 Tao Zhang1,2Jingshan Bai3,4Xinyi Zhang1,2Xiaowei Zheng1,2Nan Lu5Zhongyin Liang6Ling Lin2,7*
Tao Zhang1,2Jingshan Bai3,4Xinyi Zhang1,2Xiaowei Zheng1,2Nan Lu5Zhongyin Liang6Ling Lin2,7* Yongsong Chen2,8*
Yongsong Chen2,8*- 1Department of Ophthalmology, The First Affiliated Hospital of Shantou University Medical College, Shantou, China
- 2The Clinical Research Center of the First Affiliated Hospital of Shantou University Medical College, Shantou, China
- 3Department of Ophthalmology, The Second People's Hospital of Dongying, Dongying, China
- 4Department of Ophthalmology, The Dawang Hospital of Guangrao of Dongying, Dongying, China
- 5Department of Cardiology, The First Affiliated Hospital of Shantou University Medical College, Shantou, China
- 6Department of Bioinformatics, Berry Genomics Co., Ltd., Beijing, China
- 7Department of Rheumatology, The First Affiliated Hospital of Shantou University Medical College, Shantou, China
- 8Department of Endocrinology, The First Affiliated Hospital of Shantou University Medical College, Shantou, China
The small nuclear ribonucleoprotein 200 kDa (SNRNP200) gene plays a key role in the maturation of pre-message RNA (pre-mRNA) splicing with the indication for the etiology of retinitis pigmentosa (RP). Gene recognition can facilitate the diagnosis of these patients for better clinical management, treatment and counseling. This study aimed to outline the causative mutation in a Chinese family and the pathogenic mechanism of this SNRNP200 mutation in RP. Eighteen individuals from the affected family underwent a complete ophthalmic examination. Whole exome sequencing (WES) was conducted to identify the pathogenic variant in the proband, which was then confirmed by Sanger sequencing. Expression of the SNRNP200 transcript in zebrafish was identified via whole mount in situ hybridization. Morpholino oligonucleotide (MO) and SNRNP200 wild and mutant mRNA were injected into zebrafish embryos followed by analyses of the systemic changes and retinal phenotypes using immunofluorescence. Heterozygous SNRNP200c.C6088T (p.Arg2030Cys) mutation was ascertained in two members of this family: the proband and his father (II-2). Overexpression of SNRNP200Arg2030Cys, but not SNRNP200WT caused systemic deformities in the wild-type zebrafish embryos with the retina primarily injured, and significantly increased death rates in the morphant embryos, in which the orthologous zebrafish SNRNP200 gene was blocked. In conclusion, this study reports a novel heterozygous SNRNP200c.C6088T mutation, which is evidenced to cause RP via a dominant-negative effect.
Introduction
Retinitis pigmentosa (RP) is reported as the most regular form of inherited degenerative retinal dystrophy, with a prevalence ranging between 1/3,500 to 1/5,000 among different countries worldwide (1, 2). Nyctalopia is one of the earliest and most common symptoms of RP, followed by subsequent constricted visual fields (VFs), and eventual loss of central vision caused by the degeneration of photoreceptor and retinal pigment epithelium (RPE) (3, 4). The fundus in RP is characterized by peripheral bone-spicule pigmentary deposits, attenuation of the artery, and waxy pallor of the optic nerve head. Outer nuclear layer attenuation and the loss of outer/inner segments of RPE in the macular area of the retina are the typical characteristics. The inheritance of RP could be in three modes, autosomal dominant RP (adRP), autosomal recessive RP, and X-linked RP. Thus far, 307 genes and gene loci have been shown to be involved in retina degeneration [Retnet database: https://sph.uth.edu/retnet/; reviewed in Daiger et al. (5)]. The majority of these genes are specifically expressed in the retina. Interestingly, 6 of 22 adRP-related genes code for universally expressed pre-mRNA splicing proteins that are essential splicing factors, called the small nuclear ribonucleoprotein particles (snRNPs). These genes include PRPF6 (MIM 613979) (6), PRPF31(MIM 606419) (7), PRPF8 (MIM 607300) (8), PRPF3 (MIM 607301) (9), PIM1-associated protein [RP9 (MIM 607331)] (10), and small nuclear ribonucleoprotein 200 kDa (SNRNP200) (11, 12).
For most eukaryotic genes, the primarily transcribed RNA from the gene's DNA must be edited through a process called splicing before it becomes mature, and only then can it guide the synthesis of proteins. During the process of primary RNA editing, the sequence of the introns will be removed and the sequence of the exons will be connected together, through the actions of the spliceosome, primarily comprising U1, U2, U4/U6, and U5 snRNPs. The complex of U4/U6–U5 tri-snRNP is essential for installing and the catalytic process of the spliceosome structural rearrangements. Thus, any defect in the complex could possibly contribute to the pathogenesis of RP (13).
SNRNP200 encodes hBrr2, which is one of the U5 snRNP-specific proteins (NP_054733.2) (14), containing 2,136 amino acids (15) and catalyzing the U4/U6 unwinding (16). It has been reported in the literature that the mutation of SNRNP200 can compromise the U4/U6 unwinding (11) and when blocked, could cause the demorphogenesis of rod photoreceptors in a zebrafish model (17). However, the exact pathogenic mechanism of the SNRNP200 mutations has never been demonstrated.
Herein, we report one naturally occurring heterozygous mutation in SNRNP200, c.C6088T (p.Arg2030Cys), which associates with adRP in a Chinese family and investigate the pathogenic mechanism of this SNRNP200 mutation.
Materials and Methods
Study Subjects and Clinical Examinations
The methods in this study were approved by the Dawang hospital of Guangrao of Dongying Institutional Review Board and complied with the local laws (Certificate NO. DWZWSY2019-05). The study was performed following the Declaration of Helsinki and the standards of the Institutional Animal Care and Use Committee, Institutional Animal Ethics Committee. Written informed consent was obtained from all the study participants.
