 Caroline Bouter
Caroline Bouter Yvonne Bouter
Yvonne Bouter
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 18 April 2019
Sec. Nuclear Medicine
Volume 6 - 2019 | https://doi.org/10.3389/fmed.2019.00071
This article is part of the Research TopicPerspectives in Small Animal Radionuclide ImagingView all 6 articles
Suitable animal models and in vivo biomarkers are essential for development and evaluation of new therapeutic strategies in Alzheimer's disease (AD). 18F-Fluorodeoxyglucose (18F-FDG)-positron-emission tomography (PET) is an imaging biomarker that allows the assessment of cerebral glucose metabolism in vivo. While 18F-FDG-PET/CT is an established tool in the evaluation of AD patients, its role in preclinical studies with AD mouse models remains unclear. Here, we want to review available studies on 18F-FDG-PET/CT in AD mouse models in order to evaluate the method and its impact in preclinical AD research. Only a limited number of studies using 18F-FDG-PET in AD mice were carried out so far showing contradictory findings in cerebral FDG uptake. Methodological differences as well as underlying pathological features of used mouse models seem to be accountable for those varying results. However, 18F-FDG-PET can be a valuable tool in longitudinal in vivo therapy monitoring with a lot of potential for future studies.
Assessment of neuronal dysfunction with the glucose analog 18F-Fluorodeoxyglucose (18F-FDG) is a well-established imaging method in the differential diagnosis of neurodegenerative diseases. 18F-FDG-PET displays the rate of cerebral glucose metabolism which is mainly determined by synaptic activity (1, 2). Impairment of synaptic function and neuron loss are major pathological hallmarks of AD and other dementias. 18F-FDG-PET can be used to identify specific patterns of neuronal dysfunction characteristic for AD with high sensitivity and specificity due to reduced glucose metabolism in certain cerebral areas. Patterns of decreased glucose metabolism mainly include parietal, temporal and frontal cortical areas (3).
Using small animal PET scanners, the same molecular processes can be measured in animal models. In order to develop and evaluate new therapeutic strategies longitudinal assessment of a tested animal is essential. Non-invasive imaging with PET displays a useful tool for therapy monitoring. However, different mouse models for AD show contradictory findings in 18F-FDG-PET.
In this study we review previous use of 18F-FDG-PET in different AD mouse models in order to evaluate the impact of PET imaging in preclinical AD research (Tables 1, 2).
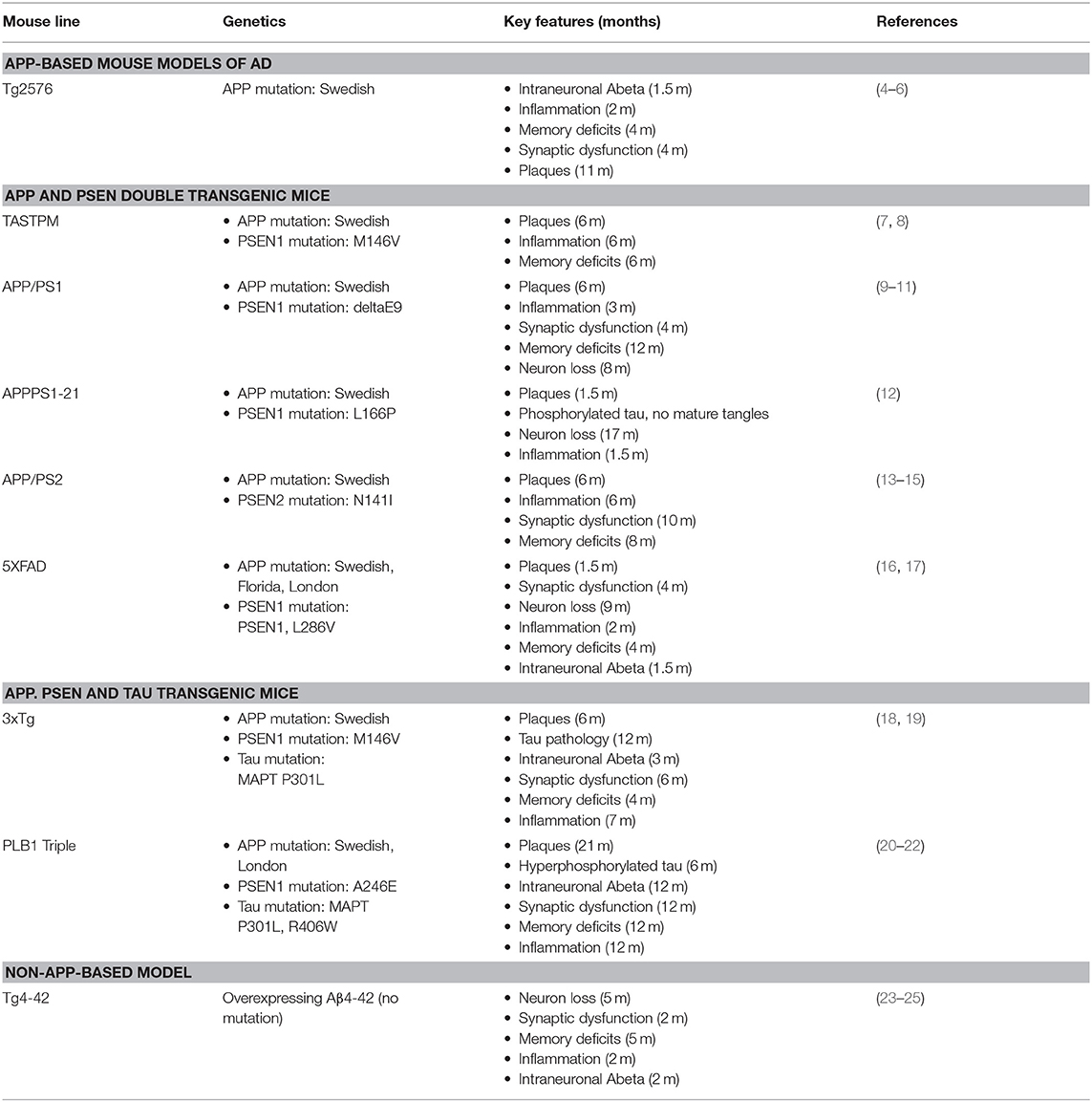
Table 1. Characteristics of AD mouse models used in 18F-FDG-PET studies.
