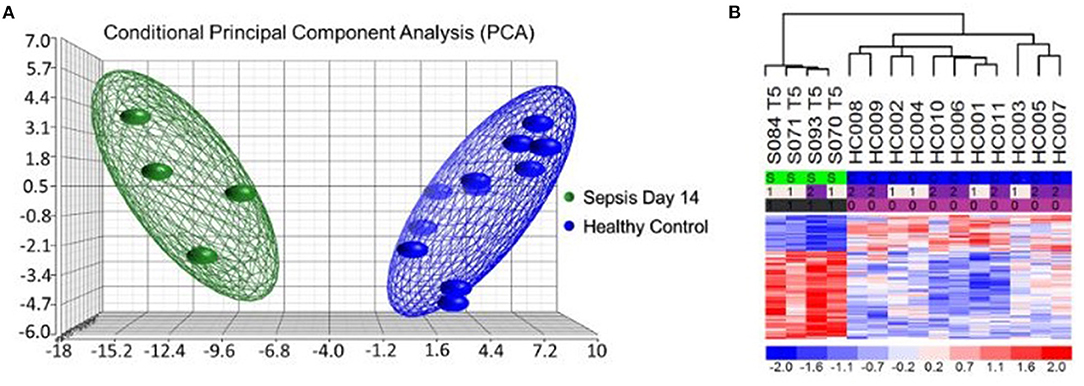
Figure 4. Microarray Transcriptomic Analysis of MDSCs from Patients 14 days after Sepsis and Healthy Control Subjects. The genomic response of bulk isolated MDSC RNA in healthy controls and septic patients 14 days after initial infectious onset. (A) Conditional principal component analysis of septic (day 14) and healthy control MDSC gene expression patterns. (B) Heat map of the hierarchical clustering of MDSC gene expression patterns and variation between septic patients (S) from day 14 and healthy (H) control subjects. Modified from Mathias et al. (67).
Although G-MDSCs (granulocyte-like MDSCs) comprise the largest subpopulation of MDSCs in sepsis, expansion of M-MDSCs (monocytic MDSCs) and E-MDSCs (early MDSCs) is also observed. Importantly, different subpopulations of MDSCs are immunosuppressive through different mechanisms, and can, therefore, have different targets for intervention (53). To understand the rich “landscape” of blood MDSCs late after sepsis (day 21), single-cell RNA sequencing and Cellular Indexing of Transcriptomes and Epitopes by Sequencing (scRNA-seq and CITE-seq) was conducted on enriched MDSCs obtained from peripheral blood mononuclear cells (PBMCs) (Figure 5). This was conducted to identify individual populations of MDSCs (G-, M-, and E-MDSCs) and their transcriptomic profiles in healthy and septic patients. In this case, samples were obtained at day 21 from two sepsis survivors with CCI, and samples were also obtained from two age and sex matched healthy, control subjects. Samples were first isolated on a Ficoll gradient, and then CD11b+CD33+HLA-DRdim cells were mixed 3:1 with original PBMCs to assure inclusion with all cell populations. sc-RNAseq of over 150,000 cells were conducted.
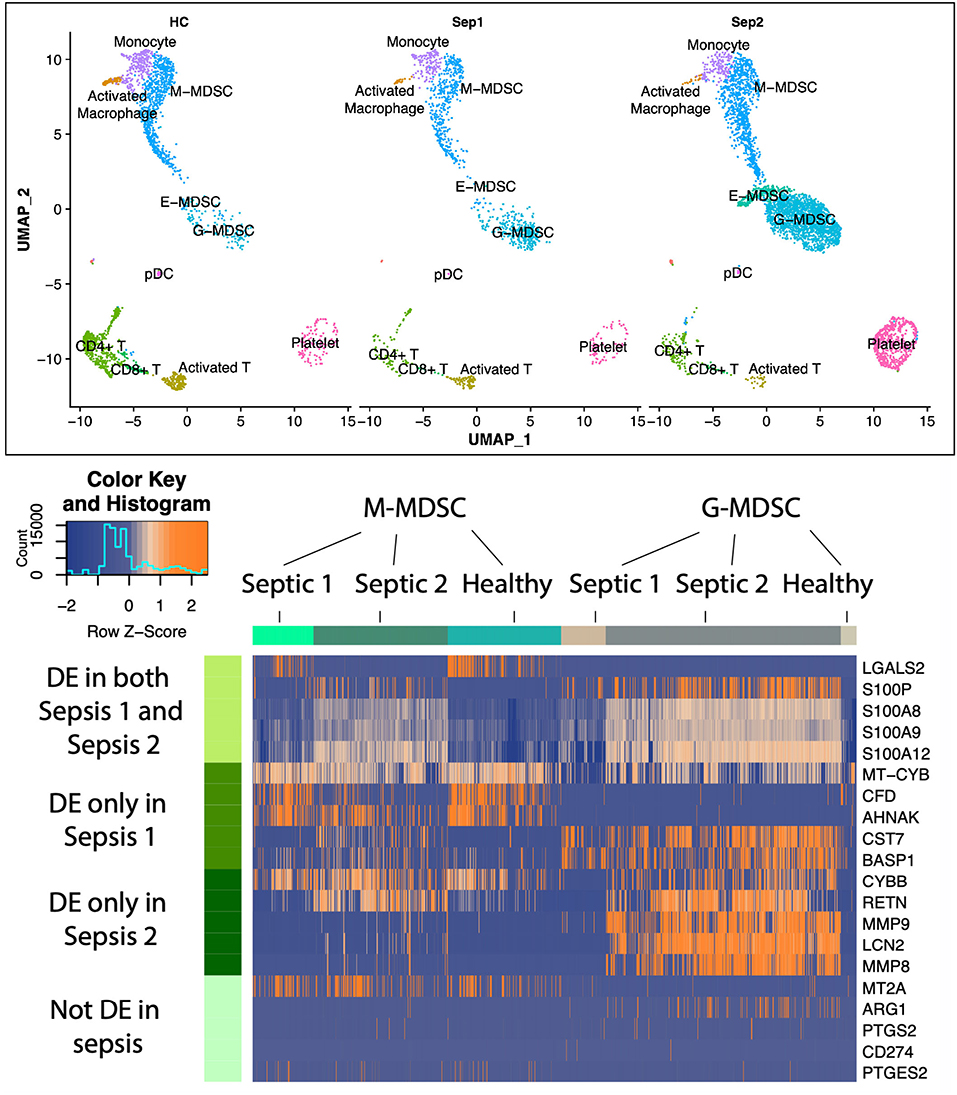
Figure 5. Uniform manifold approximation and projection (UMAP) plots of cell clusters identified in healthy patients (n = 2,340 cells) vs. sepsis 1 (bacteremia, sepsis; n = 1,544 cells) and sepsis 2 (fungemia, septic shock; n = 5,587 cells) showing three distinct MDSC subsets. Heatmap illustrating expression patterns of MDSC subsets at 21 days post-sepsis vs. healthy control subjects. Rows represent the specific genes of interest differentially expressed in both sepsis patients; only sepsis patient 1; only sepsis patient 2; and genes not differentially expressed in this study, but previously determined to be important to MDSC function in cancer and autoimmunity. Number of columns represent number of cells analyzed in each group. DE, differentially expressed genes in sepsis vs. healthy controls. Colors represent mean normalized relative expression with blue representing reduced expression and orange, increased expression. Modified from Darden et al. (68).
As displayed in Figure 5, there was a dramatic expansion of the G-MDSC subpopulation and a less dramatic expansion of M-MDSCs in the sepsis patients. E-MDSCs, which were not detectable from healthy human subjects, were modest in sepsis survivors with CCI (detectable in only one of the sepsis subjects). G-MDSCs showed not only the greatest expansion, but also the most dramatic changes in their transcriptome. Interestingly, we did not see an increase in expression among genes that are associated with immunosuppression in cancer (such as ARG1, CD274, COX2, PGE2, and NOS2). While this was a small pilot study, it suggests that MDSCs present in sepsis may be inherently different from those seen in cancer-associated immunosuppression.