A Chinese family (Figure 1) diagnosed with RP consisting of three affected patients and 15 unaffected family members was recruited from Dawang hospital of Guangrao of Dongying. Venous blood samples were collected for whole exome sequencing (WES) from family members and 400 unrelated Chinese individuals without ocular diseases serving as controls.
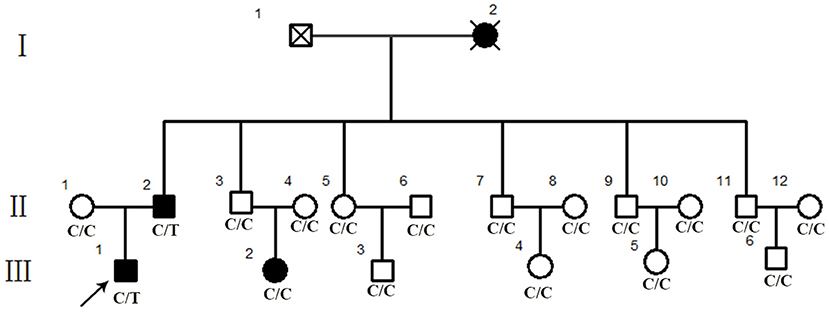
Figure 1. Pedigree of a Chinese retinitis pigmentosa families. Pedigree representing three consecutive generations of members in a Chinese family with typical phenotype of adRP. Filled symbols represent affected individuals; open symbols represent unaffected family members.
Very detailed ophthalmic examinations, including routine slit-lamp examination, visual acuity, intraocular pressure, fundus photography, visual field, and optical coherence tomography (OCT) in the macula, were conducted.
Animals
Zebrafish (Danio rerio) were maintained and reared under standard conditions-28.5 °C in a 14 h light and 10 h darkness rotation in the Model Animal Research Center. All the embryos used in experimenting were obtained by natural mating in the morning. Animal experiments were approved by the Dawang hospital of Guangrao of Dongying Animal Care commission and the local ethical review board and conformed to the Guideline for the Care and Use of laboratory animals of the National Institutes of Health.
Genetic Analysis
Whole Exome Sequencing and Sanger Sequencing
Genomic DNA was extracted from peripheral blood cells as per the protocols of the Lab-Aid DNA kit (Tiangen Biotech Co., Beijing, China). The target exomes capture was performed using the Agilent SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA), and sequencing was performed on an Illumina Hiseq2500 (Illumina, San Diego, CA, USA) following the manufacturer's protocol. The results of the selected pathogenic variants were confirmed by bidirectional Sanger sequencing.
Expression Analyses of SNRNP200
Quantitative real-time PCR (Q-PCR) and reverse-transcriptase polymerase chain reaction (RT-PCR) were conducted to investigate the expression of SNRNP200 in multiple tissues of the human cadaver eye including the cornea, iris, lens, vitreous body, retina, optic nerve and others; a total RNA extraction from these samples was performed using TRIZOL reagent. cDNA synthesis was performed using a TOYOBO RT-PCR kit (FSQ-301, TOYOBO, Japan) per the manufacturer's protocol. The expression profile of SNRNP200 was analyzed using the following primers: forward (5′-AGCTCTTTGCTGCCTGTGTC-3′), reverse (3′-TACCAAGCACCTAGCCAATG-5′); GAPDH forward (5′-TCATTGACCTCAACTACATGG−3′), reverse (3′-TCGCTCCTGGAAGATGGTG-5′).
Functional Analyses of SNRNP200 Mutation in Zebrafish
Microinjections of Zebrafish
Capped and tailed mRNAs of SNRNP200 WT (hSnrnp200 WT) and SNRNP200 c.C6088T (hSnrnp200 Arg2030Cys) were achieved from the circular plasmids cut by the endonuclease XmaI AcpxT7- SNRNP200WT and AcpxT7- SNRNP200c.C6088T with the mMESSAGE MACHINE T7 Ultra Kit (Ambion, Austin, TX, USA). RNeasy Kit (Qiagen, Hilden, Germany) was employed to erase extra nucleotides, especially DNA aiming at purifying the synthesized mRNAs.
Blocking MOs for Zebrafish
Snrnp200-MO (5′-TCAACATCAAGACAACTCACATCCT-3′) and Control-MO (5′-CCTCTTACCTCAGTTACAATTTATA-3′) were designed as previously described and purchased from Gene Tools, LLC (Philomath, Oregon, USA).
Embryos at 1 cell-stage were chosen for microinjections in the morning. For mRNA microinjection, each embryo was injected with 1 nL solution containing 200 pg SNRNP200WT or SNRNP200c.C6088T mRNA. For co-injections of MO and mRNA, zebrafish embryos were separated into four groups and were injected with 4 ng Control-MO, 4 ng Snrnp200-MO, 4 ng Snrnp200-MO plus 200 pg SNRNP200WT mRNA, and 4 ng Snrnp200-MO plus 200 pg SNRNP200c.C6088T mRNA, respectively. During the entire experiment, the embryos that died within 24 h postoperatively were eliminated, as it was likely that they died from indefinite causes. The percentage of deformation and mortality of zebrafish embryos was calculated from 2 to 3 dpf.