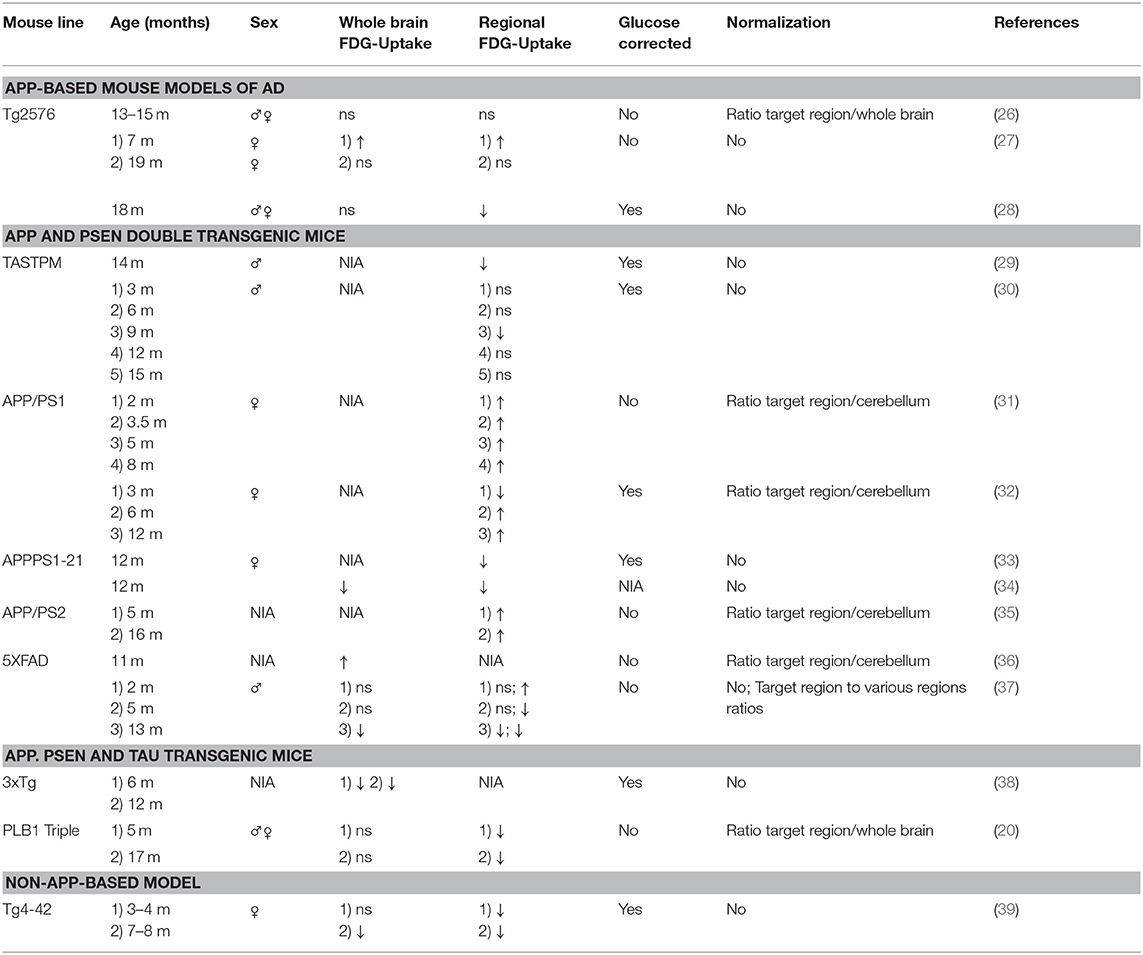
Table 2. Results of 18F-FDG-PET studies in AD mouse models.
