 Paolo Cravedi
Paolo Cravedi Marta Jarque
Marta Jarque Andrea Angeletti
Andrea Angeletti Àlex Favà
Àlex Favà Chiara Cantarelli
Chiara Cantarelli Oriol Bestard
Oriol Bestard
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 08 November 2019
Sec. Nephrology
Volume 6 - 2019 | https://doi.org/10.3389/fmed.2019.00241
This article is part of the Research Topic Kidney Transplantation and Immune-Mediated Nephropathies View all 6 articles
Primary membranous nephropathy (MN) is a glomerular disease mediated by autoreactive antibodies, being the main cause of nephrotic syndrome among adult patients. While the pathogenesis of MN is still controversial, the detection of autoantibodies against two specific glomerular antigens, phospholipase A2 receptor (PLA2R) and thrombospondin type 1 domain containing 7A (THSD7A), together with the beneficial effect of therapies targeting B cells, have highlighted the main role of autoreactive B cells driving this renal disease. In fact, the detection of PLA2R-specific IgG4 antibodies has resulted in a paradigm shift regarding the diagnosis as well as a better prediction of the progression and recurrence of primary MN. Nevertheless, some patients do not show remission of the nephrotic syndrome or do rapidly recur after immunosuppression withdrawal, regardless the absence of detectable anti-PLA2R antibodies, thus highlighting the need of other immune biomarkers for MN risk-stratification. Notably, the exclusive evaluation of circulating antibodies may significantly underestimate the magnitude of the global humoral memory immune response since it may exclude the role of antigen-specific memory B cells. Therefore, the assessment of PLA2R-specific B-cell immune responses using novel technologies in a functional manner may provide novel insight on the pathogenic mechanisms of B cells triggering MN as well as refine current immune-risk stratification solely based on circulating autoantibodies.
Primary membranous nephropathy (MN) is an autoantibody-mediated glomerular disease that represents one of the leading causes of nephrotic syndrome in adults (1). MN is characterized by the deposition of anti-podocyte targeted IgG antibodies on the subepithelial layer of the glomerular capillary wall. Autoantibodies deposition leads to the thickening of the glomerular basement membrane, complement activation, and glomerular capillary injury with consequent proteinuria. In ~25% of patients, MN is classified as “secondary,” due to a contemporary detection of a causative disease, such as malignancies, infections, drug reactions, or autoimmune diseases including systemic lupus erythematosus (2, 3). The natural history of the untreated disease is variable: spontaneous complete remission of primary MN is observed in approximately the 30–40% of patients (4, 5), whereas 30% of cases develop end-stage kidney disease (ESKD) generally over 10–15 years (6, 7). In kidney transplant recipients, MN relapses appear in 10–45% of cases (8–12) and occur as a de novo disease in about 2% of recipients (13, 14).
Current understanding of MN pathophysiology comes from studies in rodent models. In 1959, Heymann et al. (15) described a model of MN, now defined as active Heymann nephritis, which was induced by immunizing Lewis rats with intraperitoneal injections of crude kidney extracts, together with complete Freund's adjuvant. This resulted in a disease characterized by subepithelial immune complexes similar to human MN. Subsequent in vivo and in vitro studies have led to a better understanding of how subepithelial immune deposits lead to podocyte injury and proteinuria. Complement-mediated cytotoxicity plays a major role in the disease pathogenesis, especially the terminal complement complex C5b-9 (membrane attack complex—MAC), which is detectable in the urine of patients with MN and considered a marker of podocytes injury (16–20). Data suggest that in primary MN, complement cascade is firstly activated by the mannose binding lectin pathway, leading to the formation of C3 deposits in the subepithelial space along with MAC on podocyte membranes (21–23).
The identification of the cell surface protease neutral endopeptidase (NEP) as a target podocyte autoantigen in a newborn with MN represented a cornerstone in our understanding of MN pathophysiology. Pierre Ronco and Hanna Debiec described the case of a mother genetically deficient in NEP that had given birth to an infant who developed antenatal nephrotic syndrome (24). During the previous pregnancy, the mother generated circulating anti-NEP that crossed the placenta and targeted NEP on the fetal kidney during her subsequent pregnancy, leading to in situ immune deposits. Therefore, NEP represents the first podocyte protein demonstrated to be a target antigen in human MN (25).
Identification of autoantibodies reactive against M-type phospholipase A2 receptor type 1 (PLA2R) (26) and, later, against thrombospondin type 1 domain containing 7A (THSD7A) (27), two podocyte-expressed proteins, represented a further major step forward in defining the disease pathogenesis. Autoantibodies against such antigens can be detected in the 75–85% of primary MN patients (28, 29): anti-PLA2R autoantibodies are present in ~70–80% of adult cases, particularly in men (26, 30), whereas anti-THSD7A antibodies may be detected in only 3–5% of adults with primary MN, mainly in women (27, 31). Only about 1% of MN patients have both anti-PLA2R and anti-THSD7A autoantibodies detectable (32).
A 2019 study (33) showed that, in MN patients without detectable anti-PLA2R or anti-THSD7A autoantibodies, exostosin1/exostosin2 could represent target antigens. The authors performed mass spectrometry on laser microdissected glomeruli and immunohistochemistry on kidney biopsy of 22 MN patients, including 7 with anti-PLA2R antibodies and 15 without, detecting exostosin1/exostosin2 expression uniquely in five cases without detectable circulating anti-PLA2R antibodies. In a larger cohort of 209 MN patients negative for circulating anti-PLA2R antibodies, immunohistochemistry revealed bright granular glomerular basement membrane staining for exostosin 1/exostosin 2 in 16 cases (33). Eleven of the 16 cases showed signs of lupus nephritis or autoimmunity, suggesting that exostosin 1/exostosin 2 may represent a potential marker of a specific subtype of MN, most commonly associated with autoimmune diseases (33).
Altogether, these mechanistic findings have highlighted the key role of B cells in the pathogenesis of MN, both as autoantibody producing cells (34) and as antigen presenting cells (35), thus providing the basis for B-cell target therapies (36–39). However, response to such therapies remains unpredictable and the identification of subjects who would develop spontaneous remission (in whom immunosuppression could be avoided) is still very challenging. The discovery of MN-specific antigens has allowed the development of many diagnostic and prognostic serologic tests and optimal non-invasive biomarkers for monitoring disease activity. Nevertheless, while the assessment of autoantibodies provides useful information about the humoral memory immune response, other assays are needed to better immune-risk stratify patients and to tailor treatment in a personalized fashion.
According to the most recent Controversies Conference on KDIGO guidelines (39), proteinuria, and serum creatinine are still considered the gold-standard biomarkers to risk-stratify MN patients. For instance, individuals with subnephrotic proteinuria have excellent long-term renal survival, therefore, immunosuppression is not recommended (39). Conversely, in patients with proteinuria above 4–5 g/24 h, MN prognosis may range from spontaneous remission to development of ESKD.
Urinary markers of renal tubular damage, such as, Beta2 microglobulin, N-acetyl-β-D-glucosaminidase (NAG) and retinol-binding protein (RBP), kidney injury molecule 1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) have been also proposed to risk-stratify patients with MN. Yet, the levels of these biomarkers seem to not correlate with the severity of the disease (40).
Despite its invasive nature, kidney biopsy is still important for the diagnosis of MN, in particular among patients with altered kidney function and evidence of possible secondary causes (41), but the capacity of histological lesions to predict outcomes or response to therapy is limited at best. Hence, new approaches to better risk-stratify MN patients are highly needed in the clinical setting.
Over the last decade, discovery of target podocyte antigens and the development of commercial assays for the detection of serum anti-PLA2R and anti-THSD7A autoantibodies has revolutionized the traditional algorithms for diagnosis and management of MN, particularly due to their high specificity for disease diagnosis (26, 27). Such autoreactive antibodies recognize the target conformational epitopes on the membrane protein expressed on glomerular podocytes under non-reducing conditions and are predominantly of the IgG4 subclass. Importantly, both autoantibodies are emerging as clinical biomarkers to predict outcome in MN patients.