Expression Analysis of the Immunofluorescence of Zebrafish
At 3 dpf, zebrafish were randomly selected from the experiment groups. Immunostaining was performed to evaluate their ocular morphology and rod photoreceptors. The zebrafish embryo was fixed in 4% paraformaldehyde (PFA) overnight at 4°C. On the second day, the fixed ones were washed three times with phosphate-buffered saline (PBS) containing 0.01% Tween 20 (PBST) and dehydrated in 30% sucrose overnight at 4°C. Subsequently, they were embedded in an optimal cutting temperature solution (Invitrogen, Carlsbad, CA, USA) at −40°C for 2 h before being cut at −10°C using the Leica CM1860 cryostat (Leica Microsystems, Nussloch Germany). For immunostaining, the slides were dehydrated for 30 min at 37°C, rehydrated with PBST for 5 min, and blocked with PBS supplemented with 10% normal goat serum and 2% bovine serum albumin for 1 h at room temperature. The slides were then incubated with antizebrafish rhodopsin overnight at 4°C. A confocal laser scanning microscope (Nikon, Tokyo, Japan) was applied for image collection.
Results
Clinical Evaluations of the Patients With SNRNP200 Variant
Eighteen individuals from a family were recruited, following the adRP inheritance (Figure 1). The proband III:1 was first referred to an ophthalmic clinic at the age of 7 years for his night vision disorder. Afterward, his VFs and vision deteriorated further with time. At his latest visit at the age of 27 years, he demonstrated a typical RP fundus, including waxy pallor of the optic disk, attenuated artery and vein, and peripheral pigment deposit (Figures 2A vs. C). The reduced thickness of the outer nuclear layer and RPE at the macular area with partial loss of outer/inner segments (OS/IS) were demonstrated by OCT (Figures 2B vs. D), all of which corresponded to his poor visual acuity (Table 1). Patients II:2 and III:2 developed nyctalopia at 5 and 9 years of age, respectively. During the last examination at ages of 48 and 32 years, respectively, their phenotypes were similar to those of the proband, including inferior night vision, inferior central visual acuity, significantly reduced VFs (Figure 2E), and typical RP fundus appearances. The details of the clinical information of this family are presented in Table 1.
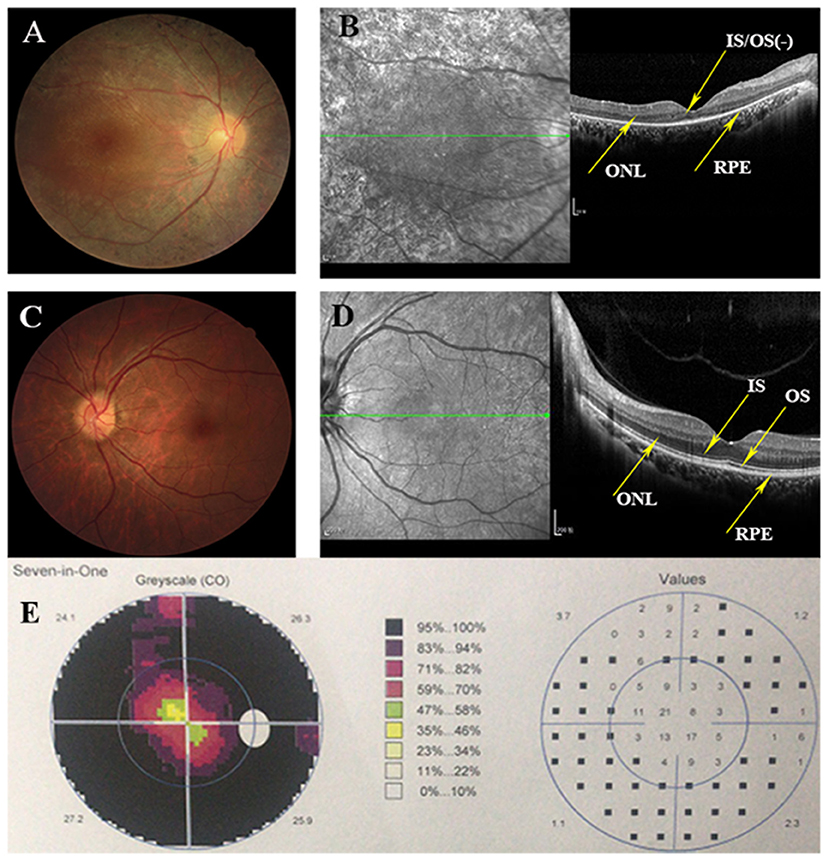
Figure 2. Clinical information of this adRP family. (A) Right eye fundus of the proband (III:1) presents characteristic RP degeneration, including a waxy pallor of optic disc, attenuated artery and vein and peripheral pigment deposit. (B) OCT scan reveals macular degeneration showing reduced thickness of outer nuclear layer (ONL) and RPE at the macular area with partial loss of outer/inner segments (OS/IS). (C,D) Fundus picture and Optical coherence tomography (OCT) scans of the left eye of the unaffected member (III:3) showing as normal controls. (E) Visual fields of the right eye of the proband indicates the tubular visual field.
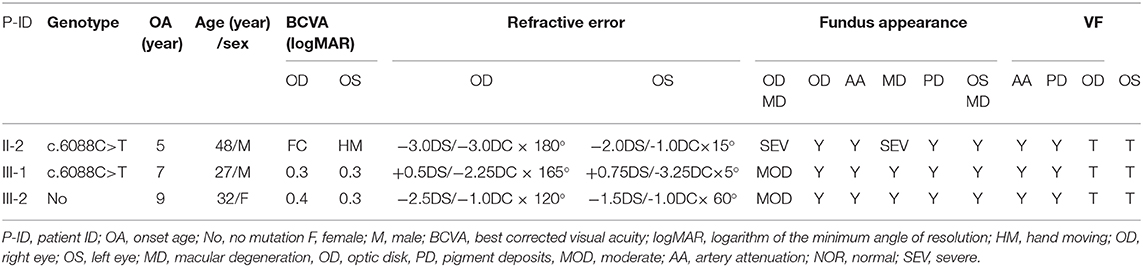
Table 1. The character of the affected patients.
