
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 14 January 2021
Sec. Intensive Care Medicine and Anesthesiology
Volume 7 - 2020 | https://doi.org/10.3389/fmed.2020.586229
This article is part of the Research Topic Emerging Technology for Monitoring and Treatment in Critical Care View all 30 articles
 Kei Hayashida1,2*
Kei Hayashida1,2* Santiago J. Miyara1,2,3,4,5
Santiago J. Miyara1,2,3,4,5 Koichiro Shinozaki1,2
Koichiro Shinozaki1,2 Ryosuke Takegawa1,2Tai Yin1,2Daniel M. Rolston2,6,7Rishabh C. Choudhary1,2Sara Guevara6
Ryosuke Takegawa1,2Tai Yin1,2Daniel M. Rolston2,6,7Rishabh C. Choudhary1,2Sara Guevara6 Ernesto P. Molmenti4,5,7Lance B. Becker1,2,7
Ernesto P. Molmenti4,5,7Lance B. Becker1,2,7Despite recent advances in the management of post–cardiac arrest syndrome (PCAS), the survival rate, without neurologic sequelae after resuscitation, remains very low. Whole-body ischemia, followed by reperfusion after cardiac arrest (CA), contributes to PCAS, for which established pharmaceutical interventions are still lacking. It has been shown that a number of different processes can ultimately lead to neuronal injury and cell death in the pathology of PCAS, including vasoconstriction, protein modification, impaired mitochondrial respiration, cell death signaling, inflammation, and excessive oxidative stress. Recently, the pathophysiological effects of inhaled gases including nitric oxide (NO), molecular hydrogen (H2), and xenon (Xe) have attracted much attention. Herein, we summarize recent literature on the application of NO, H2, and Xe for treating PCAS. Recent basic and clinical research has shown that these gases have cytoprotective effects against PCAS. Nevertheless, there are likely differences in the mechanisms by which these gases modulate reperfusion injury after CA. Further preclinical and clinical studies examining the combinations of standard post-CA care and inhaled gas treatment to prevent ischemia–reperfusion injury are warranted to improve outcomes in patients who are being failed by our current therapies.
Cardiac arrest (CA) is a significant cause of death worldwide; ~356,000 cases occur out-of-hospital (OHCA) (1), and 200,000 cases occur in-hospital (IHCA) (2) per year in the United States. In recent years, rates of layperson-initiated cardiopulmonary resuscitation (CPR) and layperson use of automated external defibrillators have increased over time (1). Despite such recent advances in social awareness and management of CA, the survival rate without neurologic sequelae after resuscitation remains very low, representing a public health challenge (1–5). To date, no specific pharmaceutical drugs are effective against post-CA syndrome (PCAS) (3–5).
Over the past decades, resuscitation guidelines have emphasized the lifesaving value of high-quality CPR methods and rapid defibrillation for achieving the return of spontaneous circulation (ROSC), as well as treatment strategies such as hypothermia for post-CA brain injury. In 2002, two randomized controlled trials (RCTs) showed that therapeutic hypothermia (TH) significantly improved long-term outcomes in patients with PCAS who presented with ventricular fibrillation (VF) as an initial rhythm, drawing attention to the multidisciplinary treatment approach for those patients (3, 4, 6, 7). However, a large European RCT conducted by Nielsen et al. in 2013, including 939 comatose patients after ROSC, showed no significant benefit of improving the neurological outcomes when TH cooled down to 33°C compared to the management at a near-normal temperature of 36°C (8). This report questioned the effectiveness of TH for PCAS, which has been recommended in the international guidelines for a decade. In light of this controversy, a large RCT conducted by Bernard et al. in 2016, including 1,198 OHCA patients, demonstrated that the induction of mild TH, using a rapid large-volume intravenous cold saline infusion during CPR, indeed decreased the rate of ROSC in adult patients with an initial shockable rhythm and did not improve the survival rate (9). Recent RCT conducted by Lascarrou et al. in 2019 has evaluated targeted temperature management (TTM) for comatose patients who had been resuscitated from CA with non-shockable rhythm. They concluded that moderate TH at 33°C for 24 h led to a higher survival with a favorable neurologic outcome at 90 days compared to targeted normothermia (10). Also, the latest American Heart Association guidelines recommended TTM for comatose adults after ROSC from OHCA and IHCA with any initial rhythm (11). Despite these evidences, many uncertainties within the topic of TTM remain, and therefore, the development of alternative approaches with or without TTM is an unmet medical need in improving the prognosis of PCAS.
Prolonged ischemia during CA results in a variety of cellular insults. After achieving ROSC, ischemia–reperfusion injury (IRI) causes oxidative stress in the reperfused tissues, leading to exacerbation of the cellular injury (12). Recently, it has been shown in several scientific publications that nitric oxide (NO) (13), molecular hydrogen (H2) (14), xenon (Xe) (15), carbon monoxide (16), argon (17), and hydrogen sulfide (18) have protective effects against organ injuries related to IRI. In general, gases are small molecules; therefore, they have excellent diffusivity and easily permeate the cell membrane, targeting different organelles including the mitochondria and the nuclei. Especially, the cytoprotective effects of NO, H2, and Xe have attracted much attention in PCAS in not only animal models but also clinical settings. Therefore, the scope of this review is to describe those selected gases that have transitioned from bench to bedside and that have been already administered in patients. Herein, we briefly introduce the pathophysiology of PCAS and present a review of recent biomedical research developments on NO, H2, and Xe that have been proposed in recent literature.
PCAS is described as a unique and complex pathophysiological condition that involves (a) systemic IRI, (b) post-CA brain injury, and (c) post-CA myocardial dysfunction (3, 4). This condition is often complicated by a fourth component: the unsolved condition that caused the CA (3).
All clinical and biological manifestations associated with PCAS are putatively attributed to the IRI in vital organs including the brain and heart (3–5). The whole-body IRI with consequent oxygen debt causes a generalized activation of the cell-mediated immunologic response, vascular endothelial damage, hypercoagulability, and immunosuppression (3, 19–21). It has been observed that sharp increases in various cytokines occur in the bloodstream as early as 3 h after CA. Several cytokines have shown greater elevations in non-survivors than in survivors (20). Accordingly, it has been proposed that the pathophysiology of PCAS has several similar features as those of sepsis (19). The causes of post-CA organ damage may include increased activation of leukocytes, upregulated cytokines production, intracellular Ca2+ overload, mitochondrial dysfunction (22), and the generation of excessive reactive oxygen species (ROS) (23, 24). Excessive ROS production leads to DNA damage and lipid peroxidation, ultimately resulting in increased necrosis, apoptosis, and necroptosis (12, 25, 26). Compelling evidence has shown that mitochondria play a crucial role as effectors and targets of IRI (27–32). In fact, mitochondria are considered as one of the most susceptible subcellular targets of brain ischemia (33–35). A dysfunctional mitochondrial electron transport chain (METC) can result in an electron “leakage” phenomenon, reduced free oxygen, and the utilization of oxygen as an ubiquitous electron donor (substrate) to produce ROS (36). A body of evidence from preclinical studies has demonstrated that post-CA normoxic therapy improves neurological impairment, histological neuronal cell death, and cerebral metabolism (37–42).
Post-CA brain injury includes anoxic neuronal degeneration due to global ischemia during CA and/or shortly after ROSC, as well as delayed neurodegeneration, which can ensue within hours or several days after CA (43, 44). In a cohort study of 187 patients who underwent brain autopsy after CA, histopathologically determined severe hypoxic–ischemic encephalopathy was observed in patients with bilaterally absent cortical somatosensory-evoked potentials, gray–white matter ratio of brain computed tomographic imaging <1.10, highly malignant electroencephalographic patterns, and serum neuron-specific enolase concentration > 67 μg/L (45). In response to the stress due to global ischemia, several cytokine/chemokines, adhesion molecules, and ROS are released by different cells, including leukocytes, endothelial cells, and activated platelets (46). Aberrant ROS generation causes damage to fatty acids in the cell membrane, leading to increased membrane permeability and disruption of the blood–brain barrier (BBB). Cell membrane damage and BBB disruption result in cell swelling and cerebral edema, which, in turn, leads to further exacerbation of brain ischemia. Hypoperfusion during CPR and/or shortly after ROSC leads to a mismatch between oxygen demand and supply, resulting in secondary hypoxia (47). The delayed neurodegeneration after ROSC involves complex and multiple mechanisms including cytotoxic free radical production, neuronal excitability, activation of apoptotic signaling pathways, intracellular Ca2+ overflow, and mitochondrial dysfunction, among others (22, 28, 30, 48). Neuronal cell damage in the brain regions that are vulnerable to ischemia, such as the hippocampus and cerebral cortex, becomes irreversible within a few hours after the onset of ischemia, thus requiring early therapeutic interventions. Notably, some evidence suggests that the brain function after ROSC could be preserved indirectly, supporting the homeostasis of damaged organs other than the brain itself (49).
Most cases of PCAS exhibit a widespread left ventricular wall motion abnormality that is transient and reversible, in cases of normal or near-normal coronary flow or non-cardiomyopathy. This phenomenon is called post-CA myocardial stunning, which has been recently recognized as a leading cause of early death after a successful ROSC (3). In one study assessing the prevalence of coronary artery disease and acute coronary artery occlusion after resuscitation for OHCA presenting with VF as an initial rhythm, significant coronary artery lesions were found in 71% (50). Approximately 30% of patients had significant coronary artery lesions even in the absence of chest pain symptoms before CA and ST-segment elevation after ROSC (50). A meta-analysis focusing on studies for OHCA patients pointed out that acute coronary angiography should be strongly considered irrespective of electrocardiographic findings, due to the high prevalence of coronary artery disease in patients without an obvious non-cardiac etiology (51). Preexisting coronary artery disease exacerbates the myocardial damage associated with PCAS. The presence of myocardium stunning prolongs the recovery of wall motion through IRI (52), which includes excessive ROS production (53) and Ca2+ overload (54, 55), resulting in hemodynamics destabilization after ROSC. In addition, clinical studies have shown that right ventricular or biventricular dysfunction can contribute to poor outcomes after ROSC (56, 57). Therefore, hemodynamic stabilization is particularly important to maintain adequate cerebral blood flow and prevent late-onset neuronal damage.