THSD7A is a large transmembrane glycoprotein expressed by podocytes. In Europe and United States only 3% of MN subjects expresses anti-THSD7A autoantibodies (predominantly IgG4), while it increases to a 9% in Japan (27, 31, 42, 43). Importantly, anti-THSD7A antibodies induce a MN-like pattern of disease when injected in mice (29). In a recent retrospective study, Zaghrini et al. (44) developed a new ELISA assay to detect THSD7A-specific antibodies: levels of anti-THSD7A autoantibodies correlated with disease activity and with response to treatment. Also, patients with high titers at baseline had a poorer clinical outcome. I has also been reported an association between anti-THSD7A autoantibodies and malignancies (42, 43, 45), but this needs to be better clarified in larger, multicenter studies.
The M-type phospholipase A2 receptor (PLA2R) is one of four members of the mannose receptor in mammals (46). PLA2R is a multifunctional receptor for soluble phospholipase A2 (sPLA2), which is described as a pro-inflammatory enzyme and PLA2R acts as a scavenger receptor to remove secreted PLA2 enzyme (47). Despite this receptor being highly expressed by human podocytes as well as by neutrophils and alveolar type II epithelial cells (26, 48, 49), autoantibodies against PLA2R exclusively induce nephrotic syndrome without apparent impairment in other organs.
The complexity of the PLA2R structure is illustrated by the identification of distinct immunogenic PLA2R epitopes, including a cysteine-rich domain (CysR), a fibronectin type II domain and eight distinct C-type lectin domains (CTLD1–8) (50), which are dependent on the protein conformation (26). Main antigenic epitopes recognized by anti-PLA2R antibodies have been recently identified and reported to be sensitive to reducing agents, thus confirming that conformational structure is of great importance in PLA2R epitopes (51, 52). A further dominant epitope of PLA2R (P28mer) was recently identified being also a dominant epitope of THSD7A in the N-terminal domain, suggesting that this shared motif could be involved in the initial B-cell activation in MN (53).
A genetic predisposition for MN was initially speculated by the associative evidence linking variants in the HLA locus and the risk of developing MN (54). Years later, family case reports of MN were also described (55).
Several genome-wide association studies (GWAS) have recently associated risk alleles in HLA genes with the increase risk of MN. Stanescu et al. (56) defined the association between HLA-DQA1 allele with MN in Caucasian individuals, suggesting that the interaction between sequence variations in immune-proteins and glomerular components may explain a trigger-target model in the disease development. Such interaction between PLA2R and HLA-DQA1 variants was also studied in an Asian cohort with similar results (57). More studies confirmed this association in different cohorts of MN patients (58–61), but the related mechanisms remain unknown.
The possible role of specific HLA alleles in MN was further investigated in two recent studies. Cui et al. (62) genotyped HLA-DRB1, DQA1, DQB1, and DPB1 genes in 261 primary MN patients and in 599 healthy controls. These investigators confirmed that risk alleles of HLA-DQA1 and PLA2R are significantly associated with the susceptibility to MN. Particularly, authors showed that these risk alleles are associated with the presence of circulating anti-PLA2R antibodies as well as to the increased expression of PLA2R in the glomeruli. Authors also detected the classical DRB1*1501 and DRB1*0301 alleles, showing significant independent effects on the risk of MN among the ethnic group of Han Chinese. Le et al. (63) sequenced HLA locus in 99 anti-PLA2R-positive MN subjects and in 100 healthy controls. Again, the association between DRB1*1501 and anti-PLA2R positive MN was demonstrated, and suggested DRB3*0202 as new risk allele for MN. These two alleles were subsequently confirmed in an independent cohort of 285 controls and 293 cases. Although DRB1*1502 was not revealed as a risk allele for MN, it was associated with significantly higher levels of anti-PLA2R autoantibodies and a significantly increased risk of progression to ESKD (64).
Altogether, GWAS has provided robust data about the genetic susceptibility to MN, suggesting that genetic tests could become a non-invasive tool to risk-stratify MN patients (65), although more data testing these associations in different ethnic groups are needed (66).
Anti-PLA2R IgG4 autoantibodies are detected in the sub-epithelial immune deposits using immunofluorescence or immunohistochemistry in patients with primary MN (67). In normal kidneys or other glomerular diseases, the PLA2R antigen is weakly expressed on the podocyte surface (67). Generally, a strong association between glomerular PLA2R staining and circulating anti-PLA2R antibodies is found (28, 60, 68), particularly when autoantibody levels are measured at the time of the biopsy assessment (69). However, glomerular PLA2R staining is not considered a diagnostic test for active disease, since the positivity of glomerular PLA2R staining with undetectable circulating anti-PLA2R autoantibodies is unlikely (28, 69, 70) and may reflect an immunologically inactive disease as a positive PLA2R antigen can persist for weeks or months after remission (67).
Western blotting was initially performed to detect anti-PLA2R (26) and anti-THSD7A (27) autoantibodies, but this test is inadequate for routine clinical use. The first commercially available assay for serum anti-PLA2R autoantibodies detection was an indirect immunofluorescence assay (CBA-IFA; Euroimmun, Luebeck, Germany), based on a semi-quantitative determination, and therefore, not ideal for monitoring therapeutic response and disease progression. Most clinical laboratories routinely use an ELISA-based assay (Euroimmun), because it is able to quantify anti-PLA2R autoantibodies, but this assay is not as sensitive as CBA-IFA assays. Conversely, the CBA-IFA anti-PLA2R immunoassays detection may be considered only when diagnosis of PLA2R-associated MN is strongly suspected, but there is a negative ELISA test. The most recent diagnostic assay is a laser bead immunoassay (ALBIA; Mitogen Advanced Diagnostics Laboratory, Calgary, Canada), that allows a sensitive and a quantitative detection of these autoantibodies. This assay allows the detection of different molecules such as antibodies, complement or cytokines. A comparison between the CBA-IFA, ELISA and ALBIA platforms, showed similar capacity across the different tests to detect anti-PLA2R autoantibodies (71).
Different groups have suggested the use of anti-PLA2R autoantibodies to predict spontaneous remission of MN. Hofstra et al. (72) reported that spontaneous remission is inversely related to high antibodies titers measured by up to 6 months after biopsy assessment. Similarly, Timmermans et al. (73) showed that, among 109 MN patients, subjects with detectable serum anti-PLA2R autoantibodies at the time of biopsy had a lower probability for spontaneous remission than seronegative patients. In a retrospective study including 68 patients with biopsy-proven MN, Jullien et al. (74), reported that spontaneous remission was correlated with low titers of anti-PLA2R autoantibody at time of biopsy. These data were recently confirmed by a prospective study involving 62 MN patients: complete spontaneous remission was more common in subjects with lower anti-PLA2R autoantibody levels at the time of diagnosis (<40 UI/mL) (75).
Beck et al. (76) evaluated the relationship between changes in serum PLA2R-specific autoantibodies levels and the response to B cell-depleting antibody rituximab therapy in 35 adult patients with MN. Circulating autoantibodies were detected in 71% of patients at baseline and levels decreased after rituximab therapy in the majority of them. The reduction of anti-PLA2R autoantibody levels anticipated the decline of proteinuria, and in one particular patient with a relapse of proteinuria, the reappearance of the autoantibody in serum preceded the recurrence of MN. However, proteinuria may persist, regardless the presence of autoreactive anti-PLA2R antibodies due to irreversible capillary wall injury thus, perpetuating albuminuria levels in absence of active autoimmunity.
More recently, Ruggenenti et al. (77) investigated the association between treatment effect, circulating anti-PLA2R autoantibodies and genetic polymorphisms predisposing to antibody production in 132 MN patients with nephrotic range proteinuria treated with rituximab. Outcome of patients with or without detectable anti-PLA2R autoantibodies at baseline were similar. However, among 81 patients with autoantibodies, lower anti-PLA2R autoantibodies titer at baseline and full depletion at 6 months post-treatment strongly predicted remission over a median follow-up period of 30.8 months. All 25 patients displaying complete remission were preceded by undetected anti-PLA2R autoantibodies in circulation, while re-emergence of circulating antibodies predicted clinical disease relapse. Accordingly, a further study involving 30 patients with MN and elevated anti-PLA2R autoantibodies (78) showed that clinical remission was heralded by a reduction in circulating autoantibodies.
