 Rita A. Moura
Rita A. Moura João Eurico Fonseca
João Eurico Fonseca
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 05 February 2021
Sec. Rheumatology
Volume 7 - 2020 | https://doi.org/10.3389/fmed.2020.607725
This article is part of the Research Topic Advance In B-cell therapies For The Treatment of Rheumatic and Musculoskeletal Diseases View all 10 articles
Rheumatoid arthritis (RA) is a chronic, systemic immune-mediated inflammatory disease that can lead to joint destruction, functional disability and substantial comorbidity due to the involvement of multiple organs and systems. B cells have several important roles in RA pathogenesis, namely through autoantibody production, antigen presentation, T cell activation, cytokine release and ectopic lymphoid neogenesis. The success of B cell depletion therapy with rituximab, a monoclonal antibody directed against CD20 expressed by B cells, has further supported B cell intervention in RA development. Despite the efficacy of synthetic and biologic disease modifying anti-rheumatic drugs (DMARDs) in the treatment of RA, few patients reach sustained remission and refractory disease is a concern that needs critical evaluation and close monitoring. Janus kinase (JAK) inhibitors or JAKi are a new class of oral medications recently approved for the treatment of RA. JAK inhibitors suppress the activity of one or more of the JAK family of tyrosine kinases, thus interfering with the JAK-Signal Transducer and Activator of Transcription (STAT) signaling pathway. To date, there are five JAK inhibitors (tofacitinib, baricitinib, upadacitinib, peficitinib and filgotinib) approved in the USA, Europe and/ or Japan for RA treatment. Evidence from the literature indicates that JAK inhibitors interfere with B cell functions. In this review, the main results obtained in clinical trials, pharmacokinetic, in vitro and in vivo studies concerning the effects of JAK inhibitors on B cell immune responses in RA are summarized.
The success of B cell depletion therapy with rituximab in autoimmune diseases such as rheumatoid arthritis (RA) has reinforced the important role that B cells have in the development of these conditions (1, 2). Indeed, B cells can be responsible for autoantibody production, antigen presentation and T cell activation and/ or cytokine and chemokine release that contribute to disease pathogenesis (3). RA is a chronic, systemic immune-mediated disease that mainly affects the small joints of hands and wrists and, though often ameliorated by treatment, can lead to bone and cartilage destruction (4, 5). Treatment options in RA include non-steroid anti-inflammatory drugs (NSAIDs), corticosteroids, synthetic and/or biologic disease modifying anti-rheumatic drugs (DMARDs). Nevertheless, despite the progresses achieved in the last decades in RA pharmacotherapy, few patients reach sustained remission and refractory disease remains a significant challenge (6–8). Janus kinase (JAK) inhibitors or JAKi are recently approved oral medications with therapeutic application in myeloproliferative disorders and inflammatory diseases such as RA. JAKi function by inhibiting the activity of one or more of the JAK family of enzymes [JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2)], thus interfering with the JAK-Signal Transducer and Activator of Transcription (STAT) signaling pathway (9, 10). There are currently five JAK inhibitors (tofacitinib, baricitinib, upadacitinib, peficitinib, and filgotinib) approved in the USA, Europe and/ or Japan for RA treatment. Furthermore, an additional JAKi (decernotinib) is under investigation for RA treatment in clinical trials (11, 12). Although the number of studies exploring the effect of JAK inhibitors on B cells in the context of RA is limited, evidence from the literature indicates that JAKi also interfere with B cell functions. In this review, we summarize the main results obtained so far in clinical trials, pharmacokinetic, in vitro and in vivo studies concerning the effects of JAK inhibitors on B cell immune responses in RA.
B cells play several important roles in the development of RA (13). B cells produce autoantibodies, such as rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA), which form immune complexes that deposit in the joints and contribute to the inflammatory process through complement and cellular activation. Furthermore, B cells act as efficient antigen presenting cells (APC) that activate T cells through the expression of costimulatory molecules. B cells also secrete cytokines and/ or chemokines that promote leukocyte infiltration in the joints and the development of ectopic lymphoid structures, thus aggravating angiogenesis, pannus formation and synovial hyperplasia. In addition, the therapeutic efficacy of rituximab, an anti-CD20 monoclonal antibody that specifically depletes B cells, in RA patients has unequivocally supported B cell targeted therapies in RA pathogenesis (1, 2, 14). Of note, previous studies by our group have demonstrated that untreated very early RA patients (with <6 weeks of disease duration) have alterations in circulating memory B cell subpopulations (15); a cytokine profile that supports an early B cell activation (16, 17); and changes in B cell gene expression levels relevant for B cell maturation and differentiation (18). These data reinforce an active role of B cells in RA pathogenesis from early disease onset. Moreover, we have recently shown that in RA, treatment with tumor necrosis factor (TNF)-inhibitors and the interleukin (IL)-6 receptor (IL-6R) antagonist tocilizumab affect B cell phenotype and IgD-CD27- memory B cells in peripheral blood (19). Importantly, clinical relapse observed in B cell depleted RA patients has been associated with B cell repopulation (20–22). In fact, the results observed in RA patients following B cell depletion therapy with rituximab suggest that alterations in the expression of B cell activating factor (BAFF)-binding receptors and an increase in class-switch recombination process, particularly in memory B cell subsets, might be associated with the re-establishment of active disease (23). Interestingly, it has also been recently demonstrated for the first time that the autoantibodies commonly found in RA patients, RF and ACPA, express the inherently autoreactive 9G4 idiotope, thus supporting an activation of autoreactive 9G4+ B cells in RA (24). Additionally, it has been recently suggested that the pattern of B cell distribution in synovial tissue from untreated early RA patients can be associated to a specific pathotype classification with cellular and molecular synovial signatures that might help to predict disease severity, radiographic progression and therapeutic response (25, 26).
Cytokines are a large family of secreted proteins that play important roles in the immune system, namely in cell differentiation, maturation and signaling. Cytokines can be produced by several types of immune cells, including macrophages, B cells, T cells and mast cells, as well as endothelial cells, fibroblasts and various stromal cells. Of note, cytokines can be major drivers of autoimmunity and inflammation. In RA, several cellular interactions and complex cytokine networks occur that contribute to disease pathogenesis (13). In fact, it has been demonstrated that cytokines including IL-1 beta (IL-1β), IL-2, IL-3, IL-6, IL-7, IL-8, IL-12, IL-15, IL-17, IL-18, IL-19, IL-20, IL-21, IL-23, IL-32, IL-33, IL-35, TNF, interferon-alpha/gamma (IFN-α/γ) and granulocyte-macrophage colony-stimulating factor (GM-CSF) have important roles in RA physiopathology as they contribute to the induction and maintenance of inflammation (13, 27–30). The inflammatory process that develops in RA leads to a cellular infiltration of the synovial membrane, angiogenesis, pannus formation, swelling, and pain. The interactions between B and T cells result in the activation and differentiation of plasma cells, which are responsible for the production of autoantibodies (RF, ACPA). These autoantibodies form immune complexes that can activate complement and stimulate cells such as monocytes by binding to their Fc-gamma receptors (FcγR), triggering cytokine and/ or chemokine release that cause inflammation. Indeed, activated monocytes, neutrophils, and fibroblasts can release high levels of cytokines such as IL-1, IL-6, and TNF, that further activate not only B and T cells, but also chondrocytes and osteoclasts, thus contributing to cartilage and bone destruction (13). Furthermore, cytokines directly related with B cell activation and survival such as A proliferation-inducing ligand (APRIL) and BAFF (31–35), which can be produced by activated monocytes and neutrophils, have been shown to contribute to RA development from an early phase in disease onset (17). Moreover, increased serum levels of BAFF have been suggested to have an important role in B cell triggering during clinical relapse after B cell depletion therapy (23). Previous studies developed by our group have demonstrated that untreated very early RA (VERA) patients (with <6 weeks of disease duration) have a cytokine pattern in circulation that supports an early activation of not only B cells, but also neutrophils and Th17 cells (16, 17) (Figure 1). Indeed, we have found that VERA patients have higher serum levels of APRIL and BAFF when compared to other very early arthritis (non-RA) patients, established RA and healthy controls (17). We also observed that established RA patients have significantly increased synovial fluid levels of APRIL, BAFF and IL-21, a cytokine important for plasma cell differentiation (17) (Figure 1A). Additionally, we found that VERA patients have increased serum levels of cytokines that promote neutrophil recruitment and activation (IL-8), Th17 cells polarization (IL-1β and IL-6) and Th17 cells-derived cytokines (IL-17A and IL-22) (16) (Figure 1B). Also, the elevated IL-1β, IL-6, IL-8, and IL-17A levels observed in the synovial fluid of established RA patients support a local role for these cytokines in synovial inflammation and bone erosion (16) (Figures 1B,C). In fact, IL-17 has been shown to induce osteoclastogenesis, thus contributing for bone resorption (36, 37). Moreover, IL-6 can support the activation and recruitment of autoreactive B cells toward RA synovium (38, 39), leading to an exacerbation of inflammation through autoantibody production and immune complex deposition (40, 41) (Figure 1C). Of note, treatment of VERA patients with corticosteroids and methotrexate (MTX), although effective in clinical improvement had no impact on the cytokine pattern in circulation (16, 17). Importantly, the success of biological therapies that directly target key cytokines such as TNF inhibitors (adalimumab, infliximab, etanercept, golimumab and certolizumab); tocilizumab (an IL-6R antagonist) and anakinra (an IL-1R antagonist) in RA further reinforce the relevance of these small proteins in disease development (42–46).
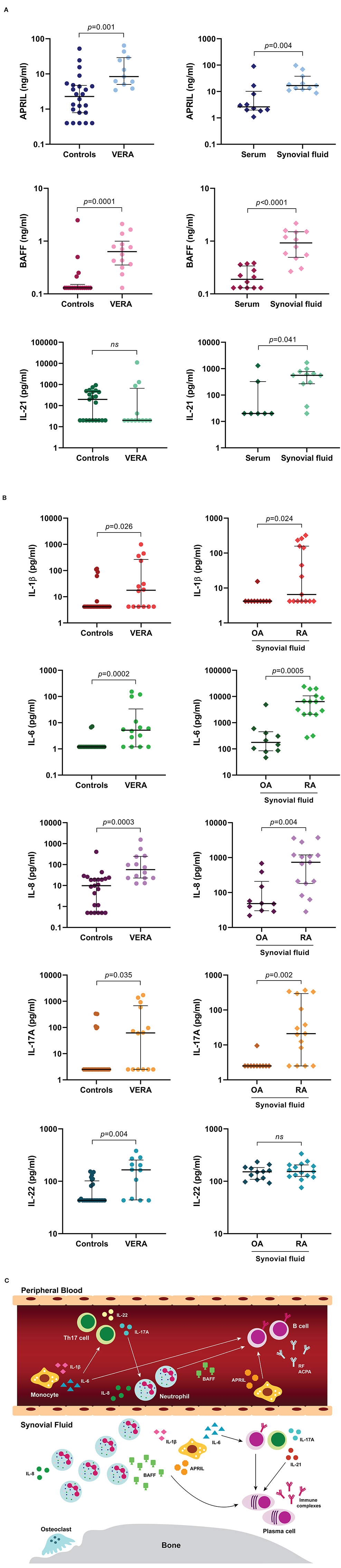
Figure 1. Cytokine profile present in peripheral blood from very early rheumatoid arthritis (VERA) patients and synovial fluid from established RA. A group of cytokines directly related with B cell activation, differentiation and survival was quantified in serum samples from untreated very early rheumatoid arthritis (VERA) patients with <6 weeks of disease duration when compared to healthy controls (A). In addition, serum and synovial fluid samples from established treated RA patients were also analyzed for comparison (A). Cytokines related with neutrophil and Th17 cells activation were also quantified in serum samples from VERA patients and healthy individuals (B). Furthermore, synovial fluid from established treated RA and osteoarthritis (OA) patients was analyzed for comparison (B). Statistical analysis of data was performed with GraphPad Prism (GraphPad Software, San Diego, CA, USA). Lines in graphs represent median values with interquartile range. Non-parametric Mann-Whitney test was used for comparisons between two independent groups. Differences were considered statistically significant for p < 0.05. Data represented in Figures 1A,B were adapted from previous published studies by our group (16–18), according to the terms of the Creative Commons license (http://creativecommons.org/licenses/by/4.0/). Figure 1C is an illustration representative of the cytokine profile present in peripheral blood from VERA patients and synovial fluid from established RA supported by previous published studies by our group (16–18). To sum up, RA patients have a cytokine profile in peripheral blood that favors B cells, neutrophils and Th17 cells activation since the first weeks of disease development. In a chronic phase of the disease, the cytokine pattern present locally in the joints supports the intervention of activated monocytes, neutrophils, T and B cells and plasma cell differentiation (C). ACPA, anti-citrullinated protein antibodies; APRIL, a proliferation-inducing ligand; BAFF, B cell activating factor; IL, interleukin; ns, non-significant; OA, osteoarthritis; RA, rheumatoid arthritis; RF, rheumatoid factor; Th17, T helper 17; VERA, very early rheumatoid arthritis.
