 Alessio Gerussi1,2
Alessio Gerussi1,2 Martina Lucà1,2Laura Cristoferi1,2
Martina Lucà1,2Laura Cristoferi1,2 Vincenzo Ronca1,2,3
Vincenzo Ronca1,2,3 Clara Mancuso1,2
Clara Mancuso1,2 Chiara Milani1,2Daphne D'Amato1,2Sarah Elizabeth O'Donnell1,2
Chiara Milani1,2Daphne D'Amato1,2Sarah Elizabeth O'Donnell1,2 Marco Carbone1,2
Marco Carbone1,2 Pietro Invernizzi1,2*
Pietro Invernizzi1,2*- 1Division of Gastroenterology and Center for Autoimmune Liver Diseases, Department of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy
- 2European Reference Network on Hepatological Diseases (ERN RARE-LIVER), San Gerardo Hospital, Monza, Italy
- 3National Institute of Health Research Liver Biomedical Research Centre Birmingham, Centre for Liver Research, Institute of Immunology and Immunotherapy, University of Birmingham, Birmingham, United Kingdom
Primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are autoimmune cholangiopathies characterized by limited treatment options. A more accurate understanding of the several pathways involved in these diseases has fostered the development of novel and promising targeted drugs. For PBC, the characterization of the role of farnesoid X receptor (FXR) and perixosome-proliferator activated receptor (PPAR) has paved the way to several clinical trials including different molecules with choleretic and antinflammatory action. Conversely, different pathogenetic models have been proposed in PSC such as the “leaky gut” hypothesis, a dysbiotic microbiota or a defect in mechanisms protecting against bile acid toxicity. Along these theories, new treatment approaches have been developed, ranging from drugs interfering with trafficking of lymphocytes from the gut to the liver, fecal microbiota transplantation or new biliary acids with possible immunomodulatory potential. Finally, for both diseases, antifibrotic agents are under investigation. In this review, we will illustrate current understanding of molecular mechanisms in PBC and PSC, focusing on actionable biological pathways for which novel treatments are being developed.
Introduction
Autoimmune diseases of the biliary tract include primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC). PBC and PSC are rare diseases of unknown etiology, immune-mediated pathogenesis and limited treatment options. Recently, there has been an increasing attention toward these rare diseases and novel agents are under investigation in clinical trials.
This review outlines the most promising novel agents for the treatment of PBC and PSC, adopting a target-driven approach: the biological target of each class of molecules is briefly summarized in the context of the pathogenesis of the disease and then preclinical and clinical results are presented (Figure 1 and Table 1).
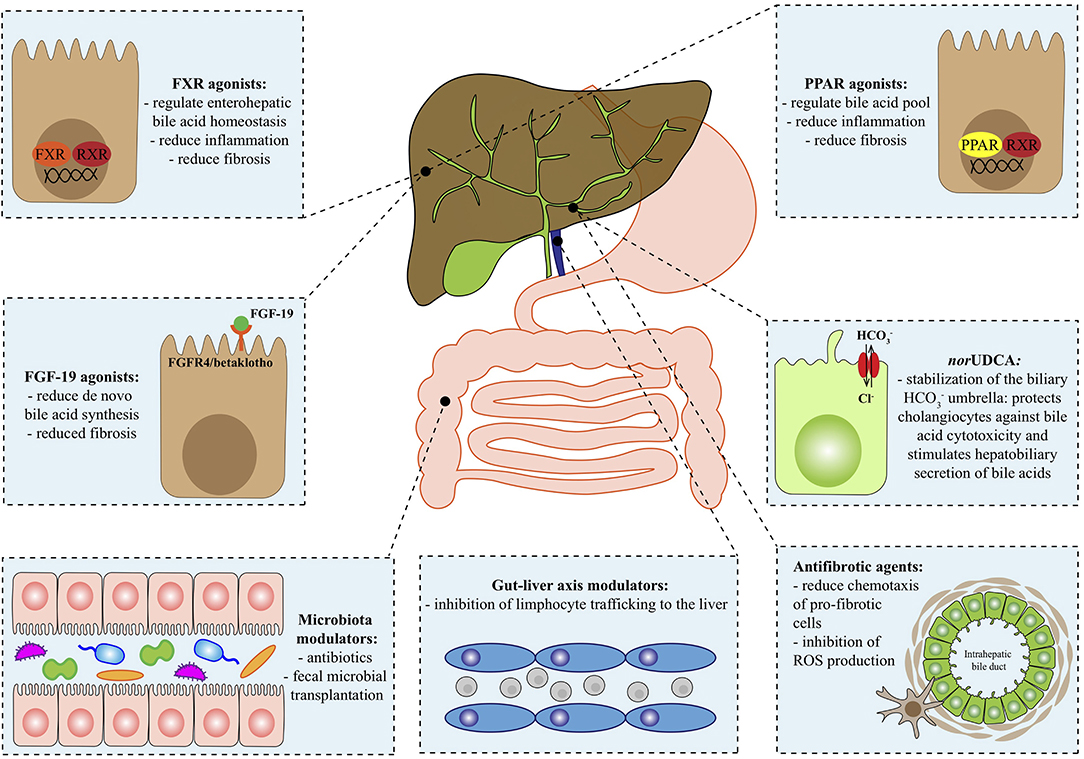
Figure 1. Therapeutical targets in autoimmune cholangiopathies. FGF-19, fibroblast growth factor 19; FGFR4, fibroblast growth factor receptor 4; FXR, farnesoid X receptor; HCO, bicarbonate; norUDCA, norursodeoxycholic acid; PPAR, Peroxisome proliferator-activated receptor; ROS, reactive oxygen species; RXR, retinoid x receptor.

Table 1. Novel pharmacological agents in autoimmune cholangiopathies.
Farnesoid X Receptor Agonists
Hepatocytes generate bile acids, i.e., cholic acid and chenodeoxycholic acid, from cholesterol through two pathways: most of the BAs are produced by the classical pathway, which involves the rate-limiting cholesterol 7alphahydroxylase (CYP7A1); the cytochrome (CYP) P450 27 alpha hydroxylase (CYP27A1) generates the remaining fractions. More recently, it has been reported that bile acids can also be generated by gut microbiota: these new bile acids are the phenylalanocholic acid, tyrosocholic acid and leucocholic acid (1).
Bile acids can exert different activities by binding to nuclear receptors, such as the farnesoid X receptor (FXR; NR1H4), pregnane X receptor, vitamin D receptor and Takeda G-protein-coupled receptor 5 (TGR5). FXR acts as transcription factor binding to FXR response elements in the DNA; it can operate as a monomer or together with retinoid X receptor (RXR; NR2B1) (2). FXR is mainly expressed in the liver and the gut, but can also be found in the kidney and adrenal gland, and its action exerts effects on the metabolism of bile acids, carbohydrates and lipids.