Identification of Putative Variants in SNRNP200
We performed WES using the genomic DNA of proband III-1 (Figure 1). A total of 51,767 variants were initially identified in the proband, including 47,875 single nucleotide variations, and 3,892 insert/deletions (Indels). The coverage of the targeted region was 96% at least 20 ×. Finally, only one putative pathogenic variant, heterozygous SNRNP200 (NM_014014.4) c.C6088T, which cosegregated with the disease phenotype in the family was identified (Figures 1, 3B). This variant was not present in all single nucleotide polymorphism databases and 400 unrelated controls as demonstrated by Sanger sequencing.
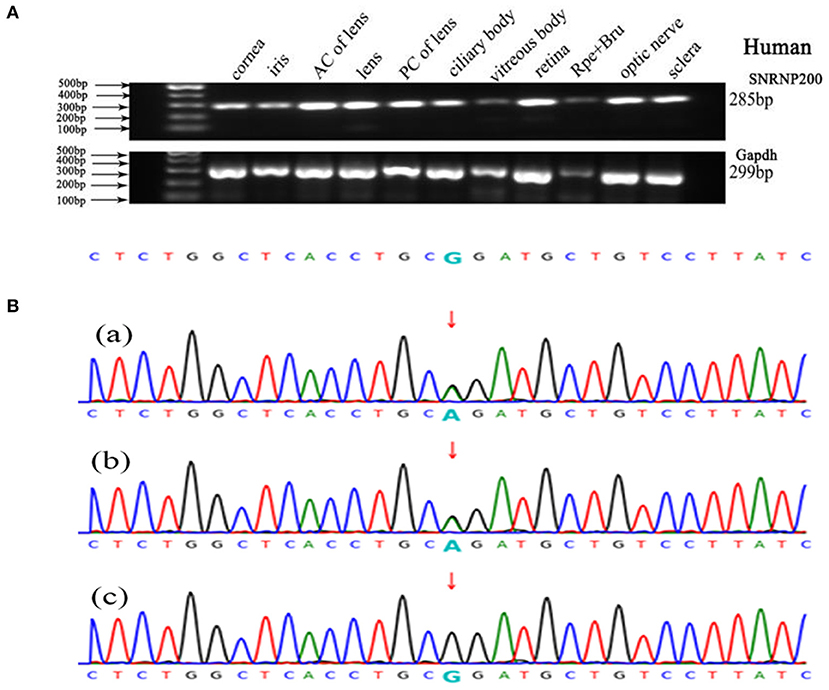
Figure 3. Genetic analyses of variants identified in the SNRNP200 Gene and its expression of SNRNP200 in cadaver eye tissues. (A) Expression of SNRNP200 in multiple cadaver eye tissues including cornea, iris, anterior capsule of lens (AC of lens), lens, posterior capsule of lens (PC of lens), ciliary body, vitreous body and neural retina and so on. retinal pigmented epithelium (RPE) is shown. A 285 bp PCR product of the SNRNP200 was detected in all tissues. A 299 bp PCR products of the human Gapdh were selected in parallel as control. (B) Sanger sequencing confirmation of the identified SNRNP200 mutations from the whole exome sequencing analysis in the recruited Chinese retinitis pigmentosa families. Sanger sequencing validation of the SNRNP200 mutations (c.6088C>T) identified by the WES analysis in this family.
Ubiquitous Expression of SNRNP200 in the Human Eye and Zebrafish
SNRNP200 has been previously investigated showing a wide expression in multiple tissues of humans and mice by reverse-transcriptase polymerase chain reaction (11). Here, we evaluated the expression pattern of SNRNP200 (NM_014014.4) in a panel of human cadaver eye tissues. As predicted, the SNRNP200 was expressed in many tissues, including the cornea, iris, the anterior capsule of the lens, lens, posterior capsule of the lens, ciliary body, vitreous body, neural retina, and so on (Figure 3A). Moreover, the expression of SNRNP200 in zebrafish was revealed by whole mount in situ hybridization among several developmental phases (Figures 4A–E). From 3 dpf, its expression was found to be enriched at the ciliary marginal zone (CMZ) (Figures 4F,G).
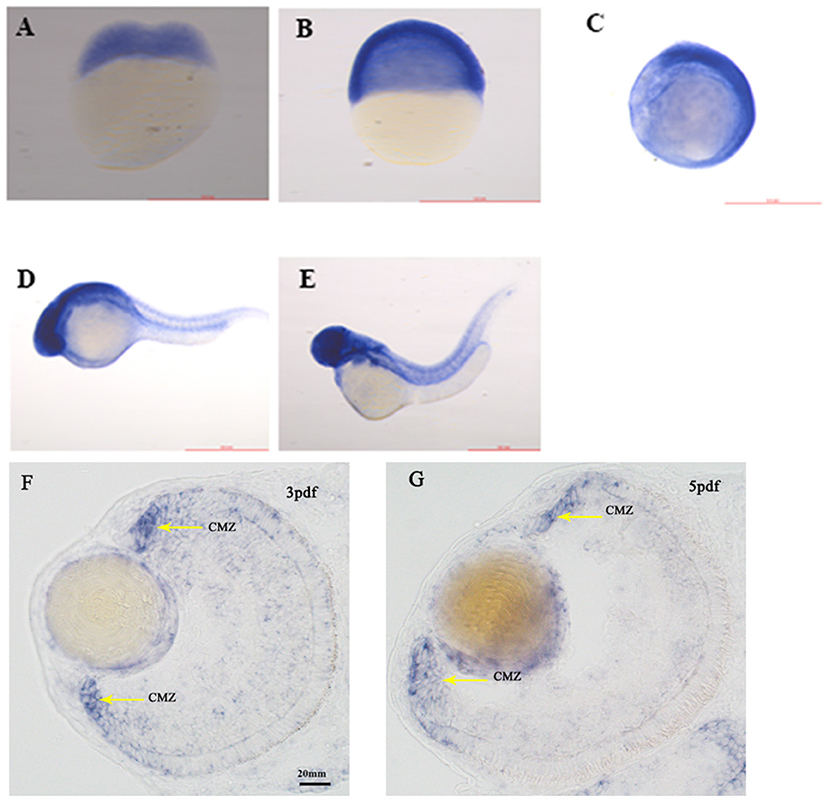
Figure 4. The expression of SNRNP200 in zebrafish. Whole mount in situ hybridization revealed the expression of the Snrnp200 gene of zebrafish at the two-cell stage (A), 50%-epiboly (B), 8-somite stage (C), 1pdf (D), and 2pdf (E). In situ hybridization (ISH) of zebrafish retinal cryosections exhibit its expression is enriched at the ciliary marginal zone (CMZ) at 3dpf (F) and 5dpf (G).
