 Xingjia Mao1†
Xingjia Mao1† Linlin Wang
Linlin Wang Chuan Xiang
Chuan Xiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 26 November 2020
Sec. Translational Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fmed.2020.581402
This article is part of the Research Topic New Trends in Osteoarthritis Treatment View all 12 articles
Osteoarthritis (OA) is a common and disabling joint disorder that is mainly characterized by cartilage degeneration and narrow joint spaces. The role of mitochondrial dysfunction in promoting the development of OA has gained much attention. Targeting endogenous molecules to improve mitochondrial function is a potential treatment for OA. Moreover, research on exogenous drugs to improve mitochondrial function in OA based on endogenous molecular targets has been accomplished. In addition, stem cells and exosomes have been deeply researched in the context of cartilage regeneration, and these factors both reverse mitochondrial dysfunctions. Thus, we hypothesize that biomedical approaches will be applied to the treatment of OA. Furthermore, we have summarized the global status of mitochondria and osteoarthritis research in the past two decades, which will contribute to the research field and the development of novel treatment strategies for OA.
Osteoarthritis (OA), a chronic and progressive cartilage degeneration disease (1) with a high morbidity and disability rate (2), is characterized by cartilage degeneration, osteophyte formation, thickening of subchondral bone, synovial inflammation, and meniscal injuries (3). As the global population ages and the proportion of obese people increases, the morbidity of OA continues to rise. At present, ~250 million people suffering from OA worldwide bear a tremendous economic burden as does society (4). OA tends to occur in the elderly population; cellular senescence is a contributor to age-related diseases (5), and studies have shown that OA is typical representatives of age-related diseases (6). Alleviating pain is the main purpose of non-surgical treatment, but this treatment does not alleviate the progression of OA (7).
Chondrocytes are the only cell type present in mature cartilage and change pathologically when OA occurs (8). Multiple factors can lead to OA, including inflammatory cytokines, mechanical stress, ageing, metabolic factors, and other pathological changes, which could increase reactive oxygen species (ROS) (9), induce oxidative stress in mitochondria, cause mitochondrial DNA (mtDNA) damage, result in mitochondrial damage, and shorten the life span of chondrocytes (10). The loss of mitochondrial membrane potential (MMP) leads to a reduction in energy production, an increase in the permeability of the mitochondrial membrane (11), and the release of apoptotic factors such as cytochrome C (Cyt-C), apoptosis-inducing factor, and procaspases from the mitochondria into the cytoplasm. Obvious changes in the morphology and function of mitochondria have been shown in ageing cells, and mitochondrial dysfunction is a key factor in cellular senescence (5, 12), demonstrating that mitochondria may be a therapeutic target for anti-ageing treatment and reduce the morbidity of OA in the elderly population (13). In addition, mitochondrial genetics are indispensable in the pathogenesis of OA. The accumulation of somatic mutations in mtDNA is a major contributor to human ageing and degenerative diseases (14). Reducing mtDNA damage, including the integrity of mtDNA4977, could optimize mitochondrial function, and maintain the homeostasis of chondrocytes. Furthermore, the mitochondrial apoptotic pathway has been implicated in chondrocyte apoptosis in OA (15). More specific therapeutic strategies on the basis of an in-depth molecular understanding of OA are thus essential (16).
With the research and application of stem cells and exosomes in cartilage repair, biomedical approaches to optimize mitochondrial function will be the preferred method for the thorough treatment of OA. Furthermore, gene therapy is also booming, and we therefore think that biological measures to modify the disease will be the major approach for OA treatment. In the present article, we have reviewed mitochondrial dysfunction mainly in the context of OA chondrocytes and summarized the endogenous molecular targets related to mitochondrial function. Moreover, research progress on exogenous drugs for the treatment of OA by restoring mitochondrial function in chondrocytes has been reviewed. In addition, we have described the global status of mitochondrial and OA research, which may contribute to predicting the trend in mitochondrial research regarding the treatment of OA. Furthermore, these findings will be instructive for mechanistic research on mitochondrial functions in OA, contributing to fundamental research on the treatment of OA through the mitochondrial pathway and providing novel strategies for the clinical treatment of OA.
Mitochondria, encapsulated by bilayer membranes, are remarkably dynamic organelles and considered as the “powerhouse” of eukaryote cells. Mitochondria not only generate the energy required for cellular metabolism by oxidative phosphorylation (OXPHOS), but they also produce heat in certain specialized cell types, such as brown adipocytes (17). Approximately 2,000 mitochondria within a eukaryotic cell occupy ~20% of the cell volume (12). There are protein complexes in the inner mitochondrial membrane that transfer and pump protons through the mitochondrial respiratory chain (MRC) for ATP production, such as NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), Cyt-C reductase (complex III), and Cyt-C oxidase (complex IV). Pyruvate and fatty acids could be converted to acetyl CoA by mitochondria, and CoA is metabolized by the citric acid cycle to produce NADH (18) where energy electrons are used to produce ATP (19). In addition to ATP production, intermediate metabolites for biosynthesis, protein modifications, signal transduction, programmed cell death, bioenergetic metabolism, the redox state, calcium homeostasis, innate immunity, stem cell reprogramming, and ageing-related responses (20–22) occur in the mitochondria (17) (Figure 1).
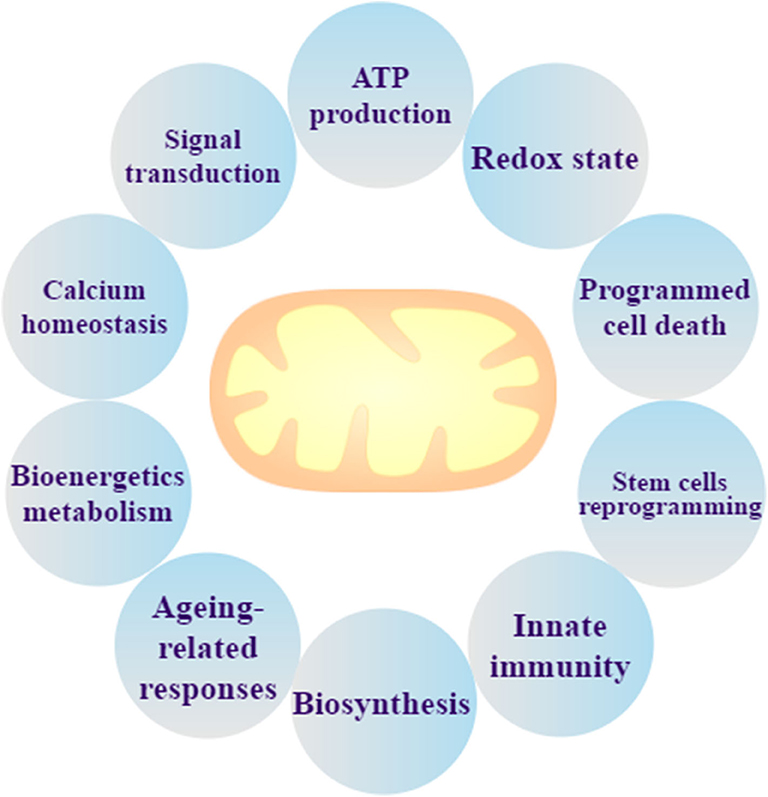
Figure 1. Mitochondrial biological function. Mitochondria are not only the organelle for ATP production and signal transduction, but they can also maintain the redox state and calcium homeostasis, regulate programmed cell death, and perform bioenergetics metabolism, stem cells reprogramming, ageing-related responses, innate immunity, and biosynthesis.
Recently, more research has focused on mitochondrial dynamics. The dynamic characteristics consist of mitochondrial fusion, mitochondrial fission and mitophagy (36), which are crucial for normal mitochondrial function and are critically associated with mitochondrial biogenesis and mitophagy (37). Mitofusins 1 (Mfn1) and Mitofusins 2 (Mfn2) mediate the fusion of the outer membrane, and optic atrophy 1 (OPA1) mediates the fusion of the inner membrane (38). Dynamin-related protein 1 (Drp1) and classical dynamin 2 (Dnm2) are the main mediators of mitochondrial fission (39) (Figure 2). When mitochondrial fission becomes increasingly dominant, damaged mitochondria undergo mitophagy in chondrocytes in the context of OA (40, 41), which could cause mitochondria to fail to produce sufficient bioenergy, regulate calcium and maintain the redox state. In contrast, mitochondrial fusion could enhance the biological function of mitochondria, which could make chondrocytes energetic and inhibit apoptosis.
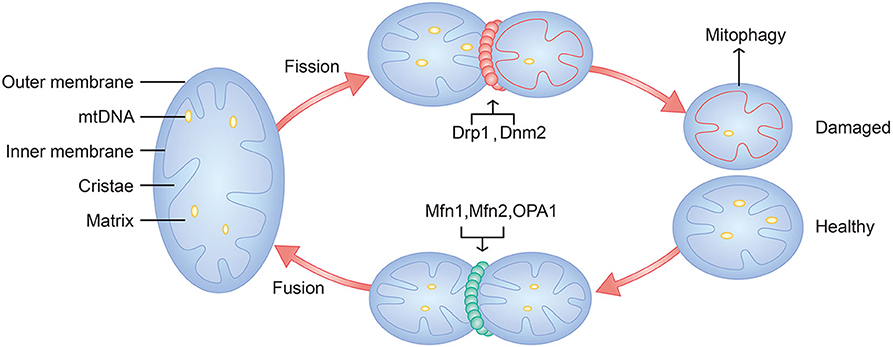
Figure 2. Schematic diagram of mitochondria and mitochondrial dynamics. Major components of mitochondria include outer membrane, inner membrane, cristae, matrix, and mtDNA. Mitochondrial fusion is mediated by Mfn1, Mfn2, and OPA1. Mitochondrial fission is mediated by Drp1, Dnm2. Damaged mitochondria will undergo mitophagy.
In normal chondrocytes, mitochondria play a role in regulating signaling by modulating the redox state, supplying cofactors for biochemical reactions, such as molecular chaperones to facilitate protein folding, and generating ligands for signal transduction, such as AMPK signaling and calcium signaling (17, 42, 43). Calcium stored in mitochondria is helpful for maintaining calcium homeostasis in cells, and mitochondria are dedicated to transport extracellular matrix (ECM) calcium (12, 44). The mineralization of cartilage has been confirmed to involve calcium phosphate-containing granules, which are known as “matrix vesicles” (45). Moreover, Professor Alexandra E. Porter and colleagues found that mitochondrial granules contribute to the transport of clusters of calcium and phosphate ions to the ECM to facilitate mineralization, and Professor Lehninger AL suggested that mitochondria could release calcium phosphate to the ECM to take part in bone formation (46, 47). In addition, Professor Brian Glancy and colleagues showed that calcium activated nearly every step within the electron transport chain (ETC) (48) and activated enzymes, such as NADH, Cyt-C, complex III, and complex IV, in the pathways of oxidative metabolism in mitochondria (49, 50). Furthermore, mitochondria could regulate and balance the apoptosis by initiating cell death (17).
Mitochondrial dysfunction mainly manifests as decreased ATP production, increased oxidative stress, calcium dysregulation, increased permeability of the mitochondrial membrane, and mtDNA alternations, which result in cartilage degeneration. Chondrocyte damage occurs and is mainly reflected in the increases in MMP-3, MMP-13, NO, and inflammatory injury with an imbalance between catabolism and anabolism of extracellular matrix (51), including reductions in aggrecan and collagen II, which eventually induce OA (Figure 3).