In the liver, FXR regulates bile acid synthesis, preventing their toxic accumulation. The bile salt export pump (BSEP) is expressed on the canalicular membranes of hepatocytes and promotes biliary excretion of bile acids. Its expression is dependent on FXR, while other transporters [e.g., multidrug resistance-associated protein (MRP) 3 and MRP4] are independent from FXR.
OSTα-OSTβ (SLC51A and SLC51B) is an heteromeric transporter expressed mainly in the distal portions of the gut and bile ducts, localized to the basolateral membrane of ileal enterocytes and biliary epithelial cells, respectively. Its main role is to transport bile acids across the membrane, but OSTα-OSTβ is also involved in steroids transport (3–5). FXR controls gene expression of OSTα-OSTβ, as proven by the marked reduction in OSTα and OSTβ expression in the ileum of Fxr-/- mice (6).
FXR induces the expression of the small heterodimer protein (SHP), also known as NR0B2 nuclear receptor subfamily 0, group B, member 2. SHP acts as transcription factor despite lacking a DNA binding domain, and inhibits CYP7A1 thanks to the recruitment of other proteins (mSin3A-Swi/Snf complex, G9a methyltransferase, the corepressor subunit GPS2) (7–10). CYP7A1 inhibition translates into a negative feedback inhibition of bile acid synthesis (2).
In pre-clinical setting, the activation of hepatic FXR is beneficial in reducing hepatic fibrosis, since it prevents toxicity due to accumulation of bile acids (11).
Among the FXR-agonists the main distinction is based on whether they are steroidal (i.e., bile acids) or non-steroidal agents. Limitations of steroidal agents are due to the intrinsic lipophilicity of FXR ligand binding site, which reduces solubility and bioavailability. Moreover, one of the most common side effects of steroidal FXR agonists is pruritus, which depends on the TGR5 agonistic properties (12). Nonetheless, also non-steroidal agents have shown mild pruritogen action despite not binding the TGR5 receptor. Interestingly, there is preliminary evidence that support the concept that non-steroidal FXR-agonists might have more anti-fibrotic effects (13).
Obeticholic acid (OCA) belongs to the class of steroidal FXR agonists and is a semi-synthetic bile acid. Currently, OCA is the only registered option for patients with PBC and incomplete response after 12 months of treatment with UDCA or intolerant to UDCA (14). The main side effect is itching, which is typically mild and seldom requires treatment withdrawal. Another side effect of OCA is the increase in low-density lipoprotein (LDL) cholesterol; the long-term significance of this side effect on cardiovascular risk is still unknown. OCA has been tested also in patients with PSC (AESOP trial), however final results are still waited.
Non-bile Acid FXR Agonists
Tropifexor has been evaluated in a double-blind, randomized, placebo-controlled, phase 2 study in PBC, but only the interim analysis is available. Interestingly, to avoid confounding due to the possible FXR-mediated alkaline phosphatase (ALP) gene induction, gamma glutamyl transpeptidase (GGT) reduction was chosen instead of ALP reduction as primary endpoint of this trial.
The non-steroidal FXR agonist Cilofexor has been tested in PSC in a randomized, double-blind, placebo-controlled phase 2 trial, and although the primary endpoint was safety, it can be noticed that it significantly reduced ALP levels, especially in the 100 mg arm (15). A randomized, double-blind, placebo-controlled phase 2 trial in PBC and a phase 3 trial in PSC are currently undergoing.
EDP-305 is another non-steroidal FXR agonist being evaluated in a randomized, double-blind, placebo-controlled phase 2 trial in PBC. In murine models of fibrosis EDP-305 reduced the extent of fibrotic areas assessed by morphometric quantification (16).
Fibroblast Growth Factor 19 Agonists
During cholestasis, high levels of bile acids favor fibroblast growth factor 19 (FGF-19) expression. The increased concentration of FGF-19 in the gut stimulates activation of the FGFR4/betaklotho receptor in the liver. FGF19 then migrates to the liver where reduces CYP7A1 gene expression (7, 17). FGFR4 and betaklotho forms a cell surface receptor complex and represent the effectors of the liver activity of FGF19. FGF19 is also induced by FXR activation in the enterocytes.
Preclinical data have shown a higher risk of hepatocellular carcinoma (HCC) in transgenic mice with ectopic expression of FGF19 in the skeletal muscle of transgenic mice (18). Furthermore, 15% of human HCC cancers show co-amplification of FGF19 and cyclin D1 on 11q13.3 (19).
A synthetic analog NGM282 was developed without carcinogenic potential and its safety and efficacy is under investigation in clinical trials. NGM282 has been tested in two randomized, double-blind, placebo-controlled trials in PBC and PSC, showing conflicting results. The PBC study lasted for 28 days while the design of the PSC study scheduled treatment for 12 weeks.
In PBC, NGM282 achieved a reduction of >15% of ALP levels in 50% of patients treated compared to 7% of the placebo group. Similar proportions were found between the 0.3 mg and the 3 mg arms. Main side effect was non severe diarrhea (20).
Conversely, in the PSC study of NGM282, ALP levels did not significantly drop in the 1 and 3 mg arms compared to placebo (21). We do acknowledge that the clinical significance of ALP in PBC and PSC is different, considering that PBC has a fairly stable and slowly progressing disease course where ALP is a good prognostic marker, compared to the erratic disease course of PSC, where ALP levels can intermittently rise and fall due to episodic cholangitis (22).
Interestingly, in both studies, NGM282 decreased the α-Hydroxy-4-cholesten-3-one (C4) levels in treatment groups confirming that this drug acts by directly inhibiting the de-novo bile acid synthesis through the classical pathway.
Peroxisome Proliferator- Activated Receptors Agonists
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors of a family containing three isotypes: PPARα (NR1C1), PPARβ/δ (NR1C2) and PPARγ (NR1C3). After engagement with their ligand, PPARs form a heterodimer with the retinoid X receptor, binding to specific DNA sequences in the regulatory regions of target genes.
PPARα is highly expressed in tissues with marked fatty acid oxidation activity, including liver, heart and skeletal muscle, brown adipose tissue and kidney. Beyond its ability to regulate fatty acid catabolism in different conditions of food intake and starvation, animal models of atherosclerosis and non-alcoholic steatohepatitis have shown that PPARα has also anti-inflammatory properties. Moreover, PPARα agonism determines inhibition of bile acids synthesis by acting on CYP7A1 and cytochrome sterol 27-hydroxylase (CYP27A1). There is also evidence that PPAR-α abrogates the uptake of bile acids in hepatocytes through the inhibition of the basolateral transporter sodium-taurocholate-cotransporting polypeptide and up-regulates expression of human MDR3 gene favoring canalicular export of phospholipids (23).