The discovery of mutations in the amyloid precursor protein (APP) and presenilin (PSEN) genes in patients with familiar AD led to the generation of a variety of AD mouse models. However, available data on 18F-FDG-PET in AD mouse models is scarce.
The Tg2576 model is one of the first transgenic mouse model based on mutant APP overexpressing human APP with the double Swedish mutation (K670N/M671L) under the control of the hamster prion promoter (40). Tg2576 mice show a distinct Aβ plaque pathology with increased inflammation by 12 monthsof age. Dendritic spine loss was detected starting at 4.5 months. However, neuron loss is very limited. Noticeably, these mice develop cognitive impairments prior to a significant plaque pathology (41, 42).
Studies with 18F-FDG-PET show varying results. The first study using 18F-FDG-PET in Tg2576 mice could not show any significant differences of cerebral glucose metabolism compared to WT (26). Animals were scanned on a microPET Focus220 (Siemens Medical Solutions, Knoxville, TN, USA) after injection of 18F-FDG to anesthetized mice. Quantification was performed using manual volumes of interest (VOIs) with the help of a 3D digital mouse phantom.
Another study by Luo et al. (27) detected an increase of FDG uptake in 7 months old Tg2576 mice while older mice (19 months) did not show differences compared to WT. In this study 18F-FDG was injected to anesthetized mice. Mice were fasted for at least 6 h prior to the study. For dynamic measuring of FDG uptake, images were acquired immediately after injection and continued for 60 min on an Inveon microPET/CT (Siemens Medical Solutions, Knoxville, TN, USA). Quantification was done using whole brain VOIs as well as 7 anatomic regions (cortex, cerebellum, thalamus, hippocampus, striatum, perirhinal cortex and entorhinal cortex) according to a mouse brain atlas. PET results were also compared to cerebral blood volume measured by functional MRI but Tg2576 mice did not show hemodynamic differences compared to age-matched WT mice (27).
In contrast, a more recent study by Coleman et al. (28) detected reduced FDG uptake in 18 months old Tg2576 mice when blood glucose levels were taken into consideration. In this study mice were fasted for 24 h, sampled for blood glucose levels and FDG was injected intraperitoneally while awake. Images were acquired on an Inveon microPET/CT (Siemens Medical Solutions, Knoxville, TN, USA). Results were also confirmed by autoradiography studies of selected animals that were used for ex vivo analyses after PET scanning (28). The authors emphasize the importance of blood glucose corrections of SUV as blood glucose levels can affect brain FDG uptake and transgenic mice are known for a significantly greater decline of blood glucose levels after fasting compared to WT (29).
In order to create a more prominent AD pathology mouse models with APP and PSEN or a combination of multiple mutations were generated. Generally speaking, double transgenic mice expressing mutated PSEN and APP develop an earlier and more aggressive pathology with severe plaque pathology, behavior impairments and increased inflammation (41, 43, 44).
APPPS1-21 mice co-express the human Swedish mutation KM670/671NL and PSEN1 L166P under the control of the neuron-specific Thy-1 promoter (44). Mice show age-dependent Aβ plaque depositions in the hippocampus starting as early as 6 weeks of age accompanied by increased astrogliosis and microgliosis. Dendritic spine loss occurs around plaques beginning 4 weeks after plaque formation, continuing for several months. Modest neuron loss could be detected in the dentate gyrus in 17 months old mice. Cognitive impairment is reported at 7 months (45).
Two studies used 18F-FDG-PET in APPPS1-21 mice so far (33, 34). Twelve months old APPPS1-21 mice showed significantly reduced FDG uptake in the thalamus and striatum. Anesthetized animals were scanned after a conscious uptake period of 45 min on a microPET/CT (Siemens Medical Solutions, Knoxville, TN, USA). Animals were fasted overnight for 8–12 h and blood glucose was monitored. Quantification was performed in predefined VOIs with the help of a mouse brain MRI template and glucose-corrected uptake values were calculated. Ex vivo autoradiography was performed confirming in vivo findings. Furthermore, FDG uptake was compared to amyloid burden but did not show a correlation (33). The second study of Takkinen et al. (34) confirmed those findings. Twelve months old APPPS1-21 mice showed significantly decreased FDG uptake in the whole brain and in several regions including cortex, hippocampus, striatum, thalamus and cerebellum. Mice were fasted for 90 min and anesthetized 30 min prior tracer injection. Dynamic images were acquired on an Inveon PET/CT scanner (Siemens Medical Solutions, Knoxville, TN, USA) for 60 min. VOIs were defined with the help of an MRI mouse brain template and SUVs were calculated. Furthermore, FDG uptake was compared to imaging of neuroinflammation using the tracer 18F-DPA-714. FDG uptake correlated positively with 18F-DPA-714 in the cortex and hippocampus of 6 months old mice (34).
The APP/PS1 model co-expresses the Swedish mutation KM670/671NL and PSEN1 delta E9. Mice show age-dependent extracellular Aβ deposition starting at 6 months of age. Mice show age-dependent cognitive impairment starting at 12 months.
First signs of astrocytosis can be detected at 3 months of age. However, severe gliosis starts around 6 months, especially in close proximity to plaques (9–11, 46).