Cytokines act by binding to cell surface receptors and subsequently activate intracellular signaling cascades, such as the JAK-STAT signaling pathway. JAK-STAT signaling pathway is an evolutionarily conserved pathway that regulates many cellular processes including innate and adaptive immune responses, cell proliferation, differentiation and apoptosis. Activation of this pathway is initiated by binding of a ligand (such as interleukins, interferons, hormones and growth factors) to specific transmembrane receptors (cytokine receptors, G protein-coupled receptors, receptor tyrosine kinases and homodimeric hormone receptors) and culminates in the transcription of target genes (9, 10, 47–49) (Figure 2). JAKs, STATs and cell-surface receptors are the main key players of this signal-transduction pathway. JAKs are a family of four members of tyrosine kinases (JAK1, JAK2, JAK3, and TYK2) that selectively associate with the intracellular domains of cell receptors (50, 51) (Figure 3). JAK1, JAK2, and TYK2 are ubiquitously expressed, whereas JAK3 expression is mainly restricted to hematopoietic cells (52). Binding of a ligand to a cell surface receptor triggers the receptor dimerization and induces the autophosphorylation and activation of the receptor-associated JAKs. Activated JAKs then phosphorylate critical tyrosine residues on the receptor, which leads to recruitment of specific STATs (49, 51, 53) (Figure 2). STATs are a family of proteins named for their dual roles of transducing signals and promoting transcription of specific genes. There are seven members of the STAT family in mammals: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6 (49, 54–57). After binding to the phosphorylated tyrosine residues on the receptor, STATs are phosphorylated by JAKs, which leads to their dissociation from the receptor. STATs form homo- or heterodimers and translocate into the cell nucleus via importins, where they bind to specific DNA regions and activate the transcription of target genes (Figure 2). STATs can be dephosphorylated by nuclear protein tyrosine phosphatases (N-PTPs), which leads to the inactivation of STATs. The unphosphorylated STATs associate with exportins to exit the nucleus and return to the cytoplasm where they can be reactivated for further rounds of gene transcription (10, 47, 49, 56). Overall, signaling via the JAK–STAT signaling pathway is a dynamic process that involves the rapid transmission of signal from the cell membrane to the nucleus followed by a highly organized response and subsequent controlled downregulation and attenuation of the initial signal (47–49, 54). Thus, negative regulators of the JAK-STAT signaling pathway also play an essential role. These include protein tyrosine phosphatases (PTPs), which remove phosphate groups from receptors, JAKs and STATs (58); protein inhibitor of activated STAT (PIAS), that prevent the DNA-binding activity of STATs (59, 60); and suppressor of cytokine signaling proteins (SOCS), which form a classical negative feedback loop that switches off the activity of JAKs (61, 62) (Figure 2). Disturbances in JAK-STAT signaling pathway, mostly associated with mutations (gain or loss of function) and polymorphisms in JAK and/ or STAT genes (9, 63), have been implicated in the pathogenesis of several diseases including inflammatory skin conditions (psoriasis, atopic dermatitis, alopecia areata, vitiligo) (64–71); cancers (myeloproliferative neoplasms, leukemia) (72, 73); immunodeficiencies (severe combined immune deficiency) (74); and autoimmune disorders such as RA (75–79); psoriatic arthritis (80, 81); systemic lupus erythematosus (82, 83); ankylosing spondylitis (84, 85); systemic sclerosis (86, 87); giant cell arteritis (88); sarcoidosis (89–91) and inflammatory bowel diseases (ulcerative colitis, Crohn's disease) (92, 93). Therefore, targeting JAKs and/ or STATs can be a safe and efficacious strategy for treating these diseases (94).
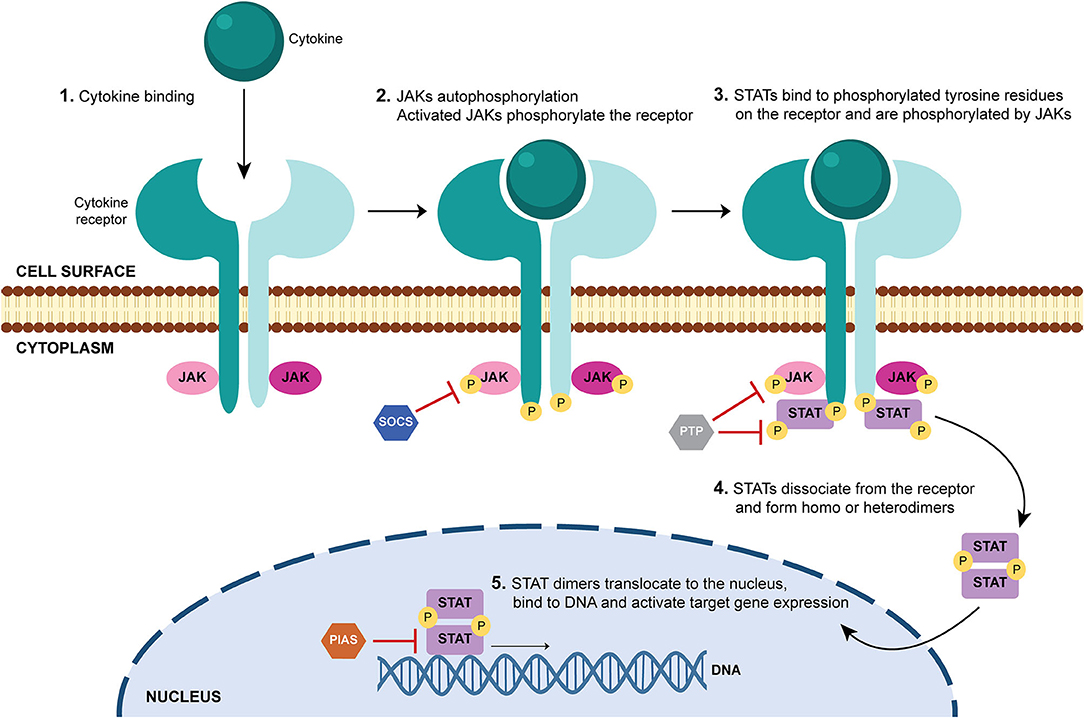
Figure 2. JAK-STAT signaling pathway. When a ligand (usually a cytokine) binds to its receptor in a cell, it triggers the autophosphorylation of the receptor-associated Janus kinases (JAKs). Activated JAKs phosphorylate the intracellular tail of the receptor on critical tyrosine residues, which leads to the recruitment and binding of signal transducer and activator of transcription (STAT) proteins. STATs are phosphorylated by JAKs, which induces their dissociation from the receptor. STATs form homo- or heterodimers and translocate into the cell nucleus, where they bind to specific DNA regions and activate target gene expression. Negative regulators of the JAK-STAT signaling pathway include protein tyrosine phosphatases (PTPs), which remove phosphate groups from receptors, JAKs and STATs; protein inhibitor of activated STAT (PIAS), that prevent the DNA-binding activity of STATs; and suppressor of cytokine signaling proteins (SOCS), which inhibit the activity of JAKs. DNA, deoxyribonucleic acid; JAK, Janus kinase; P, phosphate; PIAS, protein inhibitor of activated STAT; PTP, protein tyrosine phosphatase; SOCS, suppressor of cytokine signaling proteins; STAT, signal transducer and activator of transcription.
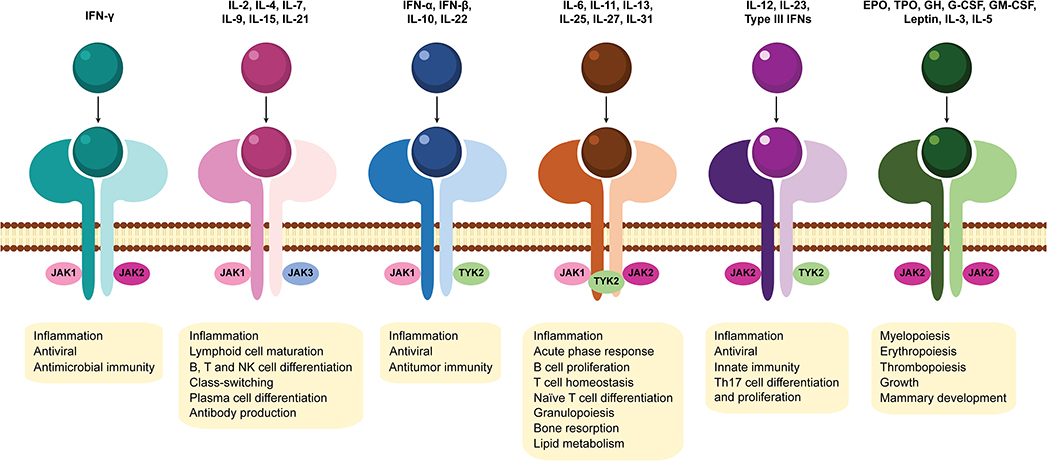
Figure 3. Association of Janus kinases with cytokine receptors and downstream effects of JAK-STAT signaling pathway activation. Janus kinase (JAK) family members include JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). Different JAK combinations with their subsequent downstream effects, each mediated by a specific subset of cytokines are represented. EPO, erythropoietin; GH, growth hormone; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; JAK, Janus kinase; NK, natural killer; Th17, T helper 17; TPO, thrombopoietin; TYK2, tyrosine kinase 2.
JAK-STAT signaling pathway has a critical role in the signal transduction of many pivotal cytokines involved in RA pathogenesis (12, 95, 96) as well as other inflammatory disorders (97). Due to their central role in the immune responses and their association with several cytokine receptors (Figure 3), the inhibition of JAKs appeared to be a promising therapeutic strategy in autoimmune diseases (94). JAK inhibitors (JAKi) represent a new class of oral drugs developed in the last decade that directly suppress the enzymatic activity of JAK family members, blocking JAK-STAT signaling pathway (12, 96). Despite the efficacy of biological DMARD treatments that target individual cytokines, biologics are large proteins that may cause immunogenicity and require either intravenous infusion or subcutaneous injection for dosing (98). In contrast, JAK inhibitors are small molecules, orally administered, that can simultaneously suppress the action of multiple cytokines. To date, five JAK inhibitors (tofacitinib, baricitinib, upadacitinib, peficitinib, and filgotinib) have been approved for the treatment of RA.
Tofacitinib is an oral JAK inhibitor with selectivity for JAK1 and JAK3 and, to a lesser extent, JAK2 and TYK2. Tofacitinib was the first JAK inhibitor approved by the United States (US) Food and Drug Administration (FDA) (November 2012) and European Medicines Agency (EMA) (March 2017) for the treatment of moderate to severe active RA patients who had had an inadequate response or intolerance to MTX (76, 78, 99–112). Data from human clinical trial studies have demonstrated the effectiveness of the use of tofacitinib in RA patients not only as a monotherapy (at a dosage of 5 mg twice daily), but also in combination with MTX and the clinical responses have proven to be at least similar to TNF antagonists (78, 103, 105, 107, 109, 112–114). Indeed, tofacitinib has demonstrated efficacy in active RA patients by significantly improving disease activity, physical functioning, health-related quality of life as well as preventing bone erosions and structural joint damage (99, 103, 114–117). Furthermore, safety reports indicate that tofacitinib is generally well-tolerated, has a consistent safety profile (as monotherapy or combination therapy) and sustained efficacy in RA patients. However, adverse events have been described in RA patients after tofacitinib treatment with mild to moderate severity that included nausea, anemia, lymphopenia, neutropenia, lipid profile changes, increase in liver enzymes, cardiovascular events, lower respiratory tract infections, herpes zoster virus (HZV) reactivation, venous thromboembolism, and development of malignancies (76, 78, 109, 112, 114, 118–125). Nevertheless, the overall risk of infection (including serious infection) and mortality rates in RA patients treated with tofacitinib is similar to those observed in RA patients treated with biologic agents (12, 120).
Baricitinib was the second JAK inhibitor approved for clinical use in RA (in February 2017 by the EMA and in June 2018 by the FDA). Baricitinib is an oral JAK1/JAK2 inhibitor, with moderate activity against TYK2 and significantly less activity against JAK3. Approved dosages (2 and 4 mg once daily) are administered to moderate to severe active RA in adult patients who are intolerant or unresponsive to one or more DMARDs (75, 126–132). Treatment of RA patients with baricitinib monotherapy, or when baricitinib was combined with conventional synthetic DMARDs (csDMARDs) such as MTX showed efficacy and had an acceptable safety profile in early active naïve csDMARD-treated RA patients who had exhibited an inadequate response to conventional synthetic or biologic DMARDs (126, 129, 131, 132). Moreover, it has been demonstrated that baricitinib had a similar or improved efficacy when compared to TNF antagonists such as adalimumab (129, 131–134). Of note, treatment of RA patients with baricitinib was associated not only with clinical improvement, but also with inhibition of radiographic joint damage (135, 136). Overall, baricitinib is considered a safe and effective treatment in RA, although some adverse events have been described similarly to what has been observed in tofacitinib treated RA patients (132, 137–139).