The global population is rapidly aging (70, 71) with increasing healthcare resources and costs devoted to this group. The frequency of hospitalizations for sepsis in patients over 50 has increased, most dramatically in patients aged 65 years or greater (72). Advanced age is also associated with more severe organ failure, infectious complications, increased ventilator days, a longer ICU LOS, an increased 28-day mortality, and an increased likelihood of discharge to skilled nursing or long-term care facilities (73).
As seen in younger populations, the in-hospital mortality from sepsis in the elderly is decreasing, but still remains significantly higher. The risk of CCI and discharge to a non-home destination is also increased in the elderly population and is multifactorial in nature (33, 72, 74). Contributing factors include senescence (normal aging), inflammaging (chronic, subclinical inflammation), comorbidities, lack of physiologic reserve, pre-existing disability, and epigenetic changes. These factors prevent older individuals from readily returning to homeostasis following critical illness and contribute to the increased risk of morbidity and mortality following sepsis (70, 71, 75, 76).
Aging has a profound role on the immune system. Immunosenescence is a state of age-associated changes in the immune system which is characterized by decreased ability to mount an effective response to pathogens (77, 78), decreased competency of the adaptive immune system (as evidenced by decreases in naïve peripheral T cells, repertoire diversity, and immunocompetent B cells) (77, 79), and dysfunctional myelopoietic effector cells (i.e., PMNs, monocytes/Mϕ, DCs, and NK cells) (78). Furthermore, the aged host's HSCs preferentially induce myelopoiesis, contributing to the substantial increase in MDSCs seen in this population as a response to the initial insult (78). “Inflammaging” is defined as chronic, low-grade inflammation that occurs with physiologic aging. It is a unique response seen in aged mammals and differs from the responses seen in the young. The “cytokine storm” seen in the younger population can be markedly attenuated or absent in the aged population, whereas immunosuppression appears to dominate (73). In these cases, early mortality is due, instead, to failure of host protective immune mechanisms to adequate microbial control.
In murine models, sepsis induces a rapid release of mature and immature myeloid cell populations from the bone marrow in response to endogenous and exogenous danger signals (55, 60). This creates niches in the bone marrow, which stimulate emergency myelopoiesis (80). Myelopoiesis predominates at the expense of lymphopoiesis and erythropoiesis (64, 80). Interestingly, elderly HSCs have this phenotype and function prior to critical illness, with myeloid-skewed cell production and a decreased ability to produce lymphoid cells (81, 82). These HSCs are also functionally inferior to their younger counterparts, with a lower functional frequency, delayed proliferative response, and reduced efficiency for short term homing (81, 82). These baseline dysfunctions are exacerbated by acute critical illness.
Skeletal muscle serves as the largest protein reserve in the body, which can be mobilized for metabolic substrates in times of stress. Critical illness is characterized by marked protein catabolism, which results from increased muscle breakdown, decreased protein synthesis, and the release of potential pro-inflammatory degradation products (83, 84). In patients who progress to CCI, this is a self-perpetuating cycle that results in profound cachexia. The exact mechanism has not been fully elucidated, but likely involves inflammation- and oxidation-associated direct mitochondrial and myocyte injury (85). Not surprisingly, muscle catabolism can result in the release of DAMPs (including mtDNA, HMGB1, and TFAM) into the systemic circulation, driving persistent inflammation (83, 86–88). In animal models, the mtDNA-TLR9-RAGE pathway, which can be activated by mtDNA or TFAM, has been shown to be involved in sepsis-induced cardiac inflammation (86, 89). As with other endogenous alarmins, these increases in both local tissue damage and systemic inflammation drives ongoing functional immunosuppression at both the level of the bone marrow (enhanced myelopoiesis) and functional lymphocyte populations (Figure 6). The role of MDSCs in cancer cachexia has been investigated for years, and several therapies are targeted at altering their function and have shown promise (90, 91).
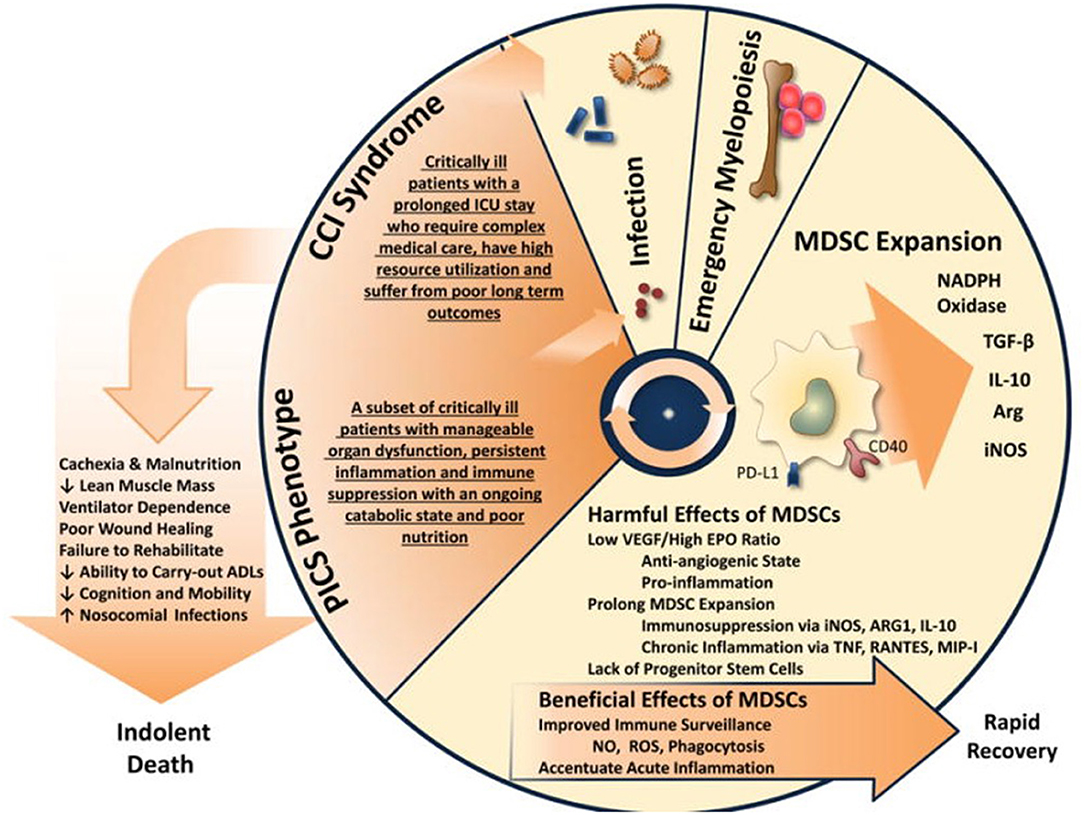
Figure 6. The proposed self-perpetuating cycle by which PICS drives muscle loss and inflammation. Modified from Mira et al. (69).
Muscle wasting not only contributes to the ongoing inflammatory state seen in CCI, but also leads to a substantial functional disability (92, 93). Loss of skeletal muscle mass is associated with profound functional deficits which are most notable in the aging due to their baseline declining muscle mass (94). In these populations, even small functional changes can cause a shift from independent to dependent living (95). Decreased skeletal muscle mass is also associated with increased falls in the elderly, which are an independent predictor of 1-year mortality (96). Functional declines aren't limited to the aging population—over 80% of critical illness survivors report a reduction in physical ability (97). Physical limitations preclude patients from returning to work and contribute substantially to their reduced healthcare-related quality of life (97, 98). As seen with the self-perpetuating cycle of PICS in CCI patients, reductions in physical capabilities propagate ongoing mental health issues, economic hardship, and burden on the healthcare system.