So far two studies scanned APP/PS1 mice with 18F-FDG-PET showing an age-dependent increase of glucose metabolism. Poisnel et al. (32) detected a significantly higher FDG uptake in the cortex and hippocampus of 12 months old animals compared to WT scanning 3, 6, and 12 months old APP/PS1 mice. Mice were scanned on a microPET Focus 220 (Siemens Medical Solutions, Knoxville, TN, USA). FDG was injected to anesthetized mice followed by a 60 min uptake period maintaining anesthesia. Blood glucose was measured and levels were in normal range. Four VOIs were defined (cortex, hippocampus, striatum and cerebellum) with the help of representative MRI scans and SUV values were calculated in each VOI. SUVs were normalized by cerebellum uptake. Furthermore, functional MRI showed cortical hypoperfusion in 12 months old APP/PS1 mice (32).
Another study by Li et al. (31) showed increased FDG uptake in several brain regions in 2, 3.5, 5, and 8 months old APP/PS1 mice. Mice were scanned on an Inveon microPET/CT (Siemens Medical Solutions, Knoxville, TN, USA). Mice were fasted at least 6 h prior to scanning. During injection and the uptake period, mice were awake. VOIs were defined manually on MRI slices of each animal and SUV values were calculated in each VOI and were normalized by cerebellum uptake (31).
The PS2APP model co-expresses human presenilin 2 (N141I mutation) and human APP (K670N/M671L mutation). PS2APP mice display age-dependent Aβ plaque pathology accompanied by gliosis starting at 6 months of age. Cognitive impairment was detected starting at 8 months (13–15).
A triple tracer study using 18F-FDG, the amyloid tracer 18F-Florbetaben and the neuroinflammation tracer 18F-GE180 showed increased FDG uptake in 5 and 16 months old PS2APP mice. Images were acquired on an Inveon microPET/CT (Siemens Medical Solutions, Knoxville, TN, USA). VOIs were defined using an MRI mouse brain atlas and SUV ratios using the cerebellum for reference were calculated. SUVr (forebrain/cerebellum) was higher in PS2APP mice compared to WT. Furthermore, SUVr correlated with microgliosis and amyloid load while microgliosis and amyloid load strongly correlated (35).
The double transgenic model TASTPM carries the Swedish mutation in APP and the M146V mutation in PSEN1. While sporadic Aβ deposits can be detected in 3 months old TASTPM mice, extracellular Aβ plaque depositions are not evident before 6 months of age. Astrogliosis and microgliosis around amyloid plaques could be detected by 6 months of age. Age-dependent cognitive impairment is described starting at 6 months (7, 8).
Three studies using 18F-FDG-PET in TASTPM mice performed by the same group showed significant regional decreased FDG uptake in 9 and 14 months old animals compared to WT. 18F-FDG-PET/CT was performed on an Inveon microPET/CT (Siemens Medical Solutions, Knoxville, TN, USA). Mice were fastened for 8–12 h before PET. 18F-FDG was injected to awake mice and images were acquired after a conscious uptake period of 45 min. Blood glucose levels were measured and used for glucose normalization of SUV. Uptake values were measured in predefined VOIs using a mouse MRI atlas. Furthermore, these studies emphasized the blood glucose normalization in order to compensate effects on FDG uptake and differences in fasting durations. However, decrease of FDG uptake did not progress over age and did not correlate with Aβ plaque pathology (29, 30, 47).
The widely used 5XFAD model co-expresses five mutations of familial AD overexpressing human APP and PSEN-1. 5XFAD mice develop plaque pathology accompanied by astrocytosis and microgliosis starting at 2 months of age with massive increase over age. Starting at 9 months of age, 5XFAD mice exhibit significant synaptic loss. Neuron loss is detectable in cortical layer 5 and subiculum. Mice show cognitive impairment starting at 4 months (16, 17).
Studies with 18F-FDG-PET show varying results. Rojas et al. (36) detected an increased uptake of 18F-FDG in 11 months old 5XFAD mice compared to WT. In this study 18F-FDG was injected while mice were anesthetized with isoflurane and images were acquired after an awake uptake period of 50 min on a Concorde Microsystems microPET (Concorde Microsystems, TN, USA). Mice were not fasted in advance to PET. Manually defined VOIs were used to calculate whole brain to cerebellum uptake ratios. The authors assumed that those findings might be explained by reactive changes of microglia and astrocytes according to inflammatory processes in the brain due to excessive brain amyloidosis (36).
Another study by Macdonald et al. (37) reported a significant decrease of 18F-FDG uptake in 13 months old 5XFAD mice. Younger mice tested at 2 and 5 months did not show significant differences in whole brain SUV compared to WT. In this study 18F-FDG was injected to conscious mice. During the uptake period mice interacted with a mechanical mouse. Images were acquired after an uptake period of 30 min on a LabPET4 preclinical PET/CT scanner (Trifoil Imaging, CA, USA). SUV values were measured in 8 predefined VOIs (amygdala, basal forebrain, basal ganglia, cerebellum, hippocampus, hypothalamus, neocortex, and thalamus) with the help of an MRI mouse brain atlas and standard uptake values (SUVs) were calculated. Thirteen months old 5XFAD mice showed significantly lower SUV in all brain regions. Furthermore, relative metabolic activity was calculated by using ratios between different brain regions. Young 5XFAD mice showed differences to WT in the basal forebrain, hippocampus, hypothalamus and thalamus relative to the neocortex as a possible early indicator of regional changes (37). Unpublished data of our own group also show significant decreases of 18F-FDG uptake in 12 months old 5XFAD mice.