Upadacitinib is a JAK1-selective inhibitor approved by the FDA (in August 2019) and EMA (in December 2019) for the treatment of RA. Upadacitinib is indicated for the treatment of adults with moderately to severely active RA who fail to adequately respond to, or are intolerant to one or more DMARDs (77, 140–146). Upadacitinib may be used as monotherapy (15 mg or 30 mg once daily) or in combination with MTX as an effective treatment for active RA patients with an inadequate response to conventional or biological DMARDs, with an acceptable safety profile (77, 143–147). Furthermore, it has been demonstrated that upadacitinib was more effective than adalimumab treatment in ameliorating disease activity in RA patients who were concomitantly receiving MTX and significantly prevented radiographic progression (148). In addition, despite being a selective JAK1 inhibitor, upadacitinib has a similar safety profile to less-selective JAKi (139, 143, 146, 147, 149). Nevertheless, longer-term safety data are necessary.
Peficitinib is a pan-JAK inhibitor with a moderate selectivity for JAK3. It was approved for the treatment of RA in Japan in 2019 and Korea in 2020; and is currently being evaluated by the US FDA to treat adult patients with moderately to severely active RA who show inadequate response to or are intolerant of MTX (150–158). Peficitinib has been tested in RA either as monotherapy (150) or in combination with MTX (151) or csDMARDs (152) and it has been shown to significantly improve disease severity in RA patients who have an inadequate response to conventional therapies. Of note, it has been demonstrated that Peficitinib 50, 100, and 150 mg dosages administered once daily were effective in treating active RA patients, without a significant risk for adverse events (159). Overall, peficitinib has an acceptable safety and tolerability profile with similarly described adverse events as the ones reported with other JAK inhibitors (139, 153–155, 158, 160–162).
Filgotinib is a JAK1-selective inhibitor recently approved by EMA and in Japan (in September 2020) for the treatment of RA (163–170). Filgotinib is indicated for the treatment of moderate to severe active RA in adults who have responded inadequately to, or who are intolerant to one or more DMARDs. Filgotinib may be used as monotherapy (100 mg or 200 mg once daily) or in combination with MTX (168–170). Of note, similarly to upadacitinib, another selective JAK1 inhibitor, it has been demonstrated that the risks of serious adverse events did not differ between filgotinib and less-selective JAKi such as tofacitinib (168–171).
In addition to these compounds, another JAK inhibitor, decernotinib, an oral JAK3-inhibitor in Phase IIb studies (172–175), is currently under investigation for the treatment of RA. Overall, results from clinical trials with JAK inhibitors in RA are encouraging (12, 125). JAKi have shown a rapid onset of action and, in case of an adverse event, their short half-life supports a rapid reversal of immunosuppressive effects (176–178). Of note, JAK inhibitors proved efficacious when administered as monotherapy and have demonstrated a comparable or superior efficacy and safety profile to those of biologic agents (179, 180). Importantly, due to the evidence of superiority or non-inferiority of JAK inhibitors when compared to adalimumab emerging from randomized clinical trials (114, 134, 181), the 2020 updated EULAR therapeutic guidelines have recommended the use of JAK inhibitors as an alternative to biologics in RA patients refractory to cDMARDs and having poor prognostic factors, as well as in those failing a previous synthetic or biologic DMARD (182).
Studies of the effects of JAK inhibitors on circulating immune cells that play important roles in the pathogenesis of autoimmune diseases may provide insights into immunologic mechanisms associated with clinical outcomes. Due to differences in JAK targeting, JAK inhibitors may also exert distinct immunologic effects. While JAK1, JAK2, and TYK2 are ubiquitously expressed, JAK3 expression is predominantly restricted to hematopoietic cells (50, 183–186), having important roles in immune function and lymphocyte development as described in both humans (74, 187) and mice (188, 189) with JAK3 deficiencies. JAK3 mediates signaling through cytokine receptors that contain the common gamma chain (γc) or IL-2R subunit gamma (IL-2RG) including IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 receptors (51). Also, it has been shown that JAK3 is constitutively associated with CD40, an important B cell co-receptor whose signaling has a wide range of effects on B cells, including cell growth, survival, differentiation, isotype switching, rescue from apoptosis and up-regulation of expression of B7 (CD80), Fas, ICAM-1, CD23 and lymphotoxin (LT)-α (190, 191). In fact, JAK3 activating mutations are found in human hematological malignancies including B-cell lymphomas (192–194). Furthermore, observations in JAK3 knockout mice confirmed JAK3 essential role in B cell division, immunoglobulin gene rearrangement, differentiation and survival (195). Taken together, these data support that the regulation of JAK3 expression and activity is important in B cell development and function (196). Therefore, the use of JAK3 inhibitors such as tofacitinib in autoimmune diseases such as RA might have important consequences in B cell activation and function. Previous studies have shown that the primary targets of tofacitinib during pathological processes in RA are dendritic cells, CD4+ T cells such as Th1 and Th17 and activated B cells, leading to multi-cytokine targeting, decreased synovial inflammation and structural joint damage (117, 197–202). Changes in lymphocyte subsets have been documented with tofacitinib treatment (116, 176, 200, 203, 204). Indeed, phase II and phase III clinical trials involving patients with RA treated with tofacitinib showed a transient increase in total lymphocytes early in treatment, with a gradual decrease over time (204–206). In phase II RA clinical trials, variable changes in T cells were observed with short-term tofacitinib treatment, while B cells and natural killer (NK) cells increased and decreased from baseline, respectively (204, 205). Importantly, no strong association between CD4+ T cell, CD8+ T cell, B cell, or NK cell counts and serious infection incidence rates was observed (204). Although the number of studies exploring the effect of tofacitinib on B cells in the context of RA is limited, results so far indicate that tofacitinib interferes with B cell functions. In fact, it has been suggested that tofacitinib suppresses B cell activation, differentiation and class-switching, but maintains B cell regulatory function (202, 207). Moreover, tofacitinib reduces IgG and RF circulating levels in RA patients, which correlates with disease activity amelioration (200). Additionally, it was shown that tofacitinib severely impaired in vitro plasmablast development, immunoglobulin secretion and induction of B-cell fate determining transcription factors from naïve B cells isolated from umbilical cord blood (208). Similar, but less pronounced results were obtained with peripheral blood B cells isolated from healthy blood donors. Indeed, in vitro treatment of total peripheral blood B cells with tofacitinib resulted in reduced but not abolished plasmablast development, as well as reduced antibody secretion (208). Furthermore, recent studies developed in murine models of lupus have demonstrated that although tofacitinib treatment did not change B cell numbers, a significant reduction in anti-double stranded DNA (anti-dsDNA) and antinuclear antibodies (ANA) was observed in serum (209, 210). These observations pointed to the potential inability of tofacitinib-treated patients to respond to novel antigens, suggesting that vaccination against new antigens prior to tofacitinib treatment should be considered (208, 211–213). Moreover, in vitro activation of B cells isolated from tofacitinib treated polyarthritis patients has revealed that, in the absence of tofacitinib, B cells can be activated again and display a normal or enhanced differentiation (208). This indicates that the inhibitory effect of tofacitinib is terminated as soon as the drug is removed (176, 201, 208). Besides tofacitinib, other JAK inhibitors have been approved or are currently being tested in clinical trials as new potential treatment options for RA and/ or other autoimmune diseases and chronic inflammatory conditions. Thus, new studies concerning the effects of JAK inhibitors on innate and adaptive immune system responses are still emerging. In fact, the diversity of cytokines that trigger B cell immune responses through JAK-STAT signaling pathway activation (Figure 4) suggests that other JAK inhibitors, besides JAK3 inhibitors, might have important roles in B cell immunity (Figure 3). Changes in lymphocyte numbers (B, T, and NK cells) and subpopulations have been recently demonstrated in active RA patients after treatment with baricitinib (214). An integrated data analysis has been performed based on results from three completed phase III trials comparing placebo with baricitinib treatment (RA-BEAM, RA-BUILD, and RA-BEACON) and one ongoing long-term extension study (RA-BEYOND) in patients with active RA. Overall, a transient increase in total lymphocyte count was observed in RA patients after 4 weeks of treatment with baricitinib, returning to baseline values by week 12. Moreover, transient changes in T cells and subsets (CD3+, CD4+, CD8+, Th1, Th17, and regulatory T cells) were observed with baricitinib treatment, with cell counts remaining largely within normal reference ranges (214). Additionally, it was shown that CD19+ B cells and B cell subpopulations (including switched memory, non-switched memory, mature naïve, and immature transitional B cells) increased after 4 weeks of baricitinib treatment and remained above baseline or stabilized over time (214). Importantly, baricitinib treatment did not result in increased autoantibody (RF and ACPA) titers, suggesting that the increase in total B cell counts is unlikely to reflect a major expansion of RA antigen-specific B cells (214). Nevertheless, it is possible that some of the class-switched memory B cells, increased by baricitinib in a dose-dependent manner, are regulatory B cells, which inhibit disease progression (214). Of note, the detected changes in lymphocyte subsets were largely consistent across the baricitinib phase III RA clinical trials, which included patients with different responsiveness to prior DMARD therapies and were not associated with increased risk of serious infections (214). Recently, the in vitro effects of baricitinib were evaluated on human peripheral blood cells and it was shown that baricitinib modulates both innate and adaptive immune responses similarly to tofacitinib (88, 197, 215). Baricitinib suppressed the expression of costimulatory molecules (CD80/CD86) on monocyte-derived dendritic cells and inhibited T cell proliferation and differentiation of Th1 and Th17 cells. Furthermore, baricitinib suppressed the differentiation of human B cells into plasmablasts by B cell receptor and type-I interferon (IFN) stimuli and inhibited the production of IL-6 from B cells (215). Also, it was recently shown that baricitinib decreased BAFF expression in RA synovial fibroblasts similarly to tofacitinib, thus inhibiting B cell activation locally in the joints (216). The impact of baricitinib on B cells is further supported by studies developed in a mouse model of graft-vs.-host disease (GVHD) in which it was demonstrated that baricitinib inhibited the activation of allogeneic antigen presenting cells (APCs) and prevented GVHD progression (217). It was shown that baricitinib suppressed the expression of major histocompatibility complex (MHC)-II, costimulatory molecules CD80/86 and PD-L1 on B220+ and CD11c+ APCs. Moreover, baricitinib expanded regulatory T cells and downregulated Th1 and Th2 cell responses (217). Studies developed in RA patients and animal models of arthritis treated with upadacitinib have reported decreased circulating numbers of lymphocytes, neutrophils and NK cells (141, 142, 218). Nonetheless, no significant changes were detected in RF and ACPA levels in RA patients after upadacitinib treatment (144). Furthermore, it has been recently shown that upadacitinib has a generally similar profile of in vitro cytokine receptor inhibition observed in human leukocyte subpopulations when compared to other JAK inhibitors (219). Particularly, it was observed that upadacitinib inhibited STAT6 phosphorylation on CD19+ B cells triggered by IL-13 stimuli similarly to tofacitinib, baricitinib and filgotinib (219). However, a recent in vitro pharmacology study comparing tofacitinib, baricitinib and upadacitinib has revealed that different JAK inhibitors modulate distinct cytokine pathways to varying degrees (220). Notably, it was shown that upadacitinib and tofacitinib were the most potent inhibitors of the JAK1/3-dependent cytokines tested, including IL-4, IL-6 and IL-21, relevant for B cell activation, plasma cell differentiation and humoral immune responses (218, 220). In addition, studies with peficitinib have demonstrated an inhibitory effect of this JAK inhibitor on T cell activation using either a rat adjuvant-induced arthritis model (221) or human peripheral blood mononuclear cells (86, 222). Moreover, it was shown that peficitinib suppressed in vitro monocyte chemotactic activity and the proliferation of fibroblast-like synoviocytes from RA patients (79, 223, 224). Interestingly, decreases in neutrophil and total lymphocyte counts were observed after peficitinib treatment, but no significant changes were detected on T cell subpopulations (152–155, 158, 222, 225). Nevertheless, studies on the potential effects of peficitinib treatment on human B cells are currently lacking. Filgotinib was recently approved by EMA for the treatment of RA and clinical trials with this JAK1-selective inhibitor are currently under investigation in other autoimmune diseases. Changes in leukocyte numbers, particularly increases in B cell frequencies, have been reported in RA patients after filgotinib treatment (163, 164, 226). Furthermore, studies exploring the action of this JAKi on B cells have demonstrated that filgotinib directly inhibits human B cell differentiation and IgG production (227). Recent reports in RA patients following treatment with filgotinib have shown significant reductions in markers important for B cell chemotaxis [chemokine (C-X-C motif) ligand 13, CXCL13]; activation and survival (BAFF); regulatory function (IL-10) and germinal center and plasma cell differentiation (IL-2, IL-5, IL-7, and IL-21) (226). Moreover, filgotinib has also been shown to suppress the production of BAFF in human primary salivary gland (SG) epithelial cells and SG organoids (227). Additionally, studies developed in a mouse model of Sjögren syndrome have shown a marked reduction in lymphocytic infiltration of salivary glands after filgotinib treatment, which contributed to disease amelioration (227). Decernotinib is another JAK inhibitor currently under evaluation for the treatment of RA (173–175, 228, 229). Although lymphopenia and neutropenia have been described in decernotinib trials (174, 175), the exact mechanisms of action and effects of this JAKi on B cell immune responses still need to be further clarified. Table 1 summarizes the impact of currently approved JAK inhibitors on B cell immune responses described in the literature. Overall, additional pharmacological studies of JAKi exploring the effect of different cytokine pathways and/ or JAK targeting in distinct human leukocyte populations remain of clinical importance.