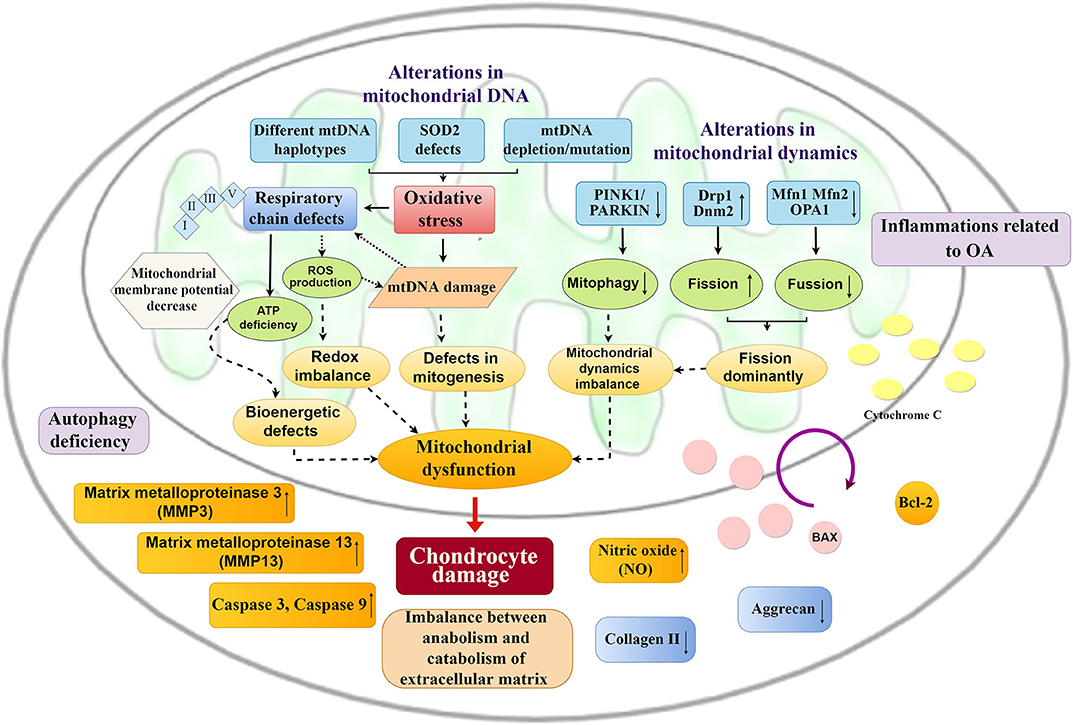
Figure 3. Mitochondrial changes and chondrocyte damage in OA. Alternations in mitochondrial DNA and mitochondrial dynamics cause a series of mitochondrial changes. Excessive oxidative stress, respiratory chain defects and imbalance of mitochondrial dynamics result in mitochondrial dysfunction, which could promote chondrocyte damage, including inflammatory injury, imbalance between anabolism and catabolism of the extracellular matrix, and an increase in apoptosis of chondrocytes.
Mitochondrial dysfunction can lead to a decrease in the activity of respiratory chain complexes I, II, III, and V, the loss of MMP, and decreases in OXPHOS in OA chondrocytes (52), which could induce chondrocytes to release interleukin-1β (IL-1β) and lead to inflammation (12). Two primary mechanisms of ATP production include substrate phosphorylation in the glycolytic pathway and in the tricarboxylic acid (TCA) cycle and OXPHOS occur at the inner membrane (53). ATP production is driven by the transmembrane proton gradient. An inflammatory response in chondrocytes with the upregulation of cyclooxygenase 2 (COX-2) and prostaglandin E2 (PGE2) production could be generated by ETC dysfunction (54). Both TNF-α and IL-1β inhibit the activity of ETC complex I (55), which induces decreases in ATP production and MMP. Inhibiting the ETC thus could decrease ATP synthesis (56).
Mitochondrial dysfunction maintains a positive regeneration circle with oxidative stress, increased ROS, and mtDNA damage, which are regarded as hallmarks of chronic degenerative diseases (57). The accumulation of ROS and mtDNA damage can activate the nuclear factor-κB (NF-κB) pathway, which is the main regulator of inflammation (57). Avascular and hypoxic tissue are always used to describe cartilage, and chondrocytes are the only cell types in articular cartilage that maintain the balance of extracellular matrix (ECM) synthesis and degradation (58). ROS, as by-products of oxidation-reduction reactions, are generated in the MRC (59). A lower level of ROS is beneficial for maintaining chondrocyte homeostasis, and a higher level of ROS induces the depolarization of mitochondrial membrane, which could lead to sustained ROS production (60). An initial theory suggested that ROS have deleterious effects on ageing and degenerative diseases (61). Accumulating evidence has demonstrated that increased oxidative stress and the overproduction of ROS, including superoxide anion, hydrogen peroxide (H2O2), and nitric oxide (NO), play pivotal roles in the pathogenesis of OA (10, 62). The overproduction and accumulation of ROS and ATP deficiency decrease mitogenesis and break the redox balance. DNA and especially mtDNA could be injured (63). Oxidative stress could damage the mitochondrial respiratory chain protein complexes in chondrocytes (12). Due to the accumulation of ROS in chondrocytes, the decrease in collagen and glycosaminoglycan synthesis and the enhancement of metalloproteinases and aggrecanases induce chondrocytes to undergo a switch from anabolic to a catabolic gene expression, which results in cartilage breakdown (34). Furthermore, the depletion of superoxide dismutase 2 (SOD2), the major mitochondrial antioxidant protein, occurs in early cartilage degradation and could exacerbate inflammation and enhance ROS, contributing to OA progression (64, 65). Mitochondria are the dominant intracellular organelles in charge of the generation of ROS (66). ROS overload induced by oxidative stress results in the loss of MMP by stimulating the mitochondrial permeability transition pore (PTP) (67). High levels of cholesterol are naturally present in the cell membrane of chondrocytes, and chondrocytes could produce their own cholesterol and synthesize all the indispensable proteins for cholesterol biosynthesis (68, 69). Hypercholesterolemia animal models with changes in cartilage have been studied by Mao et al. (69), and the researchers demonstrated the direct effect of high cholesterol on cartilage degeneration and chondrocyte hypertrophy. When exposed to the synovial fluid with raised cholesterol levels, chondrocytes could be damaged because of the changes in the fluidity of the cell membrane and activation of membrane lipid signaling pathways (70). There is a close relationship between increased cholesterol oxidation products and mitochondria-derived oxidative stress, which leads to increased production of mitochondrial ROS (69), and Mao et al. showed that the cholesterol-lowering drug and the mitochondria-specific antioxidant have protective effects on attenuating OA symptoms caused by high cholesterol, such as atorvastatin and Mito-TEMPO.
Calcium, a ubiquitous intracellular second messenger, is involved in numerous cellular processes (71, 72). Calcium overload can lead to ROS overproduction, mitochondrial depolarization, MMP damage, and apoptosis (73). The maintenance of intracellular calcium homeostasis is achieved by mitochondrial uptake of calcium through a uniport transporter and the release of calcium through the inositol-1,4,5-trisphosphate receptor (IP3R), the sodium/calcium exchanger, or through the PTP, which is stimulated by excessive calcium in the mitochondrial matrix (72, 74). The PTP is a large conductance channel in the inner membrane of mitochondria (75), and both high levels of calcium and ROS can activate the PTP opening (76). The PTP makes the membrane non-specifically permeable to any molecule up to 1.5 kDa, including protons, and the mitochondria cannot maintain a pH gradient or MMP any longer (77, 78). The PTP leads to the collapse of MMP, leading to mitochondrial swelling and release of calcium and Cyt-C, ultimately stimulating apoptosis (8, 72). Calpains are calcium-activated proteases that could destroy the sodium/calcium exchanger and result in calcium overload and cell death (79). Furthermore, calcium overload and the activation of BAX by calpains lead to mitochondrial depolarization (72, 80).
Chondrocyte apoptosis induced by inflammation, oxidative stress, and increased mitochondrial membrane permeability (81) is positively associated with the degree of cartilage damage (82, 83). The collapse of the MMP leads to mitochondrial depolarization (8), which causes mitochondrial swelling, outer mitochondrial membrane collapse, and release of Cyt-C (84, 85). The BAX/mitochondrial Cyt-C/Caspase signaling pathway is shown to be associated with chondrocyte apoptosis (31). The downregulation of Bcl-2, the increase in expression of BAX, Caspase-3, and Caspase-9, and the increase in permeability of mitochondrial membrane can promote the outflow of Cyt-C from mitochondria into the cytoplasm and the inflow of BAX from the cytoplasm into mitochondria, increasing chondrocyte apoptosis. When damaged by various oxidative stimuli, the initiation of chondrocyte apoptosis induced by increased ROS is promoted (10, 15, 86). Studies have shown that mitochondrial dysfunction with reduced MMP and increased mitochondrial membrane permeability could promote the migration of Cyt-C from the mitochondrial matrix to the cytoplasm (87), which could induce apoptosis due to the activation of caspases and increase the BAX/Bcl-2 ratio (88). Moreover, the level of ROS in mitochondria is significantly increased (89), which could induce oxidative stress, destroy cartilage homeostasis, and increase chondrocyte apoptosis (60). The balance of mitochondrial dynamics could inhibit the apoptosis induced by oxidative stress (90, 91).
In addition to mitochondrial dysfunction, the inheritance of mitochondria also acts as a pivotal role in the process of OA (92). mtDNA, a 16,569 bp circular and double-stranded molecule, encodes 13 protein subunits for the respiratory chain and 24 RNA components (22 tRNAs and 2 rRNAs) for mitochondrial protein synthesis (93). Chondrocytes from OA patients exhibit higher levels of mtDNA damage than chondrocytes from normal individuals (94). mtDNA damage could be caused by the increased ROS burden of aged chondrocytes (63, 95). At the same time, the accumulation of mtDNA mutations above a critical level could lead to dysfunction of the respiratory chain and increased ROS production, which could promote excessive chondrocyte apoptosis and enhance inflammatory responses (8). mtDNA haplogroups modulate crucial functions such as ATP production, oxygen consumption, ROS generation, and the expression of mitochondrial and nuclear genes (96). mtDNA haplotype J is associated with a lower risk of knee osteoarthritis (KOA) compared to that of mitochondrial mtDNA haplotype H (97). The mtDNA haplotype may be a biomarker for OA diagnosis and prognosis, and be closely involved in the OA phenotype (98). Therefore, the pattern of latent drugs that mimic the physiological effects of mtDNA haplotype J may be a potential treatment strategy for OA (99).
The vital role of mitochondrial changes in the development of OA has been demonstrated (8, 12, 52, 100), and endogenous molecular targets that optimize mitochondrial dynamics and morphology will turn into potential targets for OA treatment (Figure 4). AMPK, Sirtuin, PGC-1α, PINK1, PARKIN, and Nrf2 are endogenous molecules, and the activation of AMPK/SIRT1/3/PGC-1α, AMPK/SIRT3/SOD2, and AMPK/SIRT3/Parkin/PINK1 signaling could promote mitochondrial biogenesis and reduce oxidative stress, contributing to balancing mitochondrial dynamics and improving MMP. Moreover, upregulating OPA1, Mfn1, and Mfn2 and downregulating Drp1 and Dnm2 through GPS2 could promote mitochondrial fusion to enhance mitochondrial biological functions.
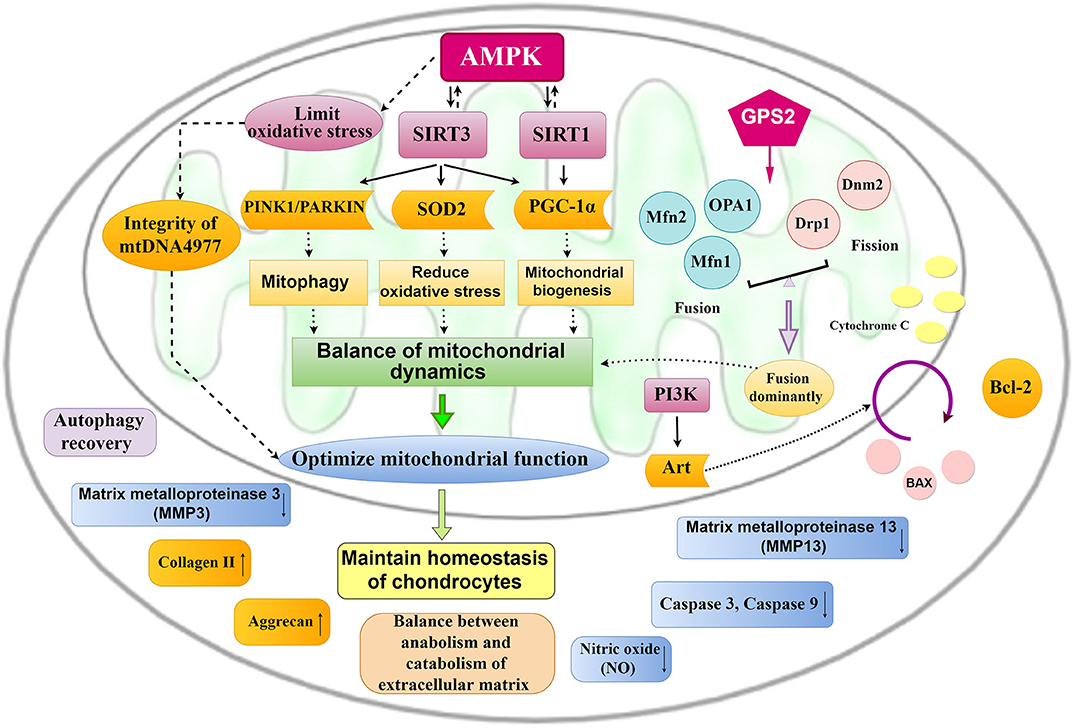
Figure 4. Mitochondrial pathways for the treatment of OA. The activation of AMPK-SIRT-3/ SIRT1-PGC-1α, AMPK-SIRT-3-SOD2, and AMPK-SIRT-3-PINK1/PARKIN signaling could promote mitochondrial biogenesis and mitochondrial fusion and reduce oxidative stress, which contribute to optimize mitochondrial function and then maintain chondrocyte homeostasis.
Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK), the serine/threonine kinase, is a key regulator to adapt to changes in energy demand (101). When in a hypoxic state, AMPK can be activated and phosphorylate multiple downstream targets, promoting the inhibition of ATP-consuming pathways and the activation of the ATP-producing pathway (101, 102). Dysregulation of AMPK has been associated with a variety of age-related diseases related to mitochondrial dysfunction and imbalance of cellular energy, including diabetes, atherosclerosis, cardiovascular disease, cancer, neurodegenerative diseases, and OA (103, 104), suggesting the translational potential of pharmacological AMPK activators to limit OA progression (52, 102). In chondrocytes, activation of AMPK suppresses NF-κB activation, oxidative stress, and multiple inflammatory and catabolic responses (104). Moreover, AMPK could regulate both mitochondrial biogenesis and mitophagy to balance mitochondrial dynamics (52).
AMPK activity regulates energy metabolism via downstream mediators, including the nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases Sirtuin1 and Sirtuin3 (SIRT1 and SIRT3, respectively). The key role of AMPK in the treatment of OA through the mitochondrial pathway and mitochondrial acetylation-induced OA has been identified, while SIRT3 is the main deacetylase in mitochondria, and SIRT3 activation can protect cells by regulating mitochondrial dynamics and mitophagy. SIRT1/3 and AMPK regulate each other (105). Increasing evidence shows that SIRT1 is significant in promoting mitochondrial dysfunction and OA progression (83). It has been proven that SIRT1 enzymatic activity is necessary for cartilage homeostasis (64, 65). The loss of SIRT1 in chondrocytes also leads to increases in MMP-13, apoptotic markers, and NF-κB, resulting in the accelerated OA development (83, 106). Upregulation of SIRT1 can inhibit the activation of COX-2, MMP-13, and NF-κB-induced TNF-α and decrease the upregulation of MMP-13 and acetylation of NF-κB p65 induced by IL-1β (83, 107). SIRT1 is a strong inducer of autophagy (108), which is reduced in OA, and therapeutic enhancement of autophagy is chondroprotective in vitro and in vivo (52, 109, 110). The NAD+-dependent deacetylase Sirtuin3 is the major deacetylase in mitochondria (111), contributing to the regulation of the mitochondrial antioxidant system and adenosine-triphosphate (ATP) production (112). Depletion of the mitochondrially localized antioxidant superoxide dismutase 2 (SOD2) promotes mitochondrial dysfunction and increased production of ROS (64, 65). A study showed that mitochondrial acetylation could promote the development of OA, while SIRT3 could enhance the antioxidant capacity of chondrocytes by enhancing the activity of SOD2 (113). Moreover, SIRT3 could activate and enhance the activity of AMPK in chondrocytes, which could reduce the loss of mtDNA4977 and maintain mtDNA integrity, thereby improving the function of mitochondria and protecting chondrocytes (28). Studies have shown that mitophagy can eliminate damaged mitochondria isolated by mitochondrial fission, which is a cytoprotective mechanism to maintain mitochondrial stability and quality (114). Moreover, the relationship between mitophagy and OA has been confirmed (60, 115). SIRT3 depletion can reduce mitophagy (116) and SIRT3 activation protects cells by regulating mitochondrial dynamics and mitophagy (117). Drugs that can activate SIRT3 may therefore be potential treatments for OA through the mitochondrial pathway.
The mitochondrial biogenesis master regulator peroxisome proliferator–activated receptor γ coactivator 1α (PGC-1α) acts by inducing the transcription of nuclear respiratory factors (NRFs) (e.g., NRF-1 and NRF-2) (52), thereby increasing the expression of mitochondrial transcription factor A (TFAM) and other nuclear-encoded mitochondrial respiratory complex subunits (118, 119). TFAM is induced to translocate to mitochondria, which stimulates mitochondrial DNA replication and mitochondrial gene expression, thus stimulating the biogenesis of mitochondria (52, 118). It is well-known that SIRT1 and its substrate PGC-1α regulate aspects of energy metabolism through mitochondria (83). PGC-1α activity is regulated by phosphorylation and NAD 1-dependent deacetylation via metabolic biosensors AMPK, SIRT1, and SIRT3 (52, 120). Furthermore, Zhao et al. showed that PGC-1α is essential for mediating AMPK activity to block catabolic responses and suppress oxidative stress in chondrocytes (118).
Autophagy is closely related to apoptosis in the pathogenesis of numerous degenerative diseases, and studies have shown that autophagy is inhibited in OA chondrocytes (121). Autophagy is a mechanism of intracellular catabolism through which cells can remove dysfunctional organelles and macromolecules to prevent the occurrence of cell stress, preventing mitochondrial dysfunction (122). Lotz et al. called the process mitophagy, which eliminates damaged mitochondria and prevents oxidative stress (123). Parkin, an E3 ubiquitin ligase and mitochondrial outer membrane (OMM) protein, operates in conjunction with PTEN-induced kinase 1 (PINK1), and phosphorylation of Parkin by PINK1 transforms it into an active phospho-ubiquitin-dependent E3 ligase, which can respond to the loss of MMP (ΔΨM) to eliminate damaged mitochondria (124). The evidence that Parkin-mediated clearance of damaged mitochondria limits the generation of ROS and prevents the induction of oxidative stress in OA chondrocytes was first demonstrated by Mohammad et al. (60).
Nuclear transcription factor erythroid-2-like factor 2 (Nrf2) plays a chondroprotective role in OA and can suppress metalloproteinase expression induced by IL-1β (125). Nrf2 is a redox-sensitive transcription factor that positively regulates the expression of antioxidant and cytoprotective enzymes, including HO-1, NQO1, GST, SOD, GPx, and CAT (35, 126). Nrf2/antioxidant response element (ARE) signal transduction is one of the crucial antioxidant systems to maintain the redox state and has been regarded as a strategy to eliminate the damage caused by excessive ROS production (99, 127). Heme oxygenase-1 (HO-1), a ARE regulated by Nrf2, has been reported to prevent diseases caused by oxidative stress as a major therapeutic target of Nrf2 (128).
The presence of the antioxidant defense system to avoid mitochondrial dysfunction and excessive chondrocyte apoptosis is extremely limited (129). Research on exogenous drugs to improve mitochondrial function in OA based on endogenous molecular targets is thus necessary (Table 1).
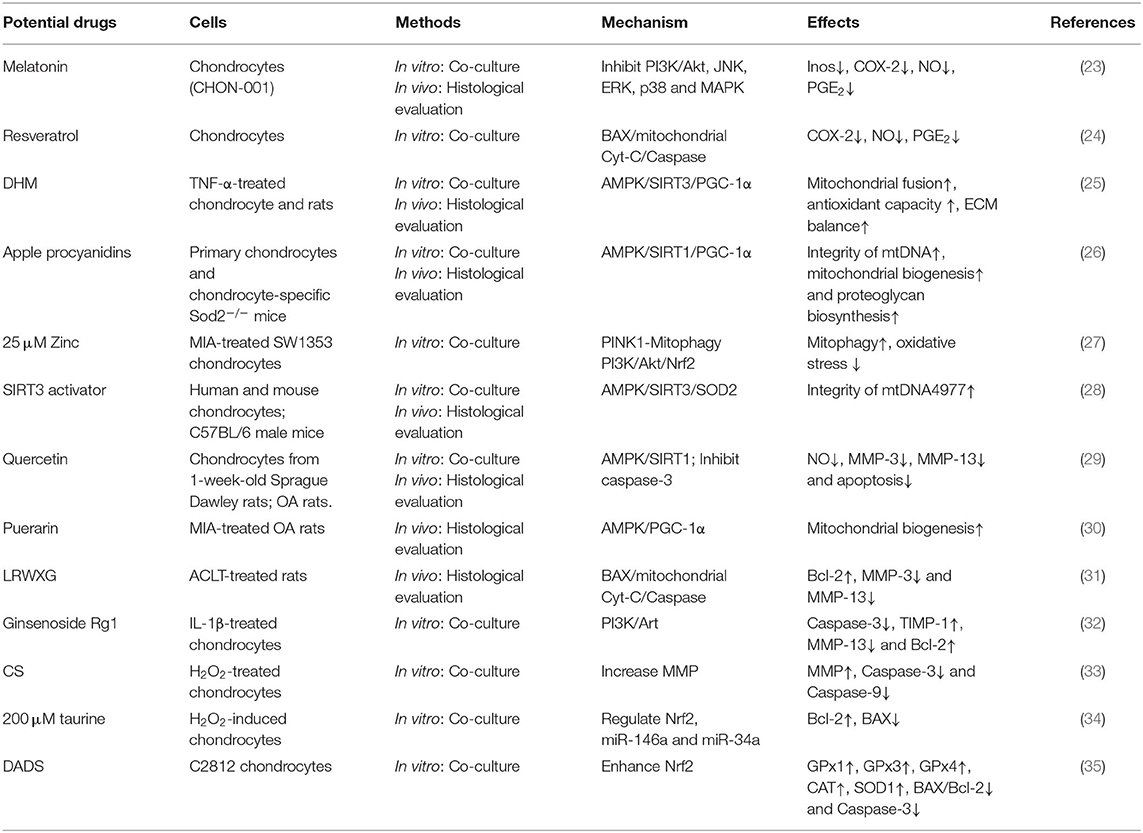
Table 1. Mitochondrial pathways for the treatment of OA.
Appropriate antioxidant strategies and the discovery of antioxidants are essential to protect chondrocytes against oxidative stress (86, 130, 131). Recent studies have shown that melatonin, dihydromyricetin, quercetin, taurine, and diallyl disulfide all act as antioxidants and are potential drugs for the treatment of OA.
Melatonin (N-acetyl-5-methoxytryptamine), an amine hormone produced by the pineal gland of mammals, is a broad-spectrum antioxidant and free radical scavenger (132). Melatonin and its metabolites can remove ROS by radical scavenging and improve the activation of antioxidant enzymes, thus regulating inflammation, proliferation, apoptosis and metastasis (133). Various experiments have demonstrated that melatonin can inhibit the phosphorylation of PI3K/Akt and MAPKs (23) and inhibit the loss of MMP and the release of mitochondrial Cyt-C (134). Kim et al. (23) demonstrated that melatonin acts as a potent inhibitor of H2O2-induced inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) gene expression while also suppressing the production of NO and PGE2 in human chondrocytes, and the researchers thought that the inhibitory effect of melatonin on cartilage degeneration may be associated with the SIRT1 pathway.
Dihydromyricetin (DHM), which is mainly composed of flavonoids, can scavenge free radicals and has anti-inflammatory and antioxidative effects (135). SIRT3 can be activated by DHM through the AMPK/SIRT3/PGC-1α signaling pathway and can enhance mitochondrial fusion, maintain mitochondrial function and the homeostasis of chondrocytes, improve the antioxidant capacity of chondrocytes, and increase aggrecan and collagen II levels (25). DHM can also promote mitophagy to protect chondrocytes by activating SIRT3, which provides a new treatment strategy for OA.