In light of the limited clinical evidence supporting TH and other conventional approaches, recent preclinical studies have been focusing on alternative strategies that could increase neuroprotection immediately after ROSC. Significant attention has been paid to the possible use of inhaled gases such as NO, H2, and Xe, which have shown cytoprotective effects on organ injuries related to PCAS (13, 58–62). The main function of the lungs is to work as a gas exchanger, which allows oxygen to diffuse from the inhaled gas in the alveolus to the blood. The blood then carries and delivers oxygen to tissues to assist in the complex process of oxidative phosphorylation (63). Inhaled gas is a unique route of drug delivery, distinct from the intravenous or oral administration of medications, which allows for inhaled gaseous molecules to pass from the lung directly into the arterial circulatory system. Alternatively, it is conceivable that circulating cells are directly exposed to the gases as they pass through the pulmonary capillaries and may interact with or “pacified,” by a certain mechanism of each inhaled gas before the cells reach the reperfused peripheral tissues including the brain and heart.
The biological effects of NO are mediated through the activation of guanylyl cyclase (GC), followed by cyclic guanosine monophosphate (cGMP) production (GC pathway) (64). The biological effects of NO are also mediated through protein S-nitrosylation (SNO), which is the covalent attachment of NO to cysteine residues of target proteins (SNO pathway), by cGMP-independent mechanisms (65, 66). Both of these mechanisms have been implicated in the bioprotective effects of NO in IR disorders. Thus, several mechanisms that are responsible for the beneficial effects of NO on PCAS have been suggested (67). Potential mechanisms responsible for the beneficial effects of NO on the outcomes of PACS are shown in Figure 1. It has been reported that the administration of NO through inhalation (13, 58, 68, 69) or with an NO-donating compound (70) improves outcomes after CA in multiple species. Additionally, in mice lacking the NO synthase 3 gene, the protective effect of TH after CA/CPR is abolished (71), suggesting that NO may play an important role in TH. Furthermore, given the well-established pulmonary vasodilating effects of inhaled NO (72), it is conceivable that inhaled NO reduces the CA-induced pulmonary vascular resistance, thus enhancing the right-sided ventricular function and improving the outcomes of PCAS. Additionally, NO inhibits leukocyte adhesion (73) and migration (74), platelet activation (75), and acute inflammation (76). It has been reported that poor survival after CA/CPR is associated with leukocyte infiltration in the brain, heart, lung, liver, and kidney in mice (77, 78). It has also been demonstrated that NO is transported from the lung to the peripheral tissues through the hemoglobin, plasma protein SNOs, and nitrite ion generation and that NO in the periphery is released in the local ischemic region that exhibits acidosis where acid–base changes produce various physiological effects (79).
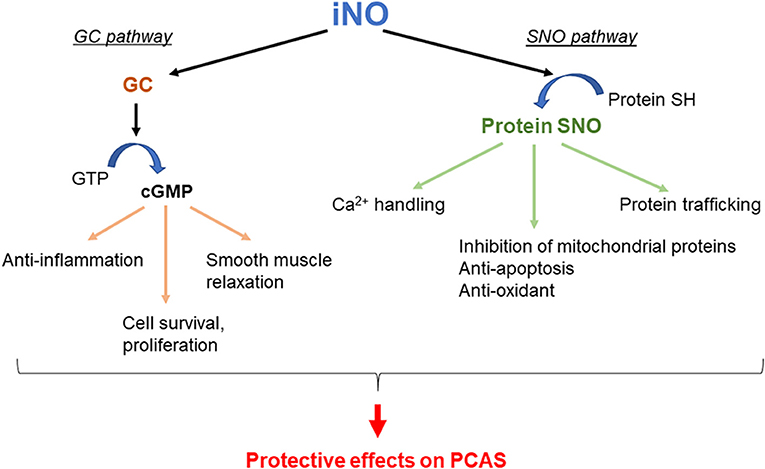
Figure 1. Potential mechanisms by which inhaled nitric oxide (iNO) improves outcomes in post–cardiac arrest syndrome (PCAS). GC, guanylyl cyclase; GTP, guanosine triphosphate; cGMP, cyclic guanosine monophosphate; SNO, S-nitrosylation.
Minamishima et al. reported that NO breathing improves the outcomes after ROSC in mice by GC-dependent mechanisms (13). Wild-type mice were subjected to 7.5 min of potassium chloride–induced CA and subsequently resuscitated. One hour after CPR, mice were extubated and breathed air alone or air supplemented with 40 ppm NO for 23 h. The post-CA mice breathing air alone (air group) exhibited a poor 10-day survival rate (4 of 13 [30.7%]), depressed neurological and left ventricular function, increased caspase-3 activation, and cytokine driven inflammation in the brain. NO breathing attenuated the neurological and cardiac dysfunction 4 days after CA/CPR and markedly improved the 10-day survival rate (11 of 13 [84.6%]; P = 0.003 vs. air group) (13). They also found that GC-1α deletion abolished the ability of inhaled NO to inhibit the production of inflammatory cytokines in the brain and to improve the neurological function and survival rate after CA (13). These observations suggest that the protective effects of inhaled NO on outcomes after ROSC are largely mediated by GC-1α-dependent mechanisms. Another research group showed that NO inhalation starting at initiation of CPR until 30 min after ROSC prevented myocardial injury and improved neurologic function and survival in rats (68). It was also shown that NO breathing, starting with the left ventricular assist device–supported CPR for 5 h, increased the transpulmonary blood flow by reducing the pulmonary artery pressure and improving neurological outcomes in pigs (69). Moreover, inhaled NO improved pulmonary artery relaxation pressure during CPR, coronary perfusion pressure during the postresuscitation phase, and short-term survival in a porcine model of CA. Interestingly, these benefits occurred despite fewer vasopressor doses and shallower chest compressions (80).
On the other hand, the protein SNO pathway has recently attracted considerable attention (65, 66, 81). Protein SNOs have demonstrated the capacity to inhibit mitochondrial proteins such as complex I in the electron transport chain, cytochrome c oxidase, and F1F0ATPase (complex V), as well as to modulate mitochondrial ROS production, influence calcium-dependent opening of the mitochondrial permeability transition pore, promote selective importation of mitochondrial proteins, and stimulate mitochondrial fission (65, 81). Furthermore, SNO proteins play a crucial role in intracellular Ca2+ handling, protein trafficking, and regulation of cellular defense against apoptosis and oxidative stress (65).
S-nitrosoglutathione (GSNO), which is the most abundant intracellular S-nitrosothiol in human tissue, plays an important role as a reservoir of NO bioactivity (82). GSNO has potent antioxidant and anti-inflammatory effects in animal models of IR (83, 84). In physiological conditions, GSNO and protein SNOs remain at equilibrium, whereas GSNO reductase (GSNOR) centrally regulates the reduction of GSNO (Figure 2) (85). GSNOR is normally expressed in all tissues including the brain, liver, vascular endothelium, and smooth muscle cells (86). As GSNOR reduces the intracellular level of protein SNO and NO bioavailability, the genetic deletion or pharmacological inhibition of GSNOR has been reported to increase the tissue levels of the protein SNO, as well as to induce vasodilation and reduce inflammation. Previous animal studies suggest that GSNOR inhibition may be beneficial for systemic and brain inflammation as well as for ischemic cardiomyopathy (87–89).
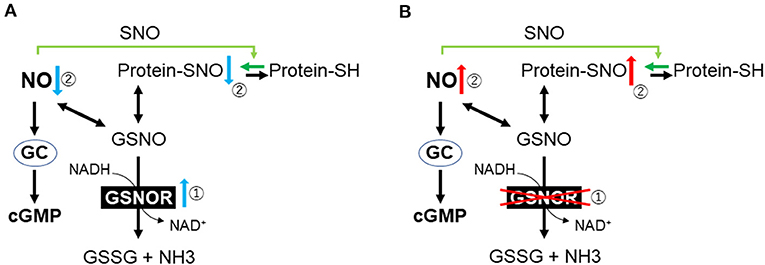
Figure 2. Outline of nitric oxide metabolism. (A) Cardiac arrest and resuscitation increase the activity of GSNOR. (B) Genetic or pharmacological inhibition of GSNOR increases the tissue levels of protein SNO and NO bioavailability. GC, guanylyl cyclase; cGMP, cyclic guanosine monophosphate; SH, cysteine thiols; GSNO, S-nitrosoglutathione; GSNOR, GSNO reductase; GSSG, glutathione disulfide; NH3, ammonia; NO, nitric oxide; SNO, S-nitrosylation.
To determine the role of GSNOR in the outcomes after CA/CPR, Hayashida et al. evaluated the effects of both GSNOR inhibitors and GSNOR gene deletion on the survival and neurological outcomes after CA in mice (90). They found that GSNOR activity increased in the plasma and brain after CA/CPR and that protein SNO levels in the brain decreased after 6 h in the placebo group, whereas GSNOR inhibitors, administered 15 min after ROSC, attenuated the upregulated GSNOR activity and restored protein SNO levels in the brain (90). Additionally, in wild-type mice after CA/CPR, GSNOR inhibitors improved the neurological deficit score and survival rate (81.8 vs. 36.4%, P = 0.031). Similarly, GSNOR-deleted mice prevented the reduction of the brain protein SNOs, suppressed neuronal damage, and improved survival. Both GSNOR inhibitor and GSNOR deletion attenuated the disruption of the BBB after CA/CPR. In PCAS patients, it was found that plasma GSNOR activity was higher than that in preoperative cardiac surgery patients or healthy volunteers (P < 0.0001) (90). In another publication, they demonstrated that plasma NO consumption in post-CA patients was 3-fold greater than in healthy volunteers (91). Overall, these observations suggest that increased GSNOR activity and the subsequent NO consumption may play an important pathogenetic role after ROSC and that the inhibition of GSNOR is a novel molecular target to improve neurological outcomes after CA/CPR (Figure 2).