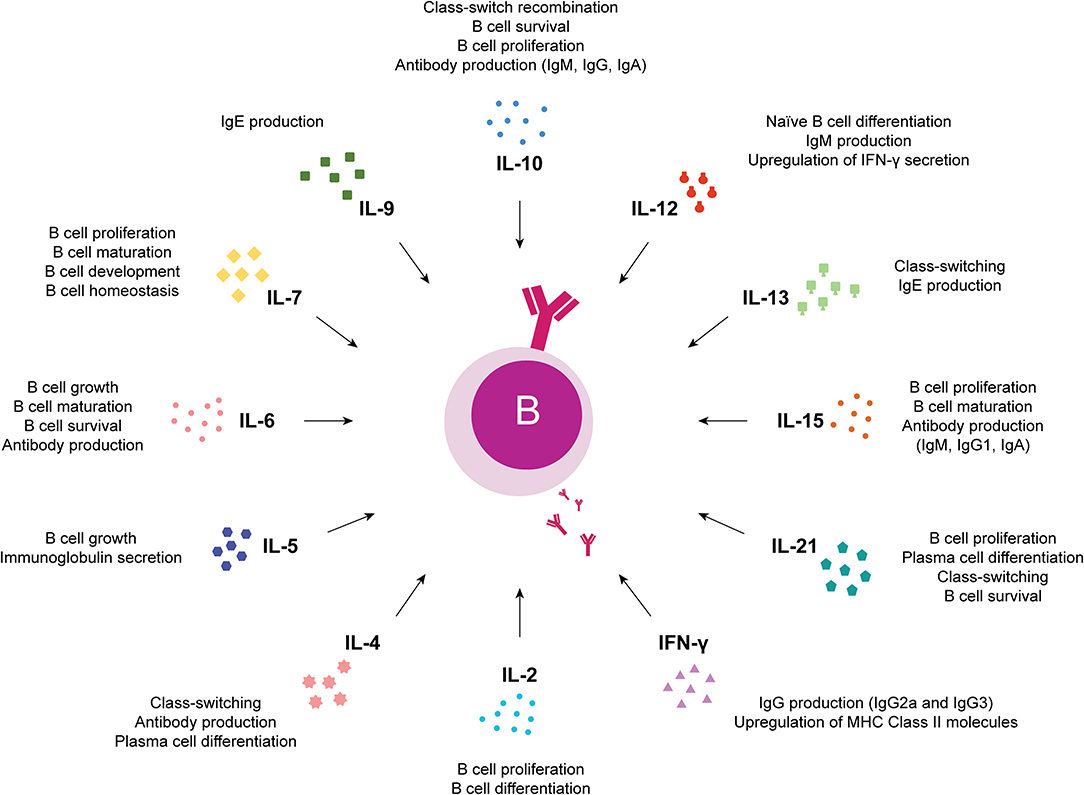
Figure 4. Cytokines that trigger B cell immune responses through JAK-STAT signaling pathway activation. Overview of the effects of cytokines relevant for B cells that trigger immune responses through JAK-STAT signaling pathway activation. IFN, interferon; IL, interleukin.
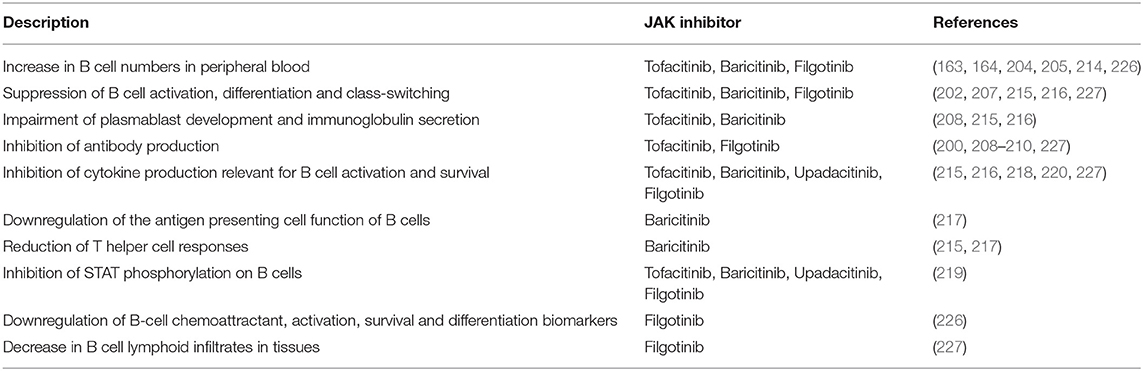
Table 1. Overview of the impact of JAK inhibitors on B cell immune responses based on pharmacokinetic, in vitro and in vivo studies.
JAK inhibitors are a new class of oral immunosuppressive drugs with proved efficacy in the treatment of chronic inflammatory conditions and autoimmune diseases such as RA. B cells play several important roles in RA pathogenesis since the first weeks of disease development. Pharmacokinetic, in vitro and in vivo studies developed so far with animal models of arthritis or other autoimmune conditions and/ or with human cells from RA patients or other chronic inflammatory disorders have demonstrated that JAK inhibitors (tofacitinib, baricitinib, upadacitinib, peficitinib, filgotinib and decernotinib) can affect B cell activation, proliferation and differentiation. Taking into consideration these B cell effects of JAKi and the relevant role of B cells since early RA onset it is likely that JAKi can have a major impact on the early phase of RA. Nevertheless, further research studies are necessary to clarify the exact mechanisms of action of JAKi on B cells and other immune cell targets not only in currently approved JAK inhibitors, but also in new JAKi under investigation.
RM and JF conceptualized the manuscript. RM reviewed the literature and wrote the manuscript. JF revised the manuscript and contributed with important intellectual input. All authors read and approved the final manuscript.
The authors would like to acknowledge Sociedade Portuguesa de Reumatologia (SPR) for funding. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Leandro MJ, Edwards JCW, Cambridge G. Clinical outcome in 22 patients with rheumatoid arthritis treated with B lymphocyte depletion. Ann Rheum Dis. (2002) 61:883–8. doi: 10.1136/ard.61.10.883
2. Edwards JCW, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. (2004) 350:2572–81. doi: 10.1056/NEJMoa032534
3. Edwards JCW, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. (2006) 6:394–403. doi: 10.1038/nri1838
4. Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet. (2010) 376:1094–108. doi: 10.1016/S0140-6736(10)60826-4
5. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
6. Polido-Pereira J, Vieira-Sousa E, Fonseca JE. Rheumatoid arthritis: what is refractory disease and how to manage it? Autoimmun Rev. (2011) 10:707–13. doi: 10.1016/j.autrev.2011.04.023
7. Romão VC, Canhão H, Fonseca JE. Old drugs, old problems: where do we stand in prediction of rheumatoid arthritis responsiveness to methotrexate and other synthetic DMARDs? BMC Med. (2013) 11:17. doi: 10.1186/1741-7015-11-17
8. Romão VC, Vital EM, Fonseca JE, Buch MH. Right drug, right patient, right time: aspiration or future promise for biologics in rheumatoid arthritis? Arthritis Res Ther. (2017) 19:239. doi: 10.1186/s13075-017-1445-3
9. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. (2017) 77:521–46. doi: 10.1007/s40265-017-0701-9
10. Lin CM, Cooles FA, Isaacs JD. Basic mechanisms of JAK Inhibition. Mediterr J Rheumatol. (2020) 31:100–4. doi: 10.31138/mjr.31.1.100
11. Westhovens R. Clinical efficacy of new JAK inhibitors under development. Just more of the same? Rheumatology. (2019) 58:i27–i33. doi: 10.1093/rheumatology/key256
12. Angelini J, Talotta R, Roncato R, Fornasier G, Barbiero G, Dal Cin L, et al. JAK-inhibitors for the treatment of rheumatoid arthritis: a focus on the present and an outlook on the future. Biomolecules. (2020) 10:1002. doi: 10.3390/biom10071002
13. Moura RA, Graca L, Fonseca JE. To B or not to B the conductor of rheumatoid arthritis orchestra. Clin Rev Allergy Immunol. (2012) 43:281–91. doi: 10.1007/s12016-012-8318-y
14. Edwards JCW, Leandro MJ, Cambridge G. B lymphocyte depletion therapy with rituximab in rheumatoid arthritis. Rheum Dis Clin North Am. (2004) 30:393–403, viii. doi: 10.1016/j.rdc.2004.01.006
15. Moura RA, Weinmann P, Pereira PA, Caetano-Lopes J, Canhão H, Sousa E, et al. Alterations on peripheral blood B-cell subpopulations in very early arthritis patients. Rheumatology. (2010) 49:1082–92. doi: 10.1093/rheumatology/keq029
16. Cascão R, Moura RA, Perpétuo I, Canhão H, Vieira-Sousa E, Mourão AF, et al. Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res Ther. (2010) 12:R196. doi: 10.1186/ar3168
17. Moura RA, Cascão R, Perpétuo I, Canhão H, Vieira-Sousa E, Mourão AF, et al. Cytokine pattern in very early rheumatoid arthritis favours B-cell activation and survival. Rheumatology. (2011) 50:278–82. doi: 10.1093/rheumatology/keq338
18. Moura RA, Canhão H, Polido-Pereira J, Rodrigues AM, Navalho M, Mourão AF, et al. BAFF and TACI gene expression are increased in patients with untreated very early rheumatoid arthritis. J Rheumatol. (2013) 40:1293–302. doi: 10.3899/jrheum.121110
19. Moura RA, Quaresma C, Vieira AR, Gonçalves MJ, Polido-Pereira J, Romão VC, et al. B-cell phenotype and IgD-CD27- memory B cells are affected by TNF-inhibitors and tocilizumab treatment in rheumatoid arthritis. PLoS ONE. (2017) 12:e0182927. doi: 10.1371/journal.pone.0182927
20. Leandro MJ, Cambridge G, Ehrenstein MR, Edwards JCW. Reconstitution of peripheral blood B cells after depletion with rituximab in patients with rheumatoid arthritis. Arthritis Rheum. (2006) 54:613–20. doi: 10.1002/art.21617
21. Cambridge G, Stohl W, Leandro MJ, Migone T-S, Hilbert DM, Edwards JCW. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum. (2006) 54:723–32. doi: 10.1002/art.21650
22. Popa C, Leandro MJ, Cambridge G, Edwards JCW. Repeated B lymphocyte depletion with rituximab in rheumatoid arthritis over 7 yrs. Rheumatology. (2007) 46:626–30. doi: 10.1093/rheumatology/kel393
23. de la Torre I, Moura RA, Leandro MJ, Edwards J, Cambridge G. B-cell-activating factor receptor expression on naive and memory B cells: relationship with relapse in patients with rheumatoid arthritis following B-cell depletion therapy. Ann Rheum Dis. (2010) 69:2181–8. doi: 10.1136/ard.2010.131326
24. Cambridge G, Moura RA, Santos T, Khawaja AA, Polido-Pereira J, Canhão H, et al. Expression of the inherently autoreactive idiotope 9G4 on autoantibodies to citrullinated peptides and on rheumatoid factors in patients with early and established rheumatoid arthritis. PLoS ONE. (2014) 9:e107513. doi: 10.1371/journal.pone.0107513
25. Humby F, Lewis M, Ramamoorthi N, Hackney JA, Barnes MR, Bombardieri M, et al. Synovial cellular and molecular signatures stratify clinical response to csDMARD therapy and predict radiographic progression in early rheumatoid arthritis patients. Ann Rheum Dis. (2019) 78:761–72. doi: 10.1136/annrheumdis-2018-214539
26. Lliso-Ribera G, Humby F, Lewis M, Nerviani A, Mauro D, Rivellese F, et al. Synovial tissue signatures enhance clinical classification and prognostic/treatment response algorithms in early inflammatory arthritis and predict requirement for subsequent biological therapy: results from the pathobiology of early arthritis cohort (PEAC). Ann Rheum Dis. (2019) 78:1642–52. doi: 10.1136/annrheumdis-2019-215751
27. Gómez-Puerta JA, Celis R, Hernández MV, Ruiz-Esquide V, Ramírez J, Haro I, et al. Differences in synovial fluid cytokine levels but not in synovial tissue cell infiltrate between anti-citrullinated peptide/protein antibody-positive and -negative rheumatoid arthritis patients. Arthritis Res Ther. (2013) 15:R182. doi: 10.1186/ar4372
28. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis—shaping the immunological landscape. Nat Rev Rheumatol. (2016) 12:63–8. doi: 10.1038/nrrheum.2015.171
29. Mateen S, Zafar A, Moin S, Khan AQ, Zubair S. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin Chim Acta. (2016) 455:161–71. doi: 10.1016/j.cca.2016.02.010
30. Noack M, Miossec P. Selected cytokine pathways in rheumatoid arthritis. Semin Immunopathol. (2017) 39:365–83. doi: 10.1007/s00281-017-0619-z
31. Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam K-P, et al. TACI and BAFF-R mediate isotype switching in B cells. J Exp Med. (2005) 201:35–9. doi: 10.1084/jem.20032000
32. Dong W, Li X, Liu H, Zhu P. Infiltrations of plasma cells in synovium are highly associated with synovial fluid levels of APRIL in inflamed peripheral joints of rheumatoid arthritis. Rheumatol Int. (2009) 29:801–6. doi: 10.1007/s00296-008-0773-7
33. Zhao J, Guo J, Wang L, Zhou W, Zhang Z. The role of a proliferation-inducing ligand (APRIL) in the pathogenesis of rheumatoid arthritis. Scand J Rheumatol. (2014) 43:462–9. doi: 10.3109/03009742.2014.905630
34. Gowhari Shabgah A, Shariati-Sarabi Z, Tavakkol-Afshari J, Ghasemi A, Ghoryani M, Mohammadi M. A significant decrease of BAFF, APRIL, and BAFF receptors following mesenchymal stem cell transplantation in patients with refractory rheumatoid arthritis. Gene. (2020) 732:144336. doi: 10.1016/j.gene.2020.144336
35. Cai X-Y, Zhu Y, Wang C, Tang X-Y, Han L, Shu J-L, t al. Etanercept inhibits B cell differentiation by regulating TNFRII/TRAF2/NF-κB signaling pathway in rheumatoid arthritis. Front Pharmacol. (2020) 11:676. doi: 10.3389/fphar.2020.00676
36. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. (1999) 103:1345–52. doi: 10.1172/JCI5703
37. Kim K-W, Kim H-R, Kim B-M, Cho M-L, Lee S-H. Th17 cytokines regulate osteoclastogenesis in rheumatoid arthritis. Am J Pathol. (2015) 185:3011–24. doi: 10.1016/j.ajpath.2015.07.017
38. Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, Van Snick J. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. (1988) 31:784–8. doi: 10.1002/art.1780310614
39. Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. (2006) 8(Suppl 2):S3. doi: 10.1186/ar1917
40. Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a growth factor for nonmalignant human plasmablasts. Blood. (2001) 97:1817–22. doi: 10.1182/blood.v97.6.1817