While APP and PSEN mouse models mirror a wide spectrum of the pathological features seen in AD patients they lack a robust neurofibrillary tangle (NFT) pathology. In order to overcome this absence familial AD mutations have been combined with tau mutations from frontotemporal dementia.
The 3xTg model is an example for such a triple transgenic mouse model as these mice show NFT pathology together with plaque depositions. 3xTg mice express three genes associated with familial AD namely human APP with the Swedish mutation, PSEN1 M146V together with mutated Tau P301L (18).
One study used 18F-FDG-PET in 3xTg mice. Sancheti et al. (38) detected significantly decreased FDG uptake in 6 months old 3xTg mice compared to WT. Mice were fasted overnight and anesthetized before PET. Images were acquired on a microPET R4 PET scanner Siemens Medical Solutions, Knoxville, TN, USA) and additional CT scans were performed. VOIs were drawn manually in co-registered PET and CT images and SUVs were calculated. In addition, the effect of lipoic acid administration on glucose uptake was evaluated and showed an increase of FDG uptake in lipoic acid fed 3xTg mice (38).
The PLB1Triple line is also a triple transgenic AD mouse line carrying human mutated APP with the London and Swedish mutation, mutated tau (P301L/R406W) as well as mutant PSEN1. PLB1Triple mice demonstrate a slow but progressive AD-like pathology. While intraneuronal Aβ can be detected in 12 months old mice Aβ plaques are not visible before 21 months of age. First memory deficits have been observed in 12 months old PLB1Triple mice (20).
Platt et al. ( 20) detected decreased bilateral FDG uptake in the occipital and parietal cortex of 5 and 17 months old PLB1 mice extending to the prefrontal cortex and cerebellum at 17 months. Mice were fasted overnight prior scanning.18F-FDG was injected intraperitoneally to conscious mice. Scans were performed on a Suinsa ARGUS dual-ring scanner (Suinsa Medical Systems, Madrid, Spain). Voxel-based analysis was performed using a digital mouse brain atlas with normalization to whole brain uptake. Regions of statistical significance were mapped on CT images (20).
Familial AD accounts only for a small fraction of all AD cases, however all of the above described AD mouse models rely on one or more FAD mutations. Most AD patients suffer from the late-onset sporadic form of the disease (LOAD) that is not linked to mutations. Therefore, development of animal models that reflect LOAD are becoming into focus.
The transgenic mouse model Tg4–42 is unique as it overexpresses Aβ4-42 without harboring mutations in the Aβ sequence. Tg4-42 mice express human Aβ4-42 fused to the murine thyrotropin-releasing hormone signal peptide, ensuring secretion through the secretory pathway, under the control of the neuronal Thy-1 promoter. Tg4-42 mice show intracellular soluble neurotoxic aggregates of Aβ4-42, without the formation of extracellular amyloid plaques. Homozygous mice show significant age-dependent neuron loss in the hippocampus starting at 5 months with associated astrogliosis and microgliosis at 2 months. Mice show age-dependent cognitive impairment starting at 5 months (23, 24).
A recent study of our group detected age-dependent reduction of 18F-FDG uptake in Tg4-42 mice. Mice were scanned as described above. Early changes in FDG uptake were detected in the hippocampus, forebrain, hypothalamus, amygdala and midbrain at 3 months of age progressing to all brain areas at 8 months. Old Tg4-42 mice showed differences in SUVGlcin all tested brain regions and whole brain compared to WT (39). Mice were scanned on a small animal PET/MRI scanner (1 Tesla nanoScan PET/MRI; Mediso, Hungary). Mice were fasted overnight and blood glucose levels were measured prior to tracer injection. 18F-FDG was injected while mice were anesthetized with isoflurane and images were acquired after an awake uptake period of 45 min. A mouse brain atlas template was used to analyze 11 different predefined brain regions (amygdala, brain stem, cerebellum, cortex, hippocampus, hypothalamus, midbrain, olfactory Bulb, septum/Basal Forebrain, striatum, and thalamus). SUV was calculated and values were corrected for blood glucose levels in order to stabilize uptake values compensating for inequalities in glucose levels and fasting durations (Figure 1).
