
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 26 October 2020
Sec. Pulmonary Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fmed.2020.589553
This article is part of the Research Topic Defining and Characterizing Respiratory Disease in an Aging Population View all 12 articles
 Ryan Brown1
Ryan Brown1 Michael C. McKelvey1
Michael C. McKelvey1 Sinéad Ryan1
Sinéad Ryan1 Shannice Creane1
Shannice Creane1 Dermot Linden1
Dermot Linden1 Joseph C. Kidney2
Joseph C. Kidney2 Daniel F. McAuley3
Daniel F. McAuley3 Clifford C. Taggart1
Clifford C. Taggart1 Sinéad Weldon1*
Sinéad Weldon1*Acute respiratory distress syndrome (ARDS) is associated with increased morbidity and mortality in the elderly population (≥65 years of age). Additionally, age is widely reported as a risk factor for the development of ARDS. However, the underlying pathophysiological mechanisms behind the increased risk of developing, and increased severity of, ARDS in the elderly population are not fully understood. This is compounded by the significant heterogeneity observed in patients with ARDS. With an aging population worldwide, a better understanding of these mechanisms could facilitate the development of therapies to improve outcomes in this population. In this review, the current clinical evidence of age as a risk factor and prognostic indicator in ARDS and the potential underlying mechanisms that may contribute to these factors are outlined. In addition, research on age-dependent treatment options and biomarkers, as well as future prospects for targeting these underlying mechanisms, are discussed.
Acute respiratory distress syndrome (ARDS) is a common yet complex syndrome that develops in critically ill patients. Clinically, ARDS presents as acute hypoxemia and the presence of bilateral pulmonary infiltrates that cannot be fully ascribed to heart failure or fluid overload (1–3). This flooding of the alveoli with protein-rich edema is associated with the breakdown of the alveolar-capillary unit and, in many cases, the influx of neutrophils and other immune cells into the air spaces. These immune cells, along with activated epithelial and endothelial cells, release numerous pro-inflammatory mediators, which propagate a profound inflammatory response, and a battery of cytotoxic species, causing damage to the lung parenchyma (4). This endothelial and alveolar cell injury with fluid and cellular exudation has been termed as diffuse alveolar damage (DAD), however, data from biopsy and autopsy studies have shown that only half of those who meet the clinical definition of ARDS present with DAD (5–7). The presence of DAD is associated with increased mortality in ARDS (7). Despite over 50 years of investigation, ARDS is still under-recognized and no specific, effective treatments are available (8, 9). Mortality due to ARDS remains high, with most estimates indicating mortality rates in the region of 30–50% (8, 10, 11). ARDS is a heterogeneous disease process that may be triggered by a variety of direct or indirect pulmonary injuries including pneumonia, aspiration, non-protective mechanical ventilation, chest trauma, sepsis, and acute pancreatitis. Although ARDS can affect people of all ages, increasing age is a widely reported risk factor and is associated with increased mortality in ARDS patients (12, 13). This relationship has been emphasized by the current Covid-19 pandemic, in which severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a causative agent of ARDS, has exhibited significant mortality among older patients (14–16).
The world's population is aging; over 65s are expected to account for 1 in 6 of the population by 2050, an increase from 1 in 11 in 2019 (17). With a strong association between aging and increased incidence of disease, including respiratory conditions such as chronic obstructive pulmonary disease and acute infection (18), a thorough understanding of age-related changes in the lung and how these impact on disease risk and severity will be important to effectively manage the health of an ever aging population. Age-associated structural changes in the lung mainly encompass increased alveolar spaces, reduced elasticity (19, 20) and a gradual decrease in lung function (21). An accumulation of senescent cells in the lung is observed in the elderly population (22). The release of soluble mediators by senescent cells contributes to persistent, low-level inflammation (23, 24), and impaired regenerative abilities (25). Similarly, the phenomenon of inflamm-aging is apparent in elderly cohorts where heightened levels of inflammatory factors are observed but several cell types including neutrophils and macrophages have attenuated phagocytic capabilities (26, 27). Thus, there are a number of pathophysiological features of the aging lung that may contribute to increased disease severity in elderly populations, which will be discussed in further detail in this review.
To date, few studies have focused primarily on assessing age per se as a risk factor for ARDS. Patients with ARDS often carry multiple risk factors and co-morbidities, some of which may be influenced by age themselves, which makes isolating the influence of age particularly challenging. Epidemiological studies frequently report the mean/median age of patients with ARDS at 55–65 years, making it a disease of late middle-age rather than exclusively old age (8, 10, 11, 28). One approach is to quantify the incidence of ARDS in the general population within different age groups. A study in the United States reported an overall incidence of 64 cases per 100,000 person-years but incidences of up to 306 cases per 100,000 person-years in the 75–84 years age group (10). Studies in Spain (29) and Taiwan (30) have shown similar age-dependent increases in incidence in the general population. The relationship between age and ARDS can also be examined by comparing cohorts of patients with another risk factor (pneumonia, trauma etc.) who did not develop ARDS with patients who had the same risk factor and did develop ARDS. Such analysis reveals that age appears to be a risk factor for ARDS in combination with certain other etiologies or risk factors. For instance, a large study of trauma patients by Johnston et al. demonstrated that the mean age of patients who developed ARDS was significantly higher than that of those who did not, and that the risk of developing ARDS increased up to 60–69 years of age (31). Likewise, patients with Covid-19 pneumonia who develop ARDS are significantly older than those who do not (16). However, in a recent study by Iriyama et al., there was no significant difference between the mean age of patients who developed ARDS following non-pulmonary sepsis and those who did not (32). Similarly, the age of patients who developed ARDS did not vary significantly from those who did not following out-of-hospital cardiac arrest (33), neutropenia after hematologic malignancy (34), or kidney transplant (35). Thus, it appears that while older age may generally be considered a risk factor for ARDS, larger studies are required to assess this relationship in ARDS with particular etiologies, especially indirect ARDS.