The Detrimental Reaction of SNRNP200 p.Arg2030Cys in Zebrafish
Overexpression of SNRNP200 p.Arg2030Cys causes deformations in a large fraction of zebrafish.
SNRNP200WT or SNRNP200c.C6088T mRNA was injected into zebrafish embryos and the one without injection was selected as control. Considering the ubiquitous expression of SNRNP200 with elementary cellular function for immature mRNA splicing, the systemic appearance of the overexpression of SNRNP200 p.Arg2030Cys and SNRNP200WT were recorded in zebrafish embryos. The group of SNRNP200c.C6088T caused severe defects in the systemic phenotype of zebrafish embryos including the curved body shape, cardiac edema, shortened body trunk, and deformed brain. In comparison with the group of SNRNP200WT, there is a significant rise of deformation of larvae in the group of SNRNP200 p.Arg2030Cys (53% vs. 9%, p < 0.0001) (Figures 5A,B). These three groups showed a low percentage of mortality without a significant statistical difference, probably because of the toxicity of exogenous mRNA.
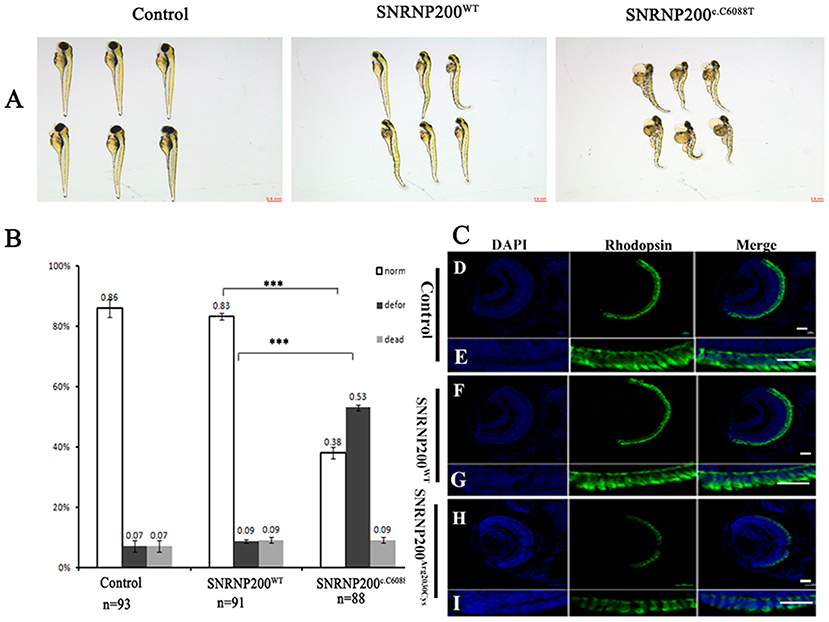
Figure 5. Detrimental effects of SNRNP200 p.Arg2030Cys in zebrafish. (A) Morphological changes at 3 dpf after injections of SNRNP200WT, SNRNP200 p.Arg2030Cys. There is a significant difference of systemic deformations among groups. (B) Statistics of normal, deformed, and dead zebrafish after injection with SNRNP200 p.Arg2030Cys, SNRNP200WT from 2 to 4 dpf. Data are shown as mean + SD from technical triplicates. n: total number of larvae for each group from triple experiment. ***P < 0.001. (C) Immunostaining analysis of the zebrafish larvae injected with different mRNAs at 3 dpf for rhodopsin (green). The normal zebrafish revealed abundant expression of rhodopsin in the IS/OS layer (D–I) by contrast, the reactivity of rhodopsin was significantly diminished in SNRNP200c.C6088T-injected larvae.
Overexpression of SNRNP200 p.Arg2030Cys affects the retina of zebrafish.
We studied the effect of overexpression of SNRNP200 p.Arg2030Cys and SNRNP200WT on the retina. Zebrafish having a normal systemic appearance from both SNRNP200WT and SNRNP200 p.Arg2030Cys groups at 3 dpf were randomly selected and their retinal phenotype was identified. Certainly, although all the zebrafish showed normal ocular size, the expression of rhodopsin in the group injected with SNRNP200c.C6088T dramatically dropped, as demonstrated by immunofluorescence staining. Conversely, the group of SNRNP200WT revealed a rich expression of SNRNP200WT with a similar amount to the normal group [Figure 5C(D–I)].
Overexpression of SNRNP200 p.Arg2030Cys significantly increased death rates in zebrafish with SNRNP200 silenced.