Organ injury contributes to the ongoing inflammation associated with CCI. The kidney plays a crucial role in both initial and long-term survival from sepsis. AKI has been associated with increased in-hospital mortality and is more prevalent in sepsis than other critical illnesses (99). This relationship is bidirectional, as patients with acute and/or chronic kidney injury are also more likely to develop sepsis (100–103). Failure to resolve AKI is associated with both increased risk of initial mortality and progression to CCI (33, 104, 105). Chronic kidney disease (CKD) is not just a marker of CCI but perpetuates the cycle as well. Renal tubule epithelial cells are highly susceptible to oxidative stress and release large quantities of DAMPs. Urinary analysis of septic patients reveals increased levels of DAMPs and over expression of several DAMP receptors (106). These DAMPs act locally to increase secretion of chemokines by renal parenchymal and dendritic cells (DCs), which further promote local inflammation (107–109). They also have systemic effects, which are mediated by PPR toll-like receptors (TLRs) (110). TLRs are also upregulated in AKI via epigenetic remodeling, priming the renal tubule epithelial cells to release increased amounts of cytokines in response to antigen stimulation (111). This “hyper-responsive state,” concomitant with decreased renal clearance, leads to amplified systemic inflammation and resultant organ injury (112, 113).
While the relationship between AKI and acute illness is well-established, the role of the kidney in CCI is more elusive. Its role in filtration exposes the kidney to over 30 times the blood volume daily (114), meaning renal DCs and lymph nodes are exposed to inflammatory mediators and pathogens significantly more than other tissues. This results in a positive feedback loop in which exposure to these stimuli result in further oxidative damage and release of additional inflammatory mediators, both as a response to the filtered pathogens and to the ongoing tubular necrosis (115). As previously discussed with sepsis-associated muscle wasting, these self-perpetuating cycles of ongoing cell death and inflammation result in development of a persistent inflammatory state rather than return to homeostasis.
Sepsis is, at its foundation, a heterogenous disease process that occurs in a diverse patient population, especially aged and those with pre-existing comorbidities. The resulting CCI in many sepsis patients is inherently intertwined with the patient's prior comorbidities and functional status. This complex pattern of seemingly infinite variables makes both clinical decision-making and systematic investigation difficult. Human studies are difficult to standardize and depend on long-term participation of patients, many of whom are overburdened with their disease process.
Animal models have been generally successful in the investigation of early sepsis responses associated with the “genomic or cytokine storm.” However, they fall short in modeling long-term processes. To better investigate these chronic mechanisms driving CCI, continued bidirectional translational research is required. Our group, along with several others, have proposed a semi-lethal cecal ligation and puncture (CLP) model with daily chronic stress to approximate the PICS endotype seen in human CCI (116–121). However, this model fails to accurately represent the sterile inflammation present in most CCI patients, and replaces it with a peritoneal abscess (122). This model also relies on young, otherwise healthy mice. It fails to capture the interplay between aging and comorbidities demonstrated in human patients. We have demonstrated that the sepsis response is notably different in aged mice when compared to their juvenile counterparts (123). Understanding these complex, interconnected mechanisms is crucial to further understanding of this disease process and development of therapeutic interventions.
Sepsis and CCI are immune dyscrasias at their foundation, and PICS is the predominant endotype behind CCI. Thus, a large majority of interventional studies have focused on the restoration of immune system homeostasis. Leukocyte growth factors (e.g., G-CSF) (124–128) immunomodulatory cytokines (e.g., IL-7, IL-15, and IFN-γ) (129–133) inhibitors of negative co-stimulatory pathways (e.g., anti-PD-1/PD-L1 Ab, anti-CTLA-4 Ab, anti-TIM3 Ab, and anti-LAG-3 Ab) (134–140) and the thymic peptide thymosin-α1 (141) have all been or are being investigated for potential benefits. These trials have been largely unsuccessful at finding a “silver bullet” cure, but some have shown promise in selective populations (124, 125). This is not surprising given the heterogenous disease process and highlights the importance of continued investigative efforts in endotyping these patients as a precision medicine approach. Oncology research has been successful in developing targeted therapies for specific cancer patients with PICS-like endotypes. These approaches may be applicable to sepsis-induced PICS, but further research is required.
Nutritional support is paramount in both the acute and chronic treatment of sepsis and CCI. As discussed before, the loss of skeletal muscle contributes to both the functional declines seen in CCI patients and the perpetuation of the PICS endotype. Early implementation of nutrition is clearly important, but the optimal protein and nutrient requirements remain undetermined. However, given their phenotypic similarities to cancer cachexia and aging sarcopenia, CCI patients likely have a daily protein requirement of roughly 1.5–2.0 g/kg/day (142–146). Arginine supplementation in sepsis remains controversial given its role as an intracellular substrate for nitric oxide. However, the upregulation of arginase-1 by MDSCs may result in a relative arginine deficiency (67, 147). Arginine is necessary for proper T-cell receptor function and wound healing (148, 149). Therefore, arginine supplementation may counteract the persistent arginine deficiency due to persistent MDSC expansion during PICS, promoting lymphocyte proliferation and improved tissue repair. Leucine is another amino acid that shows promising results, as it decreases muscle protein catabolism and induces protein synthesis (150). Leucine and other branched chain amino acids (BCAA) supplementation resulted in improved nutritional and immunologic parameters, such as nitrogen balance, prealbumin levels, and lymphocyte counts (151). It has also been shown to increase muscle protein synthesis through the mTOR pathway (152, 153). Studies in large burns have also shown promise using adjuncts such as insulin, oxandrolone, and propranolol to maintain an anabolic state (154–156).
Decreases in functional status are closely associated with decreases in health-related quality of life (QOL) among CCI patients. Maintaining, or improving, baseline functional status is the ultimate goal, but the prevention of unnecessary muscle loss is vital. Early ICU-based exercise and physical therapy programs have been associated with improved in-hospital outcomes, such as a reduction in the duration of mechanical ventilation and ICU length of stay. They are also associated with improved physical function after discharge (157). These programs, when combined with adequate nutritional support have demonstrated substantial improvements in muscle synthesis and functional outcomes (158). However, these have not been fully evaluated in the CCI population.
Technology and the field of medicine have developed rapidly over the past few decades. With the application of the Human Genome Project and the development of high throughput sequencing techniques, the development of individualized therapies has been made possible. These therapies have been remarkably successful in the treatment of cancer and congenital disease (159–161). Understanding the transcriptomic landscape of sepsis and CCI, and how they differ, is crucial to development of novel therapeutic agents for sepsis-induced CCI. Advances in technology have also made data collection and interpretation easier, making large, multicenter databases commonplace. The addition of biologic variables, in addition to clinical variables, will likely improve the prognostic power of these data sets and allow for early endotyping of patients (162–164).
“Big Data” is particularly useful in sepsis and CCI, as the disease process and patient population are increasingly heterogeneous. With the ability to quickly endotype a patient, accurate prognosis and optimal treatment is possible. We have shown recently that the leukocyte transcriptome within 48 h post-trauma is highly predictive of outcomes (165, 166). This technique, using regression-based prediction models, may be further improved by the use of machine-learning algorithms and deep-learning technologies (167, 168). Big data provides large sample sizes allowing for the identification of biomarker cutoff values with optimal sensitivity and specificity. However, these static thresholds fail to account for individual physiology; therefore, it is important that future efforts continue to improve upon precision medicine by integrating data from multicenter and multinational repositories with machine-learning and deep-learning technologies.
BF, DD, LK, PE, and LM drafted the manuscript. JR, SB, SL, and FM provided critical revisions. All authors made substantial contributions to the conception and design of the work, approved the submitted version of the manuscript, and agree to be accountable for all aspects of the work.