Accurately extrapolating the prognostic implication of age in ARDS is challenging. Observational data is derived from retrospective studies, which have inherent methodological flaws whilst elderly patients may frequently be excluded from randomized controlled trials. Consequently, the existing evidence-base may be prone to inaccuracy. Age is associated with worse ICU outcome and is embedded within several critical illness severity scoring systems, such as SAPS (simplified acute physiology score) and APACHE (acute physiology and chronic health evaluation) (36). These scores provide utility in prognostic prediction in the ICU setting, however, they lack specificity (37). There are currently no validated mortality prediction scoring systems in ARDS (36). Villar et al. prospectively evaluated a 9-point scoring tool (APPS) encompassing clinical parameters including age, PaO2/FIO2 and plateau pressure at 24 h following ARDS diagnosis (n = 600) (38). The authors stratified patients into three severity groups, assigning higher score in accordance with age group and reported a significant association with mortality (38). Subsequent external validation of the APPS score concluded that the model showed moderate-accuracy in predicting all-cause hospital mortality (39). Within adult populations, increased age is generally associated with higher disease severity (29). Several investigators have reported age as an independent predictor of mortality in ARDS (40, 41). It should be emphasized that substantial heterogeneity exists within the syndrome and non-linear associations between age and mortality have been reported within specific ARDS phenotypes [i.e., trauma; (42, 43)]. When considering the trauma-ARDS cohort, the highest burden of mortality has been shown to exist at the extremes of age (42, 43). A large retrospective review of a national trauma database (n = 1,297,190) reported the ARDS-related mortality was highest in those ≥80 and ≤4 years (42). Other investigators have reported no significant difference in mortality using a dichotomous age cut-off of 65 years, potentially supporting the notion that the relationship between age and ARDS may be more complex (44). While age is associated with ARDS mortality, there is no significant difference between ventilator or ICU free days, length of stay in ICU or length of stay in hospital between patients above 65 and those under 65 (12, 45). Indeed, a number of studies have concluded that co-morbidities rather than age primarily affect prognosis following ICU admission (46–48). Further elucidation of the impact of age upon prognosis in ARDS is required and well-designed prospective observational studies are paramount.
A number of factors influence disease severity in ARDS including features of the immune system, vasculature and structural components of the airway, as highlighted in Figure 1. Research to date has highlighted a number of mechanisms through which age may affect these systems and alter the risk of developing and/or disease severity in ARDS.
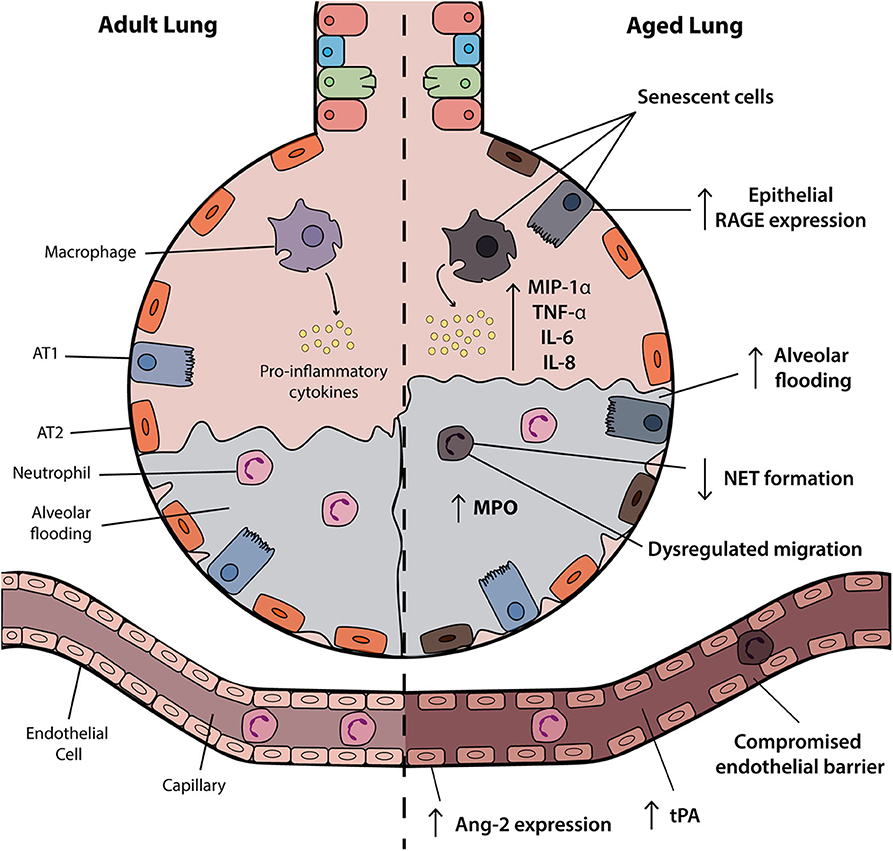
Figure 1. Age-associated changes in the ARDS lung. The aged alveolus in acute respiratory distress syndrome (ARDS) demonstrates a number of age-related changes in the alveolar epithelium, vasculature and immune cells that may contribute to disease pathogenesis in elderly patients. Vascular permeability may be affected by changes in angiopoietin-2 (Ang-2) expression and receptor for advanced glycation end products (RAGE) signaling is associated with inflammation and progression of the exudative and fibroproliferative phases of ARDS. Accumulation of senescent cells and the senescence-associated secretory phenotype (SASP) results in the release of soluble pro-inflammatory mediators such as IL-6 and IL-8. Although increased levels of inflammatory factors are observed, neutrophils and macrophages may have attenuated functional activities. AT1, alveolar type I; ATII, alveolar type II; NET, neutrophil extracellular trap; tPA, tissue plasminogen activator.