Collectively, the above studies and further published data (79–83) suggest that serial measurements of anti-PLA2R autoantibody titers in the serum may help at risk-stratifying patients, allowing to personalize treatment and to reduce the side-effects related to over-immunosuppression.
However, antigen-specific memory B cells may exist and be ready to develop a rapid and effective secondary immune response even in absence of detectable circulating autoantibodies. This suggests that the assessment of the humoral auto-immune response using other cell-based assays may significantly improve the understanding of the effector mechanisms of the disease in patients with primary MN.
Epitope spreading is a common immunopathogenic response to self-antigens: the immune response primary involves the so-defined immunodominant epitope recognized by most autoantibodies, then expands to the intramolecular epitope on the same protein (intramolecular epitope spreading) or to dominant epitopes on neighboring molecules (intermolecular epitope spreading) (84, 85). The result is an increased diversity in antibody repertoire, leading to a broader overall immune response. Epitope spreading for the CysR epitope of PLA2R has been recognized as independent risk factor for reduced renal survival (86). In the GEMRITUX (Evaluate Rituximab Treatment for Idiopathic Membranous Nephropathy) randomized controlled trial (87), including a cohort of 58 patients positive for anti-PLA2R-specific autoantibodies randomly treated with rituximab or conservative therapy, epitope spreading strongly correlated with serum titer of anti-PLA2R autoantibodies The absence of epitope spreading at onset was an independent predictor of remission at 6 months and at last follow-up (median of 23 months) (88). Of interest, 10 of the 17 patients who had epitope spreading at baseline and were treated with rituximab, showed reversal of epitope spreading at 6 months (88). The anti-PLA2R autoantibody titer has been shown to correlate with the degree of epitope spreading (88). Therefore, due to the lack of epitope-specific assays for anti-PLA2R autoantibodies for clinical practice, the total titer of anti-PLA2R autoantibodies could be considered a surrogate of epitope spreading (88).
A few studies have investigated the immune phenotype of MN patients and its changes in relation to treatment (Table 1). Some investigators reported an increase of the CD4+/CD8+ T cell ratio in MN patients with or without nephrotic proteinuria (89, 90). Some evidence has shown a reduction of CD8+ T cells in patients with MN and nephrotic syndrome when compared to healthy subjects (91). This broad phenotype seems to be associated with a more favorable prognostic response to classical immunosuppressive therapy (92), but not to anti-CD20 depletion (93). MN is characterized by a predominance of IgG4 subclass autoantibodies, thus suggesting an involvement of a Th2 immune response, which has been described in some series (94–96).
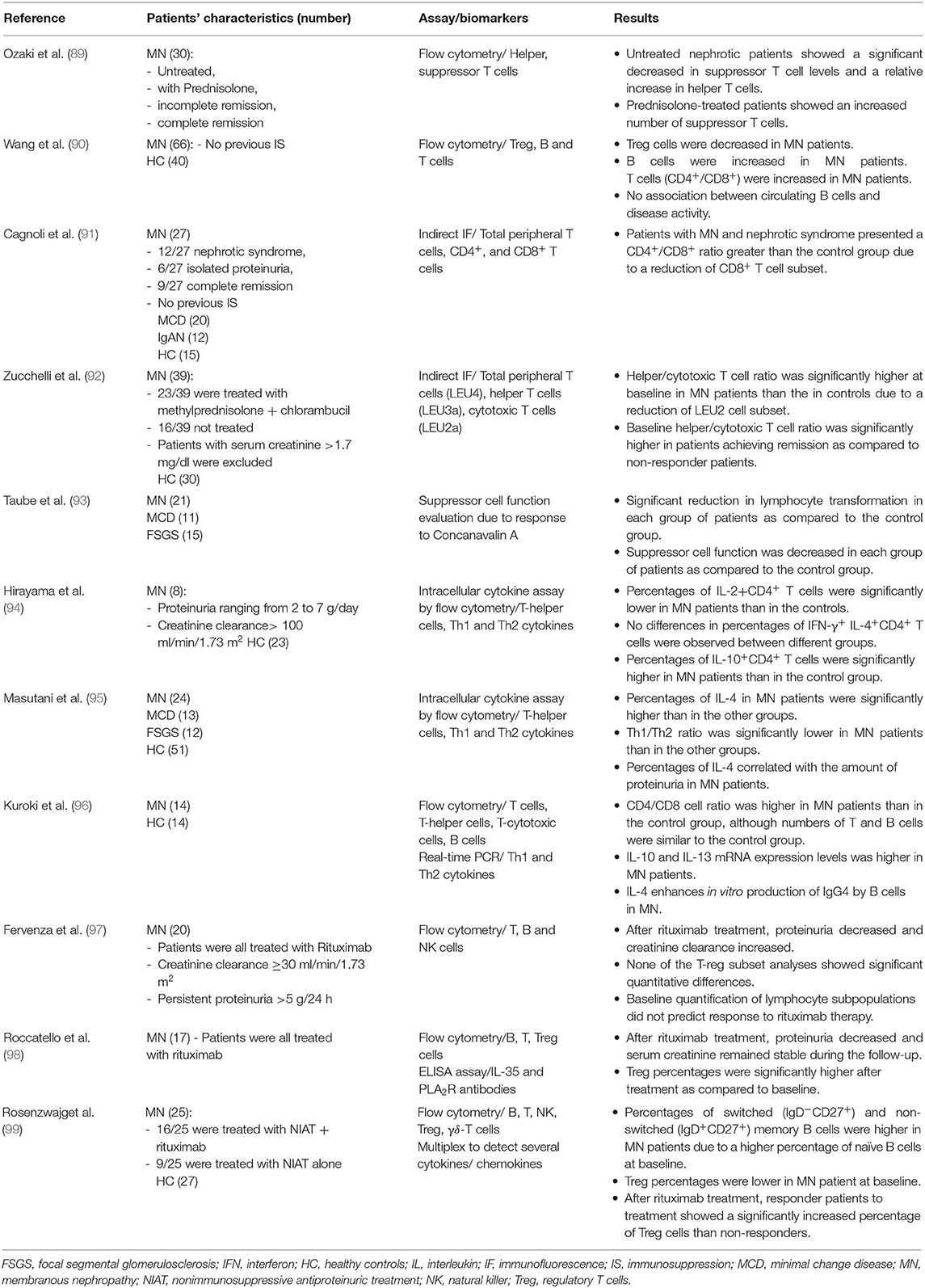
Table 1. Studies on the immune phenotype of patients with membranous nephropathy.
Interestingly, despite the well-reported role of regulatory T-cells (Treg) in autoimmune diseases (100, 101), limited studies have investigated the role and impact of Tregs in primary MN, with controversial results (97, 98). Recently, Rosenzwajg et al. (99) measured 33 lymphocyte subpopulations and also 27 serum cytokines/chemokines in 25 MN patients and 27 healthy subjects at the time of biopsy. After rituximab treatment, responder patients to treatment showed a significantly increased percentage of Tregs than non-responders concluding that monitoring T-cell subset could be a potential biomarker of MN activity.
The discovery of anti-PLA2R and anti-THSD7A autoantibodies represented a paradigm shift for the diagnosis and management of MN patients. Taking into account the putative pathogenic role of anti-PLA2R autoantibodies and the efficacy of B cell depleting therapies (77, 102–104), it is reasonable to speculate that autoreactive memory B cells play a fundamental pathogenic role in MN by fueling a persistent IgG4-specific humoral immune response. However, levels of anti-PLA2R autoantibodies fluctuate over time despite persistent renal injury, suggesting that the evaluation of anti-PLA2R autoantibodies alone may not capture the global humoral immune response taking place in patients with primary MN (69, 79–81). Once B cells recognize the target antigen through the help of autoreactive T Follicular Helper (TFH) cells, B cells can differentiate into short-lived plasmablasts (secreting manly low-affinity IgM antibodies) or into memory B cells (mBC) and long-lived plasma cells after undergoing somatic hypermutation and immunoglobulin isotype class switching in the germinal center. In case of persistence of the priming antigen and T-cell help, auto-reactive mBC can rapidly differentiate into antibody-secreting cells and produce the effector antibodies against the specific target antigen and may finally occupy empty bone marrow niches after secondary activation replenishing plasma cell pool (105, 106). Noteworthy, autoreactive memory B cells can be detected in absence of autoantibody levels in serum and its rapid differentiation and production of antibodies can be of great importance for a subsequent humoral response (Figure 1) (107, 108). Recent works in kidney transplantation have shown the value of measuring circulating allospecific mBC in a functional manner, especially in the absence of detectable alloantibodies in the serum (109–111).