PPARβ/δ is expressed in hepatocytes, cholangiocytes, and non-parenchymal cells (Kuppfer cells, hepatic stellate cells). PPARδ is utilized by biliary epithelial cells to control bile components transporters (24) and favors the timely removal of apoptotic cells by Kuppfer cells, in order to prevent potential autoimmune phenomena to arise (25). PPAR-γ expression is mostly restricted to Kupffer cells (25). In cultured human biliary epithelial cells from patients with PBC there is a down-regulation of PPAR-γ and the activation of PPAR-γ is associated with reduced inflammation (26). A PPAR-γ agonist reduced portal inflammation in murine models of PBC (27).
Natural ligands of PPAR-α are derivatives of fatty acids generated during lipolysis, lipogenesis or fatty acids catabolism. Synthetic PPAR-α agonists belong to the group of fibrates, derivatives of fibric acid: Gemfibrozil, Fenofibrate, Ciprofibrate and Bezafibrate (Figure 2). They are typically used in treatment of isolated hypertriglyceridemia or mixed dyslipidemia. Fenofibrate is 10-fold more specific for the α- isoform compared to the γ- one, while Bezafibrate is considered a pan-PPAR-agonist due to its similar affinity for the three isoforms. Most of their actions are derived from their PPARα agonism.
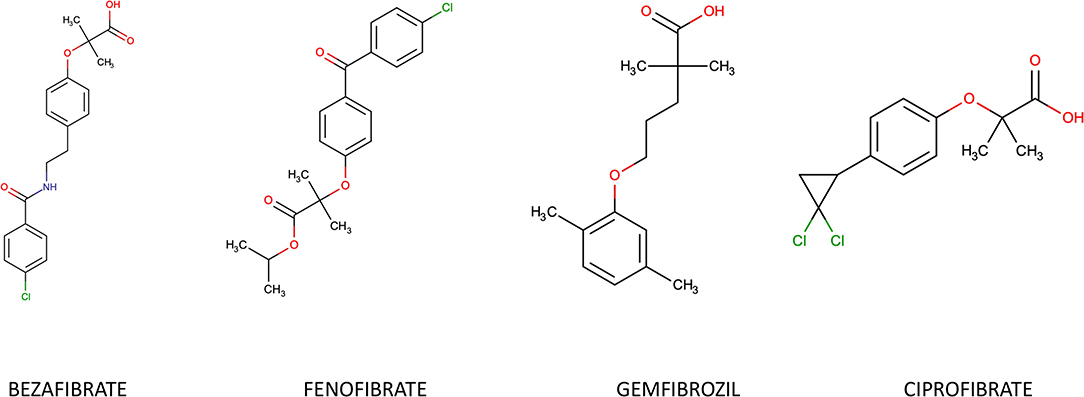
Figure 2. Chemical structure of most common fibrates.
There is growing evidence of the therapeutic efficacy of fibrates in PBC, while evidence is still limited for PSC. Bezafibrate has been evaluated in a 24 months, double-blind, randomized, placebo-controlled, phase 3 trial (BEZURSO trial), at the dosage of 400 mg per day in patients with incomplete biochemical response after 12 months of UDCA. The rate of ALP normalization was 67% in the treatment arm, compared to the 2% in placebo group. In addition, Bezafibrate did not worsen pruritus, which was even improved in a subset of cases, which is in line with available literature (28). The results from BEZURSO trial endorse the concept that fibrates reduce the production of bile acids in hepatocytes since patients in the 400 mg arm experienced a 70% drop in C4 serum levels, whereas patients in the placebo arm did not show any significant change. Conversely, experience on Fenofibrate in PBC derive from smaller cohorts (29–33). Both Bezafibrate and Fenofibrate show side effects typical of their pharmacological class, namely myalgias, transaminitis and increase in creatine kinase and creatinine. The long-term safety of these agents during treatment for cholestatic diseases is still to be ascertained. Since safety and efficacy profile of Bezafibrate and Fenofibrate seems to be similar, but Bezafibrate is supported by a randomized controlled trial, it would be reasonable to choose Bezafibrate as the fibrate of choice in PBC until new data are available.
There are now new molecules in the pipeline which show more selective PPAR-δ activity. Seladelpar is a selective PPAR-δ agonist (34), while Elafibranor is PPARα/δ.
Based on the safety information derived from two phase 2 trials (35), the potential efficacy of Seladelpar has been evaluated in the ENHANCE trial, a 52-week, double-blind, placebo-controlled, randomized, Phase 3 study. However, in November 2019, the ENHANCE trial was put on hold following the unexpected histological findings observed in the Phase 2b study of Seladelpar in subjects with non-alcoholic steatohepatitis (NASH). Atypical histological findings, including interface hepatitis and biliary injury were found in the planned interval biopsies.
Elafibranor has been recently evaluated in a multicenter randomized double-blind placebo-controlled phase 2 Study in PBC.
Literature supporting the use of fibrates in PSC is scanty. At the time of writing, no randomized controlled trials have been published, and retrospective case series come mostly from Japan (36, 37).
A French-Spanish retrospective study showed a 40% reduction in the levels of ALP after 12 weeks of treatment with fibrates [either Fenofibrate (200 mg/day) or Bezafibrate (400 mg/day)]. There were no major safety issues and authors cautiously support further studies. To us, it is conceivable to further investigate the potential benefit of fibrates in PSC, based on their inherent anticholestatic and anti-inflammatory properties.
24-norursodeoxycholic and the Biliary HCO Umbrella
The HCO umbrella hypothesis asserts that bicarbonate (HCO) ions, secreted by cholangiocytes and hepatocytes, form a defensive barrier on the apical side of the hepatocytes (38). When this system is malfunctioning, glycine-conjugated bile acids become able to cross the cholangiocyte membrane, bypassing membrane transporters. This phenomenon leads to cholangiocyte apoptosis and senescence (39). There is evidence supporting the concept that in cholangiopathies like PBC and PSC the biliary HCO umbrella is defective (40, 41).
The biliary HCO umbrella can be stabilized through 24-norursodeoxycholic (nor-UDCA), mainly thank to its ability of undergoing cholehepatic shunting. Nor-UDCA is relatively resistant to N-acyl-amidation with taurine or glycine, so that cholangiocytes can reabsorb it and it can be re-secreted into bile after getting through periductular capillary plexus and hepatocytes. The importance of this process is the ability to cause a profound stimulus to bicarbonate secretion from cholangiocytes, strengthening the impaired HCO umbrella of cholangiopathies.
Mdr2-/- mice are typically considered a surrogate in vivo model of PSC (42). The lack of Mdr2-encoded membrane protein, which is a canalicular transporter deputed to transport phospholipids from hepatocytes to the bile, causes a cholangiocyte injury, due to bile acid toxicity from increased concentration of free non-micellar bile acids. In this model nor-UDCA increases the hydrophilicity of bile acids, stimulates bile flow and induces biliary transporters and detoxification enzymes (43).