The decline in immune function with age is well-documented and is referred to as immunosenescence. Immune cell activation is a major mediator of inflammation in ARDS and immunosenescence may impact on the pathogenesis and outcomes in the aged ARDS subpopulation. Experimental murine models of ARDS induced by endotoxin indicate that aged mice display increased bronchoalveolar lavage fluid (BALF) cell counts, protein concentration, and cytokines such as the neutrophil chemoattractant CXCL1/KC in comparison to younger mice (49–51). Alterations in innate immune cell function, such as macrophage and neutrophils, may contribute to this heightened inflammatory response (52, 53). Alveolar macrophages (AMs) have increased expression of genes associated with lung injury and fibrosis (54) and defective phagocytic ability (26). In one study of lipopolysaccharide (LPS)-induced acute lung injury (ALI), expression of the M1-macrophage markers, CD80 and CD86, were increased in AMs from older mice and in vitro these cells had increased propensity to produce MIP-1α in response to LPS (50). Impairment of antigen presentation in aging macrophages has also been observed and resulting loss of bacterial and viral clearance from the lung could further promote inflammation and damage (55). In addition, aberrant neutrophil responses such as dysregulated chemotaxis (27) and the inability to form neutrophil extracellular traps (56) were observed in elderly cohorts. Studies across a breadth of ages in a small cohort of patients with ARDS suggest a correlation between age and the neutrophil biomarker myeloperoxidase (MPO) in BALF (57). Impaired neutrophil phagocytosis and microbial killing has also been observed in the elderly (58). The output of adaptive immune cells, particularly T cells, reduces over time. However, regulatory T (Treg) cells, which are responsible for regulation of immune responses, increased with age (50, 59). In experimental ARDS, aged groups show concomitant increases in both Tregs and inflammatory markers in comparison to younger groups (50). This suggests that while Treg numbers may be increased in the elderly, their anti-inflammatory function may be repressed. Clinically, there has been no consensus on the role of Tregs in ARDS with both protective (60, 61) and deleterious (62) effects observed. Furthermore, impaired adaptive immunity with decreased CD8+ T cells in advancing age may contribute to poor prognosis in ARDS (50). Recently published research brings together single cell sequencing data and tissue proteomics to document cell specific changes associated with aging (54, 63). Overall, changes in immune cell function may drive age related ARDS pathogenesis and further characterization of these changes may highlight potential targeted therapies that may be beneficial in an elderly population.
In addition to the immune system, age-related changes in structural components of the airway and vasculature may also contribute to ARDS pathogenesis. Within the vasculature, changes in the expression of angiopoietin-2 (Ang-2) have been linked to disease severity in ARDS (64). Ang-2 is a growth factor involved in angiogenesis which also plays a role in modulating endothelial permeability. Increased Ang-2 levels result in endothelial destabilization and increased vascular permeability (65). A positive correlation between Ang-2 and age has been observed in animal models and in the clinical setting (66, 67). Increases in Ang-2 may therefore be associated with ARDS severity in aging patients via increased vascular permeability and alveolar filling. Aging is also associated with an accumulation of senescent cells (68). Senescent endothelial cells (ECs) in ARDS demonstrate exacerbated permeability responses and this permeability was sustained by the upregulation of the reactive oxygen species (ROS)-generating enzyme NADPH oxidase-4 (Nox4) (69). In addition, Nox4 interacts with toll like receptor (TLR)-4 inducing NF-κB activation and inflammation (70). Taken together, these effects may contribute to increased ARDS severity, and inhibition of Nox-4 attenuated ALI in mice (71). Oxidative damage plays a significant role in age-related disease severity. Both aging and ARDS are associated with a cumulative oxidant burden and the generation of oxidized phospholipids has been linked to vascular leakage and inflammation and, as a result, the pathogenesis of ALI (72).
The structural components and mechanics of the lung also play an important role in airway pathophysiology. Aging is associated with changes in lung function and respiratory mechanics. Increased compliance and decreased small airway diameter in the elderly increase the severity of lung injury under mechanical ventilation (73). In addition to vascular ECs, senescence of airway epithelial cells and fibroblasts has also been linked to the pathogenesis of idiopathic pulmonary fibrosis through their expression of pro-inflammatory and profibrotic factors (74). This may also be of relevance in ARDS where a late fibrotic phase is associated with poor prognosis, high mortality and prolonged ventilator dependence (75). Targeting of senescent cells was beneficial in in vivo models of fibrosis, improving lung function and physical health (74). A decline in cellular autophagic function has also been observed in the aging lung (18) and induction of autophagy protected against lung permeability in models of ALI (76), highlighting the importance of this process. Another potential mechanism by which aging may contribute to increased ARDS severity is through the upregulation of receptor for advanced glycation end products (RAGE), a marker of type I alveolar epithelial cell injury (77). RAGE signaling likely promotes progression of both the exudative and fibroproliferative phases of ARDS (78, 79). Upregulation of RAGE was observed in mouse models of ALI and in ARDS patients and was associated with increased disease severity including increased mortality and fewer ventilator-free and organ failure-free days (77, 80). Treatment with recombinant soluble (s)RAGE, which acts as a decoy receptor, reduced inflammation in a murine model of LPS-induced ALI, suggesting that sRAGE participates in negative feedback after excessive inflammatory processes (81). Increased RAGE levels in the elderly may represent a potential mechanism through which aging effects ARDS severity, although a better understanding of how RAGE and its ligands impact pulmonary inflammation is needed (82).