Dezfulian et al. conducted a single-center, randomized, double-blind pilot clinical study to determine the effect of low-dose (~9.6 mg) intravenous sodium nitrate, a donor of NO, on OHCA patients (92). The patients were eligible to be enrolled in this study if the patient was successfully resuscitated from non-traumatic CA and survived to the intensive care unit (ICU) admission. Patients who had hypoxemia, hypotension, or inability to receive intravenous sodium nitrate within 12 h of onset were excluded. The results showed that there was no adverse effect on heart rate, systolic blood pressure, or blood methemoglobin level within 30 min of administration in the sodium nitrate group (n = 7) compared to the control group (n = 4). Plasma protein SNO and cGMP levels, which have protective effects on IRI (93), were elevated in the sodium nitrate group. The authors concluded that NO drug can be feasible for patients with PCAS and that further investigation is warranted (92). The same investigators are currently conducting a clinical trial to examine the effects of inhaled NO therapy on PCAS (ClinicalTrials.gov identifier: NCT04134078)1. Taken together, NO gas inhalation and NO-related drugs are currently one of the most promising pharmaceutical treatments for PCAS.
H2 is a colorless, odorless, and non-toxic gas at room temperature. H2 gas is explosive in air at a wide concentration range of 4.0–75.0% by volume, whereas in oxygen, the explosive limit is from 4.0 to 94.0% (94). The ignition point of H2 (527°C) is higher than that of gasoline (500°C), and it is difficult to ignite it spontaneously at standard conditions of pressure. These lines of evidence suggest that H2 is relatively safe in daily life when its concentration is <4% (94, 95). H2 is enzymatically metabolized as an energy source by providing electrons to METC. These enzymes catalyze the reversible redox reaction between H2 and its constituent two protons and two electrons (96). The use of inhaled H2 to diminish ischemic injury has been applied successfully in several rodent models, such as stroke (14, 97), acute myocardial infarction (MI) (98), and CA (60, 61). Consequently, clinical pilot studies have shown the beneficial effects of H2 in patients with acute MI (99) and OHCA (100).
While the mechanism of H2 protection has not been fully determined, many experts believe that its protective action is based on antioxidant properties with direct effects on ROS (101–104). Mitochondrial respiration chain, xanthine oxidase, uncoupling of NOS, and the family of nicotinamide adenine dinucleotide phosphate oxidases are significant sources of ROS (105). ROS includes superoxide anion radicals (•), hydrogen peroxide (H2O2), hydroxyl radical (•OH), peroxynitrite (ONOO−), and nitric oxide (NO•). • is putatively the primary ROS mostly generated by electron leakage from the METC (106–109). H2O2 is enzymatically converted from • by superoxide dismutase. •OH is a highly reactive, toxic ROS, and the major cause of oxidative stress (110); there is no detoxifying system for •OH in vivo. •OH is generated from H2O2 or • through the Fenton or Weiss reaction in the presence of catalytically active metals such as Fe2+ and Cu+ (111). • reacts with •NO to generate ONOO−, which is a highly active nitrogen species (112). Oxidative stress caused by H2O2 and •NO induces the production of enzymes involved in antioxidation and tolerance to protect the cells against oxidative stress, such as NF-E2–related factor 2 (113). Noteworthy, research has shown that many antioxidant supplements could not prevent cancer, MI, and atherosclerosis but rather, conversely, cause increased mortality (114–116); therefore, awareness of side effects is very important for developing an effective and safe antioxidant for ROS-related diseases. An ideal antioxidant should mitigate excessive oxidative stress without disturbing the redox homeostasis. In other words, an ideal molecule would simultaneously reduce strong oxidants such as •OH, while maintaining signaling molecules such as H2O2 (95). Preclinical studies have shown that H2 specifically quenches detrimental ROS such as •OH and ONOO−, while maintaining other less potent ROS (14). Potential mechanisms responsible for the beneficial effects of H2 on PACS are shown in Figure 3. However, more precise mechanisms of the beneficial effects of H2 remain elusive.
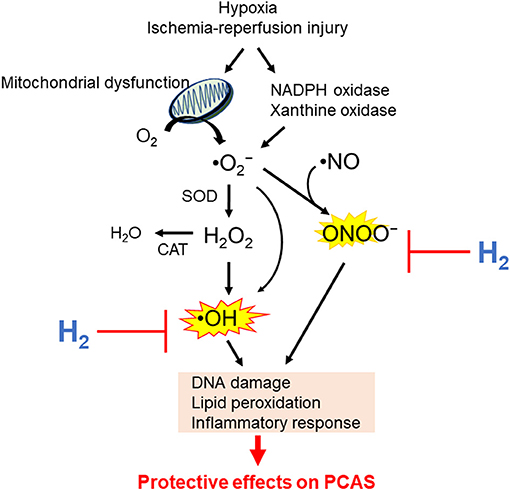
Figure 3. Potential mechanisms by which hydrogen (H2) inhalation improves outcomes in post–cardiac arrest syndrome (PCAS). NADPH, nicotinamide adenine dinucleotide phosphate; •, superoxide anion radicals; H2O2, hydrogen peroxide; •OH, hydroxyl radical; ONOO−, peroxynitrite; •NO, nitric oxide; SOD, superoxide dismutase; CAT, catalase.
In 2007, Ohsawa et al. found that H2 acts as an antioxidant by selectively reducing highly cytotoxic ROS, such as •OH and ONOO− in cultured cells, and that 2–4% of H2 has cytoprotective effects against IRI in vivo (14). Furthermore, it was demonstrated that H2 did not react with stable, lowly active ROS, such as H2O2, •, and •NO in vitro (14). In a rat model of MI, 2% H2 inhalation starting 5 min after the ligation of a coronary artery and continued for 60 min after reperfusion reduced the infarct size and inhibited the left ventricular remodeling (98). The authors confirmed that H2 diffuses into the myocardial ischemic tissues in a blood flow–independent manner, suggesting that H2 rapidly dissolved into the blood immediately after the start of inhalation and has the potential advantage of excellent diffusion even into ischemic regions (98). Another research group reported the inhibitory effect of H2 on myocardial IR damage in a dog model of acute MI (117). Moreover, the safety and efficacy of inhaled H2 for the prevention of reperfusion injury in patients with acute MI undergoing percutaneous coronary intervention have been assessed (99). In a single-center, open-label, pilot study, inhalation of 1.3% H2 did not reduce the infarct size during the acute phase after acute MI. However, the left ventricular stroke volumes assessed by magnetic resonance imaging (MRI) were improved at 6 months in comparison with 1 week after MI only in the H2 inhalation group (99). This suggests that H2 inhalation can be safely administered to patients with acute MI and can suppress adverse left ventricular remodeling at 6 months after infarction.
Hayashida et al. demonstrated that inhalation of 2% H2 starting at the beginning of CPR and administered for 2 h after ROSC significantly improves the outcomes in a rat model of CA with VF (60). H2 inhalation, but not TH, prevented an increase in the left ventricular end-diastolic pressure and myocardial injury and suppressed systemic inflammation after ROSC. The survival rate at 72 h after ROSC was 31% in the control group and 69% in both the TH and H2 groups and was even higher at 77% in the combined therapy (inhaled 2% H2 plus TH) group. Further, the same study group tested the benefit of H2 administered after ROSC under a normoxic condition, which was considered essential for clinical application (61). In this study, inhaled 1.3% H2 with 26% O2 was started 5 min after ROSC and continued for 2 h. The survival rates at 7 days were 38% in the control group, 71% in either the H2- or the TH-alone groups, and 86% in the combined therapy of H2 plus TH group. At 7 days after CA/CPR, H2 improved the motor activity and special memory assessed by the Y-maze test. Immunohistochemistry studies showed that H2 inhalation alone or in combination with TH inhibited neuronal injury in the hippocampus 7 days after ROSC. These results indicate that H2 inhalation after ROSC is as effective as TH for improving the neurological prognosis in rats with PCAS, whereas combined therapy had an additive effect (61). Further, Nemeth et al. showed that, in a hypoxic–ischemic encephalopathy piglet model, treatment with 2.1% H2 for 4 h reduced oxidative stress and improved neural recovery (118). Moreover, Cole et al. demonstrated the protective effects of inhaled H2 on neurologic injury after cardiopulmonary bypass in a porcine model of neonatal circulatory arrest (119).
In a single-center, prospective, open-label, single-arm study, Tamura et al. demonstrated the safety and feasibility of H2 inhalation after ROSC in comatose patients with a consciousness level ≤8 points on the Glasgow Coma Scale and a systolic blood pressure ≥90 mmHg (irrespective of vasopressor use) (100). In this study, the patients received 2% H2 for 18 h using a ventilator in combination with TTM of 33–36°C. The rates of survival with Cerebral Performance Category (CPC) 1–2 were assessed at 90 days after CA. The rates of survival with CPC 1–2 were assessed at 90 days after CA. One CA patient with severe pneumonia and septic shock died of respiratory deterioration 22 h after the discontinuation of H2 inhalation. An outcome of CPC 1 was achieved in 4 of all 5 eligible patients. The independent data monitoring committee concluded that no adverse event was attributable to inhaling hydrogen gas in this study. This study concluded that inhaled H2 could be feasible and performed safely in patients with PCAS. Currently, a phase II, multicenter, prospective, randomized, double-blind, placebo-controlled trial to verify the efficacy of H2 inhalation in patients with PCAS is underway (identifier: UMIN000019820) (120).