Quercetin, a flavonoid compound, is widely found in vegetables and fruits and possesses antioxidant properties. Studies have revealed that quercetin is a potent anti-atherosclerotic drug as a result of its anti-inflammatory and antioxidative capacities (136). A study showed that quercetin could be used for the treatment of OA rats and demonstrated that quercetin could reverse mitochondrial dysfunction, improving MMP, oxygen consumption, and ATP production. The induction of glutathione (GSH) and glutathione peroxidase (GPX) by quercetin eliminated excessive ROS, which reduced or even abolished oxidative stress (29). Moreover, quercetin inhibited the accumulation of nitric oxide (NO), matrix metalloproteinase 3 (MMP-3), and MMP-13 produced by inflammation through AMPK/SIRT1 signaling, playing a key role in the inhibition of extracellular matrix degeneration. Quercetin also decreased chondrocyte apoptosis by inhibiting the caspase-3 signaling pathway (7). Therefore, quercetin is a potential therapeutic drug for OA that acts through the mitochondrial pathway.
Taurine (2-aminoethane sulfonic acid), another antioxidant that is highly effective in attenuating free radical toxicity, has been identified (137). Taurine can ameliorate ROS-induced chondrocyte damage and exert chondroprotective properties, including the deposition of extracellular matrix components and proliferation of chondrocyte (138). Sara et al. (34) showed that 200 μM taurine could reduce mitochondrial superoxide anion production by activating Nrf2 and promote an increase in anti-apoptotic Bcl-2 and a reduction in proapoptotic BAX to inhibit chondrocyte apoptosis (139). In addition, the regulation of miR-146a and miR-34a expression in OA chondrocytes was first demonstrated. Taurine may be a potential drug for OA.
Diallyl disulfide (DADS), a main component of garlic with antioxidant and anti-inflammatory properties (35, 140), could reduce pro-inflammatory cytokines expression, such as TNF-α, IL-1β, inducible nitric oxide synthase (iNOS), and COX-2 (35), by inhibiting the nuclear factor-κB (NF-κB) signaling pathway (141). Moreover, the pivotal etiological role of apoptosis in cartilage degeneration and the antioxidant and anti-apoptotic properties of DADS were considered (126, 140), the mechanism of DADS in oxidative stress and consequent apoptosis induced by IL-1β in C2812 human chondrocytes was studied by Hosseinzadeh et al. (35). The findings demonstrated that DADS protected C2812 chondrocytes against oxidative stress and reduced ROS and NO production by enhancing Nrf2 nuclear translocation. In addition, DADS markedly enhanced the expression of GPx1, GPx3, GPx4, CAT, and SOD1 and decreased the ratio of BAX/Bcl-2 and Caspase-3 activation to inhibit apoptosis (35). DADS could therefore be extracted and developed a potential drug for OA, and an interesting perspective emerged that a diet rich in garlic might be beneficial to reduce both the incidence and progression of OA.
The natural polyphenolic compound resveratrol (polystilbene, C14H12O3), a non-flavonoid polyphenol compound with anti-inflammatory and antioxidative properties, is mainly derived from grape leaves, grape skin, and various fruits (142). Mitochondrial dysfunction increased the inflammatory response to cytokines in human chondrocytes and resveratrol significantly reduced the inflammatory response (143). Resveratrol alleviated the chondrocyte damage induced by interleukin-1β (IL-1β) through the NF-κB signaling pathway (144). Moreover, resveratrol has been regarded as a potent activator of SIRT1, which can prevent human chondrocyte apoptosis under cellular stresses, including nutritional stress, catabolic stress, and mechanical shear stress, by promoting Bcl-2 translocation to mitochondria and inhibiting BAX translocation to mitochondria (145). The optimization of mitochondrial function in animal models and protection against IL-1β-induced chondrocyte apoptosis can be achieved by resveratrol (24).
Xanthan gum (XG), an extracellular acidic polysaccharide, is released by the fermentation of Xanthomonas (146, 147). Studies have shown that the BCL2-associated X protein (BAX)/Cyt-C/Caspase signaling pathway contributes to cartilage degeneration (88). A low range of molecular weights of XG (LRWXG) has been applied for rabbit OA treatment (31). In this study, the inhibition of cartilage matrix destruction and the protection of subchondral bone were demonstrated. In addition, LRWXG could inhibit the formation of small pores in the mitochondrial inner membrane and inhibit the swelling and rupture of the mitochondrial outer membrane, which could stabilize membrane potential and the permeability of the mitochondrial membrane. Moreover, activation of Bcl-2 and inhibition of BAX activity were achieved by LRWXG. Both of these factors could reduce the translocation of Cyt-C from mitochondria to the cytoplasm (31). The decrease in Cyt-C in the cytoplasm downregulated Caspase-3 and Caspase-9 in chondrocytes, which reduced the formation of apoptotic bodies and decreased chondrocyte apoptosis. Xintian Shao, the author of the study, therefore thought that LRWXG could inhibit chondrocyte apoptosis by conditioning the BAX/mitochondrial Cyt-C/Caspase signaling pathway and protect chondrocytes from degeneration.
Chondroitin sulfate (CS), a glycosaminoglycan that is widely extracted from animal and fish cartilage, is an essential component of the extracellular matrix (148). A study indicated that carp chondroitin sulfate increased MMP and inhibited the levels of Caspase-3 and Caspase-9 by reducing mitochondrial fission, which decreased chondrocyte apoptosis (33). It appears that chondroitin sulfate also has the potential to treat OA through the mitochondrial pathway.
Ginsenoside Rg1 (Rg1) is one of the most active components in ginseng along with steroidal saponin (149). The therapeutic effect of Rg1 on nervous system diseases and cardiovascular diseases has been reported, which inspired Huang et al. to investigate whether Rg1 protected chondrocytes (32). Their findings showed that Rg1 could enhance Bcl-2 expression, advance tissue inhibitor of metalloproteinase-1 (TIMP-1) expression, inhibit Bax activity, inhibit MMP-13 synthesis, and inhibit Cyt C release from mitochondria to the cytosol through enhancing phosphatidylinositol 3-kinase (PI3K)/Akt signaling, which inhibited Caspase-3. The inhibition of Caspase-3 led to the inhibition of chondrocyte apoptosis and protected chondrocytes. Rg1 may thus be a potential treatment for OA treatment through the PI3K/Akt/mitochondrial signaling pathway.
Apple polyphenols from immature apples, compounds composed of several polyphenols, exert anti-allergy, anti-fatigue and life-extending effects (26, 150). Masuda et al. investigated the role of apple polyphenols in protecting chondrocytes and improving OA (26). Their findings showed that apple polyphenols could enhance mitochondrial biogenesis by promoting the integrity of mtDNA and mitochondrial fusion through AMPK/SIRT1/PGC-1α signaling. Moreover, apple polyphenols could promote proteoglycan biosynthesis. In an in vivo study, apple procyanidins protected against articular cartilage degeneration and prevented the development of knee OA in chondrocyte-specific Sod2-/- mice (26). Based on these results, we can conclude that apple polyphenols may be potential drugs for treating OA.
Puerarin, an isoflavone derivative, is isolated from the Chinese medicine Pueraria and possesses antioxidative, anti-inflammatory, anticancer and vasodilating effects (151). The ability of puerarin to restore mitochondrial dysfunction has been confirmed (152). Furthermore, puerarin could reduce mitochondrial dysfunction and damage to chondrocytes by increasing mitochondrial biogenesis and restoring mitochondrial function through the upregulation of AMPK/PGC-1α signaling, which protected chondrocytes in OA (30).
In a study of metformin for the treatment of OA, Chenzhong Wang found that metformin could improve the expression of SIRT3 in chondrocytes and activate the PINK1 (PTEN induced putative kinase 1)/Parkin signaling pathway and could ameliorate mitochondrial function and protect chondrocytes from OA by promoting mitochondrial fusion and eliminating dysfunctional mitochondria through mitophagy (89). Huang et al. showed that 25 μM zinc could protect chondrocytes injured by monosodium iodoacetate (MIA) through the PINK1-dependent selective mitophagy pathway, which indicated that 25 μM zinc was protective against OA (27).
We collected 361 papers, and the dataset from Jan. 2000 to Dec. 2019 was derived from the Web of Science (WOS) Core Collection, which is regarded as the optimum database (153). The search terms were as follows: [(TS = (mitochondria* AND osteoarthritis)] OR [TS = (mitochondrion* AND osteoarthritis))] AND (Language = English) AND (Document type = Article AND Review). The logistic growth model f(x) = a/[1 + eb−cx], where x is the year and f(x) represents the cumulative quantity of papers by year, was used to model the cumulative volume of documentation because of its great fitness and ability to predict future trends (154). VOS viewer (Leiden University, Leiden, Netherlands) were tools used to develop the co-occurrence analysis map (155).
Variations in the quantity of academic publications in a certain research field are a significant indicator of the development trend (155). Determining the number of papers within a period of time and guiding multivariate statistical analysis are conducive to the research level and future trends (155). The global status of mitochondrial and osteoarthritis research has demonstrated that research on mitochondria and OA has been a popular topic in the field. An total of 361 papers from 2000 to 2019 were obtained from the WOS database on the basis of the search formula. Over the past 20 years, there has been a growing trend in global publications, which showed that the relative research on mitochondria and OA increased and the total number of publications significantly increased (Figure 5C). Moreover, Figure 5D shows the logistic regression model meeting curves f(x) = 330/[1 + e328.2543−0.1618x] of the quantity of papers on mitochondria and OA research in the future per year. The top 20 productive countries are listed in Figure 5B due to the total quantity of papers per country. China was the largest contributor with the highest number, and Figure 5A shows the top 25 countries that made the greatest contributions to mitochondrial and OA research globally. The darker the color, the greater the quantity of papers.
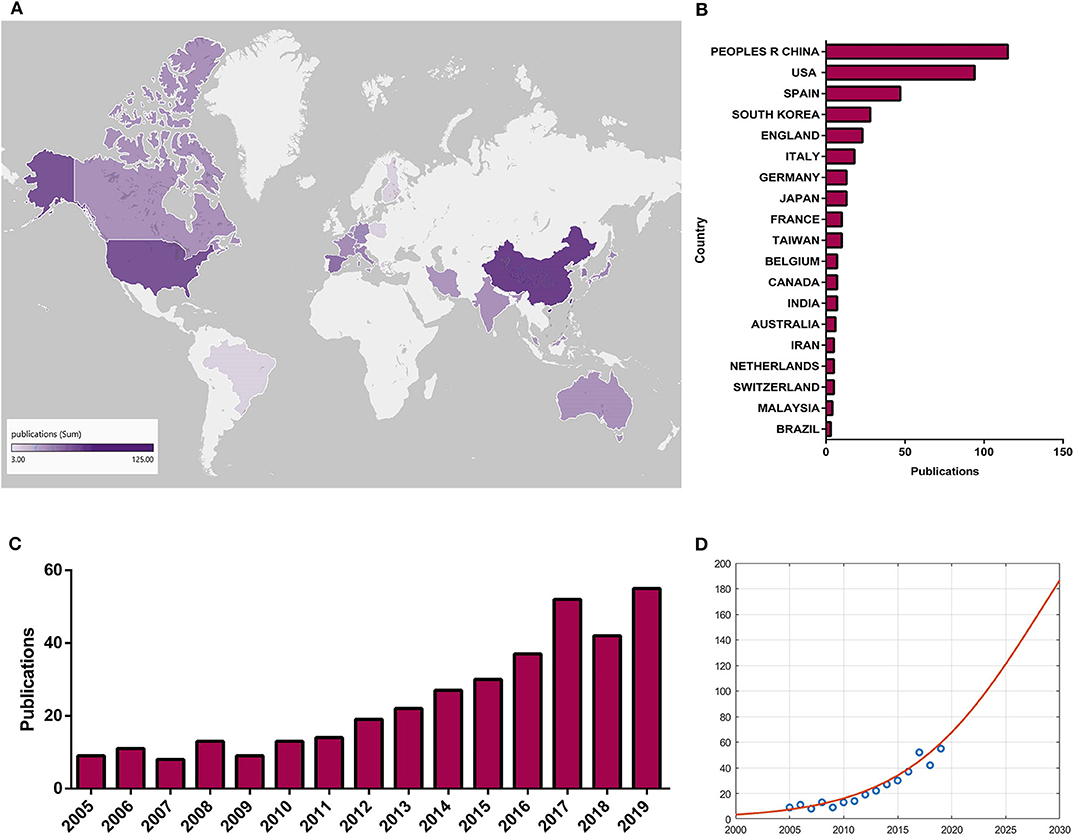
Figure 5. Global trends and contributed countries on mitochondria and OA research. (A) World map showing the distribution of mitochondria and OA research, in which the different color depths represent the different numbers of publications in different countries. (B) The sum of publications related to mitochondria and OA research from 20 countries or regions. (C) The annual number of publications related to mitochondria and OA research in the past 20 years. (D) Model fitting curves of growth trends of accumulated number of publications on mitochondria and OA research.