Biomarkers represent an important tool in diagnostics and patient stratification. However, to date, no single specific ARDS biomarker has been validated (83). Analysis of systemic inflammatory mediators and endothelial activation markers in plasma from adults with ARDS found that, compared to younger patients, elderly patients had lower levels of inflammatory mediators and endothelial activation markers (interleukin (IL)-6, IL-8, IL-10, interferon-γ, fractalkine, intracellular adhesion molecule (ICAM)-1, E-selectin) and higher levels of platelet factor-4 and tissue plasminogen activator (tPA) (12). Furthermore, advanced age was found to be independently associated with increased plasma levels of tPA and decreased plasma levels of fractalkine and E-selectin (12). However, only tPA was linked with outcome, accounting for 10% of the association between age and outcome. Although these findings suggest a possible link between aging, fibrinolysis and mortality, further work is needed to determine the role of tPA. More recently, Schouten et al. analyzed age-dependent differences in inflammatory and endothelial activation markers in BALF from ARDS patients of various ages (57). Although no difference was detected between adult and elderly cohorts, higher levels of neutrophil markers were found with increasing age and the association between increasing age and increased levels of MPO, IL-10, P-selectin, and decreased ICAM-1 remained significant after correction for severity of ARDS (57). These data suggest that plasma may be more useful in identifying age-dependent differences in ARDS patients although longitudinal analysis of BALF and plasma from the same cohort of patients may be required to shed light on the pathophysiology of pulmonary vs. systemic responses and how they interact across the spectrum of ARDS phenotypes. Like other diseases, variations in published data highlight the difficulties in identifying biomarkers that relate to disease severity and outcome in this heterogeneous syndrome, and a broader “omic” (e.g., proteomic) approach may be required. The identification of specific biomarkers would be helpful to identify those in the elderly population most at risk and to guide potential treatment options, although this may be a difficult feat.
Conclusive evidence to support age-dependent differences in treatment responses is lacking. However, there is evidence from both in vivo (84, 85) and computational studies (73, 86) to suggest that ventilator induced lung injury (VILI) may be more prevalent in the elderly population as a result of increased lung compliance and stiffness. The use of conservative fluid management and low tidal volume ventilation (LTVV) has been shown to attenuate this age-associated increase in ventilator associated mortality in vivo (87). Clinically, the benefits of LTVV are uncertain and adherence among clinicians is low (88, 89). Further study into the clinical benefits of LTVV specifically in the elderly subpopulation would be valuable. Extracorporeal membrane oxygenation (ECMO) has become a valuable therapy to support recovery in ARDS. Data on the use of ECMO in the elderly is limited, with most data coming from retrospective studies. Current data suggests that age is not a contraindication for ECMO, rather, its use should be decided on a case by case basis (90–92). Potential predictors that should be considered before initiation of ECMO support include presence of cardiogenic shock, APACHE II, and SAPS II scores (93). The accumulation of senescent cells has been suggested as a mechanism for increased mortality in elderly ARDS populations. Therefore, the use of senolytic drugs may represent an interesting therapy to improve outcomes in elderly patients with ARDS. Though senolytic drugs have not yet been evaluated clinically in ARDS, they have demonstrated benefits in preclinical models of fibrosis (74, 94). In the future, the identification of those most at risk of developing ARDS, along with the identification of the most effective treatment strategies for phenotypically and demographically distinct groups of patients, will be of paramount importance in reducing mortality in the elderly ARDS population.
Research to date has highlighted the significant analytical challenges that ARDS presents due to its complex etiology. Although aging may alter the immune system, vasculature, and airway epithelium to promote the pathogenesis of ARDS, further work is needed to elucidate these mechanisms and this will be critical to improving our understanding of the effects of aging on the risk of developing ARDS and its severity. While more remains to be learned, accumulation of senescent cells along with increased oxidative damage, defective autophagy, and increased vascular permeability due to age-related changes are likely to play a role. A better understanding of the contributions of these individual factors along with the potential impact of co-morbidities could help direct future therapeutic strategies. A clearer understanding of the treatment options that are most beneficial for elderly patients with ARDS will be vitally important to reduce mortality in this subpopulation. Cell therapies represent a promising therapeutic option in ARDS (95), and these may be of particular significance in the elderly population where senescence and immune-aging render host cells ineffective and the lungs exhibit age-related structural changes (26, 27, 69). Treatment with mesenchymal stem cells, endothelial progenitor cells, or Tregs may attenuate age-related pathology. Alternatively, slowing the process of aging to mitigate age-related functional decline may be worth exploring in ARDS. Indeed, calorie restriction slowed the rate of aging and reduced ARDS risk in a preclinical model (96). Other potential therapeutics targeting aging including rapamycin (97), mTOR inhibitors (98), and metformin (99) could also be examined in ARDS. These therapies have been discussed in a recent review (100) and represent a number of potential avenues for the development of more targeted therapies for elderly ARDS patients. The stratification of ARDS patients into hypoinflammatory and hyperinflammatory subphenotypes revolutionized the clinical perspective. Studies of several ARDS cohorts identified distinct subphenotypes based on plasma inflammatory markers, level of shock and metabolic acidosis (101–103). These subphenotypes appear to respond differently to treatment (102), and while age does not significantly differ between the subphenotypes in most of these cohorts (101, 103, 104), further studies focusing on the role of age as a determinant of ARDS subphenotypes may be a useful strategy to direct treatment. Alternatively, identification of subphenotypes within elderly ARDS patient populations allowing stratification of these patients may also prove useful.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported in part by the Department for the Economy (Northern Ireland), the Cystic Fibrosis Foundation (WELDON18G0), the Mater Hospital Young Philanthropist (YP) Trustees and the Medical Research Council (MR/T016760/1 and MR/P022847/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to colleagues whose work has not been cited due to space limitations.