41. Dienz O, Eaton SM, Bond JP, Neveu W, Moquin D, Noubade R, et al. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J Exp Med. (2009) 206:69–78. doi: 10.1084/jem.20081571
42. Choy EH, Bernasconi C, Aassi M, Molina JF, Epis OM. Treatment of rheumatoid arthritis with anti-tumor necrosis factor or Tocilizumab therapy as first biologic agent in a global comparative observational study. Arthritis Care Res. (2017) 69:1484–94. doi: 10.1002/acr.23303
43. Ramírez J, Cañete JD. Anakinra for the treatment of rheumatoid arthritis: a safety evaluation. Expert Opin Drug Saf. (2018) 17:727–32. doi: 10.1080/14740338.2018.1486819
44. Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: a review. JAMA. (2018) 320:1360–72. doi: 10.1001/jama.2018.13103
45. Köhler BM, Günther J, Kaudewitz D, Lorenz H-M. Current therapeutic options in the treatment of rheumatoid arthritis. J Clin Med. (2019) 8:938. doi: 10.3390/jcm8070938
46. Favalli EG. Understanding the role of interleukin-6 (IL-6) in the joint and beyond: a comprehensive review of IL-6 inhibition for the management of rheumatoid arthritis. Rheumatol Ther. (2020) 7:473–516. doi: 10.1007/s40744-020-00219-2
47. Mertens C, Darnell JE. SnapShot: JAK-STAT signaling. Cell. (2007) 131:612. doi: 10.1016/j.cell.2007.10.033
48. Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity. (2012) 36:503–14. doi: 10.1016/j.immuni.2012.03.013
49. Bousoik E, Montazeri Aliabadi H. “Do we know jack” about JAK? A closer look at JAK/STAT signaling pathway. Front Oncol. (2018) 8:287. doi: 10.3389/fonc.2018.00287
50. Leonard WJ, O'Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. (1998) 16:293–322. doi: 10.1146/annurev.immunol.16.1.293
51. Laurence A, Pesu M, Silvennoinen O, O'Shea J. JAK kinases in health and disease: an update. Open Rheumatol J. (2012) 6:232–44. doi: 10.2174/1874312901206010232
52. Yamaoka K, Saharinen P, Pesu M, Holt VET, Silvennoinen O, O'Shea JJ. The Janus kinases (Jaks). Genome Biol. (2004) 5:253. doi: 10.1186/gb-2004-5-12-253
53. O'Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. (2013) 368:161–70. doi: 10.1056/NEJMra1202117
54. Darnell JE. STATs and gene regulation. Science. (1997) 277:1630–5. doi: 10.1126/science.277.5332.1630
55. Bromberg J, Darnell JE. The role of STATs in transcriptional control and their impact on cellular function. Oncogene. (2000) 19:2468–73. doi: 10.1038/sj.onc.1203476
56. Abroun S, Saki N, Ahmadvand M, Asghari F, Salari F, Rahim F. STATs: an old story, yet mesmerizing. Cell J. (2015) 17:395–411. doi: 10.22074/cellj.2015.1
57. Loh C-Y, Arya A, Naema AF, Wong WF, Sethi G, Looi CY. Signal transducer and activator of transcription (STATs) proteins in cancer and inflammation: functions and therapeutic implication. Front Oncol. (2019) 9:48. doi: 10.3389/fonc.2019.00048
58. Xu D, Qu C-K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. (2008) 13:4925–32. doi: 10.2741/3051
59. Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. (2005) 5:593–605. doi: 10.1038/nri1667
60. Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. (2006) 16:196–202. doi: 10.1038/sj.cr.7310027
61. Kile BT, Alexander WS. The suppressors of cytokine signalling (SOCS). Cell Mol Life Sci. (2001) 58:1627–35. doi: 10.1007/PL00000801
62. Durham GA, Williams JJL, Nasim MT, Palmer TM. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol Sci. (2019) 40:298–308. doi: 10.1016/j.tips.2019.03.001
63. Schwartz DM, Bonelli M, Gadina M, O'Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. (2016) 12:25–36. doi: 10.1038/nrrheum.2015.167
64. Valenzuela F, Korman NJ, Bissonnette R, Bakos N, Tsai T-F, Harper MK, et al. Tofacitinib in patients with moderate-to-severe chronic plaque psoriasis: long-term safety and efficacy in an open-label extension study. Br J Dermatol. (2018) 179:853–62. doi: 10.1111/bjd.16798
65. Papp KA, Krueger JG, Feldman SR, Langley RG, Thaci D, Torii H, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: long-term efficacy and safety results from 2 randomized phase-III studies and 1 open-label long-term extension study. J Am Acad Dermatol. (2016) 74:841–50. doi: 10.1016/j.jaad.2016.01.013
66. Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. (2016) 175:902–11. doi: 10.1111/bjd.14871
67. Alves de Medeiros AK, Speeckaert R, Desmet E, Van Gele M, De Schepper S, Lambert J. JAK3 as an emerging target for topical treatment of inflammatory skin diseases. PLoS ONE. (2016) 11:e0164080. doi: 10.1371/journal.pone.0164080
68. Almutairi N, Nour TM, Hussain NH. Janus kinase inhibitors for the treatment of severe alopecia areata: an open-label comparative study. Dermatology. (2019) 235:130–6. doi: 10.1159/000494613
69. Jabbari A, Sansaricq F, Cerise J, Chen JC, Bitterman A, Ulerio G, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol. (2018) 138:1539–45. doi: 10.1016/j.jid.2018.01.032
70. Liu LY, Strassner JP, Refat MA, Harris JE, King BA. Repigmentation in vitiligo using the Janus kinase inhibitor tofacitinib may require concomitant light exposure. J Am Acad Dermatol. (2017) 77:675–82.e1. doi: 10.1016/j.jaad.2017.05.043
71. Craiglow BG, King BA. Tofacitinib citrate for the treatment of vitiligo: a pathogenesis-directed therapy. JAMA Dermatol. (2015) 151:1110–2. doi: 10.1001/jamadermatol.2015.1520
72. Kiladjian J-J, Zachee P, Hino M, Pane F, Masszi T, Harrison CN, et al. Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol. (2020) 7:e226–e237. doi: 10.1016/S2352-3026(19)30207-8
73. Braun TP, Coblentz C, Smith BM, Coleman DJ, Schonrock Z, Carratt SA, et al. Combined inhibition of JAK/STAT pathway and lysine-specific demethylase 1 as a therapeutic strategy in CSF3R/CEBPA mutant acute myeloid leukemia. Proc Natl Acad Sci USA. (2020) 117:13670–9. doi: 10.1073/pnas.1918307117
74. Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature. (1995) 377:65–8. doi: 10.1038/377065a0
75. Keystone EC, Taylor PC, Drescher E, Schlichting DE, Beattie SD, Berclaz P-Y, et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann Rheum Dis. (2015) 74:333–40. doi: 10.1136/annrheumdis-2014-206478
76. Charles-Schoeman C, Burmester G, Nash P, Zerbini CAF, Soma K, Kwok K, et al. Efficacy and safety of tofacitinib following inadequate response to conventional synthetic or biological disease-modifying antirheumatic drugs. Ann Rheum Dis. (2016) 75:1293–301. doi: 10.1136/annrheumdis-2014-207178
77. Genovese MC, Fleischmann R, Combe B, Hall S, Rubbert-Roth A, Zhang Y, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet. (2018) 391:2513–24. doi: 10.1016/S0140-6736(18)31116-4
78. van der Heijde D, Strand V, Tanaka Y, Keystone E, Kremer J, Zerbini CAF, et al. Tofacitinib in combination with methotrexate in patients with rheumatoid arthritis: clinical efficacy, radiographic, and safety outcomes from a twenty-four-month, phase III study. Arthritis Rheumatol. (2019) 71:878–91. doi: 10.1002/art.40803
79. Emori T, Kasahara M, Sugahara S, Hashimoto M, Ito H, Narumiya S, et al. Role of JAK-STAT signaling in the pathogenic behavior of fibroblast-like synoviocytes in rheumatoid arthritis: effect of the novel JAK inhibitor peficitinib. Eur J Pharmacol. (2020) 882:173238. doi: 10.1016/j.ejphar.2020.173238
80. Strand V, de Vlam K, Covarrubias-Cobos JA, Mease PJ, Gladman DD, Graham D, et al. Tofacitinib or adalimumab versus placebo: patient-reported outcomes from OPAL Broaden-a phase III study of active psoriatic arthritis in patients with an inadequate response to conventional synthetic disease-modifying antirheumatic drugs. RMD Open. (2019) 5:e000806. doi: 10.1136/rmdopen-2018-000806
81. Merola JF, Papp KA, Nash P, Gratacós J, Boehncke WH, Thaçi D, et al. Tofacitinib in psoriatic arthritis patients: skin signs and symptoms and health-related quality of life from two randomized phase 3 studies. J Eur Acad Dermatol Venereol. (2020) 34:2809–20. doi: 10.1111/jdv.16433
82. Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. (2018) 392:222–31. doi: 10.1016/S0140-6736(18)31363-1
83. Alunno A, Padjen I, Fanouriakis A, Boumpas DT. Pathogenic and therapeutic relevance of JAK/STAT signaling in systemic lupus erythematosus: integration of distinct inflammatory pathways and the prospect of their inhibition with an oral agent. Cells. (2019) 8:898. doi: 10.3390/cells8080898
84. van der Heijde D, Baraliakos X, Gensler LS, Maksymowych WP, Tseluyko V, Nadashkevich O, et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): results from a randomised, placebo-controlled, phase 2 trial. Lancet. (2018) 392:2378–87. doi: 10.1016/S0140-6736(18)32463-2
85. van der Heijde D, Song I-H, Pangan AL, Deodhar A, van den Bosch F, Maksymowych WP, et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): a multicentre, randomised, double-blind, placebo-controlled, phase 2/3 trial. Lancet. (2019) 394:2108–17. doi: 10.1016/S0140-6736(19)32534-6
86. Kitanaga Y, Imamura E, Nakahara Y, Fukahori H, Fujii Y, Kubo S, et al. In vitro pharmacological effects of peficitinib on lymphocyte activation: a potential treatment for systemic sclerosis with JAK inhibitors. Rheumatology. (2020) 59:1957–68. doi: 10.1093/rheumatology/kez526
87. Hinchcliff M, O'Reilly S. Current and potential new targets in systemic sclerosis therapy: a new hope. Curr Rheumatol Rep. (2020) 22:42. doi: 10.1007/s11926-020-00918-3
88. Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation. (2018) 137:1934–48. doi: 10.1161/CIRCULATIONAHA.117.030423
89. Damsky W, Thakral D, Emeagwali N, Galan A, King B. Tofacitinib treatment and molecular analysis of cutaneous sarcoidosis. N Engl J Med. (2018) 379:2540–6. doi: 10.1056/NEJMoa1805958
90. Damsky W, Young BD, Sloan B, Miller EJ, Obando JA, King B. Treatment of multiorgan sarcoidosis with tofacitinib. ACR Open Rheumatol. (2020) 2:106–9. doi: 10.1002/acr2.11112
91. Wang A, Singh K, Ibrahim W, King B, Damsky W. The promise of JAK inhibitors for treatment of sarcoidosis and other inflammatory disorders with macrophage activation: a review of the literature. Yale J Biol Med. (2020) 93:187–95.
92. Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. (2012) 367:616–24. doi: 10.1056/NEJMoa1112168
93. Vermeire S, Schreiber S, Petryka R, Kuehbacher T, Hebuterne X, Roblin X, et al. Clinical remission in patients with moderate-to-severe Crohn's disease treated with filgotinib (the FITZROY study): results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet. (2017) 389:266–75. doi: 10.1016/S0140-6736(16)32537-5
94. Hosseini A, Gharibi T, Marofi F, Javadian M, Babaloo Z, Baradaran B. Janus kinase inhibitors: a therapeutic strategy for cancer and autoimmune diseases. J Cell Physiol. (2020) 235:5903–24. doi: 10.1002/jcp.29593
95. Malemud CJ. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther Adv Musculoskelet Dis. (2018) 10:117–27. doi: 10.1177/1759720X18776224
96. Jamilloux Y, El Jammal T, Vuitton L, Gerfaud-Valentin M, Kerever S, Sève P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun Rev. (2019) 18:102390. doi: 10.1016/j.autrev.2019.102390
97. Fragoulis GE, McInnes IB, Siebert S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology. (2019) 58:i43–54. doi: 10.1093/rheumatology/key276
98. van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol. (2013) 9:164–72. doi: 10.1038/nrrheum.2013.4
99. Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. (2012) 367:495–507. doi: 10.1056/NEJMoa1109071
100. van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. (2012) 367:508–19. doi: 10.1056/NEJMoa1112072
101. Kremer JM, Cohen S, Wilkinson BE, Connell CA, French JL, Gomez-Reino J, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. (2012) 64:970–81. doi: 10.1002/art.33419
102. Fleischmann R, Cutolo M, Genovese MC, Lee EB, Kanik KS, Sadis S, et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. (2012) 64:617–29. doi: 10.1002/art.33383
103. van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. (2013) 65:559–70. doi: 10.1002/art.37816
104. Kremer J, Li Z-G, Hall S, Fleischmann R, Genovese M, Martin-Mola E, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. (2013) 159:253–61. doi: 10.7326/0003-4819-159-4-201308200-00006
105. Lee YH, Bae S-C, Song GG. Comparative efficacy and safety of tofacitinib, with or without methotrexate, in patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Rheumatol Int. (2015) 35:1965–74. doi: 10.1007/s00296-015-3291-4
106. Buckley F, Finckh A, Huizinga TWJ, Dejonckheere F, Jansen JP. Comparative efficacy of novel DMARDs as monotherapy and in combination with methotrexate in rheumatoid arthritis patients with inadequate response to conventional DMARDs: a network meta-analysis. J Manag Care Spec Pharm. (2015) 21:409–23. doi: 10.18553/jmcp.2015.21.5.409
107. Strand V, van Vollenhoven RF, Lee EB, Fleischmann R, Zwillich SH, Gruben D, et al. Tofacitinib or adalimumab versus placebo: patient-reported outcomes from a phase 3 study of active rheumatoid arthritis. Rheumatology. (2016) 55:1031–41. doi: 10.1093/rheumatology/kev442
108. Wallenstein GV, Kanik KS, Wilkinson B, Cohen S, Cutolo M, Fleischmann R, et al. Effects of the oral Janus kinase inhibitor tofacitinib on patient-reported outcomes in patients with active rheumatoid arthritis: results of two Phase 2 randomised controlled trials. Clin Exp Rheumatol. (2016) 34:430–42.
109. Vieira M-C, Zwillich SH, Jansen JP, Smiechowski B, Spurden D, Wallenstein GV. Tofacitinib versus biologic treatments in patients with active rheumatoid arthritis who have had an inadequate response to tumor necrosis factor inhibitors: results from a network meta-analysis. Clin Ther. (2016) 38:2628–41.e5. doi: 10.1016/j.clinthera.2016.11.004
110. Strand V, Kremer JM, Gruben D, Krishnaswami S, Zwillich SH, Wallenstein GV. Tofacitinib in combination with conventional disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: patient-reported outcomes from a phase III randomized controlled trial. Arthritis Care Res. (2017) 69:592–8. doi: 10.1002/acr.23004
111. Fleischmann R, Mease PJ, Schwartzman S, Hwang L-J, Soma K, Connell CA, et al. Efficacy of tofacitinib in patients with rheumatoid arthritis stratified by background methotrexate dose group. Clin Rheumatol. (2017) 36:15–24. doi: 10.1007/s10067-016-3436-1
112. Wollenhaupt J, Lee E-B, Curtis JR, Silverfield J, Terry K, Soma K, et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: final results of a global, open-label, long-term extension study. Arthritis Res Ther. (2019) 21:89. doi: 10.1186/s13075-019-1866-2
113. Burmester GR, Blanco R, Charles-Schoeman C, Wollenhaupt J, Zerbini C, Benda B, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. (2013) 381:451–60. doi: 10.1016/S0140-6736(12)61424-X
114. Fleischmann R, Mysler E, Hall S, Kivitz AJ, Moots RJ, Luo Z, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet. (2017) 390:457–68. doi: 10.1016/S0140-6736(17)31618-5
115. Kim JW, Choi IA, Lee EY, Song YW, Lee EB. Tofacitinib prevents radiographic progression in rheumatoid arthritis. J Korean Med Sci. (2013) 28:1134–8. doi: 10.3346/jkms.2013.28.8.1134
116. Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. (2014) 370:2377–86. doi: 10.1056/NEJMoa1310476
117. Boyle DL, Soma K, Hodge J, Kavanaugh A, Mandel D, Mease P, et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann Rheum Dis. (2015) 74:1311–6. doi: 10.1136/annrheumdis-2014-206028
118. He Y, Wong AYS, Chan EW, Lau WCY, Man KKC, Chui CSL, et al. Efficacy and safety of tofacitinib in the treatment of rheumatoid arthritis: a systematic review and meta-analysis. BMC Musculoskelet Disord. (2013) 14:298. doi: 10.1186/1471-2474-14-298
119. Wollenhaupt J, Silverfield J, Lee EB, Curtis JR, Wood SP, Soma K, et al. Safety and efficacy of tofacitinib, an oral janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J Rheumatol. (2014) 41:837–52. doi: 10.3899/jrheum.130683
120. Cohen S, Radominski SC, Gomez-Reino JJ, Wang L, Krishnaswami S, Wood SP, et al. Analysis of infections and all-cause mortality in phase II, phase III, and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:2924–37. doi: 10.1002/art.38779
121. Souto A, Salgado E, Maneiro JR, Mera A, Carmona L, Gómez-Reino JJ. Lipid profile changes in patients with chronic inflammatory arthritis treated with biologic agents and tofacitinib in randomized clinical trials: a systematic review and meta-analysis. Arthritis Rheumatol. (2015) 67:117–27. doi: 10.1002/art.38894
122. Curtis JR, Lee EB, Kaplan IV, Kwok K, Geier J, Benda B, et al. Tofacitinib, an oral Janus kinase inhibitor: analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann Rheum Dis. (2016) 75:831–41. doi: 10.1136/annrheumdis-2014-205847
123. Desai RJ, Pawar A, Weinblatt ME, Kim SC. Comparative risk of venous thromboembolism in rheumatoid arthritis patients receiving tofacitinib versus those receiving tumor necrosis factor inhibitors: an observational cohort study. Arthritis Rheumatol. (2019) 71:892–900. doi: 10.1002/art.40798
124. Sunzini F, McInnes I, Siebert S. JAK inhibitors and infections risk: focus on herpes zoster. Ther Adv Musculoskelet Dis. (2020) 12:1759720X20936059. doi: 10.1177/1759720X20936059
125. Morinobu A. JAK inhibitors for the treatment of rheumatoid arthritis. Immunol Med. (2020) 43:1–8. doi: 10.1080/25785826.2020.1770948
126. Fleischmann R, Schiff M, van der Heijde D, Ramos-Remus C, Spindler A, Stanislav M, et al. Baricitinib, methotrexate, or combination in patients with rheumatoid arthritis and no or limited prior disease-modifying antirheumatic drug treatment. Arthritis Rheumatol. (2017) 69:506–17. doi: 10.1002/art.39953
127. Dougados M, van der Heijde D, Chen Y-C, Greenwald M, Drescher E, Liu J, et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA-BUILD study. Ann Rheum Dis. (2017) 76:88–95. doi: 10.1136/annrheumdis-2016-210094
128. Schiff M, Takeuchi T, Fleischmann R, Gaich CL, DeLozier AM, Schlichting D, et al. Patient-reported outcomes of baricitinib in patients with rheumatoid arthritis and no or limited prior disease-modifying antirheumatic drug treatment. Arthritis Res Ther. (2017) 19:208. doi: 10.1186/s13075-017-1410-1
129. Smolen JS, Kremer JM, Gaich CL, DeLozier AM, Schlichting DE, Xie L, et al. Patient-reported outcomes from a randomised phase III study of baricitinib in patients with rheumatoid arthritis and an inadequate response to biological agents (RA-BEACON). Ann Rheum Dis. (2017) 76:694–700. doi: 10.1136/annrheumdis-2016-209821
130. Lee YH, Bae S-C. Comparative efficacy and safety of baricitinib 2 mg and 4 mg in patients with active rheumatoid arthritis : a Bayesian network meta-analysis of randomized controlled trials. Z Rheumatol. (2018) 77:335–42. doi: 10.1007/s00393-016-0254-4
131. Genovese MC, Kremer JM, Kartman CE, Schlichting DE, Xie L, Carmack T, et al. Response to baricitinib based on prior biologic use in patients with refractory rheumatoid arthritis. Rheumatology. (2018) 57:900–8. doi: 10.1093/rheumatology/kex489
132. Tanaka Y, Fautrel B, Keystone EC, Ortmann RA, Xie L, Zhu B, et al. Clinical outcomes in patients switched from adalimumab to baricitinib due to non-response and/or study design: phase III data in patients with rheumatoid arthritis. Ann Rheum Dis. (2019) 78:890–8. doi: 10.1136/annrheumdis-2018-214529
133. Keystone EC, Taylor PC, Tanaka Y, Gaich C, DeLozier AM, Dudek A, et al. Patient-reported outcomes from a phase 3 study of baricitinib versus placebo or adalimumab in rheumatoid arthritis: secondary analyses from the RA-BEAM study. Ann Rheum Dis. (2017) 76:1853–61. doi: 10.1136/annrheumdis-2017-211259
134. Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Del Carmen Morales L, Reyes Gonzaga J, et al. Baricitinib versus Placebo or Adalimumab in rheumatoid arthritis. N Engl J Med. (2017) 376:652–62. doi: 10.1056/NEJMoa1608345
135. Keystone EC, Genovese MC, Schlichting DE, de la Torre I, Beattie SD, Rooney TP, et al. Safety and efficacy of Baricitinib through 128 weeks in an open-label, longterm extension study in patients with rheumatoid arthritis. J Rheumatol. (2018) 45:14–21. doi: 10.3899/jrheum.161161
136. van der Heijde D, Durez P, Schett G, Naredo E, Østergaard M, Meszaros G, et al. Structural damage progression in patients with early rheumatoid arthritis treated with methotrexate, baricitinib, or baricitinib plus methotrexate based on clinical response in the phase 3 RA-BEGIN study. Clin Rheumatol. (2018) 37:2381–90. doi: 10.1007/s10067-018-4221-0
137. Smolen JS, Genovese MC, Takeuchi T, Hyslop DL, Macias WL, Rooney T, et al. Safety profile of Baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol. (2019) 46:7–18. doi: 10.3899/jrheum.171361