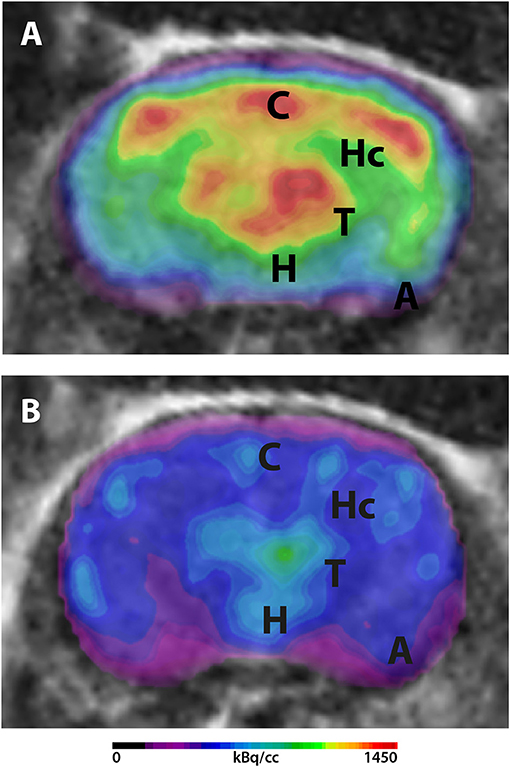
Figure 1. 18F-FDG-PET in Tg4-42 mice. Mice were fasted overnight and 18F-FDG (mean 16.2 MBq) was administered intravenously. Scans were performed after an awake uptake period of 45 min. Mice were anesthetized with isoflurane for the injection and during the scans. PET images were acquired on a small animal 1 Tesla nano scan PET/MRI (Mediso, Hungary) for 20 min. A 136 × 131 × 315 matrix with a voxel size of 0.23 × 0.3 × 0.3 mm3 was used. MRI images were used for attenuation correction (matrix 144 × 144 × 163 with a voxel size of 0.5 × 0.5 × 0.6 mm3, TR: 15 ms, TE 2.032 ms and a flip angle of 25°). Fused PET/MRI images are shown in coronal view. (A) 18F-FDG-PET/MRI of a wildtype mouse with homogenous FDG-distribution in all brain areas. (B) 18F-FDG-PET/MRI of an aged Tg4-42 mouse with distinct lower uptake compared to wildtype mice. A, amygdala; C, cortex; H, hypothalamus; Hc, hippocampus; T, thalamus.
Animal models of AD are essential in order to understand underlying pathologies of the disease and develop new therapeutic strategies. In order to study new therapies, longitudinal assessment of their effects in vivo is important and depends on disease-specific biomarkers. 18F-FDG-PET is an established imaging biomarker for neuronal dysfunction in the diagnostic workup of AD-patients. 18F-FDG-PET displays cerebral glucose metabolism which is mainly determined by synaptic activity. Changes in glucose metabolism can be detected in early stages of the disease as neuronal dysfunction precedes neuron loss. Successful translation of preclinical findings into clinical use requires animal models that display key features of the disease and allow the use of common biomarkers.
Multiple studies using 18F-FDG-PET in animal models of AD have been carried out so far and results of those studies are very inconsistent questioning whether 18F-FDG-PET is a suitable method in AD mouse models. Here we want to summarize earlier findings and discuss possible explanations of those results and their discrepancies.
In contrast to typical findings in AD patients, several studies described increased FDG uptake in transgenic AD mice including the 5XFAD, Tg2576, APPS1-21, and APP/PS1 models (27, 32, 35, 36). One study detected unaltered glucose metabolism in Tg2576 mice (26) while other studies showed decreased cerebral FDG uptake in 5XFAD, Tg2576, APPS1-21, TASTPM, Tg4-42, PLB1, and 3xTg mice (28, 30, 33, 34, 37–39).
The mainly used explanation of increased FDG uptake is the presence of inflammatory cells around amyloid plaques. High astrogliosis and microgliosis might lead to an increased glucose metabolism. However, contradicting results in the same animal models (data available for 5XFAD and Tg2576 mice) as well as in other models with known increased gliosis (APPS1-21, TASTPM, and Tg4-42 mice) showed significant cerebral hypometabolism relativizing this theory. Furthermore, AD patients also display distinct decreased brain glucose metabolism despite highly increased gliosis (48–50). Therefore, astrogliosis and microgliosis alone do not seem to be responsible for cerebral hypermetabolism.
Other explanations for high FDG uptake in the brain include vascular alterations and changes of the blood brain barrier. Next to FDG-PET, Poisnel et al. (32) used functional MRI to measure cerebral perfusion in APP/PS1 mice that showed increased FDG uptake. However, they detected an even reduced cortical perfusion showing that perfusion changes are not accountable for lower FDG uptake in this model (32). Changes in the blood brain barrier as described in histology for Tg2576 mice might lead to increased leakage of FDG into the brain (51). However, two studies on Tg2576 mice did not show a relevant effect of FDG leakage as FDG uptake was decreased or comparable to WT.