The effect of overexpression of SNRNP200WT or SNRNP200c.C6088T in a previously described zebrafish model, in which the orthologous zebrafish SNRNP200 was blocked by the injection of translational blocking morpholino oligos (MOs) into embryos (17) was further investigated. The co-injection of MO with SNRNP200WT or SNRNP200c.C6088T mRNA was implemented and compared with the pure MO-injected and control group. Our results showed that the pure MO-injected group caused a ratio of 47% deformation and 19% death at 3 dpf (Figures 6A,B). As expected, the co-injection of SNRNP200WT definitely could rescue some part of the morphant phenotypes indicated by the reduced ration of deformation and death (31 and 12%, respectively). In contrast to SNRNP200WT, the co-injection group of SNRNP200-MO and SNRNP200c.C6088T dramatically increased the frequency of dead morphants to 77% (Figures 6A,B), suggesting that p.Arg2030Cys has a dominant-negative effect, which could hinder the function of zebrafish SNRNP200 despite the loss of function of mutant SNRNP200c.C6088T.
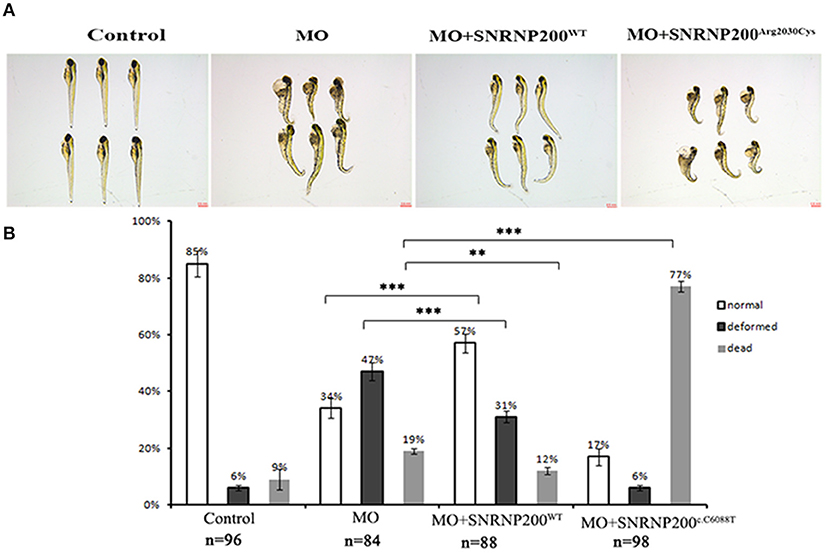
Figure 6. Phenotypic comparison among zebrafish larvae with SNRNP200 silencing, and with overexpression of SNRNP200WT, or SNRNP200c.C6088T. (A) Systemic phenotype of zebrafish larvae injected with 4 ng Control-MO (control); 4 ng SNRNP200-MO (MO); 4 ng SNRNP200-MO +200 pg SNRNP200WT mRNA (MO + SNRNP200WT); and 4 ng SNRNP200-MO+200 pg SNRNP200c.C6088T mRNA(MO + SNRNP200c.C6088T). (B) phenotype distribution in four groups injected variously as shown in (A) including normal, deformed and dead ones were calculated from 1 to 3 dpf. The percentage of those are shown in the graph. Comparing with the group injected with SNRNP200-MO, the group of injection of MO + SNRNP200WT particularly reduced the ratio of deformed and dead larvae, whereas injection of MO + SNRNP200c.C6088T significantly raise the rate of death (34 vs. 77%, P < 0.001). Data are shown as mean + SD from technical triplicates. n: total number of larvae for each group from triple experiment. **P < 0.01, ***P < 0.001.
Discussion
The key role of defects in the U4/U6–U5 tri-snRNP complex has been identified by recent genetic and functional research in the process of disease etiology and pathogenic mechanism of RP. SNRNP200, a gene encoding a crucial component of U5 snRNP known as hBrr2 (16), has long been implicated in RP. In this study, we report the correlation of RP with novel naturally occurring heterozygous variants in SNRNP200, which were absent in 400 unrelated controls. Functional analysis of this allele in zebrafish further demonstrates its pathogenic effect having a dominant-negative effect.
In our study, the novel mutation, SNRNP200c.C6088T: p.Arg2030Cys was identified in this family by WES, which caused an amino acid change from arginine into cysteine in patients II-2 and III-1. However, the same mutation of this gene was not detected in patient III-2 who had the same clinical signs of RP. Therefore, we infer that her mutation was probably from the mitochondria gene or intron of the chromosome. Next, we conducted whole gene sequencing to find the mutation of patient III-2. It is well-known that RP is characterized by the growing loss of photoreceptors and RPE, indicating a significant genetic and phenotypic heterogeneity (4). Till now, more than 70 genes have been implicated in this disorder from published studies (18). Several genes relevant to the tri-snRNP complex of the spliceosome have been demonstrated for adRP (19). Moreover, the age of onset, progress, and the eventual blindness of these patients varies according to the different genotypes. Therefore, the detection and definition of the genotypes of these patients are essential to provide prophylactic advice and available genetic therapy in the near future. Under the optimization of next-generation sequencing technology in recent years, WES analysis has been applied for determining mutation in exons and splicing sites at the genomic scale (20).