1. ARDS Definition Task Force. Acute respiratory distress syndrome. JAMA. (2012) 307:2526–33. doi: 10.1001/jama.2012.5669
2. MacSweeney R, McAuley DF. Seminar: acute respiratory distress syndrome. Lancet. (2016) 388:2416–30. doi: 10.1016/S0140-6736(16)00578-X
3. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Prim. (2019) 5:18. doi: 10.1038/s41572-019-0069-0
4. Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol. (2015) 194:855–60. doi: 10.4049/jimmunol.1402513
5. Thille AW, Esteban A, Fernández-Segoviano P, Rodriguez JM, Aramburu JA, Peñuelas O, et al. Comparison of the berlin definition for acute respiratory distress syndrome with autopsy. Am J Respir Crit Care Med. (2013) 187:761–7. doi: 10.1164/rccm.201211-1981OC
6. De Hemptinne Q, Remmelink M, Brimioulle S, Salmon I, Vincent JL. A clinicopathological confrontation. Chest. (2009) 135:944–9. doi: 10.1378/chest.08-1741
7. Kao KC, Hu HC, Chang CH, Hung CY, Chiu LC, Li SH, et al. Diffuse alveolar damage associated mortality in selected acute respiratory distress syndrome patients with open lung biopsy. Crit Care. (2015) 19:228. doi: 10.1186/s13054-015-0949-y
8. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. (2016) 315:788–800. doi: 10.1001/jama.2016.0291
9. Shaw TD, McAuley DF, O'Kane CM. Emerging drugs for treating the acute respiratory distress syndrome. Expert Opin Emerg Drugs. (2019) 24:29–41. doi: 10.1080/14728214.2019.1591369
10. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med. (2005) 353:1685–93. doi: 10.1056/NEJMoa050333
11. Villar J, Blanco J, Añón JM, Santos-Bouza A, Blanch L, Ambrós A, et al. The ALIEN study: incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive Care Med. (2011) 37:1932–41. doi: 10.1007/s00134-011-2380-4
12. Schouten LRA, Bos LDJ, Serpa Neto A, van Vught LA, Wiewel MA, Hoogendijk AJ, et al. Increased mortality in elderly patients with acute respiratory distress syndrome is not explained by host response. Intensive Care Med Exp. (2019) 7:58. doi: 10.1186/s40635-019-0270-1
13. Suchyta MR, Clemmer TP, Elliott CG, Orme JF, Morris AH, Jacobson J, et al. Increased mortality of older patients with acute respiratory distress syndrome. Chest. (1997) 111:1334–9. doi: 10.1378/chest.111.5.1334
14. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
15. Du RH, Liang LR, Yang CQ, Wang W, Cao TZ, Li M, et al. Predictors of mortality for patients with COVID-19 pneumonia caused by SARSCoV- 2: a prospective cohort study. Eur Respir J. (2020) 55:2000524. doi: 10.1183/13993003.00524-2020
16. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med. (2020) 180:1–11. doi: 10.1001/jamainternmed.2020.0994
18. Budinger GS, Kohanski RA, Gan W, Kobor MS, Amaral LA, Armanios M, et al. The intersection of aging biology and the pathobiology of lung diseases: a joint NHLBI/NIA workshop. J Gerontol Ser A Biol Sci Med Sci. (2017) 72:1492–500. doi: 10.1093/gerona/glx090
19. Weibel ER, Gomez DM. Architecture of the human lung: use of quantitative methods establishes fundamental relations between size and number of lung structures. Science. (1962) 137:577–85. doi: 10.1126/science.137.3530.577
20. Turner JM, Mead J, Wohl ME. Elasticity of human lungs in relation to age. J Appl Physiol. (1968) 25:664–71. doi: 10.1152/jappl.1968.25.6.664
21. Paulin GA, Ouriadov A, Lessard E, Sheikh K, McCormack DG, Parraga G. Noninvasive quantification of alveolar morphometry in elderly never- and ex-smokers. Physiol Rep. (2015) 3:e12583. doi: 10.14814/phy2.12583
22. Parikh P, Wicher S, Khandalavala K, Pabelick CM, Britt RD, Prakash YS. Cellular senescence in the lung across the age spectrum. Am J Physiol Lung Cell Mol Physiol. (2019) 316:826–42. doi: 10.1152/ajplung.00424.2018
23. Meyer KC, Ershler W, Rosenthal NS, Lu XG, Peterson K. Immune dysregulation in the aging human lung. Am J Respir Crit Care Med. (1996) 153:1072–9. doi: 10.1164/ajrccm.153.3.8630547
24. Rodier F, Coppé JP, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. (2009) 11:973–9. doi: 10.1038/ncb1909
25. Sueblinvong V, Neujahr DC, Todd Mills S, Roser-Page S, Ritzenthaler JD, Guidot D, et al. Predisposition for disrepair in the aged lung. Am J Med Sci. (2012) 344:41–51. doi: 10.1097/MAJ.0b013e318234c132
26. Wong CK, Smith CA, Sakamoto K, Kaminski N, Koff JL, Goldstein DR. Aging impairs alveolar macrophage phagocytosis and increases influenza-induced mortality in mice. J Immunol. (2017) 199:1060–8. doi: 10.4049/jimmunol.1700397
27. Sapey E, Patel JM, Greenwood HL, Walton GM, Hazeldine J, Sadhra C, et al. Pulmonary infections in the elderly lead to impaired neutrophil targeting, which is improved by simvastatin. Am J Respir Crit Care Med. (2017) 196:1325–36. doi: 10.1164/rccm.201704-0814OC
28. Eworuke E, Major JM, Gilbert McClain LI. National incidence rates for acute respiratory distress syndrome (ARDS) and ARDS cause-specific factors in the United States (2006–2014). J Crit Care. (2018) 47:192–7. doi: 10.1016/j.jcrc.2018.07.002
29. Manzano F, Yuste E, Colmenero M, Aranda A, García-Horcajadas A, Rivera R, et al. Incidence of acute respiratory distress syndrome and its relation to age. J Crit Care. (2005) 20:274–80. doi: 10.1016/j.jcrc.2005.05.008