Xe is one of the noble gases, which are the elements of group 18 on the periodic table. It has anesthetic properties, which were recognized ~50 years ago (121). It has the lowest blood–gas partition coefficient among anesthetic gases (122). It has the advantage of being non-flammable and non-teratogenic, and it has less cardiovascular effects and no adverse effects on cognitive function in animal models (123–126). In recent years, there has been increased interest in noble gases as novel treatments for ischemic and traumatic brain injury (127–129). Excessive activation of N-methyl-D-aspartate (NMDA)–type glutamate receptors is, in general, a key mechanism of excitotoxicity after brain injury (130, 131). During excitotoxicity, excessive glutamate release results in the activation of NMDA receptors, leading to calcium overload inside the neurons and the different types of neuroglia. This calcium overload triggers prodeath signaling pathways, ROS production, and mitochondrial damage (132–137), resulting in cell necrosis, apoptosis, and necroptosis (138). Additionally, the linkage of NMDA receptor and activation of microglia has been suggested (139, 140). Interestingly, studies have shown that NMDA-mediated excitotoxicity occurs unequally in different brain cells because neuroglia such as astrocytes do not express NMDA receptors in the same way as neurons do, making astrocytes relatively resistant to NMDA toxic effects (141, 142). Xe is an antagonist of NMDA-type glutamate receptors (143), and subsequent animal studies have reported that Xe has neuroprotective properties in animal models of stroke (144) and CA (59, 62, 145, 146). Additionally, Xe exhibits neuroprotection by inhibiting the activation of microglia and attenuating neural damage in the hippocampus after experimental subarachnoid hemorrhage (147). In a porcine model of CA, Fries et al. demonstrated that a single inhalation of Xe started 1 h after ROSC and continued for 1 h significantly improved functional recovery and reduced neuronal damage in a porcine model of CA (146). Furthermore, they showed that administration of Xe as early as 10 min after ROSC (59) and extending up to 5 h (146) did not result in additional neuroprotection. Subsequently, they demonstrated that only the combination of Xe and mild TH provided significant and persistent improvements in functional recovery in a clinically relevant, porcine model of CA/CPR. In contrast to mild TH alone, this approach also preserved cardiac output in the early postresuscitation period (62). Potential mechanisms responsible for the beneficial effects of Xe on the outcomes of PACS are shown in Figure 4.
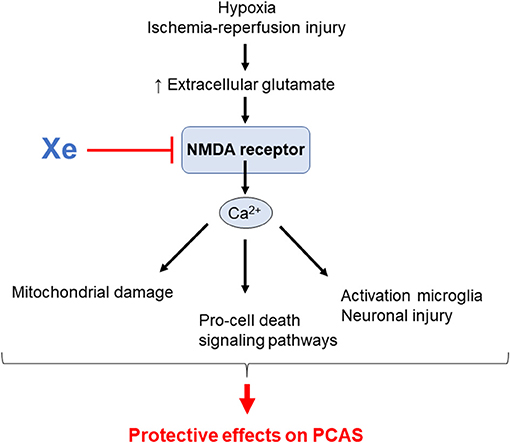
Figure 4. Potential mechanisms by which xenon (Xe) inhalation improves outcomes in post–cardiac arrest syndrome (PCAS). NMDA; N-methyl-D-aspartate.
In 2013, Arola et al. reported that Xe inhalation in combination with TH can be safely applied to patients with PCAS (148). Subsequently, Laitio et al. demonstrated that Xe had a neuroprotective effect on PCAS in a randomized, single-blind phase 2 clinical trial (149). In this study, 110 patients with PCAS admitted to the ICUs were randomly assigned to receive either TH alone (control group) or inhaled Xe in combination with TH (33°C) for 24 h (Xe group). The main inclusion criteria were the presence of a witness, initial electrocardiogram waveform VF or non-perfused ventricular tachycardia, and ROSC ≤45 min after resuscitation. The primary endpoint was the severity of ischemic white matter brain injury as evaluated by fractional anisotropy from diffusion tensor MRI, which was scheduled at 36–52 h after ROSC. Xe inhalation was started within 4 h after ROSC, and the mean end-tidal Xe concentration was 48.2%. The fractional anisotropy was significantly lower in 41.7% of the voxels in the control group than in the Xe group (i.e., 58.3% of the voxels did not significantly differ between the groups), indicating that cerebral white matter and myelin damage were suppressed in the Xe group. Specifically, the mean global fractional anisotropy value adjusted for age, sex, and site factors was 3.8% higher in the Xe group than in the control group (P = 0.006). The adjusted radial diffusivity value was 3.9% lower in the Xe group than in the control group (P = 0.03). There were no significant differences in the secondary endpoints of 6-month survival and brain function outcomes between the two groups (27.8% in the Xe group vs. 34.5% in the control group; adjusted hazard ratio, 0.49, P = 0.053) (149). Given that myelin is required for the normal functioning of the central nervous system and its damage is related to neurocognitive dysfunction (150), this study suggested that Xe may protect the cerebral white matter by preventing brain myelin injury after ROSC (149). Although there was no significant difference in survival in this study, Xe can be potentially a novel treatment for PCAS. Subsequently, Arola et al. demonstrated that among comatose survivors of OHCA, in comparison with TH alone, inhaled Xe combined with TH resulted in significantly reduced release of troponin-T, which suggests that Xe results in less severe myocardial injury, supporting its cardioprotective effects (151). These two recent clinical trials suggest the translational potential of Xe inhalation for the management of PCAS (149, 151). These studies have demonstrated that Xe inhalation in combination with TH is safe and feasible. Currently, phase III, multicenter, prospective, randomized, single-blind, placebo-controlled trial to evaluate the efficacy of Xe inhalation on neurofunctional outcomes after OHCA is underway (identifier: NCT03176186) 2.
Xe has many properties as an ideal general anesthetic, and because the noble gases emit light when an electric field is applied, they are often used as gas lasers in medical applications such as surgery (152). However, Xe has not been widely used in clinical practice as it is rare and relatively expensive (152). Therefore, as a relatively large amount of gas is expected to be used for inhalation therapy for PCAS, the feasibility in terms of the cost has been regarded as a potential problem. Hence, further investigations for the clinical application of Xe will be required.
NO is a toxic molecule (153) synthesized by NO synthases, which include three isoforms: neuronal NOS (NOS1), inducible NOS (NOS2), and endothelial NOS (NOS3) (154). In contrast to NO, mammalian cells do not have to produce intracellular Xe and H2. Although Xe is non-toxic, many of its compounds are toxic because of their strong oxidative properties. Xe readily penetrates the BBB, offering rapid onset of action, and titration of dose and response are rapid because of a low blood–gas partition coefficient (122). H2 has no known cytotoxicity even at high concentrations (155, 156).
As the primary target of NO, heme-based proteins play a central role. Integrated approaches revealed the physiological significance of NO on mitochondrial cytochrome c oxidase, a central mediator of mitochondrial respiration (157). Xe exerts neuroprotective effects by acting as an antagonist of the excitotoxic NMDA receptors (143). Excessive inflow of calcium mediated by NMDA receptors triggers complex biochemical cascades that ultimately lead to neuronal cell death (134). Although the molecular mechanisms of H2 have not yet been clarified, it has been shown that H2 does not reduce the oxidized heme of cytochrome c (14). In addition, a combined inhalation of NO plus H2 during IRI reduced the infarct size, maintained cardiac function, and reduced the generation of myocardial nitrotyrosine, which is associated with NO inhalation (158). Therefore, the primary targets of these gases seem to differ from different standpoints. Moreover, the optimal timing, concentration, and therapeutic window may differ among these gases. The exact underpinning mechanisms of these therapies remain to be unveiled in future studies. Elucidation of the mechanism of action will accelerate the translation into clinical. Summary of the past and current clinical trials investigating the effects of gases on PCAS are shown in Table 1. Because these gases are colorless, odorless, and difficult to check visually, they require a pressure regulator and flowmeter and must be handled under the local high-pressure gas safety control act, in clinical translation. It is important to clarify and establish the safety, toxicity, flammability, operability, and cost, individually, for the clinical application. However, we would like to emphasize that gas inhalation therapy may not require extensive equipment or advanced medical technology and is relatively easy to be introduced in a large number of facilities. In addition, dedicated gas cylinders can be installed in public areas or on ambulances to provide earlier therapeutic interventions during CPR or immediately after ROSC.
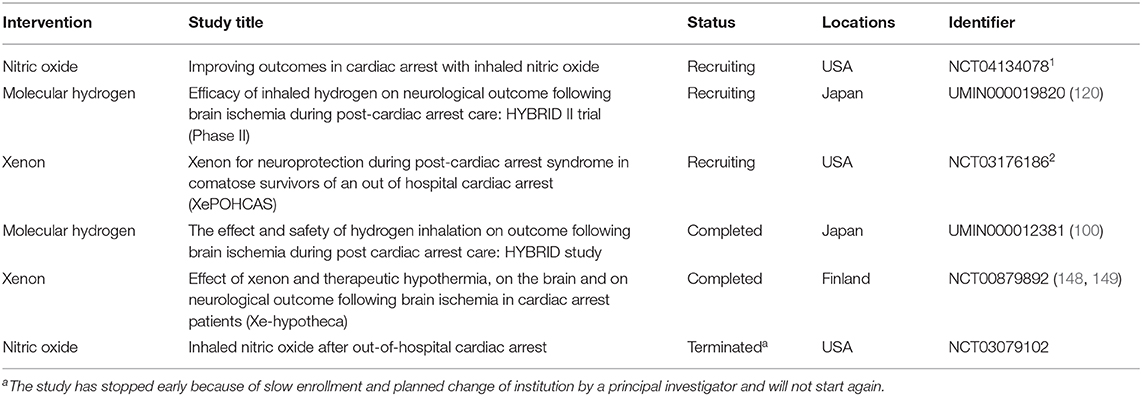
Table 1. Summary table of the past and current clinical trials on inhaled gases as therapies for PCAS (as of 1st, Dec, 2020).