The purpose of co-occurrence analysis is to determine the relevance of items according to the quantity of projects that appear together and describe the internal relationships and structure of an academic field, and reveal the research frontiers (156). The development of scientific research and programs could be monitored and followed closely as popular topics and directions were identified through co-occurrence analysis (155, 157). Keywords were analyzed by VOS viewer, and 277 identified keywords are shown in Figure 6. The larger the spheres, the greater the frequency. It was obvious that “Apoptosis,” “Chondrocytes,” “Oxidative,” “Nitric-oxide,” and “Autophagy” had the highest frequency and may be the main research themes in the past two decades. In addition, the blue color means that the keywords occurred early, and red colored keywords occurred later. We found that “Phenotype,” “SIRT3,” “PCG-1α,” “AMPK,” “FOXO transcription factors,” “Mitophagy,” “Acetylation,” “Nrf2,” and “Repair,” which were red colored, occurred recently, which may mean that research on mitochondria and osteoarthritis will focus on mechanistic studies and cartilage repair.
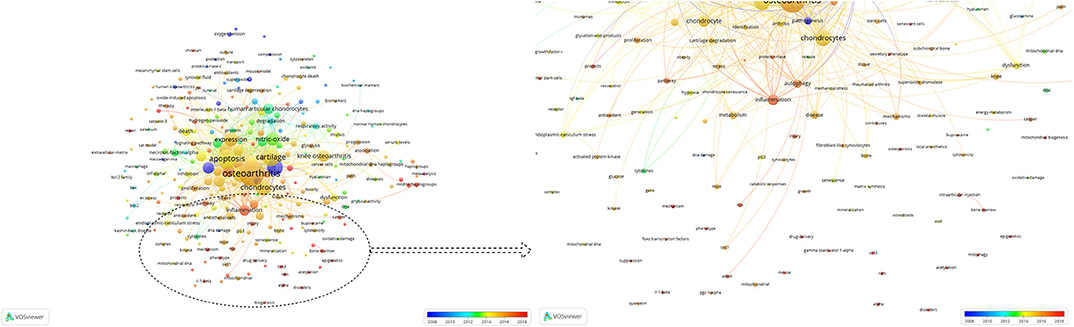
Figure 6. Co-occurrence analysis of global research about mitochondria and OA. Mapping of keywords in the research on mitochondria and OA. The size of the points represents the frequency. Distribution of keywords according to the mean frequency of appearance. Keywords in blue appeared earlier than those in yellow and red colored keywords appeared later.
Research on mitochondria and OA is currently a popular topic. Mechanistic research on the relationship between mitochondria and OA has been launched, and corresponding research on the treatment of OA has also made excellent progress. Although articular cartilage deterioration is the main pathological characteristic of OA, it is now widely accepted that the entire joint, including the synovium, is involved (158). The synovium contributes to the general physiological function of joints and the regulation of the joint microenvironment by secreting synovial fluid to supply nutrients and lubricate the cartilage (159). Fibroblast-like synoviocytes (FLSs) are highly sensitive to hypoxia and reoxygenation (H/R), and IGFBP-3 is overexpressed in cartilage and synovial fluid under H/R conditions (160). The induction of nerve growth factor-induced gene B (NGFI-B, Nur77) by IGFBP-3 has been confirmed (161). The mitochondrial membrane permeability could be enhanced by Nur77, which results in the translocation of Cyt-C from the mitochondrial matrix to the cytoplasm and initiates an intrinsic and classic apoptosis pathway: the caspase pathway (162). Therefore, improving synovitis through the mitochondrial pathway may be a potential strategy for OA treatment (163).
Strategies for OA treatment are tiered, and non-pharmacological methods, including education and self-management, exercise, weight loss if overweight or obese, and walking aids as indicated, are widely recommended and regarded as first-line treatments (3, 164). The most commonly recommended pharmacological methods in the guidelines include paracetamol and NSAIDs (3, 165). In the context of surgery, joint replacement surgery, knee osteotomy, knee joint distraction and arthroscopic knee surgery (3), and autologous chondrocyte transplantation are currently the most effective treatments (166). The current pharmacological methods used to OA treatment are largely palliative (3), thus modifying OA progression, including slowing, halting, and reversing progression, are critical.
Biotherapy and gene therapy are current research trends in disease treatment. Stem cells, including mesenchymal stem cells (BMSCs), umbilical cord stem cells, embryonic stem cells and induced pluripotent stem cells, are regarded as exceptional donor cells for mitochondrial transfer, and numerous studies have confirmed the significance of mitochondrial transfer in stem cell therapy (167), especially BMSCs (168). Moreover, transplantation of stem cells has recently become a research hotspot in treating tissue injury. Whether stem cell transplantation can optimize mitochondrial function in OA is therefore worth exploring. Exosomes, which are extracellular vesicles 30–150 nm in diameter, have similar functions as those of derived cells without apparent side effects in both healthy and diseased cells (169), and studies have shown that the therapeutic effects of mesenchymal stem cells (MSCs) can be replicated by their secreted exosomes (52, 170). MSC-derived exosomes possess the biochemical potential to restore homeostasis in bioenergetics, cell number and immunomodulation (52, 171, 172). Exosomes contain mitochondrial membrane components and mtDNA (173). Zheng et al. investigated the ability of primary chondrocyte-derived exosomes to abrogate mitochondrial dysfunction in degenerated chondrocytes (170). The results indicated that exosomes from chondrocytes could reduce the expression of inflammatory cytokines, restore mitochondrial dysfunction, and reduce macrophage polarization toward an M2 phenotype, resulting in the repair of injured chondrocytes. This finding is in accordance with the treatment of a mouse OA model with chondrocyte exosomes. Collectively, primary chondrocyte exosomes are potential disease-modifying therapeutic agents for OA. We therefore thought biomedical measures would be efficient for the treatment of OA based on optimizing mitochondrial function. CRISPR/Cas9 is the most convenient gene-editing tool so far, widely used in human embryonic stem cells (hESCs) and their derivatives for basic and clinical research (16, 174). Deng et al. showed that MSCs without DiGeorge syndrome critical region 8 (DGCR8) could alleviate human MSC senescence and mouse osteoarthritis (16, 175). More efficient and targeted gene-editing tools need to be developed, which contribute to precise genetic and epigenetic regulation, such as activation or inhibition of target genes in vivo (16). We thus predict that gene therapy will be a radical therapeutic strategy for OA treatment.
There are still numerous mechanisms that need to be further explored. Pain is the main symptom of OA patients and a major driver of clinical decisions (3, 176); therefore, whether the new strategy targeting the mitochondrial pathway for OA has an effect on pain relief is still unclear in the current study. Microvesicles are popular for research on the mechanism of OA treatment and whether microvesicles can promote mitochondrial fusion and biosynthesis to reduce chondrocyte apoptosis is not known. At the same time, how drugs that affect the mitochondrial pathway in OA work in mitochondria, which are subcellular organelles, is unclear. It is widely accepted that mitochondria and the nucleus are in two-way communication, and the way mitochondria conduct signal transduction with the nucleus after exposure to a drug effects to inhibit cell apoptosis and protect cells is worth studying. With the continuous investment in mitochondrial and OA research worldwide, a new strategy targeting the mitochondrial pathway in OA will have great breakthroughs and will make a great contribution to the treatment of OA. The day is coming when we will provide subcellular, cellular, and tissue-level mechanistic and clinical evidence for the treatment of OA to provide a more comprehensive and efficient treatment for OA patients.
XM, LW, and CX made the review article structure. CX and LW are responsible for reviewing. XM and PF finished writing. All authors contributed to the article and approved the submitted version.
This study was supported by a grant from the National Natural Science Foundation of China (No. 81972075) and the Shanxi Province Science and Technology Research Project (201803D421050).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. He BH, Christin M, Mouchbahani-Constance S, Davidova A, Sharif-Naeini R. Mechanosensitive ion channels in articular nociceptors drive mechanical allodynia in osteoarthritis. Osteoarthr Cartilage. (2017) 25:2091–9. doi: 10.1016/j.joca.2017.08.012
2. Cross M, Smith E, Hoy D, Nolte S, Ackerman I, Fransen M, et al. The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. (2014) 73:1323–30. doi: 10.1136/annrheumdis-2013-204763
3. Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. (2019) 393:1745–59. doi: 10.1016/S0140-6736(19)30417-9
4. Prieto-Alhambra D, Judge A, Javaid MK, Cooper C, Diez-Perez A, Arden NK. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: influences of age, gender and osteoarthritis affecting other joints. Ann Rheum Dis. (2014) 73:1659–64. doi: 10.1136/annrheumdis-2013-203355
5. Habiballa L, Salmonowicz H, Passos JF. Mitochondria and cellular senescence: implications for musculoskeletal ageing. Free Radic Biol Med. (2019) 132:3–10. doi: 10.1016/j.freeradbiomed.2018.10.417
6. Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, et al. Osteoarthritis. Nat Rev Dis Primers. (2016) 2:16072. doi: 10.1038/nrdp.2016.72
7. Hu Y, Gui Z, Zhou Y, Xia L, Lin K, Xu Y. Quercetin alleviates rat osteoarthritis by inhibiting inflammation and apoptosis of chondrocytes, modulating synovial macrophages polarization to M2 macrophages. Free Radic Biol Med. (2019) 145:146–60. doi: 10.1016/j.freeradbiomed.2019.09.024
8. Blanco FJ, López-Armada MJ, Maneiro E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion. (2004) 4:715–28. doi: 10.1016/j.mito.2004.07.022
9. Charlier E, Relic B, Deroyer C, Malaise O, Neuville S, Collée J, et al. Insights on molecular mechanisms of chondrocytes death in osteoarthritis. Int J Mol Sci. (2016) 17:2146. doi: 10.3390/ijms17122146
10. Lepetsos P, Papavassiliou AG. ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta. (2016) 1862:576–91. doi: 10.1016/j.bbadis.2016.01.003
11. Maneiro E, López-Armada MJ, de Andres MC, Caramés B, Martín MA, Bonilla A, et al. Effect of nitric oxide on mitochondrial respiratory activity of human articular chondrocytes. Ann Rheum Dis. (2005) 64:388–95. doi: 10.1136/ard.2004.022152
12. Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. (2011) 7:161–9. doi: 10.1038/nrrheum.2010.213
13. Birch J, Passos JF. Targeting the SASP to combat ageing: mitochondria as possible intracellular allies. Bioessays. (2017) 39:1600235. doi: 10.1002/bies.201600235
14. Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. (1989) 1:642–5. doi: 10.1016/s0140-6736(89)92145-4
15. Hwang HS, Kim HA. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int J Mol Sci. (2015) 16:26035–54. doi: 10.3390/ijms161125943
16. Zhang J, Liu GH, Qu J, Song M. Treating osteoarthritis via gene therapy with rejuvenation factors. Gene Ther. (2020) 27:309–11. doi: 10.1038/s41434-020-0149-5
17. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. (2018) 128:3716–26. doi: 10.1172/JCI120849
18. Ruiz-Romero C, López-Armada MJ, Blanco FJ. Mitochondrial proteomic characterization of human normal articular chondrocytes. Osteoarthritis Cartilage. (2006) 14:507–18. doi: 10.1016/j.joca.2005.12.004
19. Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martínez-Gomariz M, Fernández M, et al. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics. (2009) 8:172–89. doi: 10.1074/mcp.M800292-MCP200
20. Coleman MC, Goetz JE, Brouillette MJ, Seol D, Willey MC, Petersen EB, et al. Targeting mitochondrial responses to intra-articular fracture to prevent posttraumatic osteoarthritis. Sci Transl Med. (2018) 10. doi: 10.1126/scitranslmed.aan5372
21. Rambold AS, Pearce EL. Mitochondrial dynamics at the interface of immune cell metabolism and function. Trends Immunol. (2018) 39:6–18. doi: 10.1016/j.it.2017.08.006
22. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. (2018) 62:341–60. doi: 10.1042/EBC20170104
23. Lim HD, Kim YS, Ko SH, Yoon IJ, Cho SG, Chun YH, et al. Cytoprotective and anti-inflammatory effects of melatonin in hydrogen peroxide-stimulated CHON-001 human chondrocyte cell line and rabbit model of osteoarthritis via the SIRT1 pathway. J Pineal Res. (2012) 53:225–37. doi: 10.1111/j.1600-079X.2012.00991.x