30. Chen W, Chen YY, Tsai CF, Chen SCC, Lin MS, Ware LB, et al. Incidence and outcomes of acute respiratory distress syndrome a nationwide registry-based study in Taiwan, 1997 to 2011. Medicine. (2015) 94:e1849. doi: 10.1097/MD.0000000000001849
31. Johnston CJ, Rubenfeld GD, Hudson LD. Effect of age on the development of ARDS in trauma patients. Chest. (2003) 124:653–9. doi: 10.1378/chest.124.2.653
32. Iriyama H, Abe T, Kushimoto S, Fujishima S, Ogura H, Shiraishi A, et al. Risk modifiers of acute respiratory distress syndrome in patients with non-pulmonary sepsis: a retrospective analysis of the FORECAST study. J Intensive Care. (2020) 8:7. doi: 10.1186/s40560-020-0426-9
33. Johnson NJ, Caldwell E, Carlbom DJ, Gaieski DF, Prekker ME, Rea TD, et al. The acute respiratory distress syndrome after out-of-hospital cardiac arrest: incidence, risk factors, and outcomes. Resuscitation. (2019) 135:37–44. doi: 10.1016/j.resuscitation.2019.01.009
34. Rhee C, Kang J, Kim Y, Kim J, Yoon H, Kim S, et al. Risk factors for acute respiratory distress syndrome during neutropenia recovery in patients with hematologic malignancies. Crit Care. (2009) 13:R173. doi: 10.1186/cc8149
35. Shorr AF, Abbott KC, Agadoa LY. Acute respiratory distress syndrome after kidney transplantation: epidemiology, risk factors, and outcomes. Crit Care Med. (2003) 31:1325–30. doi: 10.1097/01.CCM.0000053645.38356.A6
36. Vincent JL, Moreno R. Clinical review: scoring systems in the critically ill. Crit Care. (2010) 14:207. doi: 10.1186/cc8204
37. Villar J, Kacmarek RM. The APPS: an outcome score for the acute respiratory distress syndrome. J Thorac Dis. (2016) 8:E1343–7. doi: 10.21037/jtd.2016.10.76
38. Villar J, Ambrós A, Soler JA, Martínez D, Ferrando C, Solano R, et al. Age, PaO2/FIO2, and plateau pressure score. Crit Care Med. (2016) 44:1361–9. doi: 10.1097/CCM.0000000000001653
39. Bos LD, Schouten LR, Cremer OL, Ong DSY, Schultz MJ, MARS consortium, et al. External validation of the APPS, a new and simple outcome prediction score in patients with the acute respiratory distress syndrome. Ann Intensive Care. (2016) 6:89. doi: 10.1186/s13613-016-0190-0
40. Song M, Liu Y, Lu Z, Luo H, Peng H, Chen P. Prognostic factors for ARDS: clinical, physiological and atypical immunodeficiency. BMC Pulm Med. (2020) 20:102. doi: 10.1186/s12890-020-1131-0
41. Luo L, Shaver CM, Zhao Z, Koyama T, Calfee CS, Bastarache JA, et al. Clinical predictors of hospital mortality differ between direct and indirect ARDS. Chest. (2017) 151:755–63. doi: 10.1016/j.chest.2016.09.004
42. Killien EY, Mills B, Vavilala MS, Watson RS, O'Keefe GE, Rivara FP. Association between age and acute respiratory distress syndrome development and mortality following trauma. J Trauma Acute Care Surg. (2019) 86:844–52. doi: 10.1097/TA.0000000000002202
43. Cochi SE, Kempker JA, Annangi S, Kramer MR, Martin GS. Mortality trends of acute respiratory distress syndrome in the United States from 1999 to 2013. Ann Am Thorac Soc. (2016) 13:1742–51. doi: 10.1513/AnnalsATS.201512-841OC
44. Eachempati SR, Hydo LJ, Shou J, Barie PS. Outcomes of acute respiratory distress syndrome (ARDS) in elderly patients. J Trauma Inj Infect Crit Care. (2007) 63:344–50. doi: 10.1097/TA.0b013e3180eea5a1
45. Kao KC, Hsieh MJ, Lin SW, Chuang LP, Chang CH, Hu HC, et al. Survival predictors in elderly patients with acute respiratory distress syndrome: a prospective observational cohort study. Sci Rep. (2018) 8:13459. doi: 10.1038/s41598-018-31811-w
46. Nathanson BH, Higgins TL, Brennan MJ, Kramer AA, Stark M, Teres D. Do elderly patients fare well in the ICU? Chest. (2011) 139:825–31. doi: 10.1378/chest.10-1233
47. Sacanella E, Pérez-Castejón JM, Nicolás JM, Masanés F, Navarro M, Castro P, et al. Mortality in healthy elderly patients after ICU admission. Intensive Care Med. (2009) 35:550–5. doi: 10.1007/s00134-008-1345-8
48. Bagshaw SM, Webb SAR, Delaney A, George C, Pilcher D, Hart GK, et al. Very old patients admitted to intensive care in Australia and New Zealand: a multi-centre cohort analysis. Crit Care. (2009) 13:R45. doi: 10.1186/cc7768
49. Ke Y, Karki P, Kim J, Son S, Berdyshev E, Bochkov VN, et al. Elevated truncated oxidized phospholipids as a factor exacerbating ALI in the aging lungs. FASEB J. (2019) 33:3887–900. doi: 10.1096/fj.201800981R
50. Brandenberger C, Kling KM, Vital M, Mühlfeld C. The role of pulmonary and systemic immunosenescence in acute lung injury. Aging Dis. (2018) 9:553–65. doi: 10.14336/AD.2017.0902
51. Kling KM, Lopez-Rodriguez E, Pfarrer C, Mühlfeld C, Brandenberger C. Aging exacerbates acute lung injury-induced changes of the air-blood barrier, lung function, and inflammation in the mouse. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L1–2. doi: 10.1152/ajplung.00347.2016
52. Linehan E, Fitzgerald D. Ageing and the immune system: focus on macrophages. Eur J Microbiol Immunol. (2015) 5:14–24. doi: 10.1556/EuJMI-D-14-00035
53. Drew W, Wilson DV, Sapey E. Inflammation and neutrophil immunosenescence in health and disease: targeted treatments to improve clinical outcomes in the elderly. Exp Gerontol. (2018) 105:70–7. doi: 10.1016/j.exger.2017.12.020
54. Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun. (2019) 10:963. doi: 10.1038/s41467-019-08831-9