A double-blind, randomized, placebo-controlled, phase 2 study evaluating nor-UDCA in patients with PSC has shown promising results in terms of safety of the molecule and reduction of serum alkaline phosphatase after 12 weeks of treatment (44). This trial assessed three different treatment regimens (nor-UDCA 500, 1,000 mg/day, and 1,500 mg/day) compared to a four arm with placebo. All doses showed a significant reduction in ALP levels, in a dose-dependent manner, and similar results were achieved for transaminases and gamma-glutamyl transferase levels. Pruritus did not occur more frequently in the treatment arms, and nor-UDCA showed a good safety profile.
A phase 3 clinical study is currently ongoing.
Immunological Targets
PBC and PSC are both included in the group of autoimmune diseases of the biliary tract; however, while PBC is much closer to a typical autoimmune condition, PSC is a complex disease with aspects also typical of fibrotic and preneoplastic conditions. Aside from the classificatory debate, the immune system takes part of many pathogenetic processes of both PBC and PSC. Nevertheless, many strategies targeting immune cells have failed so far (45–53). Biological agents, like Rituximab (54, 55), Ustekinumab (56), and Abatacept (57) were studied in PBC, based on preclinical promising data (rituximab and abatacept) or stimulated by results from genome-wide association studies (ustekinumab), but conflicting results have been produced and these molecules are not part of the therapeutic armamentarium.
Among the novel agents under investigation, there is Baricitinib, a small molecule which is a reversible inhibitor of Janus kinase (JAK) 1 and 2. In mammals, different receptors are bound by members of the JAK family (JAK 1-3). The receptor-ligand binding determines a cascade of activation and modifications leading to the generation of docking sites for the SH2 domain of the cytoplasmic transcription factors termed signal transducers and activators of transcription (STATs). Several downstream signals are regulated by JAK and STAT proteins, comprising interleukins (ILs), interferons (IFNs), and the switch toward T helper (Th) 1, 2, or 17 of naïve T cells is finely regulated by a JAK-mediated signaling (58). Baricitinib is already included in the available registered drugs for the treatment of rheumatoid arthritis (59), and was supposed to be evaluated in a randomized double-blind, placebo-controlled, phase 2, study in patients with PBC and partial response to UDCA. Unexpectedly, in August 2019 Lilly, the company that developed the molecule, decided to terminate the trial.
Antifibrotic Agents
Inflammation, cholestasis and fibrosis are tightly connected to determine the vicious cycle toward cirrhosis (60). Cholangiopathies show a biliary-type of fibrosis which usually spreads from the portal tract to the lobule (61). None of the registered therapies for PBC are inherently antifibrotic, but halt progression of the fibrotic process through antagonism of inflammation and/or cholestasis. However, specific antifibrotic therapies are urgently needed to potentially achieve fibrosis reversal, considered that a not negligible proportion of patients do progress despite effective treatment (62).
A novel agent located at the crossroad between inflammation and fibrosis is Cenicriviroc. Cenicriviroc targets C-C motif chemokine receptor (CCR) types 2 and 5, which are typically expressed by monocytes, Kupffer cells and hepatic stellate cells. There is mounting evidence that, upon liver injury, CCR2 and CCR5 together with their ligands favor macrophage recruitment infiltration and stellate cells activation (63, 64). In Bile-duct ligated and Mdr2-/- mice Cenicriviroc, together with another agent (all-trans retinoic acid), reduced bile acids, plasma liver enzymes and histological markers of necrosis and fibrosis (65). A randomized, double-blind, multinational, phase 2b enrolling patients with Non-alcoholic fatty liver steatohepatitis (NASH) revealed the antifibrotic capacity of Cenicriviroc, which halted fibrosis progression more frequently than placebo, without improving NASH-related liver inflammation (66). Cenicriviroc is currently under evaluation in PSC.
Setanaxib is another promising antifibrotic agent characterized by a completely novel molecular target and potentially representing a first-in-class molecule for the treatment of PBC and PSC. The main targets of Setanaxib are NADPH oxidase (NOX) proteins (67), which are enzymes over-induced in conditions of chronic stress, like chronic inflammatory and fibrosing diseases (68, 69). Activation of stellate cells to myofibroblasts is promoted by NOX1, NOX2, and NOX4 isoforms (70, 71).
In bile-duct ligation and NASH murine models, Setanaxib reduced histological markers of fibrosis through NOX-inhibition. Setanaxib has been recently evaluated in a multicenter, randomized, double-blind, placebo-controlled, phase 2 study in patients with PBC and incomplete response to UDCA. The primary endpoint was reduction in the levels of GGT, which is considered a more accurate marker of oxidative stress in the liver (72).
Modulation of the Microbiota and Gut-Liver Axis
While PBC is classically limited to the small bile ducts of the liver, PSC also involves large ducts and is often associated with inflammatory bowel disease (IBD) (53). The involvement of the distal part of the biliary tree and the frequent co-existence of IBD have suggested that PSC may derive from the disruption of some physiological process in the gut. The focus has been put onto two players: the microbiota and the gut-liver axis.
There is a rising interest in the study of microbiota changes in pathological conditions (73–76). Dysbiosis, i.e., the abnormal composition of gut microbiota, has been described in patients across several diseases, included cirrhosis (77), PBC (78), and PSC (79). Dysbiosis in the gut can also involve viral and fungal species, as recently proved in IBD (80) and PSC (81). Germ-free Mdr2-/- mice show increased hepatitis and cholestatic injury compared to conventionally-housed Mdr2-/- mice (82). Nevertheless, it is still under debate whether dysbiosis has a causal relationship with these pathological findings in humans.
From a therapeutic point of view, modulation of microbiota in PSC represents a fascinating option, since some data suggest that the progression of the disease might be due to a vicious cycle of inflammation and fibrosis driven by translocation of pathobionts from the gut to the liver (83). It is likely that tackling this process could be of benefit, and there is some clinical evidence to support this concept. The use of antibiotics (e.g., Metronidazole, Vancomycin) in patients with PSC can reduce blood markers of liver injury and cholestasis, despite data on long-term benefit are lacking (84–86).
Recently, the provoking concept that non-communicable diseases (i.e., those not caused by infectious microbes) might be communicable via the transfer of microbiota has been proposed (87). This challenging theory is supported by several line of experimental data showing that if a dysbiotic microbiota is transferred from diseased animals to healthy mice the latter will develop the disease (88). Conversely, there is also evidence that Fecal Microbial Transplantation (FMT) may be of benefit for many diseases (89, 90), in addition to the established indication for recurrent Clostridium Difficile infection (91). Indeed, a seminal open-label study has shown the capacity of FMT to augment microbiota diversity in patients with PSC. In a subset of individuals FMT improved biochemical markers of cholestasis; however the small number of individuals included in this pilot study (i.e., 10 patients) obviously prevents solid conclusions on efficacy (92).