This work was supported, in part, by National Institute of General Medical Sciences grants: R01 GM-113945 (PE), R01 GM-040586 and R01 GM-104481 (LM), R01 GM-097531 (SL) and P50 GM-111152 (FM, SB, LM, PE) awarded by the National Institute of General Medical Sciences (NIGMS). In addition, this work was supported, in part, by a post-graduate training grant T32 GM-008721 (BF, DD, LK) in burns, trauma, and perioperative injury by NIGMS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. (2013) 41:1167–74. doi: 10.1097/CCM.0b013e31827c09f8
2. Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence. (2014) 5:4–11. doi: 10.4161/viru.27372
3. Afshar M, Arain E, Ye C, Gilbert E, Xie M, Lee J, et al. Patient outcomes and cost-effectiveness of a sepsis care quality improvement program in a health system. Crit Care Med. (2019) 47:1371–9. doi: 10.1097/CCM.0000000000003919
4. Yende S, Austin S, Rhodes A, Finfer S, Opal S, Thompson T, et al. Long-term quality of life among survivors of severe sepsis: analyses of two international trials. Crit Care Med. (2016) 44:1461–7. doi: 10.1097/CCM.0000000000001658
5. Hajj J, Blaine N, Salavaci J, Jacoby D. The “centrality of sepsis”: a review on incidence, mortality, and cost of care. Healthcare (Basel). (2018) 6:90. doi: 10.3390/healthcare6030090
6. Prescott HC, Costa DK. Improving long-term outcomes after sepsis. Crit Care Clin. (2018) 34:175–88. doi: 10.1016/j.ccc.2017.08.013
7. Stortz JA, Mira JC, Raymond SL, Loftus TJ, Ozrazgat-Baslanti T, Wang Z, et al. Benchmarking clinical outcomes and the immunocatabolic phenotype of chronic critical illness after sepsis in surgical intensive care unit patients. J Trauma Acute Care Surg. (2018) 84:342–9. doi: 10.1097/TA.0000000000001758
8. Guirgis FW, Brakenridge S, Sutchu S, Khadpe JD, Robinson T, Westenbarger R, et al. The long-term burden of severe sepsis and septic shock: sepsis recidivism and organ dysfunction. J Trauma Acute Care Surg. (2016) 81:525–32. doi: 10.1097/TA.0000000000001135
9. Hotchkiss RS, Moldawer LL. Parallels between cancer and infectious disease. N Engl J Med. (2014) 371:380–3. doi: 10.1056/NEJMcibr1404664
10. Prescott HC, Angus DC. Enhancing recovery from sepsis: a review. JAMA. (2018) 319:62–75. doi: 10.1001/jama.2017.17687
11. Raith EP, Udy AA, Bailey M, McGloughlin S, MacIsaac C, Bellomo R, et al. Prognostic accuracy of the SOFA score, SIRS criteria, and qSOFA score for in-hospital mortality among adults with suspected infection admitted to the intensive care unit. JAMA. (2017) 317:290–300. doi: 10.1001/jama.2016.20328
12. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. (2017) 39:517–28. doi: 10.1007/s00281-017-0639-8
13. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med. (2011) 208:2581–90. doi: 10.1084/jem.20111354
14. Denk S, Perl M, Huber-Lang M. Damage- and pathogen-associated molecular patterns and alarmins: keys to sepsis? Eur Surg Res. (2012) 48:171–9. doi: 10.1159/000338194
15. Raymond SL, Holden DC, Mira JC, Stortz JA, Loftus TJ, Mohr AM, et al. Microbial recognition and danger signals in sepsis and trauma. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:2564–73. doi: 10.1016/j.bbadis.2017.01.013
16. Iba T, Levy JH. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J Thromb Haemost. (2018) 16:231–41. doi: 10.1111/jth.13911
17. Fani F, Regolisti G, Delsante M, Cantaluppi V, Castellano G, Gesualdo L, et al. Recent advances in the pathogenetic mechanisms of sepsis-associated acute kidney injury. J Nephrol. (2018) 31:351–9. doi: 10.1007/s40620-017-0452-4
18. Huerta LE, Rice TW. Pathologic difference between sepsis and bloodstream infections. J Appl Lab Med. (2019) 3:654–63. doi: 10.1373/jalm.2018.026245
20. Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. (2014) 20:195–203. doi: 10.1016/j.molmed.2014.01.007
21. Seeley EJ, Bernard GR. Therapeutic targets in sepsis: past, present, and future. Clin Chest Med. (2016) 37:181–9. doi: 10.1016/j.ccm.2016.01.015
22. Vincent JL, Sakr Y. Clinical trial design for unmet clinical needs: a spotlight on sepsis. Expert Rev Clin Pharmacol. (2019) 12:893–900. doi: 10.1080/17512433.2019.1643235
23. Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. (2001) 345:1368–77. doi: 10.1056/NEJMoa010307
24. Damiani E, Donati A, Serafini G, Rinaldi L, Adrario E, Pelaia P, et al. Effect of performance improvement programs on compliance with sepsis bundles and mortality: a systematic review and meta-analysis of observational studies. PLoS ONE. (2015) 10:e0125827. doi: 10.1371/journal.pone.0125827
25. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med. (2017) 45:486–552. doi: 10.1097/CCM.0000000000002255
26. Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC, Bion J, et al. The surviving sepsis campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Intensive Care Med. (2010) 36:222–31. doi: 10.1007/s00134-009-1738-3
27. Levy MM, Gesten FC, Phillips GS, Terry KM, Seymour CW, Prescott HC, et al. Mortality changes associated with mandated public reporting for sepsis. The results of the New York State Initiative. Am J Respir Crit Care Med. (2018) 198:1406–12. doi: 10.1164/rccm.201712-2545OC
28. Seymour CW, Gesten F, Prescott HC, Friedrich ME, Iwashyna TJ, Phillips GS, et al. Time to treatment and mortality during mandated emergency care for sepsis. N Engl J Med. (2017) 376:2235–44. doi: 10.1056/NEJMoa1703058
29. Ferrer R, Martin-Loeches I, Phillips G, Osborn TM, Townsend S, Dellinger RP, et al. Empiric antibiotic treatment reduces mortality in severe sepsis and septic shock from the first hour: results from a guideline-based performance improvement program. Crit Care Med. (2014) 42:1749–55. doi: 10.1097/CCM.0000000000000330
30. Kahn JM, Le T, Angus DC, Cox CE, Hough CL, White DB, et al. The epidemiology of chronic critical illness in the United States*. Crit Care Med. (2015) 43:282–7. doi: 10.1097/CCM.0000000000000710
31. Nelson JE, Cox CE, Hope AA, Carson SS. Chronic critical illness. Am J Respir Crit Care Med. (2010) 182:446–54. doi: 10.1164/rccm.201002-0210CI
32. Hawkins RB, Raymond SL, Stortz JA, Horiguchi H, Brakenridge SC, Gardner A, et al. Chronic critical illness and the persistent inflammation, immunosuppression, catabolism syndrome. Front Immunol. (2018) 9:1511. doi: 10.3389/fimmu.2018.01511
33. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. (2012) 72:1491–501. doi: 10.1097/TA.0b013e318256e000
34. Nomellini V, Kaplan LJ, Sims CA, Caldwell CC. Chronic critical illness and persistent inflammation: what can we learn from the elderly, injured, septic, and malnourished? Shock. (2018) 49:4–14. doi: 10.1097/SHK.0000000000000939
35. Prescott HC. Preventing chronic critical illness and rehospitalization: a focus on sepsis. Crit Care Clin. (2018) 34:501–13. doi: 10.1016/j.ccc.2018.06.002
36. Mira JC, Cuschieri J, Ozrazgat-Baslanti T, Wang Z, Ghita GL, Loftus TJ, et al. The epidemiology of chronic critical illness after severe traumatic injury at two level-one trauma centers. Crit Care Med. (2017) 45:1989–96. doi: 10.1097/CCM.0000000000002697