55. Herrero C, Marqués L, Lloberas J, Celada A. IFN-γ-dependent transcription of MHC class II IA is impaired in macrophages from aged mice. J Clin Invest. (2001) 107:485–493. doi: 10.1172/JCI11696
56. Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, et al. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell. (2014) 13:690–8. doi: 10.1111/acel.12222
57. Schouten LR, van Kaam AH, Kohse F, Veltkamp F, Bos LD, de Beer FM, et al. Age-dependent differences in pulmonary host responses in ARDS: a prospective observational cohort study. Ann Intensive Care. (2019) 9:55. doi: 10.1186/s13613-019-0529-4
58. Wenisch C, Patruta S, Daxböck F, Krause R, Hörl W. Effect of age on human neutrophil function. J Leukoc Biol. (2000) 67:40–45. doi: 10.1002/jlb.67.1.40
59. van der Geest KSM, Abdulahad WH, Tete SM, Lorencetti PG, Horst G, Bos NA, et al. Aging disturbs the balance between effector and regulatory CD4+ T cells. Exp Gerontol. (2014) 60:190–6. doi: 10.1016/j.exger.2014.11.005
60. Yu ZX, Ji MS, Yan J, Cai Y, Liu J, Yang HF, et al. The ratio of Th17/Treg cells as a risk indicator in early acute respiratory distress syndrome. Crit Care. (2015) 19:82. doi: 10.1186/s13054-015-0811-2
61. Halter S, Aimade L, Barbié M, Brisson H, Rouby JJ, Langeron O, et al. T regulatory cells activation and distribution are modified in critically ill patients with acute respiratory distress syndrome: a prospective single-centre observational study. Anaesth Crit Care Pain Med. (2020) 39:35–44. doi: 10.1016/j.accpm.2019.07.014
62. Adamzik M, Broll J, Steinmann J, Westendorf AM, Rehfeld I, Kreissig C, et al. An increased alveolar CD4 + CD25 + Foxp3 + T-regulatory cell ratio in acute respiratory distress syndrome is associated with increased 30-day mortality. Intensive Care Med. (2013) 39:1743–51. doi: 10.1007/s00134-013-3036-3
63. Uyar B, Palmer D, Kowald A, Escobar HM, Barrantes I, Möller S, et al. Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev. (2020) 2020:101156. doi: 10.1016/j.arr.2020.101156
64. van Der Heijden M, van Nieuw Amerongen GP, Koolwijk P, van Hinsbergh VWM, Groeneveld ABJ. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax. (2008) 63:903–9. doi: 10.1136/thx.2007.087387
65. Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. (2006) 3:356–70. doi: 10.1371/journal.pmed.0030046
66. Jeong JH, Kim KS, Lim D, Kim KH, Kim HS, Lee S, et al. Microvasculature remodeling in the mouse lower gut during inflammaging. Sci Rep. (2017) 7:39848. doi: 10.1038/srep39848
67. Lieb W, Zachariah JP, Xanthakis V, Safa R, Chen MH, Sullivan LM, et al. Clinical and genetic correlates of circulating angiopoietin-2 and soluble tie-2 in the community. Circ Cardiovasc Genet. (2010) 3:300–6. doi: 10.1161/CIRCGENETICS.109.914556
68. Miller EJ, Linge HM. Age-related changes in immunological and physiological responses following pulmonary challenge. Int J Mol Sci. (2017) 18:1294. doi: 10.3390/ijms18061294
69. Palumbo S, Shin YJ, Ahmad K, Desai AA, Quijada H, Mohamed M, et al. Dysregulated Nox4 ubiquitination contributes to redox imbalance and age-related severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L297–308. doi: 10.1152/ajplung.00305.2016
70. Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J Immunol. (2004) 173:3589–93. doi: 10.4049/jimmunol.173.6.3589
71. Cui Y, Wang Y, Li G, Ma W, Zhou X, Wang J, et al. The Nox1/Nox4 inhibitor attenuates acute lung injury induced by ischemia-reperfusion in mice. PLoS ONE. (2018) 13:e0209444. doi: 10.1371/journal.pone.0209444
72. Lichtenstern C, Hofer S, Möllers A, Snyder-Ramos S, Spies-Martin D, Martin E, et al. Lipid peroxidation in acute respiratory distress syndrome and liver failure. J Surg Res. (2011) 168:243–52. doi: 10.1016/j.jss.2009.10.028
73. Kim J, Heise RL, Reynolds AM, Pidaparti RM. Aging effects on airflow dynamics and lung function in human bronchioles. PLoS ONE. (2017) 12:e0183654. doi: 10.1371/journal.pone.0183654
74. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. (2017) 8:963. doi: 10.1038/ncomms14532
75. Burnham EL, Janssen WJ, Riches DWH, Moss M, Downey GP. The fibroproliferative response in acute respiratory distress syndrome: mechanisms and clinical significance. Eur Respir J. (2014) 43:276–85. doi: 10.1183/09031936.00196412
76. Nosaka N, Martinon D, Moreira D, Crother TR, Arditi M, Shimada K. Autophagy protects against developing increased lung permeability and hypoxemia by down regulating inflammasome activity and IL-1β in LPS plus mechanical ventilation-induced acute lung injury. Front Immunol. (2020) 11:207. doi: 10.3389/fimmu.2020.00207
77. Uchida T, Shirasawa M, Ware LB, Kojima K, Hata Y, Makita K, et al. Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am J Respir Crit Care Med. (2006) 173:1008–15. doi: 10.1164/rccm.200509-1477OC
78. Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. (2004) 113:1641–50. doi: 10.1172/JCI200418704
79. He M, Kubo H, Ishizawa K, Hegab AE, Yamamoto Y, Yamamoto H, et al. The role of the receptor for advanced glycation end-products in lung fibrosis. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L1427–36. doi: 10.1152/ajplung.00075.2007
80. Calfee CS, Ware LB, Eisner MD, Parsons PE, Thompson BT, Wickersham N, et al. Plasma receptor for advanced glycation end products and clinical outcomes in acute lung injury. Thorax. (2008) 63:1083–9. doi: 10.1136/thx.2008.095588