138. Qiu C, Zhao X, She L, Shi Z, Deng Z, Tan L, et al. Baricitinib induces LDL-C and HDL-C increases in rheumatoid arthritis: a meta-analysis of randomized controlled trials. Lipids Health Dis. (2019) 18:54. doi: 10.1186/s12944-019-0994-7
139. Xie W, Huang Y, Xiao S, Sun X, Fan Y, Zhang Z. Impact of Janus kinase inhibitors on risk of cardiovascular events in patients with rheumatoid arthritis: systematic review and meta-analysis of randomised controlled trials. Ann Rheum Dis. (2019) 78:1048–54. doi: 10.1136/annrheumdis-2018-214846
140. Mohamed M-EF, Camp HS, Jiang P, Padley RJ, Asatryan A, Othman AA. Pharmacokinetics, safety and tolerability of ABT-494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin Pharmacokinet. (2016) 55:1547–58. doi: 10.1007/s40262-016-0419-y
141. Kremer JM, Emery P, Camp HS, Friedman A, Wang L, Othman AA, et al. A phase IIb study of ABT-494, a selective JAK-1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti-tumor necrosis factor therapy. Arthritis Rheumatol. (2016) 68:2867–77. doi: 10.1002/art.39801
142. Genovese MC, Smolen JS, Weinblatt ME, Burmester GR, Meerwein S, Camp HS, et al. Efficacy and Safety of ABT-494, a Selective JAK-1 Inhibitor, in a Phase IIb Study in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate. Arthritis Rheumatol. (2016) 68:2857–66. doi: 10.1002/art.39808
143. Burmester GR, Kremer JM, Van den Bosch F, Kivitz A, Bessette L, Li Y, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. (2018) 391:2503–12. doi: 10.1016/S0140-6736(18)31115-2
144. Strand V, Pope J, Tundia N, Friedman A, Camp HS, Pangan A, et al. Upadacitinib improves patient-reported outcomes in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying antirheumatic drugs: results from SELECT-NEXT. Arthritis Res Ther. (2019) 21:272. doi: 10.1186/s13075-019-2037-1
145. Strand V, Schiff M, Tundia N, Friedman A, Meerwein S, Pangan A, et al. Effects of upadacitinib on patient-reported outcomes: results from SELECT-BEYOND, a phase 3 randomized trial in patients with rheumatoid arthritis and inadequate responses to biologic disease-modifying antirheumatic drugs. Arthritis Res Ther. (2019) 21:263. doi: 10.1186/s13075-019-2059-8
146. Song GG, Lee YH. Comparative efficacy and safety of 15 and 30 mg upadacitinib administered to patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Z Rheumatol. (2020) 79:103–11. doi: 10.1007/s00393-019-0601-3
147. Song GG, Choi SJ, Lee YH. Comparison of the efficacy and safety of tofacitinib and upadacitinib in patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Int J Rheum Dis. (2019) 22:1563–71. doi: 10.1111/1756-185X.13616
148. Fleischmann R, Pangan AL, Song I-H, Mysler E, Bessette L, Peterfy C, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol. (2019) 71:1788–800. doi: 10.1002/art.41032
149. Serhal L, Edwards CJ. Upadacitinib for the treatment of rheumatoid arthritis. Expert Rev Clin Immunol. (2019) 15:13–25. doi: 10.1080/1744666X.2019.1544892
150. Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, Kaneko Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis. (2016) 75:1057–64. doi: 10.1136/annrheumdis-2015-208279
151. Kivitz AJ, Gutierrez-Ureña SR, Poiley J, Genovese MC, Kristy R, Shay K, et al. Peficitinib, a JAK inhibitor, in the treatment of moderate-to-severe rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheumatol. (2017) 69:709–19. doi: 10.1002/art.39955
152. Genovese MC, Greenwald M, Codding C, Zubrzycka-Sienkiewicz A, Kivitz AJ, Wang A, et al. Peficitinib, a JAK inhibitor, in combination with limited conventional synthetic disease-modifying antirheumatic drugs in the treatment of moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol. (2017) 69:932–42. doi: 10.1002/art.40054
153. Takeuchi T, Tanaka Y, Tanaka S, Kawakami A, Iwasaki M, Katayama K, et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III randomised, double-blind, placebo-controlled trial (RAJ4) in Japan. Ann Rheum Dis. (2019) 78:1305–19. doi: 10.1136/annrheumdis-2019-215164
154. Tanaka Y, Takeuchi T, Tanaka S, Kawakami A, Iwasaki M, Song YW, et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to conventional DMARDs: a randomised, double-blind, placebo-controlled phase III trial (RAJ3). Ann Rheum Dis. (2019) 78:1320–32. doi: 10.1136/annrheumdis-2019-215163
155. Genovese MC, Greenwald MW, Gutierrez-Ureña SR, Cardiel MH, Poiley JE, Zubrzycka-Sienkiewicz A, et al. Two-year safety and effectiveness of peficitinib in moderate-to-severe rheumatoid arthritis: a phase IIb, open-label extension study. Rheumatol Ther. (2019) 6:503–20. doi: 10.1007/s40744-019-00167-6
156. Markham A, Keam SJ. Peficitinib: first global approval. Drugs. (2019) 79:887–91. doi: 10.1007/s40265-019-01131-y
157. Qiu Q, Feng Q, Tan X, Guo M. JAK3-selective inhibitor peficitinib for the treatment of rheumatoid arthritis. Expert Rev Clin Pharmacol. (2019) 12:547–54. doi: 10.1080/17512433.2019.1615443
158. Takeuchi T, Tanaka Y, Tanaka S, Kawakami A, Song Y-W, Chen Y-H, et al. Safety and effectiveness of peficitinib (ASP015K) in patients with rheumatoid arthritis: interim data (22.7 months mean peficitinib treatment) from a long-term, open-label extension study in Japan, Korea, and Taiwan. Arthritis Res Ther. (2020) 22:47. doi: 10.1186/s13075-020-2125-2
159. Lee YH, Song GG. Comparative efficacy and safety of Peficitinib 25, 50, 100, and 150 mg in patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Clin Drug Investig. (2020) 40:65–72. doi: 10.1007/s40261-019-00863-9
160. Lee YH, Song GG. Comparison of the efficacy and safety of tofacitinib and peficitinib in patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Int J Rheum Dis. (2020) 23:868–75. doi: 10.1111/1756-185X.13854
161. Ho Lee Y, Gyu Song G. Comparative efficacy and safety of tofacitinib, baricitinib, upadacitinib, filgotinib and peficitinib as monotherapy for active rheumatoid arthritis. J Clin Pharm Ther. (2020) 45:674–81. doi: 10.1111/jcpt.13142
162. Tanaka Y, Izutsu H. Peficitinib for the treatment of rheumatoid arthritis: an overview from clinical trials. Expert Opin Pharmacother. (2020) 21:1015–25. doi: 10.1080/14656566.2020.1739649
163. Westhovens R, Taylor PC, Alten R, Pavlova D, Enríquez-Sosa F, Mazur M, et al. Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose-finding study (DARWIN 1). Ann Rheum Dis. (2017) 76:998–1008. doi: 10.1136/annrheumdis-2016-210104
164. Kavanaugh A, Kremer J, Ponce L, Cseuz R, Reshetko OV, Stanislavchuk M, et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheum Dis. (2017) 76:1009–19. doi: 10.1136/annrheumdis-2016-210105
165. Vanhoutte F, Mazur M, Voloshyn O, Stanislavchuk M, Van der Aa A, Namour F, et al. Efficacy, safety, pharmacokinetics, and pharmacodynamics of filgotinib, a selective JAK-1 inhibitor, after short-term treatment of rheumatoid arthritis: results of two randomized phase IIa trials. Arthritis Rheumatol. (2017) 69:1949–59. doi: 10.1002/art.40186
166. Genovese M, Westhovens R, Meuleners L, Van der Aa A, Harrison P, Tasset C, et al. Effect of filgotinib, a selective JAK 1 inhibitor, with and without methotrexate in patients with rheumatoid arthritis: patient-reported outcomes. Arthritis Res Ther. (2018) 20:57. doi: 10.1186/s13075-018-1541-z
167. Genovese MC, Kalunian K, Gottenberg J-E, Mozaffarian N, Bartok B, Matzkies F, et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy: the FINCH 2 randomized clinical trial. JAMA. (2019) 322:315–25. doi: 10.1001/jama.2019.9055
168. Song GG, Lee YH. Comparative efficacy and safety of 100 mg and 200 mg filgotinib administered to patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Int J Clin Pharmacol Ther. (2020) 58:293–8. doi: 10.5414/CP203635
169. Lee YH, Song GG. Comparison of the efficacy and safety of tofacitinib and filgotinib in patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Z Rheumatol. (2020) 79:590–603. doi: 10.1007/s00393-019-00733-x
170. Sung Y-K, Lee YH. Comparative study of the efficacy and safety of tofacitinib, baricitinib, upadacitinib, and filgotinib versus methotrexate for disease-modifying antirheumatic drug-naïve patients with rheumatoid arthritis. Z Rheumatol. (2020). doi: 10.1007/s00393-020-00889-x. [Epub ahead of print].
171. Lee YH, Song GG. Relative efficacy and safety of tofacitinib, baricitinib, upadacitinib, and filgotinib in comparison to adalimumab in patients with active rheumatoid arthritis. Z Rheumatol. (2020) 79:785–96. doi: 10.1007/s00393-020-00750-1
172. Farmer LJ, Ledeboer MW, Hoock T, Arnost MJ, Bethiel RS, Bennani YL, et al. Discovery of VX-509 (Decernotinib): a potent and selective janus kinase 3 inhibitor for the treatment of autoimmune diseases. J Med Chem. (2015) 58:7195–216. doi: 10.1021/acs.jmedchem.5b00301
173. Mahajan S, Hogan JK, Shlyakhter D, Oh L, Salituro FG, Farmer L, et al. VX-509 (decernotinib) is a potent and selective janus kinase 3 inhibitor that attenuates inflammation in animal models of autoimmune disease. J Pharmacol Exp Ther. (2015) 353:405–14. doi: 10.1124/jpet.114.221176
174. Fleischmann RM, Damjanov NS, Kivitz AJ, Legedza A, Hoock T, Kinnman N. A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol. (2015) 67:334–43. doi: 10.1002/art.38949
175. Genovese MC, van Vollenhoven RF, Pacheco-Tena C, Zhang Y, Kinnman N. VX-509 (Decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol. (2016) 68:46–55. doi: 10.1002/art.39473
176. Weinhold KJ, Bukowski JF, Brennan TV, Noveck RJ, Staats JS, Lin L, et al. Reversibility of peripheral blood leukocyte phenotypic and functional changes after exposure to and withdrawal from tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin Immunol. (2018) 191:10–20. doi: 10.1016/j.clim.2018.03.002
177. Mori S, Ueki Y. Outcomes of dose reduction, withdrawal, and restart of tofacitinib in patients with rheumatoid arthritis: a prospective observational study. Clin Rheumatol. (2019) 38:3391–400. doi: 10.1007/s10067-019-04721-z
178. Kaine J, Tesser J, Takiya L, DeMasi R, Wang L, Snyder M, et al. Re-establishment of efficacy of tofacitinib, an oral JAK inhibitor, after temporary discontinuation in patients with rheumatoid arthritis. Clin Rheumatol. (2020) 39:2127–37. doi: 10.1007/s10067-020-04956-1
179. Emery P, Pope JE, Kruger K, Lippe R, DeMasi R, Lula S, et al. Efficacy of monotherapy with biologics and JAK inhibitors for the treatment of rheumatoid arthritis: a systematic review. Adv Ther. (2018) 35:1535–63. doi: 10.1007/s12325-018-0757-2
180. Doria A, Zavaglia D. Monotherapy is a relevant option in rheumatoid arthritis treatment: a literature review. Clin Exp Rheumatol. (2019) 37:862–71.