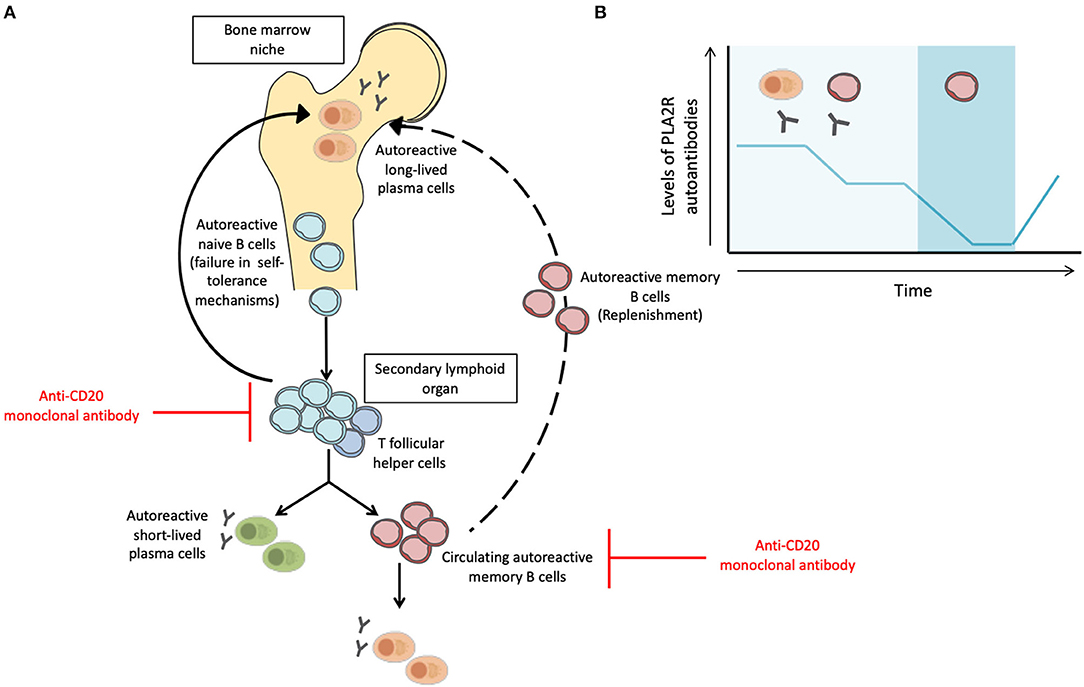
Figure 1. Activation and inhibition of autoimmune B cell responses and the influence of different B cell subsets in the fluctuation of circulating anti-PLA2R antibodies. (A) After failure in self-tolerance mechanisms, autoreactive naïve B cells may encounter the self-antigen and can be activated in the secondary lymphoid organ by helper signals from T follicular helper cells. Then, B cells can differentiate into short-lived plasma cells, which secrete mainly IgM antibodies or differentiate into memory B cells or long-lived plasma cells after somatic hypermutation and immunoglobulin isotype class switching in the germinal center. After a re-encounter with the self-antigen, memory B cells can rapidly differentiate into antibody-secreting cells, sustaining long-lasting humoral immunity. Memory B cells may occupy empty bone marrow niches after secondary activation replenishing plasma cell pool. Anti-CD20 monoclonal antibodies (Rituximab) mainly target naïve B cells and memory B cells but not long-lived plasma cells. (B) Levels of anti-PLA2R autoantibodies may fluctuate over time and may become undetectable without indicating MN remission. Memory B cells can be detected in the absence of antibody levels in serum and its rapid differentiation and production of antibodies can be of great importance for a subsequent humoral response. Such effective and rapid response of the memory-B cell population indicates that although anti-PLA2R autoantibodies may not be detected in serum, PLA2R-specific memory B cells can be a target indicator of MN relapse.
Starting from this background, our group has recently developed a new approach to functionally evaluate the PLA2R-specific mBC response in MN patients. Using a PLA2R-specific B-cell ELISPOT-based immune assay, we have been able to accurately detect circulating mBC capable of producing anti-PLA2R-specific antibodies at the time of the flare of disease activity, thus confirming the presence of an active humoral immune response (personal communication). While evaluating PLA2R-specific antibody-secreting cell frequencies using an ELISPOT-based assay allows for an accurate detection of mBC responses at the single cell level after a polyclonal mBC culture stimulation, anti-PLA2R-specific antibodies may also be detected from these cell culture supernatants using single-antigen beads immune assay. Figure 2 shows two representative patients with similar proteinuria and anti-PLA2R autoantibody levels. While the first patient with detectable autoreactive mBC is having a disease flare, the second one has no detectable autoreactive mBC and is therefore predicted to undergo remission. If properly validated, this assay may be used to differentiate patients for whom therapy is needed vs. those who will undergo spontaneous remission.
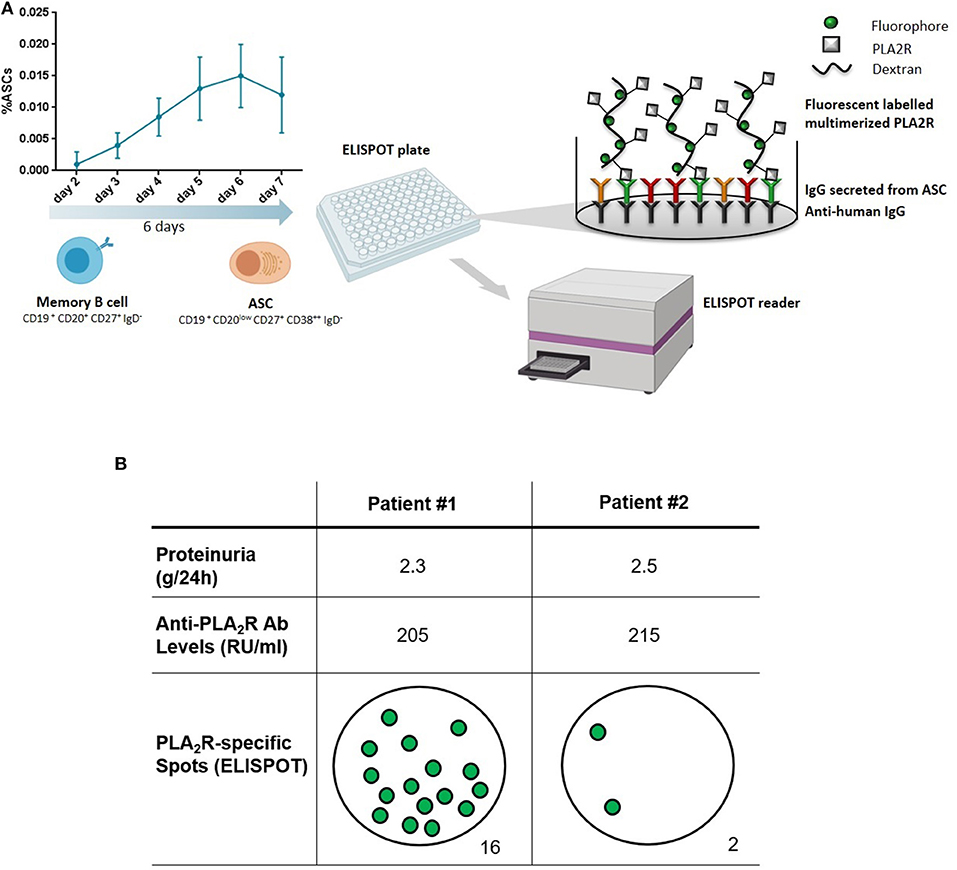
Figure 2. Measuring anti-PLA2R reactive memory B cells. (A) Peripheral blood mononuclear cells are first polyclonally activated for 6 days to expand the pool of memory B cells and antibody secreting cells (ASC). Expanded cells are next used for an enzyme-linked immune absorbent spot (ELISPOT) assay to detect cells producing antibodies against PLA2R. (B) Two representative patients with membranous nephropathy and similar levels of proteinuria and circulating anti-PLA2R antibodies. Patient #1 has a positive ELISPOT, indicating the presence of autoreactive memory B cells (sign of active disease), while Patient #2 has no detectable autoreactive memory B cells (indicative of a remission phase). Adapted from Luque et al. (112).