The knocking down of endogenous SNRNP200 in zebrafish using MO has been previously demonstrated to cause retinal degeneration, indicating that an insufficient gene quantity of SNRNP200 could result in RP in patients (17). The dominant-negative effect is probably the pathogenic mechanism associated with SNRNP200c.C6088T as indicated by the zebrafish study. The phenotype of the zebrafish model injected with SNRNP200c.C6088T mutant mRNA showing high ratios of deformation and loss of photoreceptors identified by immunofluorescence were recorded. They were not noticed in those injected with SNRNP200WT mRNA, implying that the mutation functions as an antimorph or a neomorph. Previous research showed that the zebrafish model injected with a sub-lethal dosage of the SNRNP200-MO and the retina-specific phenotype were affected with reduced or even loss of a generation of photoreceptor, which resembles the manifestation resulting from zebrafish overexpressing SNRNP200c.C6088T mRNA. These results indicate that SNRNP200c.C6088T acts as an antimorph rather than a neomorph, being expected to produce distinct phenotypes compared with a morphant model. Certainly, the co-injection of SNRNP200c.C6088T and MO overwhelmingly increased the death ratio of the morphants in our study. Given that the morphant zebrafish is not a real null allele for SNRNP200, this observation is presumably caused by the dominant-negative effect of SNRNP200c.C6088T on the sustained function of the orthologous zebrafish SNRNP200 gene, which shows 89% identity with human SNRNP200.
The mechanism of the mutations in pre-mRNA splicing genes with universal transcription in almost every cell causing retina-specific phenotypes are speculated to be via these particular mechanisms. Chen et al. (21) reported that the retina displays one of the highest rates of oxidative metabolism of all cells in the body with the consecutive renewal of OS that highly depends on the pre-mRNA splicing of many genes. Hence, the systemic defects resulting from such splicing mutations could only be compromising to the retina as supported by some data (22, 23). Moreover, the retina might be highly sensitive to the fidelity mechanisms of splicing which is influenced by the mutation of RP genes (21). As is reported, the gene SNRNP200 is essential for the proofreading of pre-mRNA splicing (24–26). More importantly, our experiment findings indicate that SNRNP200 expresses extensively in the CMZ area where retinal stem cells (RSCs) and retinal progenitor cells (RPCs) are located, which precisely coordinate proliferation and differentiation of cells in the retina of zebrafish. Therefore, we conclude that the SNRNP200c.C6088T is likely to impair the development of the retina, including the photoreceptor by the influence of RSCs and RPCs. Subsequently, we are going to investigate how the SNRNP200c.C6088T influence these cell developments by generating the zebrafish SNRNP200 mutant model.
In summary, SNRNP200c.C6088T, a novel RP-associated pre-mRNA splicing gene-locus caused the typical clinical sign of RP. Considering the genetic findings and the phenotypes of the corresponding zebrafish models, the mutation of SNRNP200 locus identified in this study, like mutations of PRPF4, is evidenced to cause RP via a dominant-negative effect. In the future, more scanning of the SNRNP200 locus in RP patients should be performed to find its function in the etiology.
Conclusion
In conclusion, this study reports a novel heterozygous SNRNP200c.C6088T mutation causing the typical adRP in a Chinese family. We found that this mutation could significantly cause the deformation of zebrash larvae with the overexpression of SNRNP200 p.Arg2030Cys mRNA and increase the death rate of zebrash when the endogenous SNRNP200 p.Arg2030Cys was blocked. All these findings indicate that this mutation causes RP via a dominant-negative effect.
Data Availability Statement
The datasets presented in this study can be found in online repository, it can be found using the accession number BioProject ID: PRJNA686229.
Ethics Statement
The studies involving human participants were reviewed and approved by First Affiliated Hospital of Shantou University Medical College (SUMC) Institutional Review Board. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by First Affiliated Hospital of Shantou University Medical College (SUMC) Institutional Review Board. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
TZ and ZL: conceptualization, data curation, formal analysis, investigation, methodology, and writing—original draft. JB: conceptualization, project administration, resources, and supervision. XZha: investigation and methodology. XZhe: investigation, methodology, and validation. NL: investigation, methodology, software, and visualization. LL and YC: conceptualization, funding acquisition, project administration, resources, supervision, and writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81672640), the Grant for Key Disciplinary Project of Clinical Medicine under the Guangdong High-level University Development Program (002-18119101), and the Special Funds for Innovation Strategy of Science and Education in Guangdong Province (2018-157). The funders had no role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
ZL was employed by company Berry Genomics Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Wenyan Li, Shuzhang Liang, Jinmin, and Ma for expert technical assistance.
References
1. Hu DN. Genetic aspects of retinitis pigmentosa in China. Am J Med Genet. (1982) 12:51–6. doi: 10.1002/ajmg.1320120107
2. Chizzolini M, Galan A, Milan E, Sebastiani A, Costagliola C, Parmeggiani F. Good epidemiologic practice in retinitis pigmentosa: from phenotyping to biobanking. Curr Genomics. (2011) 12:260–6. doi: 10.2174/138920211795860071