Methodological differences should also be taken into consideration. The most striking methodological differences between the studies are found in normalization methods of PET results. Normalization to the cerebellum is a common method as disease pathologies are expected to mainly influence cortical regions. Cerebellar glucose metabolism is relatively preserved in AD patients and normalization with the cerebellum could be shown to improve differential diagnosis of dementia (52–54). However, in mice several studies also describe lower FDG uptake in the cerebellum and therefore it seems to be somehow affected by the pathologies as well (29, 30, 39). Deleye et al. (29) described that normalization to the cerebellum yielded in higher SUVs in transgenic mice. Therefore, the cerebellum seems not appropriate as a reference region for FDG-PET results. Interestingly, all studies that normalized SUVs with the cerebellum did show increased FDG uptake compared to WT independent of the used mouse model. Unfortunately, data of non-normalized SUVs were not shown.
Another common method of normalization includes blood glucose levels. FDG uptake in the brain is highly influenced by blood glucose levels showing an inverse relationship with FDG-uptake (55). Several factors as fasting and fasting duration, stress, body temperature and anesthesia with isoflurane or ketamine influence blood glucose levels. Next to providing stable pre-imaging conditions, normalization of FDG uptake for blood glucose levels should be taken into consideration as some factors cannot always be perfectly stabilized in each individual animal. Furthermore, transgenic mice show lower blood glucose after fasting compared to WT mice (28, 29). This effect might mimic higher FDG uptake in the brain compared to WT.
Further methodological differences between studies include animal preparation and handling (e.g., fasting times), conditions during the uptake period and anesthesia. These differences can influence FDG uptake due to changes in blood glucose levels as mentioned above and some studies used glucose normalization to exactly overcome these issues. Furthermore, synaptic activity might differ between studies due to the use of anesthesia or stimulation (e.g., room conditions, mechanical companion mouse). However, these conditions should not affect results as conditions have to be stable within a study for both, WT and transgenic mice.
Furthermore, technical aspects also differ between studies including PET scanners, acquisition times and image analysis. Spatial resolution displays a major challenge for small animal imaging, especially in the brain. While autoradiography can provide a spatial resolution around 100 μm and below common modern PET scanners and reconstruction engines reach a spatial resolution of around 1.5 mm up to 700 μm in the newest systems (56). While regional differences between autoradiography and small animal PET studies can be explained by the inferiority of PET scanners in terms of spatial resolution, differences between PET scanners in the reviewed studies are not that critical and therefore varying results cannot be majorly explained by the use of different systems.
Pathological features of different mouse models might also influence FDG-PET results. Lacking cerebral hypometabolism may also be due to a limited amount of neuron loss. While atrophy and neuron loss as major hallmarks of AD mainly influence FDG uptake in humans, some transgenic APP-based animal models lack this key feature. However, FDG uptake is not only determined by neuron loss. Synaptic dysfunction also leads to lower glucose consumption in the brain. Studies on mouse models that only show minimal or even lack neuron loss as Tg2576, APPPS1-21, or TASTPM do show cerebral hypometabolism indicating that neuron loss is not the only factor driving FDG uptake.
To date, the impact of 18F-FDG-PET in preclinical studies is subordinate. However, in future studies small animal imaging with 18F-FDG-PET should become an important biomarker for therapy monitoring. In studies on new therapeutic approaches a suitable mouse model that can be monitored by 18F-FDG-PET can be beneficial, especially for monitoring early therapeutic success.
18F-FDG-PET can be a valuable tool for longitudinal in vivo monitoring of AD mice considering its strengths and limitations. Suitability of a certain animal model regarding its pathological features as well as normalization techniques should be carefully considered. Normalization with blood glucose in order to compensate for fasting and preparation inequalities that highly influence blood glucose levels in transgenic mice seems helpful whereas normalization with the cerebellum seems inappropriate.
CB and YB wrote the manuscript. All authors approved the final version.
This work was supported by the German Research Foundation (CNMPB) to YB. This study was supported by the Alzheimer Stiftung Göttingen to CB and YB. We also acknowledge support by the Open Access Publication Funds of the Georg-August-University Göttingen University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kadekaro M, Crane AM, Sokoloff L. Differential effects of electrical stimulation of sciatic nerve on metabolic activity in spinal cord and dorsal root ganglion in the rat. Proc Natl Acad Sci USA. (1985) 82:6010–3. doi: 10.1073/pnas.82.17.6010