Regarding gut-liver axis, the anatomical link between gut and liver translates also in an intimate interconnection at the molecular level, with several substances flowing to the liver through the portal vein system. When intestinal permeability is higher than normal, bacterial byproducts reaching the liver are potential drivers of inflammation and fibrogenesis. Two main barriers control this process: the epithelial barrier and the gut-vascular barrier (93). Under physiological conditions, mucosal addressin cell adhesion molecule (MAdCAM)-1 is expressed on gut endothelium and is not expressed in the liver, avoiding recruitment of T cells in the liver (94). In PSC there is evidence of aberrant expression of MAdCAM-1 in the liver (95). Despite theoretically promising, the use of Vedolizumab, which antagonize MAdCAM-1 and its receptor α4β7 and it is approved for IBD treatment, did not show efficacy in patients with PSC and IBD (a phase 3 trial was retired in 2018). A retrospective study on Vedolizumab in patients with PSC and IBD also failed to reveal a positive signal (96).
The increased expression of MAdCAM-1 in the liver is mainly due to the activation of vascular adhesion protein (VAP)-1, which is an enzyme involved in amine oxidation. Intrahepatic and circulating levels of VAP-1 are increased in patients with PSC compared to non-PSC individuals, and a single-arm, open-label, multi-center trial evaluating the efficacy of an anti-VAP-1 molecule, Timolumab, is currently ongoing (97).
Conclusions
Many novel drugs are currently under investigation for PBC and PSC. The potential future availability of many novel agents opens the challenge for the identification of the right candidate for each specific drug or combination of drugs based on the mechanism of action and safety profile.
It is key the improvement of risk stratification strategies for this purpose (98), which will require deep, longitudinal phenotyping of individuals by means of multi-omics analysis including the exposome, along with the microbiome, genome, metabolome, among the others (99–104). Then, we need to implement algorithms to proficiently integrate these big data to cluster patients across different phenotypes and trajectories of the disease (105, 106); for this, the collaboration with data science professionals and experts in artificial intelligence will be fundamental (107, 108). The last part of this process will be to put in practice clinical trials with different, multimodal treatment strategies (109).
Author Contributions
This work was done in full by AG. ML has contributed to a part of the contents. CMi and DD'A have created and processed the image. LC has created the table. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work has been supported by grants of the Italian Ministry of Health in the role of auto-reactive hepatic natural killer cells in the pathogenesis of primary biliary cholangitis (PE-2016-02363915) and in the biocompatible nano-assemblies to increase the safety and the efficacy of steroid treatment against liver inflammation (GR-2018-12367794).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors of this publication are members of the European Reference Network on Hepatological Diseases (ERN RARE-LIVER).
References
1. Quinn RA, Melnik A V, Vrbanac A, Fu T, Patras KA, Christy MP, et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature. (2020) 579:123–9. doi: 10.1038/s41586-020-2047-9
2. Han YC. Update on FXR biology: promising therapeutic target? Int J Mol Sci. (2018) 19:E2069. doi: 10.3390/ijms19072069
4. Boyer JL, Trauner M, Mennone A, Soroka CJ, Cai S-Y, Moustafa T, et al. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTα-OSTβ in cholestasis in humans and rodents. Am J Physiol Liver Physiol. (2006) 290:G1124–30. doi: 10.1152/ajpgi.00539.2005
5. Boyer JL. OSTα-OSTβ guards the ileal enterocyte from the accumulation of toxic levels of bile acids. Cell Mol Gastroenterol Hepatol. (2018) 5:649–50. doi: 10.1016/j.jcmgh.2018.01.015
6. Zollner G, Wagner M, Moustafa T, Fickert P, Silbert D, Gumhold J, et al. Coordinated induction of bile acid detoxification and alternative elimination in mice: role of FXR-regulated organic solute transporter-alpha/beta in the adaptive response to bile acids. Am J Physiol Gastrointest Liver Physiol. (2006) 290:G923–32. doi: 10.1152/ajpgi.00490.2005
7. Kliewer SA, Mangelsdorf DJ. Bile acids as hormones: the FXR-FGF15/19 pathway. Dig Dis. (2015) 33:327–31. doi: 10.1159/000371670
8. Fang S, Miao J, Xiang L, Ponugoti B, Treuter E, Kemper JK. Coordinated recruitment of histone methyltransferase G9a and other chromatin-modifying enzymes in SHP-mediated regulation of hepatic bile acid metabolism. Mol Cell Biol. (2007) 27:1407–24. doi: 10.1128/MCB.00944-06
9. Kemper JK, Kim H, Miao J, Bhalla S, Bae Y. Role of an mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Mol Cell Biol. (2004) 24:7707–19. doi: 10.1128/MCB.24.17.7707-7719.2004
10. Sanyal S, Båvner A, Haroniti A, Nilsson L-M, Lundåsen T, Rehnmark S, et al. Involvement of corepressor complex subunit GPS2 in transcriptional pathways governing human bile acid biosynthesis. Proc Natl Acad Sci USA. (2007) 104:15665–70. doi: 10.1073/pnas.0706736104
11. Chávez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. (2017) 152:1679–94. doi: 10.1053/j.gastro.2017.01.055
12. Alemi F, Kwon E, Poole DP, Lieu T, Lyo V, Cattaruzza F, et al. The TGR5 receptor mediates bile acid-induced itch and analgesia. J Clin Invest. (2013) 123:1513–30. doi: 10.1172/JCI64551
13. Verbeke L, Nevens F, Laleman W. Steroidal or non-steroidal FXR agonists - Is that the question? J Hepatol. (2017) 66:680–1. doi: 10.1016/j.jhep.2017.01.013
14. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. (2016) 375:631–43. doi: 10.1056/NEJMoa1509840
15. Trauner M, Gulamhusein A, Hameed B, Caldwell S, Shiffman ML, Landis C, et al. The nonsteroidal farnesoid x receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology. (2019) 70:788–801. doi: 10.1002/hep.30509
16. Erstad DJ, Farrar CT, Masia R, Ferreira DDS, Wei L, Choi J-K, et al. A novel farnesoid X receptor agonist: EDP-305, reduces fibrosis progression in animal models of fibrosis. J Hepatol. (2017) 66:S165. doi: 10.1016/S0168-8278(17)30608-6
17. Degirolamo C, Sabbà C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov. (2016) 15:51–69. doi: 10.1038/nrd.2015.9