24. Dave M, Attur M, Palmer G, Al-Mussawir HE, Kennish L, Patel J, et al. The antioxidant resveratrol protects against chondrocyte apoptosis via effects on mitochondrial polarization and ATP production. Arthritis Rheum. (2008) 58:2786–97. doi: 10.1002/art.23799
25. Wang J, Wang K, Huang C, Lin D, Zhou Y, Wu Y, et al. SIRT3 Activation by Dihydromyricetin Suppresses Chondrocytes Degeneration via Maintaining Mitochondrial Homeostasis, Int J Biol Sci. (2018) 14:1873–82. doi: 10.7150/ijbs.27746
26. Masuda I, Koike M, Nakashima S, Mizutani Y, Ozawa Y, Watanabe K, et al. Apple procyanidins promote mitochondrial biogenesis and proteoglycan biosynthesis in chondrocytes. Sci Rep. (2018) 8:7229. doi: 10.1038/s41598-018-25348-1
27. Huang LW, Huang TC, Hu YC, Hsieh BS, Chiu PR, Cheng HL, et al. Zinc protects chondrocytes from monosodium iodoacetate-induced damage by enhancing ATP and mitophagy. Biochem Biophys Res Commun. (2020) 521:50–6. doi: 10.1016/j.bbrc.2019.10.066
28. Chen LY, Wang Y, Terkeltaub R, Liu-Bryan R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthritis Cartilage. (2018) 26:1539–50. doi: 10.1016/j.joca.2018.07.004
29. Qiu L, Luo Y, Chen X. Quercetin attenuates mitochondrial dysfunction and biogenesis via upregulated AMPK/SIRT1 signaling pathway in OA rats. Biomed Pharmacother. (2018) 103:1585–91. doi: 10.1016/j.biopha.2018.05.003
30. Wang L, Shan H, Wang B, Wang N, Zhou Z, Pan C, et al. Puerarin attenuates osteoarthritis via upregulating AMP-activated protein kinase/proliferator-activated receptor-γ coactivator-1 signaling pathway in osteoarthritis rats. Pharmacology. (2018) 102:117–25. doi: 10.1159/000490418
31. Shao X, Chen Q, Dou X, Chen L, Wu J, Zhang W, et al. Lower range of molecular weight of xanthan gum inhibits cartilage matrix destruction via intrinsic bax-mitochondria cytochrome c-caspase pathway. Carbohydr Polym. (2018) 198:354–63. doi: 10.1016/j.carbpol.2018.06.108
32. Huang Y, Wu D, Fan W. Protection of ginsenoside Rg1 on chondrocyte from IL-1β-induced mitochondria-activated apoptosis through PI3K/Akt signaling. Mol Cell Biochem. (2014) 392:249–57. doi: 10.1007/s11010-014-2035-1
33. Liu Q, Wang J, Sun Y, Han S. Chondroitin sulfate from sturgeon bone protects chondrocytes via inhibiting apoptosis in osteoarthritis. Int J Biol Macromol. (2019) 134:1113–9. doi: 10.1016/j.ijbiomac.2019.05.110
34. Cheleschi S, De Palma A, Pascarelli NA, Giordano N, Galeazzi M, Tenti S, et al. Could oxidative stress regulate the expression of MicroRNA-146a and MicroRNA-34a in human osteoarthritic chondrocyte cultures. Int J Mol Sci. (2017) 18:2660. doi: 10.3390/ijms18122660
35. Hosseinzadeh A, Jafari D, Kamarul T, Bagheri A, Sharifi AM. Evaluating the protective effects and mechanisms of diallyl disulfide on interlukin-1β-induced oxidative stress and mitochondrial apoptotic signaling pathways in cultured chondrocytes. J Cell Biochem. (2017) 118:1879–88. doi: 10.1002/jcb.25907
36. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
37. Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov. (2013) 12:465–83. doi: 10.1038/nrd4023
38. Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu B, et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature. (2017) 542:372–6. doi: 10.1038/nature21077
39. Pagliuso A, Cossart P, Stavru F. The ever-growing complexity of the mitochondrial fission machinery. Cell Mol Life Sci. (2018) 75:355–74. doi: 10.1007/s00018-017-2603-0
40. Blanco FJ, Rego-Pérez I. Mitochondria and mitophagy: biosensors for cartilage degradation and osteoarthritis. Osteoarthritis Cartilage. (2018) 26:989–91. doi: 10.1016/j.joca.2018.05.018
41. Blanco FJ, Fernández-Moreno M. Mitochondrial biogenesis: a potential therapeutic target for osteoarthritis. Osteoarthritis Cartilage. (2020) 28:1003–6. doi: 10.1016/j.joca.2020.03.018
42. Scorrano L. Keeping mitochondria in shape: a matter of life and death. Eur J Clin Invest. (2013) 43:886–93. doi: 10.1111/eci.12135
43. Raimundo N. Mitochondrial pathology: stress signals from the energy factory. Trends Mol Med. (2014) 20:282–92. doi: 10.1016/j.molmed.2014.01.005
44. Shapiro IM, Golub EE, Kakuta S, Hazelgrove J, Havery J, Chance B, et al. Initiation of endochondral calcification is related to changes in the redox state of hypertrophic chondrocytes. Science. (1982) 217:950–2. doi: 10.1126/science.7112108
45. Anderson HC. Electron microscopic studies of induced cartilage development and calcification. J Cell Biol. (1967) 35:81–101. doi: 10.1083/jcb.35.1.81
46. Lehninger AL. Mitochondria and biological mineralization processes: an exploration. Horiz Biochem Biophys. (1977) 4:1–30.
47. Boonrungsiman S, Gentleman E, Carzaniga R, Evans ND, McComb DW, Porter AE, et al. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc Natl Acad Sci USA. (2012) 109:14170–5. doi: 10.1073/pnas.1208916109
48. Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. (2013) 52:2793–809. doi: 10.1021/bi3015983
49. Murphy AN, Kelleher JK, Fiskum G. Submicromolar Ca2+ regulates phosphorylating respiration by normal rat liver and AS-30D hepatoma mitochondria by different mechanisms. J Biol Chem. (1990) 265:10527–34.
50. Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. (2000) 466:130–4. doi: 10.1016/s0014-5793(99)01773-1
51. Cheleschi S, Cantarini L, Pascarelli NA, Collodel G, Lucherini OM, Galeazzi M, et al. Possible chondroprotective effect of canakinumab: an In vitro study on human osteoarthritic chondrocytes. Cytokine. (2015) 71:165–72. doi: 10.1016/j.cyto.2014.10.023
52. Wang Y, Zhao X, Lotz M, Terkeltaub R, Liu-Bryan R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. (2015) 67:2141–53. doi: 10.1002/art.39182
53. Terkeltaub R, Johnson K, Murphy A, Ghosh S. Invited review: the mitochondrion in osteoarthritis. Mitochondrion. (2002) 1:301–19. doi: 10.1016/s1567-7249(01)00037-x
54. Cillero-Pastor B, Mateos J, Fernández-López C, Oreiro N, Ruiz-Romero C, Blanco FJ. Dimethylarginine dimethylaminohydrolase 2, a newly identified mitochondrial protein modulating nitric oxide synthesis in normal human chondrocytes. Arthritis Rheum. (2012) 64:204–12. doi: 10.1002/art.30652
55. Caramés B, López-Armada MJ, Cillero-Pastor B, Lires-Dean M, Vaamonde C, Galdo F, et al. Differential effects of tumor necrosis factor-alpha and interleukin-1beta on cell death in human articular chondrocytes. Osteoarthritis Cartilage. (2008) 16:715–22. doi: 10.1016/j.joca.2007.10.006
56. Cannon B, Shabalina IG, Kramarova TV, Petrovic N, Nedergaard J. Uncoupling proteins: a role in protection against reactive oxygen species–or not. Biochim Biophys Acta. (2006) 1757:449–58. doi: 10.1016/j.bbabio.2006.05.016
57. Minguzzi M, Cetrullo S, D'Adamo S, Silvestri Y, Flamigni F, Borzì RM. Emerging players at the intersection of chondrocyte loss of maturational arrest, oxidative stress, senescence and low-grade inflammation in osteoarthritis. Oxid Med Cell Longev. (2018) 2018:3075293. doi: 10.1155/2018/3075293
58. Melrose J, Shu C, Whitelock JM, Lord MS. The cartilage extracellular matrix as a transient developmental scaffold for growth plate maturation. Matrix Biol. (2016) 52–54:363–83. doi: 10.1016/j.matbio.2016.01.008
59. Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. (1998) 78:547–81. doi: 10.1152/physrev.1998.78.2.547
60. Ansari MY, Khan NM, Ahmad I, Haqqi TM. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthritis Cartilage. (2018) 26:1087–97. doi: 10.1016/j.joca.2017.07.020
61. Trifunovic A, Larsson NG. Mitochondrial dysfunction as a cause of ageing. J Intern Med. (2008) 263:167–78. doi: 10.1111/j.1365-2796.2007.01905.x
62. Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. (2011) 7:33–42. doi: 10.1038/nrrheum.2010.196
63. Akhmedov AT, Marín-García J. Mitochondrial DNA maintenance: an appraisal. Mol Cell Biochem. (2015) 409:283–305. doi: 10.1007/s11010-015-2532-x
64. Scott JL, Gabrielides C, Davidson RK, Swingler TE, Clark IM, Wallis GA, et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann Rheum Dis. (2010) 69:1502–10. doi: 10.1136/ard.2009.119966
65. Gavriilidis C, Miwa S, von Zglinicki T, Taylor RW, Young DA. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. (2013) 65:378–87. doi: 10.1002/art.37782
66. Mammucari C, Rizzuto R. Signaling pathways in mitochondrial dysfunction and aging. Mech Ageing Dev. (2010) 131:536–43. doi: 10.1016/j.mad.2010.07.003
67. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
68. Arkill KP, Winlove CP. Fatty acid transport in articular cartilage. Arch Biochem Biophys. (2006) 456:71–8. doi: 10.1016/j.abb.2006.09.014
69. Farnaghi S, Prasadam I, Cai G, Friis T, Du Z, Crawford R, et al. Protective effects of mitochondria-targeted antioxidants and statins on cholesterol-induced osteoarthritis. FASEB J. (2017) 31:356–67. doi: 10.1096/fj.201600600R
70. Oliviero F, Lo Nigro A, Bernardi D, Giunco S, Baldo G, Scanu A, et al. A comparative study of serum and synovial fluid lipoprotein levels in patients with various arthritides. Clin Chim Acta. (2012) 413:303–7. doi: 10.1016/j.cca.2011.10.019
71. Bouron A, Chauvet S, Dryer S, Rosado JA. Second messenger-operated calcium entry through TRPC6. Adv Exp Med Biol. (2016) 898:201–49. doi: 10.1007/978-3-319-26974-0_10
72. Huser CA, Davies ME. Calcium signaling leads to mitochondrial depolarization in impact-induced chondrocyte death in equine articular cartilage explants. Arthritis Rheum. (2007) 56:2322–34. doi: 10.1002/art.22717
73. Yin S, Zhang L, Ding L, Huang Z, Xu B, Li X, et al. Transient receptor potential ankyrin 1 (trpa1) mediates il-1β-induced apoptosis in rat chondrocytes via calcium overload and mitochondrial dysfunction. J Inflamm (Lond). (2018) 15:27. doi: 10.1186/s12950-018-0204-9
74. Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J Physiol. (1999) 516 (Pt 1):1–17. doi: 10.1111/j.1469-7793.1999.001aa.x
75. Antoniel M, Giorgio V, Fogolari F, Glick GD, Bernardi P, Lippe G. The oligomycin-sensitivity conferring protein of mitochondrial ATP synthase: emerging new roles in mitochondrial pathophysiology. Int J Mol Sci. (2014) 15:7513–36. doi: 10.3390/ijms15057513
76. Bauer TM, Murphy E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ Res. (2020) 126:280–93. doi: 10.1161/CIRCRESAHA.119.316306
77. Singh BK, Tripathi M, Pandey PK, Kakkar P. Nimesulide aggravates redox imbalance and calcium dependent mitochondrial permeability transition leading to dysfunction. In vitro. Toxicology. (2010) 275:1–9. doi: 10.1016/j.tox.2010.05.001
78. Singh BK, Tripathi M, Pandey PK, Kakkar P. Alteration in mitochondrial thiol enhances calcium ion dependent membrane permeability transition and dysfunction In vitro: a cross-talk between mtThiol, Ca(2+), and ROS. Mol Cell Biochem. (2011) 357:373–85. doi: 10.1007/s11010-011-0908-0
79. Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, et al. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. (2005) 120:275–85. doi: 10.1016/j.cell.2004.11.049
80. Oh SH, Lee BH, Lim SC. Cadmium induces apoptotic cell death in WI 38 cells via caspase-dependent Bid cleavage and calpain-mediated mitochondrial Bax cleavage by Bcl-2-independent pathway. Biochem Pharmacol. (2004) 68:1845–55. doi: 10.1016/j.bcp.2004.06.021
81. Toh WS, Brittberg M, Farr J, Foldager CB, Gomoll AH, Hui JH, et al. Cellular senescence in aging and osteoarthritis. Acta Orthop. (2016) 87:6–14. doi: 10.1080/17453674.2016.1235087
82. Thomas CM, Fuller CJ, Whittles CE, Sharif M. Chondrocyte death by apoptosis is associated with cartilage matrix degradation. Osteoarthritis Cartilage. (2007) 15:27–34. doi: 10.1016/j.joca.2006.06.012
83. Ma CH, Chiua YC, Wu CH, Jou IM, Tu YK, Hung CH, et al. Homocysteine causes dysfunction of chondrocytes and oxidative stress through repression of SIRT1/AMPK pathway: a possible link between hyperhomocysteinemia and osteoarthritis. Redox Biol. (2018) 15:504–12. doi: 10.1016/j.redox.2018.01.010
84. Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. (1999) 341 (Pt 2):233–49.