We reviewed the developments in research on basic and clinical applications of NO, H2, and Xe for PCAS. The discussed studies provide insights on new frontiers regarding the fact that gas therapy may bring promising improvements in the prognosis of patients after ROSC. Nevertheless, there are substantial differences in the mechanisms by which these gases modulate IRI after ROSC. Further preclinical and clinical studies examining the combinations of standard post-CA care plus inhaled gas treatment to prevent IRI are warranted to improve outcomes in patients who are being failed by our current therapies.
KH: concept, design, and drafting manuscript. SM, KS, RT, TY, DR, RC, SG, EM, and LB: critical revision of the manuscript for important intellectual content. All authors have read and approved the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Koichi Saga, PhD (Sumitomo Seika Chemicals Co., Ltd., Japan) for his contributions to the literature search.
1. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: a report from the American heart association. Circulation. (2019) 139:e56–28. doi: 10.1161/CIR.0000000000000659
2. Merchant RM, Yang L, Becker LB, Berg RA, Nadkarni V, Nichol G, et al. Incidence of treated cardiac arrest in hospitalized patients in the United States. Critic Care Med. (2011) 39:2401–6. doi: 10.1097/CCM.0b013e3182257459
3. Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Bottiger BW, et al. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, Inter-American Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation. (2008) 118:2452–83. doi: 10.1161/CIRCULATIONAHA.108.190652
4. Callaway CW, Donnino MW, Fink EL, Geocadin RG, Golan E, Kern KB, et al. Part 8: post-cardiac arrest care: 2015 American Heart Association guidelines update for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. (2015) 132(18 Suppl. 2):S465–82. doi: 10.1161/CIR.0000000000000262
5. Patil KD, Halperin HR, Becker LB. Cardiac arrest: resuscitation and reperfusion. Circ Res. (2015) 116:2041–9. doi: 10.1161/CIRCRESAHA.116.304495
6. Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. New Engl J Med. (2002) 346:557–63. doi: 10.1056/NEJMoa003289
7. Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. New Engl J Med. (2002) 346:549–56. doi: 10.1056/NEJMoa012689
8. Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, et al. Targeted temperature management at 33 degrees C versus 36 degrees C after cardiac arrest. New Engl J Med. (2013) 369:2197–206. doi: 10.1056/NEJMoa1310519
9. Bernard SA, Smith K, Finn J, Hein C, Grantham H, Bray JE, et al. Induction of therapeutic hypothermia during out-of-hospital cardiac arrest using a rapid infusion of cold saline: the RINSE trial (rapid infusion of cold normal saline). Circulation. (2016) 134:797–805. doi: 10.1161/CIRCULATIONAHA.116.021989
10. Lascarrou JB, Merdji H, Le Gouge A, Colin G, Grillet G, Girardie P, et al. Targeted temperature management for cardiac arrest with non-shockable rhythm. New Engl J Med. (2019) 381:2327–37. doi: 10.1056/NEJMoa1906661
11. Berg KM, Cheng A, Panchal AR, Topjian AA, Aziz K, Bhanji F, et al. Part 7: systems of care: 2020 American Heart association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. (2020) 142:S580–604. doi: 10.1161/CIR.0000000000000918
12. Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. (2015) 6:524–51. doi: 10.1016/j.redox.2015.08.020
13. Minamishima S, Kida K, Tokuda K, Wang H, Sips PY, Kosugi S, et al. Inhaled nitric oxide improves outcomes after successful cardiopulmonary resuscitation in mice. Circulation. (2011) 124:1645–53. doi: 10.1161/CIRCULATIONAHA.111.025395
14. Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. (2007) 13:688–94. doi: 10.1038/nm1577
15. Preckel B, Mullenheim J, Moloschavij A, Thamer V, Schlack W. Xenon administration during early reperfusion reduces infarct size after regional ischemia in the rabbit heart in vivo. Anesth Analg. (2000) 91:1327–32. doi: 10.1097/00000539-200012000-00003
16. Wu J, Li Y, Yang P, Huang Y, Lu S, Xu F. Novel role of carbon monoxide in improving neurological outcome after cardiac arrest in aged rats: involvement of inducing mitochondrial autophagy. J Am Heart Assoc. (2019) 8:e011851. doi: 10.1161/JAHA.118.011851
17. Brucken A, Cizen A, Fera C, Meinhardt A, Weis J, Nolte K, et al. Argon reduces neurohistopathological damage and preserves functional recovery after cardiac arrest in rats. Br J Anaesth. (2013) 110(Suppl. 1):i106–12. doi: 10.1093/bja/aes509
18. Wei X, Duan L, Bai L, Tian M, Li W, Zhang B. Effects of exogenous hydrogen sulfide on brain metabolism and early neurological function in rabbits after cardiac arrest. Intensive Care Med. (2012) 38:1877–85. doi: 10.1007/s00134-012-2714-x
19. Adrie C, Adib-Conquy M, Laurent I, Monchi M, Vinsonneau C, Fitting C, et al. Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis-like” syndrome. Circulation. (2002) 106:562–8. doi: 10.1161/01.CIR.0000023891.80661.AD
20. Adrie C, Laurent I, Monchi M, Cariou A, Dhainaou JF, Spaulding C. Postresuscitation disease after cardiac arrest: a sepsis-like syndrome? Curr Opin Critic Care. (2004) 10:208–12. doi: 10.1097/01.ccx.0000126090.06275.fe
21. Beurskens CJ, Horn J, de Boer AM, Schultz MJ, van Leeuwen EM, Vroom MB, et al. Cardiac arrest patients have an impaired immune response, which is not influenced by induced hypothermia. Critical Care. (2014) 18:R162. doi: 10.1186/cc14002
22. Wiberg S, Stride N, Bro-Jeppesen J, Holmberg MJ, Kjaergaard J, Larsen S, et al. Mitochondrial dysfunction in adults after out-of-hospital cardiac arrest. Eur Heart J Acute Cardiovasc Care. 2019:2048872618814700. doi: 10.1177/2048872618814700
23. Ferrari R, Ceconi C, Curello S, Cargnoni A, Pasini E, Visioli O. The occurrence of oxidative stress during reperfusion in experimental animals and men. Cardiovasc Drugs Ther. (1991) 5(Suppl. 2):277–87. doi: 10.1007/BF00054749
24. Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. (2004) 61:461–70. doi: 10.1016/j.cardiores.2003.10.025
25. Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem. (1992) 267:5317–23.
26. Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. (2016) 1863:2977–92. doi: 10.1016/j.bbamcr.2016.09.012
27. Kilbaugh TJ, Sutton RM, Karlsson M, Hansson MJ, Naim MY, Morgan RW, et al. Persistently altered brain mitochondrial bioenergetics after apparently successful resuscitation from cardiac arrest. J Am Heart Assoc. (2015) 4:e002232. doi: 10.1161/JAHA.115.002232
28. Ayoub IM, Radhakrishnan J, Gazmuri RJ. Targeting mitochondria for resuscitation from cardiac arrest. Critical Care Med. (2008) 36(Suppl. 11):S440–6. doi: 10.1097/CCM.0b013e31818a89f4
29. Kim J, Villarroel JP, Zhang W, Yin T, Shinozaki K, Hong A, et al. The responses of tissues from the brain, heart, kidney, and liver to resuscitation following prolonged cardiac arrest by examining mitochondrial respiration in rats. Oxid Med Cell Longev. (2016) 2016:7463407. doi: 10.1155/2016/7463407
30. Han F, Da T, Riobo NA, Becker LB. Early mitochondrial dysfunction in electron transfer activity and reactive oxygen species generation after cardiac arrest. Critic Care Med. (2008) 36(Suppl. 11):S447–53. doi: 10.1097/CCM.0b013e31818a8a51
31. Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, Becker LB. Reperfusion injury on cardiac myocytes after simulated ischemia. Am J Physiol. (1996) 270:H1334–41. doi: 10.1152/ajpheart.1996.270.4.H1334
32. Anderson TC, Li CQ, Shao ZH, Hoang T, Chan KC, Hamann KJ, et al. Transient and partial mitochondrial inhibition for the treatment of postresuscitation injury: getting it just right. Critic Care Med. (2006) 34(Suppl. 12):S474–82. doi: 10.1097/01.CCM.0000246014.19486.A1
33. Almeida A, Delgado-Esteban M, Bolanos JP, Medina JM. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem. (2002) 81:207–17. doi: 10.1046/j.1471-4159.2002.00827.x
34. Anderson MF, Sims NR. Mitochondrial respiratory function and cell death in focal cerebral ischemia. J Neurochem. (1999) 73:1189–99. doi: 10.1046/j.1471-4159.1999.0731189.x
35. Sims NR, Zaidan E. Biochemical changes associated with selective neuronal death following short-term cerebral ischaemia. Int J Biochem Cell Biol. (1995) 27:531–50. doi: 10.1016/1357-2725(95)00026-L
36. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
37. Vereczki V, Martin E, Rosenthal RE, Hof PR, Hoffman GE, Fiskum G. Normoxic resuscitation after cardiac arrest protects against hippocampal oxidative stress, metabolic dysfunction, and neuronal death. J Cereb Blood Flow Metab. (2006) 26:821–35. doi: 10.1038/sj.jcbfm.9600234
38. Liu Y, Rosenthal RE, Haywood Y, Miljkovic-Lolic M, Vanderhoek JY, Fiskum G. Normoxic ventilation after cardiac arrest reduces oxidation of brain lipids and improves neurological outcome. Stroke. (1998) 29:1679–86. doi: 10.1161/01.STR.29.8.1679
39. Richards EM, Fiskum G, Rosenthal RE, Hopkins I, McKenna MC. Hyperoxic reperfusion after global ischemia decreases hippocampal energy metabolism. Stroke. (2007) 38:1578–84. doi: 10.1161/STROKEAHA.106.473967
40. Brucken A, Kaab AB, Kottmann K, Rossaint R, Nolte KW, Weis J, et al. Reducing the duration of 100% oxygen ventilation in the early reperfusion period after cardiopulmonary resuscitation decreases striatal brain damage. Resuscitation. (2010) 81:1698–703. doi: 10.1016/j.resuscitation.2010.06.027
41. Hazelton JL, Balan I, Elmer GI, Kristian T, Rosenthal RE, Krause G, et al. Hyperoxic reperfusion after global cerebral ischemia promotes inflammation and long-term hippocampal neuronal death. J Neurotrauma. (2010) 27:753–62. doi: 10.1089/neu.2009.1186
42. Danilov CA, Fiskum G. Hyperoxia promotes astrocyte cell death after oxygen and glucose deprivation. Glia. (2008) 56:801–8. doi: 10.1002/glia.20655
43. Colbourne F, Corbett D. Delayed and prolonged post-ischemic hypothermia is neuroprotective in the gerbil. Brain Res. (1994) 654:265–72. doi: 10.1016/0006-8993(94)90488-X
44. Che D, Li L, Kopil CM, Liu Z, Guo W, Neumar RW. Impact of therapeutic hypothermia onset and duration on survival, neurologic function, and neurodegeneration after cardiac arrest. Critic Care Med. (2011) 39:1423–30. doi: 10.1097/CCM.0b013e318212020a
45. Endisch C, Westhall E, Kenda M, Streitberger KJ, Kirkegaard H, Stenzel W, et al. Hypoxic-ischemic encephalopathy evaluated by brain autopsy and neuroprognostication after cardiac arrest. JAMA Neurol. (2020) 77:1–10. doi: 10.1001/jamaneurol.2020.2340
46. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
47. Hossmann KA. Reperfusion of the brain after global ischemia: hemodynamic disturbances. Shock. (1997) 8:95–101. doi: 10.1097/00024382-199708000-00004
48. Neumar RW. Molecular mechanisms of ischemic neuronal injury. Ann Emerg Med. (2000) 36:483–506. doi: 10.1067/mem.2000.110995
49. Safar P. Resuscitation of the ischemic brain. In: Albin MS, editor. Textbook of Neuroanesthesia with Neurosurgical and Neuroscience Perspectives. New York, NY: McGraw-Hill (1977). p. 557–93.