18. Nicholes K, Guillet S, Tomlinson E, Hillan K, Wright B, Frantz GD, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol. (2002) 160:2295–307. doi: 10.1016/S0002-9440(10)61177-7
19. Sawey ET, Chanrion M, Cai C, Wu G, Zhang J, Zender L, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell. (2011) 19:347–58. doi: 10.1016/j.ccr.2011.01.040
20. Mayo MJ, Wigg AJ, Leggett BA, Arnold H, Thompson AJ, Weltman M, et al. NGM282 for treatment of patients with primary biliary cholangitis: a multicenter, randomized, double-blind, placebo-controlled trial. Hepatol Commun. (2018) 2:1037–50. doi: 10.1002/hep4.1209
21. Hirschfield GM, Chazouillères O, Drenth JP, Thorburn D, Harrison SA, Landis CS, et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: a multicenter, randomized, double-blind, placebo-controlled phase II trial. J Hepatol. (2019) 70:483–93. doi: 10.1016/j.jhep.2018.10.035
22. Gerussi A, Invernizzi P. Better end points needed in primary sclerosing cholangitis trials. Nat Rev Gastroenterol Hepatol. (2019) 16:143–4. doi: 10.1038/s41575-019-0110-5
23. Ghonem NS, Assis DN, Boyer JL. Fibrates and cholestasis. Hepatology. (2015) 62:635–43. doi: 10.1002/hep.27744
24. Xia X, Jung D, Webb P, Zhang A, Zhang B, Li L, et al. Liver X receptor β and peroxisome proliferator-activated receptor δ regulate cholesterol transport in murine cholangiocytes. Hepatology. (2012) 56:2288–96. doi: 10.1002/hep.25919
25. Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-δ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. (2009) 15:1266–72. doi: 10.1038/nm.2048
26. Harada K, Isse K, Kamihira T, Shimoda S, Nakanuma Y. Th1 cytokine-induced downregulation of PPARγ in human biliary cells relates to cholangitis in primary biliary cirrhosis. Hepatology. (2005) 41:1329–38. doi: 10.1002/hep.20705
27. Nozaki Y, Harada K, Sanzen T, Nakanuma Y. PPARγ ligand attenuates portal inflammation in the MRL-lpr mouse: a new strategy to restrain cholangiopathy in primary biliary cirrhosis. Med Mol Morphol. (2013) 46:153–9. doi: 10.1007/s00795-013-0017-0
28. Reig A, Sesé P, Parés A. Effects of bezafibrate on outcome and pruritus in primary biliary cholangitis with suboptimal ursodeoxycholic acid response. Am J Gastroenterol. (2018) 113:49–55. doi: 10.1038/ajg.2017.287
29. Hegade VS, Khanna A, Walker LJ, Wong L-L, Dyson JK, Jones DEJ. Long-term fenofibrate treatment in primary biliary cholangitis improves biochemistry but not the UK-PBC risk score. Dig Dis Sci. (2016) 61:3037–44. doi: 10.1007/s10620-016-4250-y
30. Han XF, Wang QX, Liu Y, You ZR, Bian ZL, Qiu DK, et al. Efficacy of fenofibrate in Chinese patients with primary biliary cirrhosis partially responding to ursodeoxycholic acid therapy. J Dig Dis. (2012) 13:219–24. doi: 10.1111/j.1751-2980.2012.00574.x
31. Liberopoulos EN, Florentin M, Elisaf MS, Mikhailidis DP, Tsianos E. Fenofibrate in primary biliary cirrhosis: a pilot study. Open Cardiovasc Med J. (2010) 4:120–6. doi: 10.2174/1874192401004010120
32. Cheung AC, Lapointe-Shaw L, Kowgier M, Meza-Cardona J, Hirschfield GM, Janssen HLA, et al. Combined ursodeoxycholic acid (UDCA) and fenofibrate in primary biliary cholangitis patients with incomplete UDCA response may improve outcomes. Aliment Pharmacol Ther. (2016) 43:283–93. doi: 10.1111/apt.13465
33. Levy C, Peter JA, Nelson DR, Keach J, Petz J, Cabrera R, et al. Pilot study: fenofibrate for patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Aliment Pharmacol Ther. (2011) 33:235–42. doi: 10.1111/j.1365-2036.2010.04512.x
34. Haczeyni F, Wang H, Barn V, Mridha AR, Yeh MM, Haigh WG, et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol Commun. (2017) 1:663–74. doi: 10.1002/hep4.1072
35. Jones D, Boudes PF, Swain MG, Bowlus CL, Galambos MR, Bacon BR, et al. Seladelpar (MBX-8025), a selective PPAR-δ agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: a double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol Hepatol. (2017) 2:716–26. doi: 10.1016/S2468-1253(17)30246-7
36. Mizuno S, Hirano K, Isayama H, Watanabe T, Yamamoto N, Nakai Y, et al. Prospective study of bezafibrate for the treatment of primary sclerosing cholangitis. J Hepatobiliary Pancreat Sci. (2015) 22:766–70. doi: 10.1002/jhbp.281
37. Mizuno S, Hirano K, Tada M, Yamamoto K, Yashima Y, Yagioka H, et al. Bezafibrate for the treatment of primary sclerosing cholangitis. J Gastroenterol. (2010) 45:758–62. doi: 10.1007/s00535-010-0204-x
38. Beuers U, Hohenester S, de Buy Wenniger LJM, Kremer AE, Jansen PLM, Elferink RPJO. The biliary HCO umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology. (2010) 52:1489–96. doi: 10.1002/hep.23810
39. Hohenester S, Maillette de Buy Wenniger L, Paulusma CC, van Vliet SJ, Jefferson DM, Oude Elferink RP, et al. A biliary HCO3– umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology. (2012) 55:173–83. doi: 10.1002/hep.24691
40. Banales JM, Sáez E, Úriz M, Sarvide S, Urribarri AD, Splinter P, Tietz Bogert PS, et al. Up-regulation of microRNA 506 leads to decreased Cl–/HCO3– anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology. (2012) 56:687–97. doi: 10.1002/hep.25691
41. Rodrigues PM, Perugorria MJ, Santos-Laso A, Bujanda L, Beuers U, Banales JM. Primary biliary cholangitis: a tale of epigenetically-induced secretory failure? J Hepatol. (2018) 69:1371–83. doi: 10.1016/j.jhep.2018.08.020
42. Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. (2004) 127:261–74. doi: 10.1053/j.gastro.2004.04.009
43. Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. (2006) 130:465–81. doi: 10.1053/j.gastro.2005.10.018
44. Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Färkkilä M, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol. (2017) 67:549–58. doi: 10.1016/j.jhep.2017.05.009
45. Gong Y, Christensen E, Gluud C. Azathioprine for primary biliary cirrhosis. Cochrane Database Syst Rev. (2007) 18:CD006000. doi: 10.1002/14651858.CD006000.pub2
46. Wolfhagen FHJ, van Hoogstraten HJF, va Buuren HR, van Berge-Henegouwen GP, ten Kate FJW, Hop WCJ, et al. Triple therapy with ursodeoxycholic acid, prednisone and azathioprine in primary biliary cirrhosis: a 1-year randomized, placebo-controlled study. J Hepatol. (1998) 29:736–42. doi: 10.1016/S0168-8278(98)80254-7
47. Treiber G, Malfertheiner P. Mycophenolate mofetil for the treatment of primary biliary cirrhosis in patients with an incomplete response to ursodeoxycholic acid. J Clin Gastroenterol. (2005) 39:168–71. doi: 10.1097/01.mcg.0000177235.36640.21