37. Rosenthal MD, Kamel AY, Rosenthal CM, Brakenridge S, Croft CA, Moore FA. Chronic critical illness: application of what we know. Nutr Clin Pract. (2018) 33:39–45. doi: 10.1002/ncp.10024
38. Loss SH, Nunes DSL, Franzosi OS, Salazar GS, Teixeira C, Vieira SRR. Chronic critical illness: are we saving patients or creating victims? Rev Bras Ter Intensiva. (2017) 29:87–95. doi: 10.5935/0103-507X.20170013
39. Gardner AK, Ghita GL, Wang Z, Ozrazgat-Baslanti T, Raymond SL, Mankowski RT, et al. The development of chronic critical illness determines physical function, quality of life, and long-term survival among early survivors of sepsis in surgical ICUs. Crit Care Med. (2019) 47:566–73. doi: 10.1097/CCM.0000000000003655
40. Unroe M, Kahn JM, Carson SS, Govert JA, Martinu T, Sathy SJ, et al. One-year trajectories of care and resource utilization for recipients of prolonged mechanical ventilation: a cohort study. Ann Intern Med. (2010) 153:167–75. doi: 10.7326/0003-4819-153-3-201008030-00007
41. Cox CE, Martinu T, Sathy SJ, Clay AS, Chia J, Gray AL, et al. Expectations and outcomes of prolonged mechanical ventilation. Crit Care Med. (2009) 37:2888–94; quiz 2904. doi: 10.1097/CCM.0b013e3181ab86ed
42. Horiguchi H, Loftus TJ, Hawkins RB, Raymond SL, Stortz JA, Hollen MK, et al. Innate immunity in the persistent inflammation, immunosuppression, and catabolism syndrome and its implications for therapy. Front Immunol. (2018) 9:595. doi: 10.3389/fimmu.2018.00595
43. Hesselink L, Hoepelman RJ, Spijkerman R, de Groot MCH, van Wessem KJP, Koenderman L, et al. Persistent inflammation, immunosuppression and catabolism syndrome (PICS) after polytrauma: a rare syndrome with major consequences. J Clin Med. (2020) 9:191. doi: 10.3390/jcm9010191
44. Russell CD, Baillie JK. Treatable traits and therapeutic targets: goals for systems biology in infectious disease. Curr Opin Syst Biol. (2017) 2:140–6. doi: 10.1016/j.coisb.2017.04.003
45. Patil NK, Bohannon JK, Sherwood ER. Immunotherapy: a promising approach to reverse sepsis-induced immunosuppression. Pharmacol Res. (2016) 111:688–702. doi: 10.1016/j.phrs.2016.07.019
46. Bouras M, Asehnoune K, Roquilly A. Contribution of dendritic cell responses to sepsis-induced immunosuppression and to susceptibility to secondary pneumonia. Front Immunol. (2018) 9:2590. doi: 10.3389/fimmu.2018.02590
47. Stieglitz D, Schmid T, Chhabra NF, Echtenacher B, Männel DN, Mostböck S. TNF and regulatory T cells are critical for sepsis-induced suppression of T cells. Immun Inflamm Dis. (2015) 3:374–85. doi: 10.1002/iid3.75
48. Fuchs A, Monlish DA, Ghosh S, Chang SW, Bochicchio GV, Schuettpelz LG, et al. Trauma induces emergency hematopoiesis through IL-1/MyD88-dependent production of G-CSF. J Immunol. (2019) 202:3020–32. doi: 10.4049/jimmunol.1801456
49. Furusawa J, Mizoguchi I, Chiba Y, Hisada M, Kobayashi F, Yoshida H, et al. Promotion of expansion and differentiation of hematopoietic stem cells by interleukin-27 into myeloid progenitors to control infection in emergency myelopoiesis. PLoS Pathog. (2016) 12:e1005507. doi: 10.1371/journal.ppat.1005507
50. Loftus TJ, Mohr AM, Moldawer LL. Dysregulated myelopoiesis and hematopoietic function following acute physiologic insult. Curr Opin Hematol. (2018) 25:37–43. doi: 10.1097/MOH.0000000000000395
51. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. (2013) 13:862–74. doi: 10.1038/nri3552
52. Maher SG, Romero-Weaver AL, Scarzello AJ, Gamero AM. Interferon: cellular executioner or white knight? Curr Med Chem. (2007) 14:1279–89. doi: 10.2174/092986707780597907
53. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
54. Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol. (2018) 35:19–28. doi: 10.1016/j.smim.2017.12.004
55. Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. (2011) 17:281–92. doi: 10.2119/molmed.2010.00178
56. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. (2016) 126:23–31. doi: 10.1172/JCI82224
57. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
58. Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol. (2015) 98:913–22. doi: 10.1189/jlb.4RI0515-204R
59. Haverkamp JM, Smith AM, Weinlich R, Dillon CP, Qualls JE, Neale G, et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity. (2014) 41:947–59. doi: 10.1016/j.immuni.2014.10.020
60. Delano MJ, Thayer T, Gabrilovich S, Kelly-Scumpia KM, Winfield RD, Scumpia PO, et al. Sepsis induces early alterations in innate immunity that impact mortality to secondary infection. J Immunol. (2011) 186:195–202. doi: 10.4049/jimmunol.1002104
61. Leliefeld PH, Wessels CM, Leenen LP, Koenderman L, Pillay J. The role of neutrophils in immune dysfunction during severe inflammation. Crit Care. (2016) 20:73. doi: 10.1186/s13054-016-1250-4
62. Hollen MK, Stortz JA, Darden D, Dirain ML, Nacionales DC, Hawkins RB, et al. Myeloid-derived suppressor cell function and epigenetic expression evolves over time after surgical sepsis. Crit Care. (2019) 23:355. doi: 10.1186/s13054-019-2628-x
63. Schrijver IT, Théroude C, Roger T. Myeloid-derived suppressor cells in sepsis. Front Immunol. (2019) 10:327. doi: 10.3389/fimmu.2019.00327
64. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. (2007) 204:1463–74. doi: 10.1084/jem.20062602
65. Cuenca AG, Cuenca AL, Winfield RD, Joiner DN, Gentile L, Delano MJ, et al. Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J Immunol. (2014) 192:6111–9. doi: 10.4049/jimmunol.1302895
66. Uhel F, Azzaoui I, Grégoire M, Pangault C, Dulong J, Tadié JM, et al. Early expansion of circulating granulocytic myeloid-derived suppressor cells predicts development of nosocomial infections in patients with sepsis. Am J Respir Crit Care Med. (2017) 196:315–27. doi: 10.1164/rccm.201606-1143OC
67. Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, et al. The sepsis, human myeloid-derived suppressor cells are associated with chronic immune suppression after severe sepsis/septic shock. Ann Surg. (2017) 265:827–34. doi: 10.1097/SLA.0000000000001783
68. Darden DB, Bacher R, Brusko MA, Knight P, Hawkins RB, Cox MC, et al. Single cell RNA-SEQ of human myeloid derived suppressor cells in late sepsis reveals multiple subsets with unique transcriptional responses: a pilot study. Shock. (2020). doi: 10.1097/SHK.0000000000001671. [Epub ahead of print].
69. Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, et al. Sepsis pathophysiology, chronic critical illness, and persistent inflammation-immunosuppression and catabolism syndrome. Crit Care Med. (2017) 45:253–62. doi: 10.1097/CCM.0000000000002074
70. Brummel NE, Balas MC, Morandi A, Ferrante LE, Gill TM, Ely EW. Understanding and reducing disability in older adults following critical illness. Crit Care Med. (2015) 43:1265–75. doi: 10.1097/CCM.0000000000000924
71. Baldwin MR. Measuring and predicting long-term outcomes in older survivors of critical illness. Minerva Anestesiol. (2015) 81:650–61.