85. Maneiro E, Martín MA, de Andres MC, López-Armada MJ, Fernández-Sueiro JL, del Hoyo P, et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. (2003) 48:700–8. doi: 10.1002/art.10837
86. Park C, Hong SH, Shin SS, Lee DS, Han MH, Cha HJ, et al. Activation of the Nrf2/HO-1 signaling pathway contributes to the protective effects of sargassum serratifolium extract against oxidative stress-induced DNA damage and apoptosis in SW1353 human chondrocytes. Int J Environ Res Public Health. (2018) 15:1173. doi: 10.3390/ijerph15061173
87. Wenz T. PGC-1alpha activation as a therapeutic approach in mitochondrial disease. IUBMB Life. (2009) 61:1051–62. doi: 10.1002/iub.261
88. Liang S, Sun K, Wang Y, Dong S, Wang C, Liu L, et al. Role of Cyt-C/caspases-9,3, Bax/Bcl-2 and the FAS death receptor pathway in apoptosis induced by zinc oxide nanoparticles in human aortic endothelial cells and the protective effect by alpha-lipoic acid. Chem Biol Interact. (2016) 258:40–51. doi: 10.1016/j.cbi.2016.08.013
89. Wang C, Yang Y, Zhang Y, Liu J, Yao Z, Zhang C. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci Trends. (2019) 12:605–12. doi: 10.5582/bst.2018.01263
90. Zhang T, Ikejima T, Li L, Wu R, Yuan X, Zhao J, et al. Impairment of mitochondrial biogenesis and dynamics involved in isoniazid-induced apoptosis of HepG2 Cells Was Alleviated by p38 MAPK Pathway. Front Pharmacol. (2017) 8:753. doi: 10.3389/fphar.2017.00753
91. Cheng QQ, Wan YW, Yang WM, Tian MH, Wang YC, He HY, et al. Gastrodin protects H9c2 cardiomyocytes against oxidative injury by ameliorating imbalanced mitochondrial dynamics and mitochondrial dysfunction. Acta Pharmacol Sin. (2020) 41:1314–27. doi: 10.1038/s41401-020-0382-x
92. Cortés-Pereira E, Fernández-Tajes J, Fernández-Moreno M, Vázquez-Mosquera ME, Relaño S, Ramos-Louro P, et al. Differential association of mitochondrial dna haplogroups j and h with the methylation status of articular cartilage: potential role in apoptosis and metabolic and developmental processes. Arthritis Rheumatol. (2019) 71:1191–200. doi: 10.1002/art.40857
93. Larsson NG, Clayton DA. Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet. (1995) 29:151–78. doi: 10.1146/annurev.ge.29.120195.001055
94. Grishko VI, Ho R, Wilson GL, Pearsall AW IV. Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthritis Cartilage. (2009) 17:107–13. doi: 10.1016/j.joca.2008.05.009
95. McCulloch K, Litherland GJ, Rai TS. Cellular senescence in osteoarthritis pathology, Aging Cell. (2017) 16:210–8. doi: 10.1111/acel.12562
96. Blanco FJ, Valdes AM, Rego-Pérez I. Mitochondrial DNA variation and the pathogenesis of osteoarthritis phenotypes. Nat Rev Rheumatol. (2018) 14:327–40. doi: 10.1038/s41584-018-0001-0
97. Blanco FJ, June RK II. Cartilage metabolism, mitochondria, and osteoarthritis. J Am Acad Orthop Surg. (2019) 28:e242–e244. doi: 10.5435/JAAOS-D-19-00442
98. Rego-Pérez I, Blanco FJ, Roemer FW, Guermazi A, Ran D, Ashbeck EL, et al. Mitochondrial DNA haplogroups associated with MRI-detected structural damage in early knee osteoarthritis. Osteoarthritis Cartilage. (2018) 26:1562–9. doi: 10.1016/j.joca.2018.06.016
99. Collins JA, Diekman BO, Loeser RF. Targeting aging for disease modification in osteoarthritis. Curr Opin Rheumatol. (2018) 30:101–7. doi: 10.1097/BOR.0000000000000456
100. Liu H, Li Z, Cao Y, Cui Y, Yang X, Meng Z, et al. Effect of chondrocyte mitochondrial dysfunction on cartilage degeneration: A possible pathway for osteoarthritis pathology at the subcellular level. Mol Med Rep. (2019) 20:3308–16. doi: 10.3892/mmr.2019.10559
101. Witczak CA, Sharoff CG, Goodyear LJ. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci. (2008) 65:3737–55. doi: 10.1007/s00018-008-8244-6
102. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. (2009) 89:1025–78. doi: 10.1152/physrev.00011.2008
103. Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1β and tumor necrosis factor α. Arthritis Rheum. (2011) 63:1928–37. doi: 10.1002/art.30333
104. Petursson F, Husa M, June R, Lotz M, Terkeltaub R, Liu-Bryan R. Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res Ther. (2013) 15:R77. doi: 10.1186/ar4254
105. Cantó C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. (2009) 20:98–105. doi: 10.1097/MOL.0b013e328328d0a4
106. Fang H, Liu X, Shen L, Li F, Liu Y, Chi H, et al. Role of mtDNA haplogroups in the prevalence of knee osteoarthritis in a southern Chinese population. Int J Mol Sci. (2014) 15:2646–59. doi: 10.3390/ijms15022646
107. Moon MH, Jeong JK, Lee YJ, Seol JW, Jackson CJ, Park SY. SIRT1, a class III histone deacetylase, regulates TNF-α-induced inflammation in human chondrocytes. Osteoarthritis Cartilage. (2013) 21:470–80. doi: 10.1016/j.joca.2012.11.017
108. Srinivas V, Bohensky J, Shapiro IM. Autophagy: a new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs. (2009) 189:88–92. doi: 10.1159/000151428
109. Caramés B, Kiosses WB, Akasaki Y, Brinson DC, Eap W, Koziol J, et al. Glucosamine activates autophagy In vitro and In vivo. Arthritis Rheum. (2013) 65:1843–52. doi: 10.1002/art.37977
110. Caramés B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. (2012) 71:575–81. doi: 10.1136/annrheumdis-2011-200557
111. Zhang M, Tang J, Li Y, Xie Y, Shan H, Chen M, et al. Curcumin attenuates skeletal muscle mitochondrial impairment in COPD rats: PGC-1α/SIRT3 pathway involved. Chem Biol Interact. (2017) 277:168–75. doi: 10.1016/j.cbi.2017.09.018
112. Ansari A, Rahman MS, Saha SK, Saikot FK, Deep A, Kim KH. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell. (2017) 16:4–16. doi: 10.1111/acel.12538
113. Fu Y, Kinter M, Hudson J, Humphries KM, Lane RS, White JR, et al. Aging promotes sirtuin 3-dependent cartilage superoxide dismutase 2 acetylation and osteoarthritis. Arthritis Rheumatol. (2016) 68:1887–98. doi: 10.1002/art.39618
114. Eiyama A, Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol. (2015) 33:95–101. doi: 10.1016/j.ceb.2015.01.002
115. Sasaki H, Takayama K, Matsushita T, Ishida K, Kubo S, Matsumoto T, et al. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. (2012) 64:1920–8. doi: 10.1002/art.34323
116. Feng J, Lu C, Dai Q, Sheng J, Xu M. SIRT3 facilitates amniotic fluid stem cells to repair diabetic nephropathy through protecting mitochondrial homeostasis by modulation of mitophagy. Cell Physiol Biochem. (2018) 46:1508–24. doi: 10.1159/000489194
117. Tseng AH, Shieh SS, Wang DL. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med. (2013) 63:222–34. doi: 10.1016/j.freeradbiomed.2013.05.002
118. Zhao X, Petursson F, Viollet B, Lotz M, Terkeltaub R, Liu-Bryan R. Peroxisome proliferator-activated receptor γ coactivator 1α and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheumatol. (2014) 66:3073–82. doi: 10.1002/art.38791
119. Kang C, Li Ji L. Role of PGC-1α signaling in skeletal muscle health and disease. Ann N Y Acad Sci. (2012) 1271:110–7. doi: 10.1111/j.1749-6632.2012.06738.x
120. Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network, Ageing Res Rev. (2012) 11:230–41. doi: 10.1016/j.arr.2011.12.005
121. Cetrullo S, D'Adamo S, Guidotti S, Borzì RM, Flamigni F. Hydroxytyrosol prevents chondrocyte death under oxidative stress by inducing autophagy through sirtuin 1-dependent and -independent mechanisms. Biochim Biophys Acta. (2016) 1860:1181–91. doi: 10.1016/j.bbagen.2016.03.002
122. López de Figueroa P, Lotz MK, Blanco FJ, Caramés B. Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes, Arthritis Rheumatol. (2015) 67:966–76. doi: 10.1002/art.39025
123. Caramés B, Olmer M, Kiosses WB, Lotz MK. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. (2015) 67:1568–76. doi: 10.1002/art.39073
124. Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. (2013) 496:372–6. doi: 10.1038/nature12043
125. Poulet B, Staines KA. New developments in osteoarthritis and cartilage biology. Curr Opin Pharmacol. (2016) 28:8–13. doi: 10.1016/j.coph.2016.02.009
126. Shan Y, Wei Z, Tao L, Wang S, Zhang F, Shen C, et al. Prophylaxis of diallyl disulfide on skin carcinogenic model via p21-dependent Nrf2 stabilization. Sci Rep. (2016) 6:35676. doi: 10.1038/srep35676
127. Marchev AS, Dimitrova PA, Burns AJ, Kostov RV, Dinkova-Kostova AT, Georgiev MI. Oxidative stress and chronic inflammation in osteoarthritis: can NRF2 counteract these partners in crime. Ann N Y Acad Sci. (2017) 1401:114–35. doi: 10.1111/nyas.13407
128. Ndisang JF. Synergistic interaction between heme oxygenase (HO) and nuclear-factor e2- related factor-2 (nrf2) against oxidative stress in cardiovascular related diseases. Curr Pharm Des. (2017) 23:1465–70. doi: 10.2174/1381612823666170113153818
129. Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. (2016) 12:412–20. doi: 10.1038/nrrheum.2016.65
130. Portal-Núñez S, Esbrit P, Alcaraz MJ, Largo R. Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem Pharmacol. (2016) 108:1–10. doi: 10.1016/j.bcp.2015.12.012
131. Chin KY, Pang KL. Therapeutic effects of olive and its derivatives on osteoarthritis: from bench to bedside. Nutrients. (2017) 9:1060. doi: 10.3390/nu9101060
132. Hosseinzadeh A, Kamrava SK, Joghataei MT, Darabi R, Shakeri-Zadeh A, Shahriari M, et al. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J Pineal Res. (2016) 61:411–25. doi: 10.1111/jpi.12362
133. Borin TF, Arbab AS, Gelaleti GB, Ferreira LC, Moschetta MG, Jardim-Perassi BV, et al. Melatonin decreases breast cancer metastasis by modulating Rho-associated kinase protein-1 expression. J Pineal Res. (2016) 60:3–15. doi: 10.1111/jpi.12270
134. Rodella LF, Favero G, Rossini C, Foglio E, Bonomini F, Reiter RJ, et al. Aging and vascular dysfunction: beneficial melatonin effects. Age (Dordr). (2013) 35:103–15. doi: 10.1007/s11357-011-9336-z