48. Wiesner RH, Ludwig J, Lindor KD, Jorgensen RA, Baldus WP, Homburger HA, et al. A controlled trial of cyclosporine in the treatment of primary biliary cirrhosis. N Engl J Med. (1990) 322:1419–24. doi: 10.1056/NEJM199005173222003
49. Combes B, Emerson SS, Flye NL, Munoz SJ, Luketic VA, Mayo MJ, et al. Methotrexate (MTX) plus ursodeoxycholic acid (UDCA) in the treatment of primary biliary cirrhosis. Hepatology. (2005) 42:1184–93. doi: 10.1002/hep.20897
50. Leuschner M, Maier K, Schlichting J, Strahl S, Herrmann G, Dahm HH, et al. Oral budesonide and ursodeoxycholic acid for treatment of primary biliary cirrhosis: results of a prospective double-blind trial. Gastroenterology. (1999) 117:918–25. doi: 10.1016/S0016-5085(99)70351-3
51. Rautiainen H, Kärkkäinen P, Karvonen A-L, Nurmi H, Pikkarainen P, Nuutinen H, et al. Budesonide combined with UDCA to improve liver histology in primary biliary cirrhosis: A three-year randomized trial. Hepatology. (2005) 41:747–52. doi: 10.1002/hep.20646
52. Angulo P, Jorgensen RA, Keach JC, Dickson ER, Smith C, Lindor KD. Oral budesonide in the treatment of patients with primary biliary cirrhosis with a suboptimal response to ursodeoxycholic acid. Hepatology. (2000) 31:318–23. doi: 10.1002/hep.510310209
53. Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis - a comprehensive review. J Hepatol. (2017) 67:1298–323. doi: 10.1016/j.jhep.2017.07.022
54. Tsuda M, Moritoki Y, Lian ZX, Zhang W, Yoshida K, Wakabayashi K, et al. Biochemical and immunologic effects of rituximab in patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Hepatology. (2012) 55:512–21. doi: 10.1002/hep.24748
55. Myers RP, Swain MG, Lee SS, Shaheen AAM, Burak KW. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid. Am J Gastroenterol. (2013) 108:933–41. doi: 10.1038/ajg.2013.51
56. Hirschfield GM, Gershwin ME, Strauss R, Mayo MJ, Levy C, Zou B, et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: a proof-of-concept study. Hepatology. (2016) 64:189–99. doi: 10.1002/hep.28359
57. Bowlus CL, Yang G-X, Liu CH, Johnson CR, Dhaliwal SS, Frank D, et al. Therapeutic trials of biologics in primary biliary cholangitis: an open label study of abatacept and review of the literature. J Autoimmun. (2019) 101:26–34. doi: 10.1016/j.jaut.2019.04.005
58. Villarino A V, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. (2017) 18:374–84. doi: 10.1038/ni.3691
59. Smolen JS, Landewé R, Breedveld FC, Buch M, Burmester G, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. (2014) 73:492–509. doi: 10.1136/annrheumdis-2013-204573
60. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. (2017) 127:55–64. doi: 10.1172/JCI88881
61. Pinzani M, Luong TV. Pathogenesis of biliary fibrosis. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1279–83. doi: 10.1016/j.bbadis.2017.07.026
62. Schuppan D, Ashfaq-Khan M, Yang AT, Kim YO. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. (2018) 68–9:435–51. doi: 10.1016/j.matbio.2018.04.006
63. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1310–21. doi: 10.1152/ajpgi.00365.2011
64. Berres M-L, Koenen RR, Rueland A, Zaldivar MM, Heinrichs D, Sahin H, et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J Clin Invest. (2010) 120:4129–40. doi: 10.1172/JCI41732
65. Yu D, Cai S-Y, Mennone A, Vig P, Boyer JL. Cenicriviroc, a cytokine receptor antagonist, potentiates all-trans retinoic acid in reducing liver injury in cholestatic rodents. Liver Int. (2018) 38:1128–38. doi: 10.1111/liv.13698
66. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. (2018) 67:1754–67. doi: 10.1002/hep.29477
67. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. (2004) 4:181–9. doi: 10.1038/nri1312
68. Campbell EL, Colgan SP. Control and dysregulation of redox signalling in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol. (2018) 16:106–20. doi: 10.1038/s41575-018-0079-5
69. Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. (2011) 79:944–56. doi: 10.1038/ki.2010.516
70. Cui W, Matsuno K, Iwata K, Ibi M, Matsumoto M, Zhang J, et al. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology. (2011) 54:949–58. doi: 10.1002/hep.24465
71. Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schröder K, et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. (2012) 53:289–96. doi: 10.1016/j.freeradbiomed.2012.05.007
72. Kunutsor SK. Gamma-glutamyltransferase-friend or foe within? Liver Int. (2016) 36:1723–34. doi: 10.1111/liv.13221
73. Lynch S V, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med. (2016) 375:2369–79. doi: 10.1056/NEJMra1600266
74. Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol. (2018) 16:35–56. doi: 10.1038/s41575-018-0061-2
75. Clemente JC, Manasson J, Scher JU. The role of the gut microbiome in systemic inflammatory disease. BMJ. (2018) 360:j5145. doi: 10.1136/bmj.j5145
76. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. Erratum to: the gut-liver axis and the intersection with the microbiome (Nature Reviews Gastroenterology & Hepatology, (2018), 15, 7, (397-411), Nat Rev Gastroenterol Hepatol. (2018) 15:785. doi: 10.1038/s41575-018-0011-z
77. Woodhouse CA, Patel VC, Singanayagam A, Shawcross DL. Review article: the gut microbiome as a therapeutic target in the pathogenesis and treatment of chronic liver disease. Aliment Pharmacol Ther. (2018) 47:192–202. doi: 10.1111/apt.14397
78. Tang R, Wei Y, Li Y, Chen W, Chen H, Wang Q, et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut. (2018) 67:534–41. doi: 10.1136/gutjnl-2016-313332
79. Sabino J, Vieira-Silva S, Machiels K, Joossens M, Falony G, Ballet V, et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut. (2016) 65:1681–9. doi: 10.1136/gutjnl-2015-311004
80. Ungaro F, Massimino L, D'Alessio S, Danese S. The gut virome in inflammatory bowel disease pathogenesis: from metagenomics to novel therapeutic approaches. United Eur Gastroenterol J. (2019) 7:999–1007. doi: 10.1177/2050640619876787
81. Lemoinne S, Kemgang A, Ben Belkacem K, Straube M, Jegou S, Corpechot C, et al. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut. (2019) 69:92–102. doi: 10.1136/gutjnl-2018-317791
82. Tabibian JH, O'Hara SP, Trussoni CE, Tietz PS, Splinter PL, Mounajjed T, et al. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology. (2016) 63:185–96. doi: 10.1002/hep.27927