50. Spaulding CM, Joly LM, Rosenberg A, Monchi M, Weber SN, Dhainaut JF, et al. Immediate coronary angiography in survivors of out-of-hospital cardiac arrest. New Engl J Med. (1997) 336:1629–33. doi: 10.1056/NEJM199706053362302
51. Larsen JM, Ravkilde J. Acute coronary angiography in patients resuscitated from out-of-hospital cardiac arrest–a systematic review and meta-analysis. Resuscitation. (2012) 83:1427–33. doi: 10.1016/j.resuscitation.2012.08.337
52. Duncker DJ, Schulz R, Ferrari R, Garcia-Dorado D, Guarnieri C, Heusch G, et al. Myocardial stunning remaining questions. Cardiovasc Res. (1998) 38:549–58. doi: 10.1016/S0008-6363(98)00061-3
53. Bolli R. Oxygen-derived free radicals and myocardial reperfusion injury: an overview. Cardiovasc Drugs Ther. (1991) 5(Suppl. 2):249–68. doi: 10.1007/BF00054747
54. Ehring T, Bohm M, Heusch G. The calcium antagonist nisoldipine improves the functional recovery of reperfused myocardium only when given before ischemia. J Cardiovasc Pharmacol. (1992) 20:63–74. doi: 10.1097/00005344-199207000-00009
55. Opie L. Myocardial stunning: a role for calcium antagonists during reperfusion? Cardiovasc Res. (1992) 26:20–4. doi: 10.1093/cvr/26.1.20
56. Ramjee V, Grossestreuer AV, Yao Y, Perman SM, Leary M, Kirkpatrick JN, et al. Right ventricular dysfunction after resuscitation predicts poor outcomes in cardiac arrest patients independent of left ventricular function. Resuscitation. (2015) 96:186–91. doi: 10.1016/j.resuscitation.2015.08.008
57. Wardi G, Blanchard D, Dittrich T, Kaushal K, Sell R. Right ventricle dysfunction and echocardiographic parameters in the first 24h following resuscitation in the post-cardiac arrest patient: a retrospective cohort study. Resuscitation. (2016) 103:71–4. doi: 10.1016/j.resuscitation.2016.03.009
58. Kida K, Shirozu K, Yu B, Mandeville JB, Bloch KD, Ichinose F. Beneficial effects of nitric oxide on outcomes after cardiac arrest and cardiopulmonary resuscitation in hypothermia-treated mice. Anesthesiology. (2014) 120:880–9. doi: 10.1097/ALN.0000000000000149
59. Fries M, Coburn M, Nolte KW, Timper A, Kottmann K, Kuru TH, et al. Early administration of xenon or isoflurane may not improve functional outcome and cerebral alterations in a porcine model of cardiac arrest. Resuscitation. (2009) 80:584–90. doi: 10.1016/j.resuscitation.2009.02.019
60. Hayashida K, Sano M, Kamimura N, Yokota T, Suzuki M, Maekawa Y, et al. H(2) gas improves functional outcome after cardiac arrest to an extent comparable to therapeutic hypothermia in a rat model. J Am Heart Assoc. (2012) 1:e003459. doi: 10.1161/JAHA.112.003459
61. Hayashida K, Sano M, Kamimura N, Yokota T, Suzuki M, Ohta S, et al. Hydrogen inhalation during normoxic resuscitation improves neurological outcome in a rat model of cardiac arrest independently of targeted temperature management. Circulation. (2014) 130:2173–80. doi: 10.1161/CIRCULATIONAHA.114.011848
62. Fries M, Brucken A, Cizen A, Westerkamp M, Lower C, Deike-Glindemann J, et al. Combining xenon and mild therapeutic hypothermia preserves neurological function after prolonged cardiac arrest in pigs. Critic Care Med. (2012) 40:1297–303. doi: 10.1097/CCM.0b013e31823c8ce7
63. Hatefi Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem. (1985) 54:1015–69. doi: 10.1146/annurev.bi.54.070185.005055
64. Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res. (2003) 93:907–16. doi: 10.1161/01.RES.0000100390.68771.CC
65. Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circ Res. (2010) 106:285–96. doi: 10.1161/CIRCRESAHA.109.209452
66. Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. (2010) 106:633–46. doi: 10.1161/CIRCRESAHA.109.207381
67. Miyazaki Y, Ichinose F. Nitric oxide in post-cardiac arrest syndrome. J Cardiovasc Pharmacol. (2020) 75:508–15. doi: 10.1097/FJC.0000000000000765
68. Brucken A, Derwall M, Bleilevens C, Stoppe C, Gotzenich A, Gaisa NT, et al. Brief inhalation of nitric oxide increases resuscitation success and improves 7-day-survival after cardiac arrest in rats: a randomized controlled animal study. Critic Care. (2015) 19:408. doi: 10.1186/s13054-015-1128-x
69. Derwall M, Ebeling A, Nolte KW, Weis J, Rossaint R, Ichinose F, et al. Inhaled nitric oxide improves transpulmonary blood flow and clinical outcomes after prolonged cardiac arrest: a large animal study. Critic Care. (2015) 19:328. doi: 10.1186/s13054-015-1050-2
70. Dezfulian C, Shiva S, Alekseyenko A, Pendyal A, Beiser DG, Munasinghe JP, et al. Nitrite therapy after cardiac arrest reduces reactive oxygen species generation, improves cardiac and neurological function, and enhances survival via reversible inhibition of mitochondrial complex I. Circulation. (2009) 120:897–905. doi: 10.1161/CIRCULATIONAHA.109.853267
71. Nishida T, Yu JD, Minamishima S, Sips PY, Searles RJ, Buys ES, et al. Protective effects of nitric oxide synthase 3 and soluble guanylate cyclase on the outcome of cardiac arrest and cardiopulmonary resuscitation in mice. Critic Care Med. (2009) 37:256–62. doi: 10.1097/CCM.0b013e318192face
72. Gladwin MT, Raat NJ, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, et al. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol. (2006) 291:H2026–35. doi: 10.1152/ajpheart.00407.2006
73. Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. (1991) 88:4651–5. doi: 10.1073/pnas.88.11.4651
74. Elferink JG, De Koster BM. The involvement of protein kinase G in stimulation of neutrophil migration by endothelins. Eur J Pharmacol. (1998) 350:285–91. doi: 10.1016/S0014-2999(98)00265-9
75. Mellion BT, Ignarro LJ, Ohlstein EH, Pontecorvo EG, Hyman AL, Kadowitz PJ. Evidence for the inhibitory role of guanosine 3', 5'-monophosphate in ADP-induced human platelet aggregation in the presence of nitric oxide and related vasodilators. Blood. (1981) 57:946–55. doi: 10.1182/blood.V57.5.946.bloodjournal575946
76. Kang JL, Park W, Pack IS, Lee HS, Kim MJ, Lim CM, et al. Inhaled nitric oxide attenuates acute lung injury via inhibition of nuclear factor-kappa B and inflammation. J Appl Physiol. (1985) 92:795-801. doi: 10.1152/japplphysiol.00202.2001
77. Deng G, Carter J, Traystman RJ, Wagner DH, Herson PS. Pro-inflammatory T-lymphocytes rapidly infiltrate into the brain and contribute to neuronal injury following cardiac arrest and cardiopulmonary resuscitation. J Neuroimmunol. (2014) 274:132–40. doi: 10.1016/j.jneuroim.2014.07.009
78. Menzebach A, Bergt S, von Waldthausen P, Dinu C, Noldge-Schomburg G, Vollmar B. A comprehensive study of survival, tissue damage, and neurological dysfunction in a murine model of cardiopulmonary resuscitation after potassium-induced cardiac arrest. Shock. (2010) 33:189–96. doi: 10.1097/SHK.0b013e3181ad59a3
79. Nagasaka Y, Fernandez BO, Garcia-Saura MF, Petersen B, Ichinose F, Bloch KD, et al. Brief periods of nitric oxide inhalation protect against myocardial ischemia-reperfusion injury. Anesthesiology. (2008) 109:675–82. doi: 10.1097/ALN.0b013e318186316e
80. Morgan RW, Sutton RM, Karlsson M, Lautz AJ, Mavroudis CD, Landis WP, et al. Pulmonary vasodilator therapy in shock-associated cardiac arrest. Am J Respir Critic Care Med. (2018) 197:905–12. doi: 10.1164/rccm.201709-1818OC
81. Piantadosi CA. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim Biophys Acta. (2012) 1820:712–21. doi: 10.1016/j.bbagen.2011.03.008
82. Maron BA, Tang SS, Loscalzo J. S-nitrosothiols and the S-nitrosoproteome of the cardiovascular system. Antioxid Redox Signal. (2013) 18:270–87. doi: 10.1089/ars.2012.4744
83. Konorev EA, Tarpey MM, Joseph J, Baker JE, Kalyanaraman B. S-nitrosoglutathione improves functional recovery in the isolated rat heart after cardioplegic ischemic arrest-evidence for a cardioprotective effect of nitric oxide. J Pharmacol Exp Ther. (1995) 274:200–6.