Primary MN is the main cause of nephrotic syndrome in adults and is caused by the formation of autoimmune complexes in the glomeruli. Since the identification of different podocyte antigenic targets, the diagnostic strategies and treatment options for MN have significantly improved. The efficacy of rituximab treatment in MN patients has highlighted the importance of B cells in the pathogenesis of the disease (113); therefore a more accurate investigation of autoreactive mBC using new technology may refine current immune-monitoring largely based on the measurement of circulating anti-PLA2R or anti-THSD7A autoantibodies.
PC, MJ, AA, ÀF, CC, and OB conceived the article contents, prepared the manuscript, and endorsed the final draft submitted.
This work was supported by 2 Spanish competitive grants from the Instituto de Salud Carlos III [ICI14/00242; PI16/01321] a FEDER funding way to build Europe. Also, this work was partly supported by the SLT002/16/00183 grant, from the Department of Health of the Generalitat de Catalunya by the call Acció instrumental de programes de recerca orientats en l'àmbit de la recerca i la innovació en salut. We thank the CERCA Programme/Generalitat de Catalunya for the institutional support. OB was awarded with an intensification grant from the Instituto de Salud Carlos III [INT15/00112]. MJ received a research fellowship grant from the Instituto de Salud Carlos III [FI17/00233].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Glassock RJ. The pathogenesis of idiopathic membranous nephropathy: a 50-years odyssey. Am J Kidney Dis. (2010) 56:157–67. doi: 10.1053/j.ajkd.2010.01.008
2. Jennette JC, Iskandar SS, Dalldorf FG. Pathologic differentiation between lupus and non-lupus membranous glomerulopathy. Kidney Int. (1983) 24:377–85. doi: 10.1038/ki.1983.170
3. Hofstra JM, Fervenza FC, Wetzels JF. Treatment of idiopathic membranous nephropathy. Nat Rev Nephrol. (2013) 9:443–58. doi: 10.1038/nrneph.2013.125
4. Tran TH, Hughes GJ, Greenfeld C, Pham JT. Overview of current and alternative therapies for idiopathic membranous nephropathy. Pharmacotherapy. (2015) 35:396–411. doi: 10.1002/phar.1575
5. Huh H, Lee H, Lee JP, Kim DK, Oh S, Oh YK, et al. Factors affecting the long-term outcomes of idiopathic membranous nephropathy. BMC Nephrol. (2017) 18:104. doi: 10.1186/s12882-017-0525-6
6. Erwin DT, Donadio JV Jr, Holley KE. The clinical course of idiopathic membranous nephropathy. Mayo Clin Proc. (1973) 48:697–712.
7. Schieppati A, Mosconi L, Perna A, Mecca G, Bertani T, Garattini S, Remuzzi G. Prognosis of untreated patients with idiopathic membranous nephropathy. N Engl J Med. (1993) 329:85–9. doi: 10.1056/NEJM199307083290203
8. Josephson MA, Spargo B, Hollandsworth D, Thistlethwaite JR. The recurrence of recurrent membranous glomerulopathy in a renal transplant recipient: case report and literature review. Am J Kidney Dis. (1994) 24:873–8. doi: 10.1016/S0272-6386(12)80685-8
9. Briganti EM, Russ GR, McNeil JJ, Atkins RC, Chadban SJ. Risk of renal allograft loss from recurrent glomerulonephritis. N Engl J Med. (2002) 347:103–9. doi: 10.1056/NEJMoa013036
10. Rodriguez EF, Cosio FG, Nasr SH, Sethi S, Fidler ME, Stegall MD, et al. The pathology and clinical features of early recurrent membranous glomerulonephritis. Am J Transplant. (2012) 12:1029–38. doi: 10.1111/j.1600-6143.2011.03903.x
11. Cosio FG, Cattran DC. Recent advances in our understanding of recurrent primary glomerulonephritis after kidney transplantation. Kidney Int. (2017) 91:304–14. doi: 10.1016/j.kint.2016.08.030
12. Uffing A, Perez-Saez MJ, La Manna G, Comai G, Fischman C, Farouk S, et al. A large, international study on post-transplant glomerular diseases: the TANGO project. BMC Nephrol. (2018) 19:229. doi: 10.1186/s12882-018-1025-z
13. Mirza MK, Kim L, Kadambi PV, Chang A, Meehan SM. Membranous nephropathy transplanted in the donor kidney: observations of resolving glomerulopathy in serial allograft biopsies. Nephrol Dial Transplant. (2014) 29:2343–7. doi: 10.1093/ndt/gfu333
14. Filippone EJ, Farber JL. Membranous nephropathy in the kidney allograft. Clin Transplant. (2016) 30:1394–402. doi: 10.1111/ctr.12847
15. Heymann W, Hackel DB, Harwood S, Wilson SG, Hunter JL. Production of nephrotic syndrome in rats by Freund's adjuvants and rat kidney suspensions. Proc Soc Exp Biol Med. (1959) 100:660–4. doi: 10.3181/00379727-100-24736
16. Salant DJ, Belok S, Madaio MP, Couser WG. A new role for complement in experimental membranous nephropathy in rats. J Clin Invest. (1980) 66:1339–50. doi: 10.1172/JCI109987
17. Doi T, Mayumi M, Kanatsu K, Suehiro F, Hamashima Y. Distribution of IgG subclasses in membranous nephropathy. Clin Exp Immunol. (1984) 58:57–62.
18. Kusunoki Y, Itami N, Tochimaru H, Takekoshi Y, Nagasawa S, Yoshiki T. Glomerular deposition of C4 cleavage fragment (C4d) and C4-binding protein in idiopathic membranous glomerulonephritis. Nephron. (1989) 51:17–9. doi: 10.1159/000185234
19. Quigg RJ, Holers VM, Morgan BP, Sneed AE III. Crry and CD59 regulate complement in rat glomerular epithelial cells and are inhibited by the nephritogenic antibody of passive Heymann nephritis. J Immunol. (1995) 154:3437–43.
20. Ma H, Sandor DG, Beck LH Jr. The role of complement in membranous nephropathy. Semin Nephrol. (2013) 33:531–42. doi: 10.1016/j.semnephrol.2013.08.004
21. Mellors RC, Ortega LG. Analytical pathology. III. New observations on the pathogenesis of glomerulonephritis, lipid nephrosis, periarteritis nodosa, and secondary amyloidosis in man. Am J Pathol. (1956) 32:455–99.