83. Hov J, Karlsen T. The microbiome in primary sclerosing cholangitis: current evidence and potential concepts. Semin Liver Dis. (2017) 37:314–31. doi: 10.1055/s-0037-1608801
84. Tabibian JH, Weeding E, Jorgensen RA, Petz JL, Keach JC, Talwalkar JA, et al. Randomised clinical trial: vancomycin or metronidazole in patients with primary sclerosing cholangitis - a pilot study. Aliment Pharmacol Ther. (2013) 37:604–12. doi: 10.1111/apt.12232
85. Silveira MG, Torok NJ, Gossard AA, Keach JC, Jorgensen RA, Petz JL, et al. Minocycline in the treatment of patients with primary sclerosing cholangitis: results of a pilot study. Am J Gastroenterol. (2009) 104:83–8. doi: 10.1038/ajg.2008.14
86. Rahimpour S, Nasiri-Toosi M, Khalili H, Daryani NE, Taromlou MKN, Azizi Z. A triple blinded, randomized, placebo-controlled clinical trial to evaluate the efficacy and safety of oral vancomycin in primary sclerosing cholangitis: a pilot study. J Gastrointest Liver Dis. (2016) 25:457–64. doi: 10.15403/jgld.2014.1121.254.rah
87. Finlay BB. Are noncommunicable diseases communicable? Science (80-). (2020) 367:250–1. doi: 10.1126/science.aaz3834
88. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. (2011) 145:745–57. doi: 10.1016/j.cell.2011.04.022
89. Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med. (2018) 24:1804–8. doi: 10.1038/s41591-018-0238-9
90. D'Haens GR, Jobin C. Fecal microbial transplantation for diseases beyond recurrent clostridium difficile infection. Gastroenterology. (2019) 157:624–36. doi: 10.1053/j.gastro.2019.04.053
91. Hvas CL, Dahl Jørgensen SM, Jørgensen SP, Storgaard M, Lemming L, Hansen MM, et al. Fecal microbiota transplantation is superior to fidaxomicin for treatment of recurrent clostridium difficile infection. Gastroenterology. (2019) 156:1324–32.e3. doi: 10.1053/j.gastro.2018.12.019
92. Allegretti JR, Kassam Z, Carrellas M, Mullish BH, Marchesi JR, Pechlivanis A, et al. Fecal microbiota transplantation in patients with primary sclerosing cholangitis: a pilot clinical trial. Am J Gastroenterol. (2019) 114:1071–9. doi: 10.14309/ajg.0000000000000115
93. Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science (80-). (2015) 350:830–4. doi: 10.1126/science.aad0135
94. Habtezion A, Nguyen LP, Hadeiba H, Butcher EC. Leukocyte trafficking to the small intestine and colon. Gastroenterology. (2016) 150:340–54. doi: 10.1053/j.gastro.2015.10.046
95. Grant AJ, Lalor PF, Hübscher SG, Briskin M, Adams DH. MAdCAM-1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium (MAdCAM-1 in chronic inflammatory liver disease). Hepatology. (2001) 33:1065–72. doi: 10.1053/jhep.2001.24231
96. Lynch KD, Chapman RW, Keshav S, Montano-Loza AJ, Mason AL, Kremer AE, et al. Effects of vedolizumab in patients with primary sclerosing cholangitis and inflammatory bowel diseases. Clin Gastroenterol Hepatol. (2019) 18:179–87.e6. doi: 10.1016/j.cgh.2019.05.013
97. Trivedi PJ, Tickle J, Vesterhus MN, Eddowes PJ, Bruns T, Vainio J, et al. Vascular adhesion protein-1 is elevated in primary sclerosing cholangitis, is predictive of clinical outcome and facilitates recruitment of gut-tropic lymphocytes to liver in a substrate-dependent manner. Gut. (2018) 67:1135–45. doi: 10.1136/gutjnl-2016-312354
98. Bossen L, Gerussi A, Lygoura V, Mells GF, Carbone M, Invernizzi P. Support of precision medicine through risk-stratification in autoimmune liver diseases - histology, scoring systems, and non-invasive markers. Autoimmun Rev. (2018) 17:854–65. doi: 10.1016/j.autrev.2018.02.013
99. Karczewski KJ, Snyder MP. Integrative omics for health and disease. Nat Rev Genet. (2018) 19:299–310. doi: 10.1038/nrg.2018.4
100. Sia D, Llovet JM. Liver cancer: translating “-omics” results into precision medicine for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. (2017) 14:571–2. doi: 10.1038/nrgastro.2017.103
101. Schüssler-Fiorenza Rose SM, Contrepois K, Moneghetti KJ, Zhou W, Mishra T, Mataraso S, et al. A longitudinal big data approach for precision health. Nat Med. (2019) 25:792–804. doi: 10.1038/s41591-019-0414-6
102. Menche J, Guney E, Sharma A, Branigan PJ, Loza MJ, Baribaud F, et al. Integrating personalized gene expression profiles into predictive disease-associated gene pools. NPJ Syst Biol Appl. (2017) 3:10. doi: 10.1038/s41540-017-0009-0
103. Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, et al. Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature. (2019) 569:663–71. doi: 10.1038/s41586-019-1236-x
104. Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HYK, Chen R, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. (2012) 148:1293–307. doi: 10.1016/j.cell.2012.02.009
105. Seymour CW, Kennedy JN, Wang S, Chang CH, Elliott CF, Xu Z, et al. Derivation, validation, and potential treatment implications of novel clinical phenotypes for sepsis. JAMA. (2019) 321:2003–17. doi: 10.1001/jama.2019.5791
106. Ahlqvist E, Storm P, Käräjämäki A, Martinell M, Dorkhan M, Carlsson A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. (2018) 6:361–9. doi: 10.1016/S2213-8587(18)30051-2
107. Topol EJ. High-performance medicine: the convergence of human and artificial intelligence. Nat Med. (2019) 25:44–56. doi: 10.1038/s41591-018-0300-7
108. Littmann M, Selig K, Cohen-Lavi L, Frank Y, Hönigschmid P, Kataka E, et al. Validity of machine learning in biology and medicine increased through collaborations across fields of expertise. Nat Mach Intell. (2020) 38:991–4. doi: 10.1038/s42256-019-0139-8
Keywords: primary biliary cholangitis, primary sclerosing cholangitis, liver, FXR agonists, fibrates, microbiome, gut-liver axis
Citation: Gerussi A, Lucà M, Cristoferi L, Ronca V, Mancuso C, Milani C, D'Amato D, O'Donnell SE, Carbone M and Invernizzi P (2020) New Therapeutic Targets in Autoimmune Cholangiopathies. Front. Med. 7:117. doi: 10.3389/fmed.2020.00117
Received: 04 February 2020; Accepted: 18 March 2020;
Published: 07 April 2020.
Edited by:
Gianfranco Danilo Alpini, Indiana University, United StatesReviewed by:
Debjyoti Kundu, Indiana University School of Medicine, United StatesGrace L. Guo, Rutgers, The State University of New Jersey, United States
Copyright © 2020 Gerussi, Lucà, Cristoferi, Ronca, Mancuso, Milani, D'Amato, O'Donnell, Carbone and Invernizzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pietro Invernizzi, cGlldHJvLmludmVybml6emlAdW5pbWliLml0