181. Fleischmann RM, Genovese MC, Enejosa JV, Mysler E, Bessette L, Peterfy C, et al. Safety and effectiveness of upadacitinib or adalimumab plus methotrexate in patients with rheumatoid arthritis over 48 weeks with switch to alternate therapy in patients with insufficient response. Ann Rheum Dis. (2019) 78:1454–62. doi: 10.1136/annrheumdis-2019-215764
182. Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. (2020) 79:685–99. doi: 10.1136/annrheumdis-2019-216655
183. Kawamura M, McVicar DW, Johnston JA, Blake TB, Chen YQ, Lal BK, et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc Natl Acad Sci USA. (1994) 91:6374–78. doi: 10.1073/pnas.91.14.6374
184. Gurniak CB, Berg LJ. Murine JAK3 is preferentially expressed in hematopoietic tissues and lymphocyte precursor cells. Blood. (1996) 87:3151–60.
185. Verbsky JW, Bach EA, Fang YF, Yang L, Randolph DA, Fields LE. Expression of Janus kinase 3 in human endothelial and other non-lymphoid and non-myeloid cells. J Biol Chem. (1996) 271:13976–80. doi: 10.1074/jbc.271.24.13976
186. Cornejo MG, Boggon TJ, Mercher T. JAK3: a two-faced player in hematological disorders. Int J Biochem Cell Biol. (2009) 41:2376–9. doi: 10.1016/j.biocel.2009.09.004
187. Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. (1995) 270:797–800. doi: 10.1126/science.270.5237.797
188. Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. (1995) 270:794–7. doi: 10.1126/science.270.5237.794
189. Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, et al. Defective lymphoid development in mice lacking Jak3. Science. (1995) 270:800–2. doi: 10.1126/science.270.5237.800
190. Hanissian SH, Geha RS. Jak3 is associated with CD40 and is critical for CD40 induction of gene expression in B cells. Immunity. (1997) 6:379–87. doi: 10.1016/s1074-7613(00)80281-2
191. Klaus GG, Choi MS, Lam EW, Johnson-Léger C, Cliff J. CD40: a pivotal receptor in the determination of life/death decisions in B lymphocytes. Int Rev Immunol. (1997) 15:5–31. doi: 10.3109/08830189709068169
192. Uckun FM, Pitt J, Qazi S. JAK3 pathway is constitutively active in B-lineage acute lymphoblastic leukemia. Expert Rev Anticancer Ther. (2011) 11:37–48. doi: 10.1586/era.10.203
193. Ge Y, Wang C, Song S, Huang J, Liu Z, Li Y, et al. Identification of highly potent BTK and JAK3 dual inhibitors with improved activity for the treatment of B-cell lymphoma. Eur J Med Chem. (2018) 143:1847–57. doi: 10.1016/j.ejmech.2017.10.080
194. Chi F, Chen L, Wang C, Li L, Sun X, Xu Y, et al. JAK3 inhibitors based on thieno[3,2-d]pyrimidine scaffold: design, synthesis and bioactivity evaluation for the treatment of B-cell lymphoma. Bioorg Chem. (2020) 95:103542. doi: 10.1016/j.bioorg.2019.103542
195. Dillon SR, Schlissel MS. Partial restoration of B cell development in Jak-3(-/-) mice achieved by co-expression of IgH and E(mu)-myc transgenes. Int Immunol. (2002) 14:893–904. doi: 10.1093/intimm/dxf052
196. Tortolani PJ, Lal BK, Riva A, Johnston JA, Chen YQ, Reaman GH, et al. Regulation of JAK3 expression and activation in human B cells and B cell malignancies. J Immunol. (1995) 155:5220–6.
197. Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol. (2011) 186:4234–43. doi: 10.4049/jimmunol.1003668
198. Maeshima K, Yamaoka K, Kubo S, Nakano K, Iwata S, Saito K, et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon-γ and interleukin-17 production by human CD4+ T cells. Arthritis Rheum. (2012) 64:1790–8. doi: 10.1002/art.34329
199. Kubo S, Yamaoka K, Kondo M, Yamagata K, Zhao J, Iwata S, et al. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann Rheum Dis. (2014) 73:2192–8. doi: 10.1136/annrheumdis-2013-203756
200. Sonomoto K, Yamaoka K, Kubo S, Hirata S, Fukuyo S, Maeshima K, et al. Effects of tofacitinib on lymphocytes in rheumatoid arthritis: relation to efficacy and infectious adverse events. Rheumatology. (2014) 53:914–8. doi: 10.1093/rheumatology/ket466
201. Piscianz E, Valencic E, Cuzzoni E, De Iudicibus S, De Lorenzo E, Decorti G, et al. Fate of lymphocytes after withdrawal of tofacitinib treatment. PLoS ONE. (2014) 9:e85463. doi: 10.1371/journal.pone.0085463
202. Wang S-P, Iwata S, Nakayamada S, Sakata K, Yamaoka K, Tanaka Y. Tofacitinib, a JAK inhibitor, inhibits human B cell activation in vitro. Ann Rheum Dis. (2014) 73:2213–5. doi: 10.1136/annrheumdis-2014-205615
203. Gertel S, Mahagna H, Karmon G, Watad A, Amital H. Tofacitinib attenuates arthritis manifestations and reduces the pathogenic CD4 T cells in adjuvant arthritis rats. Clin Immunol. (2017) 184:77–81. doi: 10.1016/j.clim.2017.04.015
204. van Vollenhoven R, Lee EB, Strengholt S, Mojcik C, Valdez H, Krishnaswami S, et al. Evaluation of the short-, mid-, and long-term effects of Tofacitinib on lymphocytes in patients with rheumatoid arthritis. Arthritis Rheumatol. (2019) 71:685–95. doi: 10.1002/art.40780
205. Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez J-B, Dowty ME, et al. The mechanism of action of tofacitinib—an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. (2016) 34:318–28.
206. Schulze-Koops H, Strand V, Nduaka C, DeMasi R, Wallenstein G, Kwok K, et al. Analysis of haematological changes in tofacitinib-treated patients with rheumatoid arthritis across phase 3 and long-term extension studies. Rheumatology. (2017) 56:46–57. doi: 10.1093/rheumatology/kew329
207. Martina MN, Ramirez Bajo MJ, Bañon-Maneus E, Moya Rull D, Hierro-Garcia N, Revuelta I, et al. Inhibition of JAK3 and PKC via immunosuppressive drugs Tofacitinib and Sotrastaurin inhibits proliferation of human B lymphocytes in vitro. Transplant Proc. (2016) 48:3046–52. doi: 10.1016/j.transproceed.2016.07.052
208. Rizzi M, Lorenzetti R, Fischer K, Staniek J, Janowska I, Troilo A, et al. Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J Autoimmun. (2017) 77:55–66. doi: 10.1016/j.jaut.2016.10.005
209. Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, et al. Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol. (2017) 69:148–60. doi: 10.1002/art.39818
210. Ikeda K, Hayakawa K, Fujishiro M, Kawasaki M, Hirai T, Tsushima H, et al. JAK inhibitor has the amelioration effect in lupus-prone mice: the involvement of IFN signature gene downregulation. BMC Immunol. (2017) 18:41. doi: 10.1186/s12865-017-0225-9
211. Winthrop KL, Silverfield J, Racewicz A, Neal J, Lee EB, Hrycaj P, et al. The effect of tofacitinib on pneumococcal and influenza vaccine responses in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:687–95. doi: 10.1136/annrheumdis-2014-207191
212. Winthrop KL, Wouters AG, Choy EH, Soma K, Hodge JA, Nduaka CI, et al. The safety and immunogenicity of live zoster vaccination in patients with rheumatoid arthritis before starting tofacitinib: a randomized phase II trial. Arthritis Rheumatol. (2017) 69:1969–77. doi: 10.1002/art.40187
213. Calabrese LH, Abud-Mendoza C, Lindsey SM, Lee S-H, Tatulych S, Takiya L, et al. Live zoster vaccine in patients with rheumatoid arthritis treated with tofacitinib with or without methotrexate, or adalimumab with methotrexate: a post hoc analysis of data from a phase IIIb/IV randomized study. Arthritis Care Res. (2020) 72:353–9. doi: 10.1002/acr.24010
214. Tanaka Y, McInnes IB, Taylor PC, Byers NL, Chen L, de Bono S, et al. Characterization and changes of lymphocyte subsets in baricitinib-treated patients with rheumatoid arthritis: an integrated analysis. Arthritis Rheumatol. (2018) 70:1923–32. doi: 10.1002/art.40680
215. Kubo S, Nakayamada S, Sakata K, Kitanaga Y, Ma X, Lee S, et al. Janus kinase inhibitor baricitinib modulates human innate and adaptive immune system. Front Immunol. (2018) 9:1510. doi: 10.3389/fimmu.2018.01510
216. Bonelli M, Dalwigk K, Platzer A, Olmos Calvo I, Hayer S, Niederreiter B, et al. IRF1 is critical for the TNF-driven interferon response in rheumatoid fibroblast-like synoviocytes : JAKinibs suppress the interferon response in RA-FLSs. Exp Mol Med. (2019) 51:75. doi: 10.1038/s12276-019-0267-6
217. Choi J, Cooper ML, Staser K, Ashami K, Vij KR, Wang B, et al. Baricitinib-induced blockade of interferon gamma receptor and interleukin-6 receptor for the prevention and treatment of graft-versus-host disease. Leukemia. (2018) 32:2483–94. doi: 10.1038/s41375-018-0123-z
218. Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. (2018) 2:23. doi: 10.1186/s41927-018-0031-x
219. Dowty ME, Lin TH, Jesson MI, Hegen M, Martin DA, Katkade V, et al. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol Res Perspect. (2019) 7:e00537. doi: 10.1002/prp2.537
220. McInnes IB, Byers NL, Higgs RE, Lee J, Macias WL, Na S, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther. (2019) 21:183. doi: 10.1186/s13075-019-1964-1
221. Ito M, Yamazaki S, Yamagami K, Kuno M, Morita Y, Okuma K, et al. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J Pharmacol Sci. (2017) 133:25–33. doi: 10.1016/j.jphs.2016.12.001
222. Shibata M, Hatta T, Saito M, Toyoshima J, Kaneko Y, Oda K, et al. Pharmacokinetics, pharmacodynamics, and safety of peficitinib (ASP015K) in healthy male caucasian and japanese subjects. Clin Drug Investig. (2020) 40:469–84. doi: 10.1007/s40261-020-00910-w
223. Ikari Y, Isozaki T, Tsubokura Y, Kasama T. Peficitinib inhibits the chemotactic activity of monocytes via proinflammatory cytokine production in rheumatoid arthritis fibroblast-like synoviocytes. Cells. (2019) 8:561. doi: 10.3390/cells8060561
224. Diller M, Hasseli R, Hülser M-L, Aykara I, Frommer K, Rehart S, et al. Targeting activated synovial fibroblasts in rheumatoid arthritis by peficitinib. Front Immunol. (2019) 10:541. doi: 10.3389/fimmu.2019.00541
225. Cao YJ, Sawamoto T, Valluri U, Cho K, Lewand M, Swan S, et al. Pharmacokinetics, pharmacodynamics, and safety of ASP015K (Peficitinib), a new janus kinase inhibitor, in healthy subjects. Clin Pharmacol Drug Dev. (2016) 5:435–49. doi: 10.1002/cpdd.273
226. Tarrant JM, Galien R, Li W, Goyal L, Pan Y, Hawtin R, et al. Filgotinib, a JAK1 inhibitor, modulates disease-related biomarkers in rheumatoid arthritis: results from two randomized, controlled phase 2b trials. Rheumatol Ther. (2020) 7:173–90. doi: 10.1007/s40744-019-00192-5
227. Lee J, Lee J, Kwok S-K, Baek S, Jang SG, Hong S-M, et al. JAK-1 inhibition suppresses interferon-induced BAFF production in human salivary gland: potential therapeutic strategy for primary Sjögren's syndrome. Arthritis Rheumatol. (2018) 70:2057–66. doi: 10.1002/art.40589
228. Genovese MC, Yang F, Østergaard M, Kinnman N. Efficacy of VX-509 (decernotinib) in combination with a disease-modifying antirheumatic drug in patients with rheumatoid arthritis: clinical and MRI findings. Ann Rheum Dis. (2016) 75:1979–83. doi: 10.1136/annrheumdis-2015-208901
Keywords: JAK-STAT pathway, JAK inhibitors, B cells, cytokines, rheumatoid arthritis
Citation: Moura RA and Fonseca JE (2021) JAK Inhibitors and Modulation of B Cell Immune Responses in Rheumatoid Arthritis. Front. Med. 7:607725. doi: 10.3389/fmed.2020.607725
Received: 18 September 2020; Accepted: 18 December 2020;
Published: 05 February 2021.
Edited by:
Md Yuzaiful Md Yusof, University of Leeds, United KingdomReviewed by:
Felice Rivellese, Queen Mary University of London, United KingdomCopyright © 2021 Moura and Fonseca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rita A. Moura, cml0YWFndWlhcm1vdXJhQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.