84. Khan M, Sekhon B, Giri S, Jatana M, Gilg AG, Ayasolla K, et al. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab. (2005) 25:177–92. doi: 10.1038/sj.jcbfm.9600012
85. Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. (2001) 410:490–4. doi: 10.1038/35068596
86. Chen Q, Sievers RE, Varga M, Kharait S, Haddad DJ, Patton AK, et al. Pharmacological inhibition of S-nitrosoglutathione reductase improves endothelial vasodilatory function in rats in vivo. J Appl Physiol. (2013) 114:752–60. doi: 10.1152/japplphysiol.01302.2012
87. Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. (2004) 116:617–28. doi: 10.1016/S0092-8674(04)00131-X
88. Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci USA. (2009) 106:6297–302. doi: 10.1073/pnas.0901043106
89. Elphinstone RE, Besla R, Shikatani EA, Lu Z, Hausladen A, Davies M, et al. S-Nitrosoglutathione reductase deficiency confers improved survival and neurological outcome in experimental cerebral malaria. Infect Immun. (2017) 85:e00371-17. doi: 10.1128/IAI.00371-17
90. Hayashida K, Bagchi A, Miyazaki Y, Hirai S, Seth D, Silverman MG, et al. Improvement in outcomes after cardiac arrest and resuscitation by inhibition of S-nitrosoglutathione reductase. Circulation. (2019) 139:815–27. doi: 10.1161/CIRCULATIONAHA.117.032488
91. Hayashida K, Miyazaki Y, Yu B, Silverman MG, Pinciroli R, Berra L, et al. Depletion of vascular nitric oxide contributes to poor outcomes after cardiac arrest. Am J Respir Critic Care Med. (2019) 199:1288–90. doi: 10.1164/rccm.201812-2377LE
92. Dezfulian C, Olsufka M, Fly D, Scruggs S, Do R, Maynard C, et al. Hemodynamic effects of IV sodium nitrite in hospitalized comatose survivors of out of hospital cardiac arrest. Resuscitation. (2018) 122:106–12. doi: 10.1016/j.resuscitation.2017.11.055
93. Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. (2007) 101:1155–63. doi: 10.1161/CIRCRESAHA.107.155879
94. Ohta S. Molecular hydrogen as a novel antioxidant: overview of the advantages of hydrogen for medical applications. Methods Enzymol. (2015) 555:289–317. doi: 10.1016/bs.mie.2014.11.038
95. Ohta S. Recent progress toward hydrogen medicine: potential of molecular hydrogen for preventive and therapeutic applications. Curr Pharm Design. (2011) 17:2241–52. doi: 10.2174/138161211797052664
96. van Berkel-Arts A, Dekker M, van Dijk C, Grande HJ, Hagen WR, Hilhorst R, et al. Application of hydrogenase in biotechnological conversions. Biochimie. (1986) 68:201–9. doi: 10.1016/S0300-9084(86)81084-7
97. Ono H, Nishijima Y, Ohta S, Sakamoto M, Kinone K, Horikosi T, et al. Hydrogen gas inhalation treatment in acute cerebral infarction: a randomized controlled clinical study on safety and neuroprotection. J Stroke Cerebrovasc Dis. (2017) 26:2587–94. doi: 10.1016/j.jstrokecerebrovasdis.2017.06.012
98. Hayashida K, Sano M, Ohsawa I, Shinmura K, Tamaki K, Kimura K, et al. Inhalation of hydrogen gas reduces infarct size in the rat model of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun. (2008) 373:30–5. doi: 10.1016/j.bbrc.2008.05.165
99. Katsumata Y, Sano F, Abe T, Tamura T, Fujisawa T, Shiraishi Y, et al. The effects of hydrogen gas inhalation on adverse left ventricular remodeling after percutaneous coronary intervention for ST-elevated myocardial infarction- first pilot study in humans. Circ J. (2017) 81:940–7. doi: 10.1253/circj.CJ-17-0105
100. Tamura T, Hayashida K, Sano M, Suzuki M, Shibusawa T, Yoshizawa J, et al. Feasibility and safety of hydrogen gas inhalation for post-cardiac arrest syndrome- first-in-human pilot study. Circ J. (2016) 80:1870–3. doi: 10.1253/circj.CJ-16-0127
101. Ohta S. Molecular hydrogen as a preventive and therapeutic medical gas: initiation, development and potential of hydrogen medicine. Pharmacol Ther. (2014) 144:1–11. doi: 10.1016/j.pharmthera.2014.04.006
102. Iketani M, Ohsawa I. Molecular hydrogen as a neuroprotective agent. Curr Neuropharmacol. (2017) 15:324–31. doi: 10.2174/1570159X14666160607205417
103. Shen M, Zhang H, Yu C, Wang F, Sun X. A review of experimental studies of hydrogen as a new therapeutic agent in emergency and critical care medicine. Med Gas Res. (2014) 4:17. doi: 10.1186/2045-9912-4-17
104. Huang CS, Kawamura T, Toyoda Y, Nakao A. Recent advances in hydrogen research as a therapeutic medical gas. Free Radic Res. (2010) 44:971–82. doi: 10.3109/10715762.2010.500328
105. Dikalov SI, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol. (2013) 305:H1417–27. doi: 10.1152/ajpheart.00089.2013
106. Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. (2004) 10:S18–25. doi: 10.1038/nrn1434
107. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. (2000) 408:239–47. doi: 10.1038/35041687
108. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. (2003) 552:335–44. doi: 10.1113/jphysiol.2003.049478
109. Indo HP, Yen HC, Nakanishi I, Matsumoto K, Tamura M, Nagano Y, et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J Clin Biochem Nutr. (2015) 56:1–7. doi: 10.3164/jcbn.14-42
110. Lipinski B. Hydroxyl radical and its scavengers in health and disease. Oxid Med Cell Longev. (2011) 2011:809696. doi: 10.1155/2011/809696
111. Halliwell B, Gutteridge JM. Biologically relevant metal ion-dependent hydroxyl radical generation. An update. FEBS Lett. (1992) 307:108–12. doi: 10.1016/0014-5793(92)80911-Y
112. Radi R. Peroxynitrite, a stealthy biological oxidant. J Biol Chem. (2013) 288:26464–72. doi: 10.1074/jbc.R113.472936
113. Jazwa A, Cuadrado A. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr Drug Targets. (2010) 11:1517–31. doi: 10.2174/1389450111009011517
114. Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol. (2008) 101:14D−9. doi: 10.1016/j.amjcard.2008.02.003
115. Brambilla D, Mancuso C, Scuderi MR, Bosco P, Cantarella G, Lempereur L, et al. The role of antioxidant supplement in immune system, neoplastic, and neurodegenerative disorders: a point of view for an assessment of the risk/benefit profile. Nutr J. (2008) 7:29. doi: 10.1186/1475-2891-7-29
116. Hackam DG. Review: antioxidant supplements for primary and secondary prevention do not decrease mortality. ACP J Club. (2007) 147:4. doi: 10.7326/ACPJC-2007-147-1-004
117. Yoshida A, Asanuma H, Sasaki H, Sanada S, Yamazaki S, Asano Y, et al. H(2) mediates cardioprotection via involvements of K(ATP) channels and permeability transition pores of mitochondria in dogs. Cardiovasc Drugs Ther. (2012) 26:217–26. doi: 10.1007/s10557-012-6381-5
118. Nemeth J, Toth-Szuki V, Varga V, Kovacs V, Remzso G, Domoki F. Molecular hydrogen affords neuroprotection in a translational piglet model of hypoxic-ischemic encephalopathy. J Physiol Pharmacol. (2016) 67:677–89. doi: 10.14232/phd.4072
119. Cole AR, Perry DA, Raza A, Nedder AP, Pollack E, Regan WL, et al. Perioperatively inhaled hydrogen gas diminishes neurologic injury following experimental circulatory arrest in Swine. JACC Basic Transl Sci. (2019) 4:176–87. doi: 10.1016/j.jacbts.2018.11.006
120. Tamura T, Hayashida K, Sano M, Onuki S, Suzuki M. Efficacy of inhaled HYdrogen on neurological outcome following BRain Ischemia During post-cardiac arrest care (Phase II, multicenter, prospective, randomized, double-blinded, placebo-controlled trial). Trials. (2020) 18:488. doi: 10.1186/s13063-017-2246-3
121. Cullen SC, Gross EG. The anesthetic properties of xenon in animals and human beings, with additional observations on krypton. Science. (1951) 113:580–2. doi: 10.1126/science.113.2942.580
122. Nakata Y, Goto T, Ishiguro Y, Terui K, Kawakami H, Santo M, et al. Minimum alveolar concentration (MAC) of xenon with sevoflurane in humans. Anesthesiology. (2001) 94:611–4. doi: 10.1097/00000542-200104000-00014