3. Mendes HF, Van Der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. (2005) 11:177–85. doi: 10.1016/j.molmed.2005.02.007
4. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. (2006) 368:1795–809. doi: 10.1016/S0140-6736(06)69740-7
5. Daiger SP, Sullivan LS, Bowne SJ, Rossiter BJF. (2020). RetNet. Available online at: https://sph.uth.edu/retnet/
6. Tanackovic G, Ransijn A, Ayuso C, Harper S, Berson EL, Rivolta C. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am J Hum Genet. (2011) 88:643–9. doi: 10.1016/j.ajhg.2011.04.008
7. Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, Chakarova C, et al. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4. (RP11). Mol. Cell. (2001) 8:375–81. doi: 10.1016/S1097-2765(01)00305-7
8. Mckie AB, Mchale JC, Keen TJ, Tarttelin EE, Goliath R, Van Lith-Verhoeven JJ, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum Mol Genet. (2001) 10:1555–62. doi: 10.1093/hmg/10.15.1555
9. Chakarova CF, Hims MM, Bolz H, Abu-Safieh L, Patel RJ, Papaioannou MG, et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. (2002) 11:87–92. doi: 10.1093/hmg/11.1.87
10. Keen TJ, Hims MM, Mckie AB, Moore AT, Doran RM, Mackey DA, et al. Mutations in a protein target of the Pim-1 kinase associated with the RP9 form of autosomal dominant retinitis pigmentosa. Eur J Hum Genet. (2002) 10:245–9. doi: 10.1038/sj.ejhg.5200797
11. Zhao C, Bellur DL, Lu S, Zhao F, Grassi MA, Bowne SJ, et al. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet. (2009) 85:617–27. doi: 10.1016/j.ajhg.2009.09.020
12. Li N, Mei H, Macdonald IM, Jiao X, Hejtmancik JF. Mutations in ASCC3L1 on 2q11.2 are associated with autosomal dominant retinitis pigmentosa in a Chinese family. Invest Ophthalmol Vis Sci. (2010) 51:1036–43. doi: 10.1167/iovs.09-3725
13. Laggerbauer B, Liu S, Makarov E, Vornlocher HP, Makarova O, Ingelfinger D, et al. The human U5 snRNP 52K protein. (CD2BP2) interacts with U5-102K. (hPrp6), a U4/U6.U5 tri-snRNP bridging protein, but dissociates upon tri-snRNP formation. RNA. (2005) 11:598–608. doi: 10.1261/rna.2300805
14. Liu T, Jin X, Zhang X, Yuan H, Cheng J, Lee J, et al. A novel missense SNRNP200 mutation associated with autosomal dominant retinitis pigmentosa in a Chinese family. PLoS ONE. (2012) 7:e45464. doi: 10.1371/journal.pone.0045464
15. Hahn D, Beggs JD. Brr2p RNA helicase with a split personality: insights into structure and function. Biochem Soc Trans. (2010) 38:1105–9. doi: 10.1042/BST0381105
16. Lauber J, Fabrizio P, Teigelkamp S, Lane WS, Hartmann E, Luhrmann R. The HeLa 200 kDa U5 snRNP-specific protein and its homologue in Saccharomyces cerevisiae are members of the DEXH-box protein family of putative RNA helicases. EMBO J. (1996) 15:4001–15. doi: 10.1002/j.1460-2075.1996.tb00774.x
17. Liu Y, Chen X, Qin B, Zhao K, Zhao Q, Staley JP, et al. Knocking down Snrnp200 initiates demorphogenesis of rod photoreceptors in zebrafish. J Ophthalmol. (2015) 2015:816329. doi: 10.1155/2015/816329
18. Zhang Q. Retinitis pigmentosa: progress and perspective. Asia Pac J Ophthalmol. (2016) 5:265–71. doi: 10.1097/APO.0000000000000227
19. Benaglio P, San Jose PF, Avila-Fernandez A, Ascari G, Harper S, Manes G, et al. Mutational screening of splicing factor genes in cases with autosomal dominant retinitis pigmentosa. Mol Vis. (2014) 20:843–51.
20. Petersen BS, Fredrich B, Hoeppner MP, Ellinghaus D, Franke A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. (2017) 18:14. doi: 10.1186/s12863-017-0479-5
21. Chen X, Liu Y, Sheng X, Tam PO, Zhao K, Chen X, et al. PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum Mol Genet. (2014) 23:2926–39. doi: 10.1093/hmg/ddu005
22. Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. (2008) 133:585–600. doi: 10.1016/j.cell.2008.03.031
23. Huranova M, Ivani I, Benda A, Poser I, Brody Y, Hof M, et al. The differential interaction of snRNPs with pre-mRNA reveals splicing kinetics in living cells. J Cell Biol. (2010) 191:75–86. doi: 10.1083/jcb.201004030
24. Van Nues RW, Beggs JD. Functional contacts with a range of splicing proteins suggest a central role for Brr2p in the dynamic control of the order of events in spliceosomes of Saccharomyces cerevisiae. Genetics. (2001) 157:1451–67.
25. Liu S, Rauhut R, Vornlocher HP, Luhrmann R. The network of protein-protein interactions within the human U4/U6.U5 tri-snRNP. RNA. (2006) 12:1418–30. doi: 10.1261/rna.55406
26. Nguyen TH, Li J, Galej WP, Oshikane H, Newman AJ, Nagai K. Structural basis of Brr2-Prp8 interactions and implications for U5 snRNP biogenesis and the spliceosome active site. Structure. (2013) 21:910–9. doi: 10.1016/j.str.2013.04.017
Nomenclature
RP, retinitis pigmentosa
pre-mRNA, pre-message RNA
SNRNP200, small nuclear ribonucleoprotein 200 kDa
WES, Whole exome sequencing
MO, Morpholino oligonucleotide
VFs, visual fields
RPE, retinal pigment epithelium
adRP, autosomal dominant RP
snRNPs, small nuclear ribonucleoprotein particles
OCT, optical coherence tomography
Q-PCR, Quantitative real-time PCR
OS/IS, outer/inner segments.
Keywords: retinitis pigmentosa, SNRNP200, whole exome sequencing, mutation, morpholino oligonucleotide
Citation: Zhang T, Bai J, Zhang X, Zheng X, Lu N, Liang Z, Lin L and Chen Y (2021) SNRNP200 Mutations Cause Autosomal Dominant Retinitis Pigmentosa. Front. Med. 7:588991. doi: 10.3389/fmed.2020.588991
Received: 30 July 2020; Accepted: 18 December 2020;
Published: 21 January 2021.
Edited by:
Gemmy Cheung, Singapore National Eye Center, SingaporeReviewed by:
Ravirajsinh Jadeja, Augusta University, United StatesSe Joon Woo, Seoul National University Bundang Hospital, South Korea
Copyright © 2021 Zhang, Bai, Zhang, Zheng, Lu, Liang, Lin and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Lin, bGxpbmNAMTYzLm5ldA==; Yongsong Chen, eW9uZ3NvbmdjaGVuQDEyNi5jb20=