72. Kumar G, Kumar N, Taneja A, Kaleekal T, Tarima S, McGinley E, et al. Nationwide trends of severe sepsis in the 21st century (2000-2007). Chest. (2011) 140:1223–31. doi: 10.1378/chest.11-0352
73. Vanzant EL, Hilton RE, Lopez CM, Zhang J, Ungaro RF, Gentile LF, et al. Advanced age is associated with worsened outcomes and a unique genomic response in severely injured patients with hemorrhagic shock. Crit Care. (2015) 19:77. doi: 10.1186/s13054-015-0788-x
74. Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. (2003) 348:1546–54. doi: 10.1056/NEJMoa022139
75. Hazeldine J, Lord JM, Hampson P. Immunesenescence and inflammaging: a contributory factor in the poor outcome of the geriatric trauma patient. Ageing Res Rev. (2015) 24:349–57. doi: 10.1016/j.arr.2015.10.003
76. Olivieri F, Rippo MR, Monsurrò V, Salvioli S, Capri M, Procopio AD, et al. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res Rev. (2013) 12:1056–68. doi: 10.1016/j.arr.2013.05.001
77. Weng NP. Aging of the immune system: how much can the adaptive immune system adapt? Immunity. (2006) 24:495–9. doi: 10.1016/j.immuni.2006.05.001
78. Solana R, Pawelec G, Tarazona R. Aging and innate immunity. Immunity. (2006) 24:491–4. doi: 10.1016/j.immuni.2006.05.003
79. Suzuki K, Inoue S, Kametani Y, Komori Y, Chiba S, Sato T, et al. Reduced immunocompetent B cells and increased secondary infection in elderly patients with severe sepsis. Shock. (2016) 46:270–8. doi: 10.1097/SHK.0000000000000619
80. Scumpia PO, Kelly-Scumpia KM, Delano MJ, Weinstein JS, Cuenca AG, Al-Quran S, et al. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol. (2010) 184:2247–51. doi: 10.4049/jimmunol.0903652
81. Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. (2011) 208:2691–703. doi: 10.1084/jem.20111490
82. Guerrettaz LM, Johnson SA, Cambier JC. Acquired hematopoietic stem cell defects determine B-cell repertoire changes associated with aging. Proc Natl Acad Sci USA. (2008) 105:11898–902. doi: 10.1073/pnas.0805498105
83. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Bossola M, et al. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res. (2018) 21:350–9. doi: 10.1089/rej.2017.1989
84. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Landi F, et al. Fueling Inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int J Mol Sci. (2017) 18:933. doi: 10.3390/ijms18050933
85. Argilés JM, Busquets S, Stemmler B, López-Soriano FJ. Cachexia and sarcopenia: mechanisms and potential targets for intervention. Curr Opin Pharmacol. (2015) 22:100–6. doi: 10.1016/j.coph.2015.04.003
86. Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP, et al. Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS ONE. (2015) 10:e0139416. doi: 10.1371/journal.pone.0139416
87. Zhang W, Lavine KJ, Epelman S, Evans SA, Weinheimer CJ, Barger PM, et al. Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J Am Heart Assoc. (2015) 4:e001993. doi: 10.1161/JAHA.115.001993
88. Laitano O, Robinson GP, Garcia CK, Mattingly AJ, Sheikh LH, Murray KO, et al. Skeletal muscle INTERLEUKIN-6 contributes to the innate immune response in septic MICE. Shock (Augusta, Ga.). (2020) doi: 10.1097/SHK.0000000000001641. [Epub ahead of print].
89. Gong Y, Zou L, Feng Y, Li D, Cai J, Chen D, et al. Importance of toll-like receptor 2 in mitochondrial dysfunction during polymicrobial sepsis. Anesthesiology. (2014) 121:1236–47. doi: 10.1097/ALN.0000000000000470
90. Assi M, Rébillard A. The janus-faced role of antioxidants in cancer cachexia: new insights on the established concepts. Oxid Med Cell Longev. (2016) 2016:9579868. doi: 10.1155/2016/9579868
91. Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, et al. Myeloid-derived suppressor cell survival and function are regulated by the transcription factor Nrf2. J Immunol. (2016) 196:3470–8. doi: 10.4049/jimmunol.1501785
92. Batt J, dos Santos CC, Cameron JI, Herridge MS. Intensive care unit-acquired weakness: clinical phenotypes and molecular mechanisms. Am J Respir Crit Care Med. (2013) 187:238–46. doi: 10.1164/rccm.201205-0954SO
93. Puthucheary Z, Harridge S, Hart N. Skeletal muscle dysfunction in critical care: wasting, weakness, rehabilitation strategies. Crit Care Med. (2010) 38:S676–82. doi: 10.1097/CCM.0b013e3181f2458d
94. Larsson L, Degens H, Li M, Salviati L, Lee YI, Thompson W, et al. Sarcopenia: aging-related loss of muscle mass and function. Physiol Rev. (2019) 99:427–511. doi: 10.1152/physrev.00061.2017
95. Fiatarone MA, Marks EC, Ryan ND, Meredith CN, Lipsitz LA, Evans WJ. High-intensity strength training in nonagenarians. Effects on skeletal muscle. JAMA. (1990) 263:3029–34. doi: 10.1001/jama.263.22.3029
96. Berková M, Berka Z. Falls: a significant cause of morbidity and mortality in elderly people. Vnitr Lek. (2018) 64:1076–83.
97. Fonsmark L, Rosendahl-Nielsen M. Experience from multidisciplinary follow-up on critically ill patients treated in an intensive care unit. Dan Med J. (2015) 62:A5062.