123. Derwall M, Timper A, Kottmann K, Rossaint R, Fries M. Neuroprotective effects of the inhalational anesthetics isoflurane and xenon after cardiac arrest in pigs. Critic Care Med. (2008) 36(Suppl. 11):S492–5. doi: 10.1097/CCM.0b013e31818a904a
124. Hobbs C, Thoresen M, Tucker A, Aquilina K, Chakkarapani E, Dingley J. Xenon and hypothermia combine additively, offering long-term functional and histopathologic neuroprotection after neonatal hypoxia/ischemia. Stroke. (2008) 39:1307–13. doi: 10.1161/STROKEAHA.107.499822
125. Chakkarapani E, Thoresen M, Liu X, Walloe L, Dingley J. Xenon offers stable haemodynamics independent of induced hypothermia after hypoxia-ischaemia in newborn pigs. Intensive Care Med. (2012) 38:316–23. doi: 10.1007/s00134-011-2442-7
126. Moro F, Fossi F, Magliocca A, Pascente R, Sammali E, Baldini F, et al. Efficacy of acute administration of inhaled argon on traumatic brain injury in mice. Br J Anaesth. (2020) S0007-0912(20)30686-3. doi: 10.1016/j.bja.2020.08.027
127. Dickinson R, Franks NP. Bench-to-bedside review: molecular pharmacology and clinical use of inert gases in anesthesia and neuroprotection. Critic Care. (2010) 14:229. doi: 10.1186/cc9051
128. Maze M. Preclinical neuroprotective actions of xenon and possible implications for human therapeutics: a narrative review. Can J Anaesth. (2016) 63:212–6. doi: 10.1007/s12630-015-0507-8
129. Hollig A, Schug A, Fahlenkamp AV, Rossaint R, Coburn M, Argon Organo-Protective N. Argon: systematic review on neuro- and organoprotective properties of an “inert” gas. Int J Mol Sci. (2014) 15:18175–96. doi: 10.3390/ijms151018175
130. Vyklicky V, Korinek M, Smejkalova T, Balik A, Krausova B, Kaniakova M, et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol Res. (2014) 63(Suppl. 1):S191–203. doi: 10.33549/physiolres.932678
131. Wu QJ, Tymianski M. Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol Brain. (2018) 11:15. doi: 10.1186/s13041-018-0357-8
132. Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, et al. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. (2009) 29:9330–43. doi: 10.1523/JNEUROSCI.2212-09.2009
133. Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, et al. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. (1999) 19:5910–8. doi: 10.1523/JNEUROSCI.19-14-05910.1999
134. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. (2012) 298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7
135. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. (1999) 22:391–7. doi: 10.1016/S0166-2236(99)01401-0
136. Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke. (1998) 29:705–18. doi: 10.1161/01.STR.29.3.705
137. Marsh JD, Smith TS. Calcium overload and ischemic myocardial injury. Circulation. (1991) 83:709–11. doi: 10.1161/01.CIR.83.2.709
138. Hernandez DE, Salvadores NA, Moya-Alvarado G, Catalan RJ, Bronfman FC, Court FA. Axonal degeneration induced by glutamate excitotoxicity is mediated by necroptosis. J Cell Sci. (2018) 131:jcs214684. doi: 10.1242/jcs.214684
139. Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. (2001) 166:7527–33. doi: 10.4049/jimmunol.166.12.7527
140. Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G, et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol. (2012) 72:536–49. doi: 10.1002/ana.23626
141. Shuaib A, Sochocka E, Code W, Hertz L. Hypothermia protects astrocytes during ischemia in cell culture. Neurosci Lett. (1992) 146:69–71. doi: 10.1016/0304-3940(92)90174-6
142. Montes de Oca Balderas P, Gonzalez Hernandez JR. NMDA receptors in astroglia: chronology, controversies and contradictions from a complex molecule. In: Gentile MT, editor. Physiology and Pathology. London: IntechOpen (2018).
143. Franks NP, Dickinson R, de Sousa SL, Hall AC, Lieb WR. How does xenon produce anaesthesia? Nature. (1998) 396:324. doi: 10.1038/24525
144. Homi HM, Yokoo N, Ma D, Warner DS, Franks NP, Maze M, et al. The neuroprotective effect of xenon administration during transient middle cerebral artery occlusion in mice. Anesthesiology. (2003) 99:876–81. doi: 10.1097/00000542-200310000-00020
145. Ma D, Yang H, Lynch J, Franks NP, Maze M, Grocott HP. Xenon attenuates cardiopulmonary bypass-induced neurologic and neurocognitive dysfunction in the rat. Anesthesiology. (2003) 98:690–8. doi: 10.1097/00000542-200303000-00017
146. Fries M, Nolte KW, Coburn M, Rex S, Timper A, Kottmann K, et al. Xenon reduces neurohistopathological damage and improves the early neurological deficit after cardiac arrest in pigs. Critic Care Med. (2008) 36:2420–6. doi: 10.1097/CCM.0b013e3181802874
147. Veldeman M, Coburn M, Rossaint R, Clusmann H, Nolte K, Kremer B, et al. Xenon reduces neuronal hippocampal damage and alters the pattern of microglial activation after experimental subarachnoid hemorrhage: a randomized controlled animal trial. Front Neurol. (2017) 8:511. doi: 10.3389/fneur.2017.00511
148. Arola OJ, Laitio RM, Roine RO, Gronlund J, Saraste A, Pietila M, et al. Feasibility and cardiac safety of inhaled xenon in combination with therapeutic hypothermia following out-of-hospital cardiac arrest. Critic Care Med. (2013) 41:211624. doi: 10.1097/CCM.0b013e31828a4337
149. Laitio R, Hynninen M, Arola O, Virtanen S, Parkkola R, Saunavaara J, et al. Effect of inhaled xenon on cerebral white matter damage in comatose survivors of out-of-hospital cardiac arrest: a randomized clinical trial. JAMA. (2016) 315:1120–8. doi: 10.1001/jama.2016.1933
150. Stadelmann C, Timmler S, Barrantes-Freer A, Simons M. Myelin in the central nervous system: structure, function, and pathology. Physiol Rev. (2019) 99:1381–431. doi: 10.1152/physrev.00031.2018
151. Arola O, Saraste A, Laitio R, Airaksinen J, Hynninen M, Backlund M, et al. Inhaled xenon attenuates myocardial damage in comatose survivors of out-of-hospital cardiac arrest: the xe-hypotheca trial. J Am Coll Cardiol. (2017) 70:2652–60. doi: 10.1016/j.jacc.2017.09.1088
152. Lynch C, 3rd, Baum J, Tenbrinck R. Xenon anesthesia. Anesthesiology. (2000) 92:865–8. doi: 10.1097/00000542-200003000-00031
153. Tuteja N, Chandra M, Tuteja R, Misra MK. Nitric oxide as a unique bioactive signaling messenger in physiology and pathophysiology. J Biomed Biotechnol. (2004) 2004:227–37. doi: 10.1155/S1110724304402034
155. Lillo RS, Parker EC. Mixed-gas model for predicting decompression sickness in rats. J Appl Physiol. (2000) 89:2107–16. doi: 10.1152/jappl.2000.89.6.2107
156. Abraini JH, Gardette-Chauffour MC, Martinez E, Rostain JC, Lemaire C. Psychophysiological reactions in humans during an open sea dive to 500 m with a hydrogen-helium-oxygen mixture. J Appl Physiol. (1994) 76:1113–8. doi: 10.1152/jappl.1994.76.3.1113
157. Kajimura M, Fukuda R, Bateman RM, Yamamoto T, Suematsu M. Interactions of multiple gas-transducing systems: hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid Redox Signal. (2010) 13:157–92. doi: 10.1089/ars.2009.2657
Keywords: cardiac arrest, cardiopulmonary resuscitation, ischemia-reperfusion injury, neuroprotection, nitric oxide, xenon, molecular hydrogen (H2), PCAS
Citation: Hayashida K, Miyara SJ, Shinozaki K, Takegawa R, Yin T, Rolston DM, Choudhary RC, Guevara S, Molmenti EP and Becker LB (2021) Inhaled Gases as Therapies for Post–Cardiac Arrest Syndrome: A Narrative Review of Recent Developments. Front. Med. 7:586229. doi: 10.3389/fmed.2020.586229
Received: 22 July 2020; Accepted: 04 December 2020;
Published: 14 January 2021.
Edited by:
Inge Bauer, University Hospital of Düsseldorf, GermanyReviewed by:
Shoji Yokobori, Nippon Medical School, JapanCopyright © 2021 Hayashida, Miyara, Shinozaki, Takegawa, Yin, Rolston, Choudhary, Guevara, Molmenti and Becker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kei Hayashida, a2hheWFzaGlkYUBub3J0aHdlbGwuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.