135. Zhou Y, Liang X, Chang H, Shu F, Wu Y, Zhang T, et al. Ampelopsin-induced autophagy protects breast cancer cells from apoptosis through Akt-mTOR pathway via endoplasmic reticulum stress. Cancer Sci. (2014) 105:1279–87. doi: 10.1111/cas.12494
136. Huang WY, Fu L, Li CY, Xu LP, Zhang LX, Zhang WM. Quercetin, hyperin, and chlorogenic acid improve endothelial function by antioxidant, antiinflammatory, and ACE inhibitory effects. J Food Sci. (2017) 82:1239–46. doi: 10.1111/1750-3841.13706
137. Marcinkiewicz J, Kontny E. Taurine and inflammatory diseases. Amino Acids. (2014) 46:7–20. doi: 10.1007/s00726-012-1361-4
138. Liu C, Cao Y, Yang X, Shan P, Liu H. Tauroursodeoxycholic acid suppresses endoplasmic reticulum stress in the chondrocytes of patients with osteoarthritis. Int J Mol Med. (2015) 36:1081–7. doi: 10.3892/ijmm.2015.2295
139. Kim YS, Kim EK, Hwang JW, Kim JS, Shin WB, Dong X, et al. Neuroprotective effect of taurine-rich cuttlefish (Sepia officinalis) extract against hydrogen peroxide-induced oxidative stress in SH-SY5Y cells. Adv Exp Med Biol. (2017) 975 Pt 1:243–54. doi: 10.1007/978-94-024-1079-2_22
140. Lee IC, Kim SH, Baek HS, Moon C, Kang SS, Kim SH, et al. The involvement of Nrf2 in the protective effects of diallyl disulfide on carbon tetrachloride-induced hepatic oxidative damage and inflammatory response in rats. Food Chem Toxicol. (2014) 63:174–85. doi: 10.1016/j.fct.2013.11.006
141. Saud SM, Li W, Gray Z, Matter MS, Colburn NH, Young MR, et al. Diallyl Disulfide (DADS), a constituent of garlic, inactivates NF-κB and prevents colitis-induced colorectal cancer by inhibiting GSK-3β. Cancer Prev Res (Phila). (2016) 9:607–15. doi: 10.1158/1940-6207.CAPR-16-0044
142. Baolin L, Inami Y, Tanaka H, Inagaki N, Iinuma M, Nagai H. Resveratrol inhibits the release of mediators from bone marrow-derived mouse mast cells In vitro. Planta Med. (2004) 70:305–9. doi: 10.1055/s-2004-818940
143. Vaamonde-García C, Riveiro-Naveira RR, Valcárcel-Ares MN, Hermida-Carballo L, Blanco FJ, López-Armada MJ. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. (2012) 64:2927–36. doi: 10.1002/art.34508
144. Yi H, Zhang W, Cui ZM, Cui SY, Fan JB, Zhu XH, et al. Resveratrol alleviates the interleukin-1β-induced chondrocytes injury through the NF-κB signaling pathway. J Orthop Surg Res. (2020) 15:424. doi: 10.1186/s13018-020-01944-8
145. Takayama K, Ishida K, Matsushita T, Fujita N, Hayashi S, Sasaki K, et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum. (2009) 60:2731–40. doi: 10.1002/art.24864
146. Han G, Chen Q, Liu F, Cui Z, Shao H, Liu F, et al. Low molecular weight xanthan gum for treating osteoarthritis. Carbohydr Polym. (2017) 164:386–95. doi: 10.1016/j.carbpol.2017.01.101
147. Chen Q, Shao X, Ling P, Liu F, Shao H, Ma A, et al. Low molecular weight xanthan gum suppresses oxidative stress-induced apoptosis in rabbit chondrocytes. Carbohydr Polym. (2017) 169:255–63. doi: 10.1016/j.carbpol.2017.04.018
148. Sun Y, Zhang G, Liu Q, Liu X, Wang L, Wang J, et al. Chondroitin sulfate from sturgeon bone ameliorates pain of osteoarthritis induced by monosodium iodoacetate in rats. Int J Biol Macromol. (2018) 117:95–101. doi: 10.1016/j.ijbiomac.2018.05.124
149. Wu J, Pan Z, Wang Z, Zhu W, Shen Y, Cui R, et al. Ginsenoside Rg1 protection against β-amyloid peptide-induced neuronal apoptosis via estrogen receptor α and glucocorticoid receptor-dependent anti-protein nitration pathway. Neuropharmacology. (2012) 63:349–61. doi: 10.1016/j.neuropharm.2012.04.005
150. Sunagawa T, Shimizu T, Kanda T, Tagashira M, Sami M, Shirasawa T. Procyanidins from apples (Malus pumila Mill.) extend the lifespan of Caenorhabditis elegans. Planta Med. (2011) 77:122–7. doi: 10.1055/s-0030-1250204
151. Zhou YX, Zhang H, Peng C. Puerarin: a review of pharmacological effects. Phytother Res. (2014) 28:961–75. doi: 10.1002/ptr.5083
152. Song XB, Liu G, Wang ZY, Wang L. Puerarin protects against cadmium-induced proximal tubular cell apoptosis by restoring mitochondrial function. Chem Biol Interact. (2016) 260:219–31. doi: 10.1016/j.cbi.2016.10.006
153. Aggarwal A, Lewison G, Idir S, Peters M, Aldige C, Boerckel W, et al. The state of lung cancer research: a global analysis. J Thorac Oncol. (2016) 11:1040–50. doi: 10.1016/j.jtho.2016.03.010
154. Zhao J, Yu G, Cai M, Lei X, Yang Y, Wang Q, et al. Bibliometric analysis of global scientific activity on umbilical cord mesenchymal stem cells: a swiftly expanding and shifting focus. Stem Cell Res Ther. (2018) 9:32. doi: 10.1186/s13287-018-0785-5
155. Mao X, Chen C, Wang B, Hou J, Xiang C. A global bibliometric and visualized analysis in the status and trends of subchondral bone research. Medicine (Baltimore). (2020) 99:e20406. doi: 10.1097/MD.0000000000020406
156. Mao X, Guo L, Fu P, Xiang C. The status and trends of coronavirus research: A global bibliometric and visualized analysis. Medicine (Baltimore). (2020) 99:e20137. doi: 10.1097/MD.0000000000020137
157. Gao Y, Wang Y, Zhai X, He Y, Chen R, Zhou J, et al. Publication trends of research on diabetes mellitus and T cells (1997-2016): a 20-year bibliometric study. PLoS ONE. (2017) 12:e0184869. doi: 10.1371/journal.pone.0184869
158. Huang Z, Ding C, Li T, Yu SP. Current status and future prospects for disease modification in osteoarthritis. Rheumatology (Oxford). (2018) 57:iv108-108iv123. doi: 10.1093/rheumatology/kex496
159. Hui AY, McCarty WJ, Masuda K, Firestein GS, Sah RL. A systems biology approach to synovial joint lubrication in health, injury, and disease. Wiley Interdiscip Rev Syst Biol Med. (2012) 4:15–37. doi: 10.1002/wsbm.157
160. Zhou S, Wen H, Cai W, Zhang Y, Li H. Effect of hypoxia/reoxygenation on the biological effect of IGF system and the inflammatory mediators in cultured synoviocytes. Biochem Biophys Res Commun. (2019) 508:17–24. doi: 10.1016/j.bbrc.2018.11.099
161. Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. (2014) 2014:150845. doi: 10.1155/2014/150845
162. Paharkova-Vatchkova V, Lee KW. Nuclear export and mitochondrial and endoplasmic reticulum localization of IGF-binding protein 3 regulate its apoptotic properties. Endocr Relat Cancer. (2010) 17:293–302. doi: 10.1677/ERC-09-0106
163. Atukorala I, Kwoh CK, Guermazi A, Roemer FW, Boudreau RM, Hannon MJ, et al. Synovitis in knee osteoarthritis: a precursor of disease. Ann Rheum Dis. (2016) 75:390–5. doi: 10.1136/annrheumdis-2014-205894
164. Block JA. Osteoarthritis: OA guidelines: improving care or merely codifying practice. Nat Rev Rheumatol. (2014) 10:324–6. doi: 10.1038/nrrheum.2014.61
165. French SD, Bennell KL, Nicolson PJ, Hodges PW, Dobson FL, Hinman RS. What do people with knee or hip osteoarthritis need to know? An international consensus list of essential statements for osteoarthritis. Arthritis Care Res (Hoboken). (2015) 67:809–16. doi: 10.1002/acr.22518
166. Zhang W, Ouyang H, Dass CR, Xu J. Current research on pharmacologic and regenerative therapies for osteoarthritis. Bone Res. (2016) 4:15040. doi: 10.1038/boneres.2015.40
167. Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y, et al. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am J Respir Cell Mol Biol. (2014) 51:455–65. doi: 10.1165/rcmb.2013-0529OC
168. Hsu YC, Wu YT, Yu TH, Wei YH. Mitochondria in mesenchymal stem cell biology and cell therapy: from cellular differentiation to mitochondrial transfer. Semin Cell Dev Biol. (2016) 52:119–31. doi: 10.1016/j.semcdb.2016.02.011
169. Wang Y, Yu D, Liu Z, Zhou F, Dai J, Wu B, et al. Exosomes from embryonic mesenchymal stem cells alleviate osteoarthritis through balancing synthesis and degradation of cartilage extracellular matrix. Stem Cell Res Ther. (2017) 8:189. doi: 10.1186/s13287-017-0632-0
170. Zheng L, Wang Y, Qiu P, Xia C, Fang Y, Mei S, et al. Primary chondrocyte exosomes mediate osteoarthritis progression by regulating mitochondrion and immune reactivity. Nanomedicine (Lond). (2019) 14:3193–212. doi: 10.2217/nnm-2018-0498
171. Zhang B, Yin Y, Lai RC, Tan SS, Choo AB, Lim SK. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. (2014) 23:1233–44. doi: 10.1089/scd.2013.0479
172. Toh WS, Lai RC, Hui J, Lim SK. MSC exosome as a cell-free MSC therapy for cartilage regeneration: implications for osteoarthritis treatment. Semin Cell Dev Biol. (2017) 67:56–64. doi: 10.1016/j.semcdb.2016.11.008
173. Hough KP, Trevor JL, Strenkowski JG, Wang Y, Chacko BK, Tousif S, et al. Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells. Redox Biol. (2018) 18:54–64. doi: 10.1016/j.redox.2018.06.009
174. Wang S, Min Z, Ji Q, Geng L, Su Y, Liu Z, et al. Rescue of premature aging defects in Cockayne syndrome stem cells by CRISPR/Cas9-mediated gene correction. Protein Cell. (2020) 11:1–22. doi: 10.1007/s13238-019-0623-2
175. Deng L, Ren R, Liu Z, Song M, Li J, Wu Z, et al. Stabilizing heterochromatin by DGCR8 alleviates senescence and osteoarthritis. Nat Commun. (2019) 10:3329. doi: 10.1038/s41467-019-10831-8
Keywords: osteoarthritis, mitochondria, mitochondrial dynamics mtDNA, biomedical therapy, bibliometrics
Citation: Mao X, Fu P, Wang L and Xiang C (2020) Mitochondria: Potential Targets for Osteoarthritis. Front. Med. 7:581402. doi: 10.3389/fmed.2020.581402
Received: 08 July 2020; Accepted: 19 October 2020;
Published: 26 November 2020.
Edited by:
Ovidiu Constantin Baltatu, Khalifa University, United Arab EmiratesReviewed by:
Hanlin Zhang, University of California, Berkeley, United StatesCopyright © 2020 Mao, Fu, Wang and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuan Xiang, Y2h1YW54aWFuZ0BzeG11LmVkdS5jbg==; Linlin Wang, d2FuZ2xpbmxpbkB6anUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.