81. Zhang H, Tasaka S, Shiraishi Y, Fukunaga K, Yamada W, Seki H, et al. Role of soluble receptor for advanced glycation end products on endotoxin-induced lung injury. Am J Respir Crit Care Med. (2008) 178:356–62. doi: 10.1164/rccm.200707-1069OC
82. Ramasamy R, Vannucci SJ, Shi S, Yan D, Herold K, Fang Yan S, et al. REVIEW advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. (2005) 15:16–28. doi: 10.1093/glycob/cwi053
83. Isabel García-Laorden M, Lorente JA, Flores C, Slutsky AS, Villar J. Biomarkers for the acute respiratory distress syndrome: how to make the diagnosis more precise. Ann Transl Med. (2017) 5:283. doi: 10.21037/atm.2017.06.49
84. Setzer F, Oschatz K, Hueter L, Schmidt B, Schwarzkopf K, Schreiber T. Susceptibility to ventilator induced lung injury is increased in senescent rats. Crit Care. (2013) 17:R99. doi: 10.1186/cc12744
85. Nin N, Lorente JA, Paula M De, Fernández-Segoviano P, Peñuelas O, Sánchez-Ferrer A, et al. Aging increases the susceptibility to injurious mechanical ventilation. Intensive Care Med. (2008) 34:923–31. doi: 10.1007/s00134-007-0960-0
86. Med CC, Forum S, Med IC, Respir AJ, Care C, Med IC, et al. Counterpoint : is low tidal volume mechanical ventilation preferred for all patients on ventilation? No. Chest. (2011) 140:11–3. doi: 10.1378/chest.11-0827
87. Herbert JA, Valentine MS, Saravanan N, Schneck MB, Pidaparti R, Fowler AA, et al. Conservative fluid management prevents age-associated ventilator induced mortality. Exp Gerontol. (2016) 81:101–9. doi: 10.1016/j.exger.2016.05.005
88. Nadeem RN, Elhoufi AM, Soliman MA, Bon I, Obaida ZA, Hussien MM, et al. Clinical predictors of adherence to low tidal volume ventilation practice: is it different on weekend and night shifts? Cureus. (2019) 11:e4844. doi: 10.7759/cureus.4844
89. Shen Y, Cai G, Gong S, Dong L, Yan J, Cai W. Interaction between low tidal volume ventilation strategy and severity of acute respiratory distress syndrome: a retrospective cohort study. Crit Care. (2019) 23:254. doi: 10.1186/s13054-019-2530-6
90. Mendiratta P, Tang X, Collins RT, Rycus P, Brogan T V., Prodhan P. Extracorporeal membrane oxygenation for respiratory failure in the elderly. ASAIO J. (2014) 60:385–90. doi: 10.1097/MAT.0000000000000090
91. Narotsky DL, Mosca MS, Mochari-Greenberger H, Beck J, Liao M, Mongero L, et al. Short-term and longer-term survival after veno-arterial extracorporeal membrane oxygenation in an adult patient population: does older age matter? Perfus. (2016) 31:366–75. doi: 10.1177/0267659115609092
92. Volpi S, Sertic F, Valchanov K, Silva R. Use veno-venous extra corporeal membrane oxygenation in elderly patients with post-cardiotomy hypoxia: the changing paradigm of respiratory support in adult respiratory distress syndrome. J Cardiothorac Surg. (2019) 14:10. doi: 10.1186/s13019-019-0833-y
93. Yeh T-C, Chang H-H, Ger L-P, Wang J-O, Kao S, Ho S-T. Clinical risk factors of extracorporeal membrane oxygenation support in older adults. PLoS ONE. (2018) 13:e0195445. doi: 10.1371/journal.pone.0195445
94. Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J. (2017) 50:1602367. doi: 10.1183/13993003.02367-2016
95. Horie S, Gonzalez HE, Laffey JG, Masterson CH. Cell therapy in acute respiratory distress syndrome. J Thorac Dis. (2018) 10:5607–20. doi: 10.21037/jtd.2018.08.28
96. Kim EC, Kim JR. Senotherapeutics: emerging strategy for healthy aging and age-related disease. BMB Rep. (2019) 52:47–55. doi: 10.5483/BMBRep.2019.52.1.293
97. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. (2009) 460:392–5. doi: 10.1038/nature08221
98. Selman C, Tullet JMA, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. (2009) 326:140–4. doi: 10.1126/science.1177221
99. Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. (2020) 32:44–55. doi: 10.1016/j.cmet.2020.04.015
100. Partridge L, Fuentealba M, Kennedy BK. The quest to slow ageing through drug discovery. Nat Rev Drug Discov. (2020) 19:513–32. doi: 10.1038/s41573-020-0067-7
101. Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, et al. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med. (2017) 195:331–8. doi: 10.1164/rccm.201603-0645OC
102. Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, et al. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med. (2018) 6:691–8. doi: 10.1016/S2213-2600(18)30177-2
103. Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med. (2014) 2:611–20. doi: 10.1016/S2213-2600(14)70097-9
Keywords: acute respiratory distress syndrome, acute lung injury, aging, immunosenescence, biomarkers
Citation: Brown R, McKelvey MC, Ryan S, Creane S, Linden D, Kidney JC, McAuley DF, Taggart CC and Weldon S (2020) The Impact of Aging in Acute Respiratory Distress Syndrome: A Clinical and Mechanistic Overview. Front. Med. 7:589553. doi: 10.3389/fmed.2020.589553
Received: 31 July 2020; Accepted: 01 October 2020;
Published: 26 October 2020.
Edited by:
Claudia A. Staab-Weijnitz, Helmholtz Zentrum München, GermanyReviewed by:
Jennifer Casey, University of Alabama at Birmingham, United StatesCopyright © 2020 Brown, McKelvey, Ryan, Creane, Linden, Kidney, McAuley, Taggart and Weldon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sinéad Weldon, cy53ZWxkb25AcXViLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.