22. Movat HZ, Mc GD. The fine structure of the glomerulus in membranous glomerulonephritis (lipoid nephrosis) in adults. Am J Clin Pathol. (1959) 32:109–27. doi: 10.1093/ajcp/32.2.109
23. Mathern DR, Heeger PS. Molecules great and small: the complement system. Clin J Am Soc Nephrol. (2015) 10:1636–50. doi: 10.2215/CJN.06230614
24. Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med. (2002) 346:2053–60. doi: 10.1056/NEJMoa012895
25. Debiec H, Nauta J, Coulet F, van der Burg M, Guigonis V, Schurmans T, et al. Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathies. Lancet. (2004) 364:1252–9. doi: 10.1016/S0140-6736(04)17142-0
26. Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. (2009) 361:11–21. doi: 10.1056/NEJMoa0810457
27. Tomas NM, Beck LH Jr, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med. (2014) 371:2277–87. doi: 10.1056/NEJMoa1409354
28. Debiec H, Ronco P. PLA2R autoantibodies and PLA2R glomerular deposits in membranous nephropathy. N Engl J Med. (2011) 364:689–90. doi: 10.1056/NEJMc1011678
29. Tomas NM, Hoxha E, Reinicke AT, Fester L, Helmchen U, Gerth J, et al. Autoantibodies against thrombospondin type 1 domain-containing 7A induce membranous nephropathy. J Clin Invest. (2016) 126:2519–32. doi: 10.1172/JCI85265
30. Du Y, Li J, He F, Lv Y, Liu W, Wu P, et al. The diagnosis accuracy of PLA2R-AB in the diagnosis of idiopathic membranous nephropathy: a meta-analysis. PLoS ONE. (2014) 9:e104936. doi: 10.1371/journal.pone.0104936
31. Iwakura T, Ohashi N, Kato A, Baba S, Yasuda H. Prevalence of enhanced granular expression of thrombospondin type-1 domain-containing 7A in the glomeruli of japanese patients with idiopathic membranous nephropathy. PLoS ONE. (2015) 10:e0138841. doi: 10.1371/journal.pone.0138841
32. Larsen CP, Cossey LN, Beck LH. THSD7A staining of membranous glomerulopathy in clinical practice reveals cases with dual autoantibody positivity. Mod Pathol. (2016) 29:421–6. doi: 10.1038/modpathol.2016.32
33. Sethi S, Madden BJ, Debiec H, Charlesworth MC, Gross L, Ravindran A, et al. Exostosin 1/exostosin 2-associated membranous nephropathy. J Am Soc Nephrol. (2019) 30:1123–36. doi: 10.1681/ASN.2018080852
34. Ronco P, Debiec H. Pathophysiological advances in membranous nephropathy: time for a shift in patient's care. Lancet. (2015) 385:1983–92. doi: 10.1016/S0140-6736(15)60731-0
35. Cohen CD, Calvaresi N, Armelloni S, Schmid H, Henger A, Ott U, et al. CD20-positive infiltrates in human membranous glomerulonephritis. J Nephrol. (2005) 18:328–33.
36. Cravedi P, Sghirlanzoni MC, Marasa M, Salerno A, Remuzzi G, Ruggenenti P. Efficacy and safety of rituximab second-line therapy for membranous nephropathy: a prospective, matched-cohort study. Am J Nephrol. (2011) 33:461–8. doi: 10.1159/000327611
37. Barrett C, Willcocks LC, Jones RB, Tarzi RM, Henderson RB, Cai G, et al. Effect of belimumab on proteinuria and anti-phospholipase A2 receptor autoantibody in primary membranous nephropathy. Nephrol Dial Transplant. (2019) gfz086. doi: 10.1093/ndt/gfz086. [Epub ahead of print].
38. Fervenza FC, Appel GB, Barbour SJ, Rovin BH, Lafayette RA, Aslam N, et al. Rituximab or cyclosporine in the treatment of membranous nephropathy. N Engl J Med. (2019) 381:36–46. doi: 10.1056/NEJMoa1814427
39. Floege J, Barbour SJ, Cattran DC, Hogan JJ, Nachman PH, Tang SCW, et al. Management and treatment of glomerular diseases (part 1): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) controversies conference. Kidney Int. (2019) 95:268–80. doi: 10.1016/j.kint.2018.10.018
40. An C, Akankwasa G, Liu J, Wang D, Cheng G, Zhang J, et al. Urine markers of renal tubular injury in idiopathic membranous nephropathy: a cross sectional study. Clin Chim Acta. (2019) 492:7–11. doi: 10.1016/j.cca.2019.01.015
41. Bobart SA, De Vriese AS, Pawar AS, Zand L, Sethi S, Giesen C, et al. Non-invasive diagnosis of primary membranous nephropathy using phospholipase A2 receptor antibodies. Kidney Int. (2019) 95:429–38. doi: 10.1016/j.kint.2018.10.021
42. Beck LH Jr. PLA2R and THSD7A: disparate paths to the same disease? J Am Soc Nephrol. (2017) 28:2579–89. doi: 10.1681/ASN.2017020178
43. Hoxha E, Beck LH Jr, Wiech T, Tomas NM, Probst C, Mindorf S, et al. An indirect immunofluorescence method facilitates detection of thrombospondin type 1 domain-containing 7A-specific antibodies in membranous nephropathy. j Am Soc Nephrol. (2017) 28:520–31. doi: 10.1681/ASN.2016010050
44. Zaghrini C, Seitz-Polski B, Justino J, Dolla G, Payre C, Jourde-Chiche N, et al. Novel ELISA for thrombospondin type 1 domain-containing 7A autoantibodies in membranous nephropathy. Kidney Int. (2019) 95:666–79. doi: 10.1016/j.kint.2018.10.024
45. Zhang C, Zhang M, Chen D, Ren Q, Xu W, Zeng C, et al. Features of phospholipase A2 receptor and thrombospondin type-1 domain-containing 7A in malignancy-associated membranous nephropathy. J Clin Pathol. (2019) 72:705–11. doi: 10.1136/jclinpath-2019-205852
46. East L, Isacke CM. The mannose receptor family. Biochim Biophys Acta. (2002) 1572:364–86. doi: 10.1016/S0304-4165(02)00319-7
47. Zvaritch E, Lambeau G, Lazdunski M. Endocytic properties of the M-type 180-kDa receptor for secretory phospholipases A2. J Biol Chem. (1996) 271:250–7. doi: 10.1074/jbc.271.1.250
48. Yokota Y, Higashino K, Nakano K, Arita H, Hanasaki K. Identification of group X secretory phospholipase A(2) as a natural ligand for mouse phospholipase A(2) receptor. FEBS Lett. (2000) 478:187–91. doi: 10.1016/S0014-5793(00)01848-2
49. Silliman CC, Moore EE, Zallen G, Gonzalez R, Johnson JL, Elzi DJ, et al. Presence of the M-type sPLA(2) receptor on neutrophils and its role in elastase release and adhesion. Am J Physiol Cell Physiol. (2002) 283:C1102–13. doi: 10.1152/ajpcell.00608.2001
50. Ancian P, Lambeau G, Mattei MG, Lazdunski M. The human 180-kDa receptor for secretory phospholipases A2. Molecular cloning, identification of a secreted soluble form, expression, and chromosomal localization. J Biol Chem. (1995) 270:8963–70. doi: 10.1074/jbc.270.15.8963
51. Fresquet M, Jowitt TA, Gummadova J, Collins R, O'Cualain R, McKenzie EA, et al. Identification of a major epitope recognized by PLA2R autoantibodies in primary membranous nephropathy. J Am Soc Nephrol. (2015) 26:302–13. doi: 10.1681/ASN.2014050502
52. Kao L, Lam V, Waldman M, Glassock RJ, Zhu Q. Identification of the immunodominant epitope region in phospholipase A2 receptor-mediating autoantibody binding in idiopathic membranous nephropathy. J Am Soc Nephrol. (2015) 26:291–301. doi: 10.1681/ASN.2013121315
53. Fresquet M, Rhoden SJ, Jowitt TA, McKenzie EA, Roberts I, Lennon R, et al. Autoantigens PLA2R and THSD7A in membranous nephropathy share a common epitope motif in the N-terminal domain. J Autoimmun. (2019) 2019:102308. doi: 10.1016/j.jaut.2019.102308
54. Klouda PT, Manos J, Acheson EJ, Dyer PA, Goldby FS, Harris R, et al. Strong association between idiopathic membranous nephropathy and HLA-DRW3. Lancet. (1979) 2:770–1. doi: 10.1016/S0140-6736(79)92118-4
55. Scolari F, Amoroso A, Savoldi S, Borelli I, Valzorio B, Costantino E, et al. Familial membranous nephropathy. J Nephrol. (1998) 11:35–9.
56. Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med. (2011) 364:616–26. doi: 10.1056/NEJMoa1009742
57. Lv J, Hou W, Zhou X, Liu G, Zhou F, Zhao N, et al. Interaction between PLA2R1 and HLA-DQA1 variants associates with anti-PLA2R antibodies and membranous nephropathy. J Am Soc Nephrol. (2013) 24:1323–9. doi: 10.1681/ASN.2012080771
58. Bullich G, Ballarin J, Oliver A, Ayasreh N, Silva I, Santin S, et al. HLA-DQA1 and PLA2R1 polymorphisms and risk of idiopathic membranous nephropathy. Clin J Am Soc Nephrol. (2014) 9:335–43. doi: 10.2215/CJN.05310513
59. Saeed M, Beggs ML, Walker PD, Larsen CP. PLA2R-associated membranous glomerulopathy is modulated by common variants in PLA2R1 and HLA-DQA1 genes. Genes Immun. (2014) 15:556–61. doi: 10.1038/gene.2014.50
60. Ramachandran R, Kumar V, Kumar A, Yadav AK, Nada R, Kumar H, et al. PLA2R antibodies, glomerular PLA2R deposits and variations in PLA2R1 and HLA-DQA1 genes in primary membranous nephropathy in South Asians. Nephrol Dial Transplant. (2016) 31:1486–93. doi: 10.1093/ndt/gfv399
61. Sekula P, Li Y, Stanescu HC, Wuttke M, Ekici AB, Bockenhauer D, et al. Genetic risk variants for membranous nephropathy: extension of and association with other chronic kidney disease aetiologies. Nephrol Dial Transplant. (2017) 32:325–32. doi: 10.1093/ndt/gfw001
62. Cui Z, Xie LJ, Chen FJ, Pei ZY, Zhang LJ, Qu Z, et al. MHC class II risk alleles and amino acid residues in idiopathic membranous nephropathy. J Am Soc Nephrol. (2017) 28:1651–64. doi: 10.1681/ASN.2016020114
63. Le WB, Shi JS, Zhang T, Liu L, Qin HZ, Liang S, et al. HLA-DRB1*15:01 and HLA-DRB3*02:02 in PLA2R-related membranous nephropathy. J Am Soc Nephrol. (2017) 28:1642–50. doi: 10.1681/ASN.2016060644
64. Wang HY, Cui Z, Xie LJ, Zhang LJ, Pei ZY, Chen FJ, et al. HLA class II alleles differing by a single amino acid associate with clinical phenotype and outcome in patients with primary membranous nephropathy. Kidney Int. (2018) 94:974–82. doi: 10.1016/j.kint.2018.06.005
65. Bomback AS, Gharavi AG. Can genetics risk-stratify patients with membranous nephropathy? J Am Soc Nephrol. (2013) 24:1190–2. doi: 10.1681/ASN.2013060576
66. Mladkova N, Kiryluk K. Genetic complexities of the HLA region and idiopathic membranous nephropathy. J Am Soc Nephrol. (2017) 28:1331–4. doi: 10.1681/ASN.2017030283
67. De Vriese AS, Glassock RJ, Nath KA, Sethi S, Fervenza FC. A proposal for a serology-based approach to membranous nephropathy. J Am Soc Nephrol. (2017) 28:421–30. doi: 10.1681/ASN.2016070776
68. Hoxha E, Kneissler U, Stege G, Zahner G, Thiele I, Panzer U, et al. Enhanced expression of the M-type phospholipase A2 receptor in glomeruli correlates with serum receptor antibodies in primary membranous nephropathy. Kidney Int. (2012) 82:797–804. doi: 10.1038/ki.2012.209
69. Svobodova B, Honsova E, Ronco P, Tesar V, Debiec H. Kidney biopsy is a sensitive tool for retrospective diagnosis of PLA2R-related membranous nephropathy. Nephrol Dial Transplant. (2013) 28:1839–44. doi: 10.1093/ndt/gfs439
70. Ramachandran R, Kumar V, Nada R, Jha V. Serial monitoring of anti-PLA2R in initial PLA2R-negative patients with primary membranous nephropathy. Kidney Int. (2015) 88:1198–9. doi: 10.1038/ki.2015.310
71. Behnert A, Schiffer M, Muller-Deile J, Beck LH Jr, Mahler M, Fritzler MJ. Antiphospholipase A(2) receptor autoantibodies: a comparison of three different immunoassays for the diagnosis of idiopathic membranous nephropathy. J Immunol Res. (2014) 2014:143274. doi: 10.1155/2014/143274
72. Hofstra JM, Debiec H, Short CD, Pelle T, Kleta R, Mathieson PW, et al. Antiphospholipase A2 receptor antibody titer and subclass in idiopathic membranous nephropathy. J Am Soc Nephrol. (2012) 23:1735–43. doi: 10.1681/ASN.2012030242
73. Timmermans SA, Damoiseaux JG, Heerings-Rewinkel PT, Ayalon R, Beck LH Jr, Schlumberger W, et al. Evaluation of anti-PLA2R1 as measured by a novel ELISA in patients with idiopathic membranous nephropathy: a cohort study. Am J Clin Pathol. (2014) 142:29–34. doi: 10.1309/AJCP8QMOY5GLRSFP
74. Jullien P, Seitz Polski B, Maillard N, Thibaudin D, Laurent B, Ollier E, et al. Anti-phospholipase A2 receptor antibody levels at diagnosis predicts spontaneous remission of idiopathic membranous nephropathy. Clin Kidney J. (2017) 10:209–14. doi: 10.1093/ckj/sfw121
75. Rodas LM, Matas-Garcia A, Barros X, Blasco M, Vinas O, Llobell A, et al. Antiphospholipase 2 receptor antibody levels to predict complete spontaneous remission in primary membranous nephropathy. Clin Kidney J. (2019) 12:36–41. doi: 10.1093/ckj/sfy005
76. Beck LH Jr, Fervenza FC, Beck DM, Bonegio RG, Malik FA, Erickson SB, et al. Rituximab-induced depletion of anti-PLA2R autoantibodies predicts response in membranous nephropathy. J Am Soc Nephrol. (2011) 22:1543–50. doi: 10.1681/ASN.2010111125
77. Ruggenenti P, Debiec H, Ruggiero B, Chianca A, Pelle T, Gaspari F, et al. Anti-phospholipase A2 receptor antibody titer predicts post-rituximab outcome of membranous nephropathy. J Am Soc Nephrol. (2015) 26:2545–58. doi: 10.1681/ASN.2014070640
78. Ramachandran R, Yadav AK, Kumar V, Inamdar N, Nada R, Gupta KL, et al. Temporal association between PLA2R antibodies and clinical outcomes in primary membranous nephropathy. Kidney Int Rep. (2018) 3:142–7. doi: 10.1016/j.ekir.2017.09.001
79. Beck LH Jr, Salant DJ. Membranous nephropathy: recent travels and new roads ahead. Kidney Int. (2010) 77:765–70. doi: 10.1038/ki.2010.34
80. Hofstra JM, Beck LH Jr, Beck DM, Wetzels JF, Salant DJ. Anti-phospholipase A(2) receptor antibodies correlate with clinical status in idiopathic membranous nephropathy. Clin J Am Soc Nephrol. (2011) 6:1286–91. doi: 10.2215/CJN.07210810
81. Qin W, Beck LH Jr, Zeng C, Chen Z, Li S, Zuo K, et al. Anti-phospholipase A2 receptor antibody in membranous nephropathy. J Am Soc Nephrol. (2011) 22:1137–43. doi: 10.1681/ASN.2010090967
82. Radice A, Trezzi B, Maggiore U, Pregnolato F, Stellato T, Napodano P, et al. Clinical usefulness of autoantibodies to M-type phospholipase A2 receptor (PLA2R) for monitoring disease activity in idiopathic membranous nephropathy (IMN). Autoimmun Rev. (2016) 15:146–54. doi: 10.1016/j.autrev.2015.10.004
83. Qu Z, Zhang MF, Cui Z, Wang J, Wang M, Zhang YM, et al. Antibodies against M-type phospholipase A2 receptor may predict treatment response and outcome in membranous nephropathy. Am J Nephrol. (2018) 48:438–46. doi: 10.1159/000494662
84. Chen JL, Hu SY, Jia XY, Zhao J, Yang R, Cui Z, et al. Association of epitope spreading of antiglomerular basement membrane antibodies and kidney injury. Clin J Am Soc Nephrol. (2013) 8:51–8. doi: 10.2215/CJN.05140512
85. Cornaby C, Gibbons L, Mayhew V, Sloan CS, Welling A, Poole BD. B cell epitope spreading: mechanisms and contribution to autoimmune diseases. Immunol Lett. (2015) 163:56–68. doi: 10.1016/j.imlet.2014.11.001
86. Seitz-Polski B, Dolla G, Payre C, Girard CA, Polidori J, Zorzi K, et al. Epitope spreading of autoantibody response to PLA2R associates with poor prognosis in membranous nephropathy. J Am Soc Nephrol. (2016) 27:1517–33. doi: 10.1681/ASN.2014111061
87. Dahan K, Debiec H, Plaisier E, Cachanado M, Rousseau A, Wakselman L, et al. Rituximab for severe membranous nephropathy: a 6-month trial with extended follow-up. J Am Soc Nephrol. (2017) 28:348–58. doi: 10.1681/ASN.2016040449
88. Seitz-Polski B, Debiec H, Rousseau A, Dahan K, Zaghrini C, Payre C, et al. Phospholipase A2 receptor 1 epitope spreading at baseline predicts reduced likelihood of remission of membranous nephropathy. J Am Soc Nephrol. (2018) 29:401–8. doi: 10.1681/ASN.2017070734
89. Ozaki T, Tomino Y, Nakayama S, Koide H. Two-color analysis of lymphocyte subpopulations in patients with nephrotic syndrome due to membranous nephropathy. Clin Nephrol. (1992) 38:75–80.