98. Kamdar BB, Suri R, Suchyta MR, Digrande KF, Sherwood KD, Colantuoni E, et al. Return to work after critical illness: a systematic review and meta-analysis. Thorax. (2020) 75:17–27. doi: 10.1136/thoraxjnl-2019-213803
99. Skube SJ, Katz SA, Chipman JG, Tignanelli CJ. Acute kidney injury and sepsis. Surg Infect (Larchmt). (2018) 19:216–24. doi: 10.1089/sur.2017.261
100. White LE, Hassoun HT, Bihorac A, Moore LJ, Sailors RM, McKinley BA, et al. Acute kidney injury is surprisingly common and a powerful predictor of mortality in surgical sepsis. J Trauma Acute Care Surg. (2013) 75:432–8. doi: 10.1097/TA.0b013e31829de6cd
101. Bihorac A, Brennan M, Ozrazgat-Baslanti T, Bozorgmehri S, Efron PA, Moore FA, et al. National surgical quality improvement program underestimates the risk associated with mild and moderate postoperative acute kidney injury. Crit Care Med. (2013) 41:2570–83. doi: 10.1097/CCM.0b013e31829860fc
102. Bihorac A, Delano MJ, Schold JD, Lopez MC, Nathens AB, Maier RV, et al. Incidence, clinical predictors, genomics, and outcome of acute kidney injury among trauma patients. Ann Surg. (2010) 252:158–65. doi: 10.1097/SLA.0b013e3181deb6bc
103. Bihorac A, Baslanti TO, Cuenca AG, Hobson CE, Ang D, Efron PA, et al. Acute kidney injury is associated with early cytokine changes after trauma. J Trauma Acute Care Surg. (2013) 74:1005–13. doi: 10.1097/TA.0b013e31828586ec
104. Matejovic M, Chvojka J, Radej J, Ledvinova L, Karvunidis T, Krouzecky A, et al. Sepsis and acute kidney injury are bidirectional. Contrib Nephrol. (2011) 174:78–88. doi: 10.1159/000329239
105. White LE, Chaudhary R, Moore LJ, Moore FA, Hassoun HT. Surgical sepsis and organ crosstalk: the role of the kidney. J Surg Res. (2011) 167:306–15. doi: 10.1016/j.jss.2010.11.923
106. Jansen MPB, Pulskens WP, Butter LM, Florquin S, Juffermans NP, Roelofs J, et al. Mitochondrial DNA is released in urine of SIRS patients with acute kidney injury and correlates with severity of renal dysfunction. Shock. (2018) 49:301–10. doi: 10.1097/SHK.0000000000000967
107. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. (2012) 23:1375–88. doi: 10.1681/ASN.2011111077
108. Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. (2005) 115:2894–903. doi: 10.1172/JCI22832
109. Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. (2007) 117:2847–59. doi: 10.1172/JCI31008
110. Fleshner M, Crane RC. Exosomes, DAMPs and miRNA: features of stress physiology and immune homeostasis. Trends Immunol. (2017) 38:768–76. doi: 10.1016/j.it.2017.08.002
111. Zager RA, Johnson AC, Lund S, Hanson S. Acute renal failure: determinants and characteristics of the injury-induced hyperinflammatory response. Am J Physiol Renal Physiol. (2006) 291:F546–56. doi: 10.1152/ajprenal.00072.2006
112. Hoke TS, Douglas IS, Klein CL, He Z, Fang W, Thurman JM, et al. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J Am Soc Nephrol. (2007) 18:155–64. doi: 10.1681/ASN.2006050494
113. Liu KD, Altmann C, Smits G, Krawczeski CD, Edelstein CL, Devarajan P, et al. Serum interleukin-6 and interleukin-8 are early biomarkers of acute kidney injury and predict prolonged mechanical ventilation in children undergoing cardiac surgery: a case-control study. Crit Care. (2009) 13:R104. doi: 10.1186/cc7940
114. Tryggvason K, Wartiovaara J. How does the kidney filter plasma? Physiology (Bethesda). (2005) 20:96–101. doi: 10.1152/physiol.00045.2004
115. Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest. (2013) 123:236–46. doi: 10.1172/JCI63679
116. Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS. Anti-PD-L1 peptide improves survival in sepsis. J Surg Res. (2017) 208:33–9. doi: 10.1016/j.jss.2016.08.099
117. Jung E, Perrone EE, Liang Z, Breed ER, Dominguez JA, Clark AT, et al. Cecal ligation and puncture followed by methicillin-resistant Staphylococcus aureus pneumonia increases mortality in mice and blunts production of local and systemic cytokines. Shock. (2012) 37:85–94. doi: 10.1097/SHK.0b013e3182360faf
118. Gentile LF, Nacionales DC, Lopez MC, Vanzant E, Cuenca A, Cuenca AG, et al. Protective immunity and defects in the neonatal and elderly immune response to sepsis. J Immunol. (2014) 192:3156–65. doi: 10.4049/jimmunol.1301726
119. Gentile LF, Nacionales DC, Lopez MC, Vanzant E, Cuenca A, Szpila BE, et al. Host responses to sepsis vary in different low-lethality murine models. PLoS ONE. (2014) 9:e94404. doi: 10.1371/journal.pone.0094404
120. Kelly-Scumpia KM, Scumpia PO, Delano MJ, Weinstein JS, Cuenca AG, Wynn JL, et al. Type I interferon signaling in hematopoietic cells is required for survival in mouse polymicrobial sepsis by regulating CXCL10. J Exp Med. (2010) 207:319–26. doi: 10.1084/jem.20091959
121. Nacionales DC, Szpila B, Ungaro R, Lopez MC, Zhang J, Gentile LF, et al. A detailed characterization of the dysfunctional immunity and abnormal myelopoiesis induced by severe shock and trauma in the aged. J Immunol. (2015) 195:2396–407. doi: 10.4049/jimmunol.1500984
122. Halbach JL, Wang AW, Hawisher D, Cauvi DM, Lizardo RE, Rosas J, et al. Why antibiotic treatment is not enough for sepsis resolution: an evaluation in an experimental animal model. Infect Immun. (2017) 85:e00664–17. doi: 10.1128/IAI.00664-17
123. Efron PA, Mohr AM, Moore FA, Moldawer LL. The future of murine sepsis and trauma research models. J Leukoc Biol. (2015) 98:945–52. doi: 10.1189/jlb.5MR0315-127R
124. Nelson S, Belknap SM, Carlson RW, Dale D, DeBoisblanc B, Farkas S, et al. A randomized controlled trial of filgrastim as an adjunct to antibiotics for treatment of hospitalized patients with community-acquired pneumonia. CAP Study Group. J Infect Dis. (1998) 178:1075–80. doi: 10.1086/515694
125. Root RK, Lodato RF, Patrick W, Cade JF, Fotheringham N, Milwee S, et al. Multicenter, double-blind, placebo-controlled study of the use of filgrastim in patients hospitalized with pneumonia and severe sepsis. Crit Care Med. (2003) 31:367–73. doi: 10.1097/01.CCM.0000048629.32625.5D
126. Paine R, III, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL, et al. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med. (2012) 40:90–7. doi: 10.1097/CCM.0b013e31822d7bf0
127. Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. (2009) 180:640–8. doi: 10.1164/rccm.200903-0363OC
128. Bo L, Wang F, Zhu J, Li J, Deng X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: a meta-analysis. Crit Care. (2011) 15:R58. doi: 10.1186/cc10031
129. Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. The new normal: immunomodulatory agents against sepsis immune suppression. Trends Mol Med. (2014) 20:224–33. doi: 10.1016/j.molmed.2014.01.002
130. Unsinger J, Burnham CA, McDonough J, Morre M, Prakash PS, Caldwell CC, et al. Interleukin-7 ameliorates immune dysfunction and improves survival in a 2-hit model of fungal sepsis. J Infect Dis. (2012) 206:606–16. doi: 10.1093/infdis/jis383
131. Kasten KR, Prakash PS, Unsinger J, Goetzman HS, England LG, Cave CM, et al. Interleukin-7 (IL-7) treatment accelerates neutrophil recruitment through gamma delta T-cell IL-17 production in a murine model of sepsis. Infect Immun. (2010) 78:4714–22. doi: 10.1128/IAI.00456-10
132. Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. (2010) 184:1401–9. doi: 10.4049/jimmunol.0902307
133. Nalos M, Santner-Nanan B, Parnell G, Tang B, McLean AS, Nanan R. Immune effects of interferon gamma in persistent staphylococcal sepsis. Am J Respir Crit Care Med. (2012) 185:110–2. doi: 10.1164/ajrccm.185.1.110
134. Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care. (2013) 17:R85. doi: 10.1186/cc12711
135. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, e al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci USA. (2009) 106:6303–8. doi: 10.1073/pnas.0809422106
136. Inoue S, Bo L, Bian J, Unsinger J, Chang K, Hotchkiss RS. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock. (2011) 36:38–44. doi: 10.1097/SHK.0b013e3182168cce
137. Yang X, Jiang X, Chen G, Xiao Y, Geng S, Kang C, et al. T cell Ig mucin-3 promotes homeostasis of sepsis by negatively regulating the TLR response. J Immunol. (2013) 190:2068–79. doi: 10.4049/jimmunol.1202661
138. Zhao Z, Jiang X, Kang C, Xiao Y, Hou C, Yu J, e al. Blockade of the T cell immunoglobulin and mucin domain protein 3 pathway exacerbates sepsis-induced immune deviation and immunosuppression. Clin Exp Immunol. (2014) 178:279–91. doi: 10.1111/cei.12401
139. Workman CJ, Wang Y, El Kasmi KC, Pardoll DM, Murray PJ, Drake CG, et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol. (2009) 182:1885–91. doi: 10.4049/jimmunol.0800185
140. Durham NM, Nirschl CJ, Jackson CM, Elias J, Kochel CM, Anders RA, et al. Lymphocyte activation gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE. (2014) 9:e109080. doi: 10.1371/journal.pone.0109080
141. Wu J, Zhou L, Liu J, Ma G, Kou Q, He Z, et al. The efficacy of thymosin alpha 1 for severe sepsis (ETASS): a multicenter, single-blind, randomized and controlled trial. Crit Care. (2013) 17:R8. doi: 10.1186/cc11932
142. Moore FA, Phillips SM, McClain CJ, Patel JJ, Martindale RG. Nutrition support for persistent inflammation, immunosuppression, catabolism syndrome. Nutr Clin Pract. (2017) 32:121s−7s. doi: 10.1177/0884533616687502
143. Bozzetti F, Arends J, Lundholm K, Micklewright A, Zurcher G, Muscaritoli M. ESPEN guidelines on parenteral nutrition: non-surgical oncology. Clin Nutr. (2009) 28:445–54. doi: 10.1016/j.clnu.2009.04.011
144. Wolfe RR, Miller SL, Miller KB. Optimal protein intake in the elderly. Clin Nutr. (2008) 27:675–84. doi: 10.1016/j.clnu.2008.06.008
145. Morley JE, Argiles JM, Evans WJ, Bhasin S, Cella D, Deutz NE, et al. Nutritional recommendations for the management of sarcopenia. J Am Med Dir Assoc. (2010) 11:391–6. doi: 10.1016/j.jamda.2010.04.014
146. Herndon DN, Tompkins RG. Support of the metabolic response to burn injury. Lancet. (2004) 363:1895–902. doi: 10.1016/S0140-6736(04)16360-5
147. Zhu X, Pribis JP, Rodriguez PC, Morris SM, Jr., Vodovotz Y, Billiar TR, et al. The central role of arginine catabolism in T-cell dysfunction and increased susceptibility to infection after physical injury. Ann Surg. (2014) 259:171–8. doi: 10.1097/SLA.0b013e31828611f8
148. Wu G, Bazer FW, Davis TA, Kim SW, Li P, Marc Rhoads J, et al. Arginine metabolism and nutrition in growth, health and disease. Amino Acids. (2009) 37:153–68. doi: 10.1007/s00726-008-0210-y
149. Schlegel L, Coudray-Lucas C, Barbut F, Le Boucher J, Jardel A, Zarrabian S, et al. Bacterial dissemination and metabolic changes in rats induced by endotoxemia following intestinal E. coli overgrowth are reduced by ornithine alpha-ketoglutarate administration. J Nutr. (2000) 130:2897–902. doi: 10.1093/jn/130.12.2897
150. English KL, Mettler JA, Ellison JB, Mamerow MM, Arentson-Lantz E, Pattarini JM, et al. Leucine partially protects muscle mass and function during bed rest in middle-aged adults. Am J Clin Nutr. (2016) 103:465–73. doi: 10.3945/ajcn.115.112359
151. Cerra FB, Siegel JH, Coleman B, Border JR, McMenamy RR. Septic autocannibalism. A failure of exogenous nutritional support. Ann Surg. (1980) 192:570–80. doi: 10.1097/00000658-198010000-00015
152. Hernandez-García AD, Columbus DA, Manjarín R, Nguyen HV, Suryawan A, Orellana RA, et al. Leucine supplementation stimulates protein synthesis and reduces degradation signal activation in muscle of newborn pigs during acute endotoxemia. Am J Physiol Endocrinol Metab. (2016) 311:E791–801. doi: 10.1152/ajpendo.00217.2016
153. Cynober L, de Bandt JP, Moinard C. Leucine and citrulline: two major regulators of protein turnover. World Rev Nutr Diet. (2013) 105:97–105. doi: 10.1159/000341278
154. Hrynyk M, Neufeld RJ. Insulin and wound healing. Burns. (2014) 40:1433–46. doi: 10.1016/j.burns.2014.03.020
155. Herndon DN, Voigt CD, Capek KD, Wurzer P, Guillory A, Kline A, et al. Reversal of growth arrest with the combined administration of oxandrolone and propranolol in severely burned children. Ann Surg. (2016) 264:421–8. doi: 10.1097/SLA.0000000000001844
156. Diaz EC, Herndon DN, Porter C, Sidossis LS, Suman OE, Børsheim E. Effects of pharmacological interventions on muscle protein synthesis and breakdown in recovery from burns. Burns. (2015) 41:649–57. doi: 10.1016/j.burns.2014.10.010
157. Kayambu G, Boots R, Paratz J. Physical therapy for the critically ill in the ICU: a systematic review and meta-analysis. Crit Care Med. (2013) 41:1543–54. doi: 10.1097/CCM.0b013e31827ca637
158. Cermak NM, Res PT, de Groot LC, Saris WH, van Loon LJ. Protein supplementation augments the adaptive response of skeletal muscle to resistance-type exercise training: a meta-analysis. Am J Clin Nutr. (2012) 96:1454–64. doi: 10.3945/ajcn.112.037556
159. Berger MF, Mardis ER. The emerging clinical relevance of genomics in cancer medicine. Nat Rev Clin Oncol. (2018) 15:353–65. doi: 10.1038/s41571-018-0002-6
160. Aguti S, Malerba A, Zhou H. The progress of AAV-mediated gene therapy in neuromuscular disorders. Expert Opin Biol Ther. (2018) 18:681–93. doi: 10.1080/14712598.2018.1479739
161. Xiao-Jie L, Hui-Ying X, Zun-Ping K, Jin-Lian C, Li-Juan J. CRISPR-Cas9: a new and promising player in gene therapy. J Med Genet. (2015) 52:289–96. doi: 10.1136/jmedgenet-2014-102968
162. Christaki E, Giamarellos-Bourboulis EJ. The beginning of personalized medicine in sepsis: small steps to a bright future. Clin Genet. (2014) 86:56–61. doi: 10.1111/cge.12368
163. Wang L, Ko ER, Gilchrist JJ, Pittman KJ, Rautanen A, Pirinen M, et al. Human genetic and metabolite variation reveals that methylthioadenosine is a prognostic biomarker and an inflammatory regulator in sepsis. Sci Adv. (2017) 3:e1602096. doi: 10.1126/sciadv.1602096
164. Burnham KL, Davenport EE, Radhakrishnan J, Humburg P, Gordon AC, Hutton P, et al. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. Am J Respir Crit Care Med. (2017) 196:328–39. doi: 10.1164/rccm.201608-1685OC
165. Cuenca AG, Gentile LF, Lopez MC, Ungaro R, Liu H, Xiao W, et al. Development of a genomic metric that can be rapidly used to predict clinical outcome in severely injured trauma patients. Crit Care Med. (2013) 41:1175–85. doi: 10.1097/CCM.0b013e318277131c
166. Raymond SL, Hawkins RB, Wang Z, Mira JC, Stortz JA, Han F, et al. Prospective validation of a transcriptomic metric in severe trauma. Ann Surg. (2020) 271:802–10. doi: 10.1097/SLA.0000000000003204
167. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. (2015) 521:436–44. doi: 10.1038/nature14539
Keywords: inflammation, immunosuppression, aging, acute kidney injury, PICS
Citation: Fenner BP, Darden DB, Kelly LS, Rincon J, Brakenridge SC, Larson SD, Moore FA, Efron PA and Moldawer LL (2021) Immunological Endotyping of Chronic Critical Illness After Severe Sepsis. Front. Med. 7:616694. doi: 10.3389/fmed.2020.616694
Received: 13 October 2020; Accepted: 14 December 2020;
Published: 15 February 2021.
Edited by:
David Paterson, The University of Queensland, AustraliaReviewed by:
Pedro Xavier-Elsas, Federal University of Rio de Janeiro, BrazilCopyright © 2021 Fenner, Darden, Kelly, Rincon, Brakenridge, Larson, Moore, Efron and Moldawer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lyle L. Moldawer, bHlsZS5tb2xkYXdlckBzdXJnZXJ5LnVmbC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.