90. Wang B, Zuo K, Wu Y, Huang Q, Qin WS, Zeng CH, et al. Correlation between B lymphocyte abnormality and disease activity in patients with idiopathic membranous nephropathy. J Int Med Res. (2011) 39:86–95. doi: 10.1177/147323001103900111
91. Cagnoli L, Tabacchi P, Pasquali S, Cenci M, Sasdelli M, Zucchelli P. T cell subset alterations in idiopathic glomerulonephritis. Clin Exp Immunol. (1982) 50:70–6.
92. Zucchelli P, Ponticelli C, Cagnoli L, Aroldi A, Beltrandi E. Prognostic value of T lymphocyte subset ratio in idiopathic membranous nephropathy. Am J Nephrol. (1988) 8:15–20. doi: 10.1159/000167547
93. Taube D, Brown Z, Williams DG. Impaired lymphocyte and suppressor cell function in minimal change nephropathy, membranous nephropathy and focal glomerulosclerosis. Clin Nephrol. (1984) 22:176–82.
94. Hirayama K, Ebihara I, Yamamoto S, Kai H, Muro K, Yamagata K, et al. Predominance of type-2 immune response in idiopathic membranous nephropathy. Cytoplasmic cytokine analysis. Nephron. (2002) 91:255–61. doi: 10.1159/000058401
95. Masutani K, Taniguchi M, Nakashima H, Yotsueda H, Kudoh Y, Tsuruya K, et al. Up-regulated interleukin-4 production by peripheral T-helper cells in idiopathic membranous nephropathy. Nephrol Dial Transplant. (2004) 19:580–6. doi: 10.1093/ndt/gfg572
96. Kuroki A, Iyoda M, Shibata T, Sugisaki T. Th2 cytokines increase and stimulate B cells to produce IgG4 in idiopathic membranous nephropathy. Kidney Int. (2005) 68:302–10. doi: 10.1111/j.1523-1755.2005.00415.x
97. Fervenza FC, Abraham RS, Erickson SB, Irazabal MV, Eirin A, Specks U, et al. Rituximab therapy in idiopathic membranous nephropathy: a 2-year study. Clin J Am Soc Nephrol. (2010) 5:2188–98. doi: 10.2215/CJN.05080610
98. Roccatello D, Sciascia S, Di Simone D, Solfietti L, Naretto C, Fenoglio R, et al. New insights into immune mechanisms underlying response to Rituximab in patients with membranous nephropathy: a prospective study and a review of the literature. Autoimmun Rev. (2016) 15:529–38. doi: 10.1016/j.autrev.2016.02.014
99. Rosenzwajg M, Languille E, Debiec H, Hygino J, Dahan K, Simon T, et al. B- and T-cell subpopulations in patients with severe idiopathic membranous nephropathy may predict an early response to rituximab. Kidney Int. (2017) 92:227–37. doi: 10.1016/j.kint.2017.01.012
100. Noris M, Casiraghi F, Todeschini M, Cravedi P, Cugini D, Monteferrante G, et al. Regulatory T cells and T cell depletion: role of immunosuppressive drugs. J Am Soc Nephrol. (2007) 18:1007–18. doi: 10.1681/ASN.2006101143
101. Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. (2010) 11:7–13. doi: 10.1038/ni.1818
102. Remuzzi G, Chiurchiu C, Abbate M, Brusegan V, Bontempelli M, Ruggenenti P. Rituximab for idiopathic membranous nephropathy. Lancet. (2002) 360:923–4. doi: 10.1016/S0140-6736(02)11042-7
103. Bomback AS, Derebail VK, McGregor JG, Kshirsagar AV, Falk RJ, Nachman PH. Rituximab therapy for membranous nephropathy: a systematic review. Clin J Am Soc Nephrol. (2009) 4:734–44. doi: 10.2215/CJN.05231008
104. Ruggenenti P, Cravedi P, Chianca A, Perna A, Ruggiero B, Gaspari F, et al. Rituximab in idiopathic membranous nephropathy. J Am Soc Nephrol. (2012) 23:1416–25. doi: 10.1681/ASN.2012020181
105. Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B cell signalling in autoimmunity. Nat Rev Immunol. (2017) 17:421–36. doi: 10.1038/nri.2017.24
106. Lebrun C, Cohen M, Rosenthal-Allieri MA, Bresch S, Benzaken S, Marignier R, et al. Only follow-up of memory B cells helps monitor rituximab administration to patients with neuromyelitis optica spectrum disorders. Neurol Ther. (2018) 7:373–83. doi: 10.1007/s40120-018-0101-4
107. Bauer T, Jilg W. Hepatitis B surface antigen-specific T and B cell memory in individuals who had lost protective antibodies after hepatitis B vaccination. Vaccine. (2006) 24:572–7. doi: 10.1016/j.vaccine.2005.08.058
108. Han M, Rogers JA, Lavingia B, Stastny P. Peripheral blood B cells producing donor-specific HLA antibodies in vitro. Hum Immunol. (2009) 70:29–34. doi: 10.1016/j.humimm.2008.10.013
109. Heidt S, Roelen DL, de Vaal YJ, Kester MG, Eijsink C, Thomas S, et al. A NOVel ELISPOT assay to quantify HLA-specific B cells in HLA-immunized individuals. Am J Transplant. (2012) 12:1469–78. doi: 10.1111/j.1600-6143.2011.03982.x
110. Lucia M, Luque S, Crespo E, Melilli E, Cruzado JM, Martorell J, et al. Preformed circulating HLA-specific memory B cells predict high risk of humoral rejection in kidney transplantation. Kidney Int. (2015) 88:874–87. doi: 10.1038/ki.2015.205
111. Luque S, Lucia M, Melilli E, Lefaucheur C, Crespo M, Loupy A, et al. Value of monitoring circulating donor-reactive memory B cells to characterize antibody-mediated rejection after kidney transplantation. Am J Transplant. (2019) 19:368–80. doi: 10.1111/ajt.15055
112. Luque S, Lúcia M, Crespo E, Jarque M, Grinyó JM, Bestard O. A multicolour HLA-specific B-cell FluoroSpot assay to functionally track circulating HLA-specific memory B cells. J Immunol Methods. (2018) 462:23–33. doi: 10.1016/j.jim.2018.07.011
Keywords: membranous nephropathy, glomerulonephritis recurrence, PLA2R, THSD7A, autoreactive B cells
Citation: Cravedi P, Jarque M, Angeletti A, Favà À, Cantarelli C and Bestard O (2019) Immune-Monitoring Disease Activity in Primary Membranous Nephropathy. Front. Med. 6:241. doi: 10.3389/fmed.2019.00241
Received: 10 July 2019; Accepted: 14 October 2019;
Published: 08 November 2019.
Edited by:
Piergiorgio Messa, University of Milan, ItalyReviewed by:
Manuel Praga, Complutense University of Madrid, SpainCopyright © 2019 Cravedi, Jarque, Angeletti, Favà, Cantarelli and Bestard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oriol Bestard, b2Jlc3RhcmRAYmVsbHZpdGdlaG9zcGl0YWwuY2F0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.