2. Sokoloff L. The physiological and biochemical bases of functional brain imaging. Cogn Neurodyn. (2008) 2:1–5. doi: 10.1007/s11571-007-9033-x
3. Bloudek LM, Spackman DE, Blankenburg M, Sullivan SD. Review and meta-analysis of biomarkers and diagnostic imaging in Alzheimer's disease. J Alzheimers Dis. (2011) 26:627–45. doi: 10.3233/JAD-2011-110458
4. Irizarry MC, Mcnamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. (1997) 56:965–73. doi: 10.1097/00005072-199709000-00002
5. Sasaki A, Shoji M, Harigaya Y, Kawarabayashi T, Ikeda M, Naito M, et al. Amyloid cored plaques in Tg2576 transgenic mice are characterized by giant plaques, slightly activated microglia, and the lack of paired helical filament-typed, dystrophic neurites. Virchows Arch. (2002) 441:358–67. doi: 10.1007/s00428-002-0643-8
6. Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. (2006) 103:5161–6. doi: 10.1073/pnas.0600948103
7. Howlett DR, Richardson JC, Austin A, Parsons AA, Bate ST, Davies DC, et al. Cognitive correlates of Abeta deposition in male and female mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. Brain Res. (2004) 1017:130–6. doi: 10.1016/j.brainres.2004.05.029
8. Howlett DR, Bowler K, Soden PE, Riddell D, Davis JB, Richardson JC, et al. Abeta deposition and related pathology in an APP x PS1 transgenic mouse model of Alzheimer's disease. Histol Histopathol. (2008) 23:67–76. doi: 10.14670/HH-23.67
9. Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. (2004) 13:159–70. doi: 10.1093/hmg/ddh019
10. Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE. (2012) 7:e42823. doi: 10.1371/journal.pone.0042823
11. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
12. Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. (2006) 7:940–6. doi: 10.1038/sj.embor.7400784
13. Ozmen L, Albientz A, Czech C, Jacobsen H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener Dis. (2009) 6:29–36. doi: 10.1159/000170884
14. Weidensteiner C, Metzger F, Bruns A, Bohrmann B, Kuennecke B, Von Kienlin M. Cortical hypoperfusion in the B6.PS2APP mouse model for Alzheimer's disease: comprehensive phenotyping of vascular and tissular parameters by MRI. Magn Reson Med. (2009) 62:35–45. doi: 10.1002/mrm.21985
15. Poirier R, Veltman I, Pflimlin MC, Knoflach F, Metzger F. Enhanced dentate gyrus synaptic plasticity but reduced neurogenesis in a mouse model of amyloidosis. Neurobiol Dis. (2010) 40:386–93. doi: 10.1016/j.nbd.2010.06.014
16. Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. (2006) 26:10129–40. doi: 10.1523/JNEUROSCI.1202-06.2006
17. Eimer WA, Vassar R. Neuron loss in the 5XFAD mouse model of Alzheimer's disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener. (2013) 8:2. doi: 10.1186/1750-1326-8-2
18. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. (2003) 39:409–21. doi: 10.1016/S0896-6273(03)00434-3
19. Billings LM, Oddo S, Green KN, Mcgaugh JL, Laferla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. (2005) 45:675–88. doi: 10.1016/j.neuron.2005.01.040
20. Platt B, Drever B, Koss D, Stoppelkamp S, Jyoti A, Plano A, et al. Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PLoS ONE. (2011) 6:e27068. doi: 10.1371/journal.pone.0027068
21. Koss DJ, Drever BD, Stoppelkamp S, Riedel G, Platt B. Age-dependent changes in hippocampal synaptic transmission and plasticity in the PLB1Triple Alzheimer mouse. Cell Mol Life Sci. (2013) 70:2585–601. doi: 10.1007/s00018-013-1273-9
22. Jyoti A, Plano A, Riedel G, Platt B. Progressive age-related changes in sleep and EEG profiles in the PLB1Triple mouse model of Alzheimer's disease. Neurobiol Aging. (2015) 36:2768–84. doi: 10.1016/j.neurobiolaging.2015.07.001
23. Antonios G, Borgers H, Richard BC, Brauss A, Meissner J, Weggen S, et al. Alzheimer therapy with an antibody against N-terminal Abeta 4-X and pyroglutamate Abeta 3-X. Sci Rep. (2015) 5:17338. doi: 10.1038/srep17338
24. Bouter Y, Dietrich K, Wittnam JL, Rezaei-Ghaleh N, Pillot T, Papot-Couturier S, et al. N-truncated amyloid beta (Abeta) 4-42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. (2013) 126:189–205. doi: 10.1007/s00401-013-1129-2
25. Dietrich K, Bouter Y, Muller M, Bayer TA. Synaptic alterations in mouse models for Alzheimer disease-a special focus on N-truncated Abeta 4-42. Molecules. (2018) 23. doi: 10.3390/molecules23040718
26. Kuntner C, Kesner AL, Bauer M, Kremslehner R, Wanek T, Mandler M, et al. Limitations of small animal PET imaging with [18F]FDDNP and FDG for quantitative studies in a transgenic mouse model of Alzheimer's disease. Mol Imaging Biol. (2009) 11:236–40. doi: 10.1007/s11307-009-0198-z
27. Luo F, Rustay NR, Ebert U, Hradil VP, Cole TB, Llano DA, et al. Characterization of 7- and 19-month-old Tg2576 mice using multimodal in vivo imaging: limitations as a translatable model of Alzheimer's disease. Neurobiol Aging. (2012) 33:933–44. doi: 10.1016/j.neurobiolaging.2010.08.005
28. Coleman RA, Liang C, Patel R, Ali S, Mukherjee J. Brain and brown adipose tissue metabolism in transgenic Tg2576 mice models of Alzheimer disease assessed using (18)F-FDG PET imaging. Mol Imaging. (2017) 16:1536012117704557. doi: 10.1177/1536012117704557
29. Deleye S, Waldron AM, Richardson JC, Schmidt M, Langlois X, Stroobants S, et al. The effects of physiological and methodological determinants on 18F-FDG mouse brain imaging exemplified in a double transgenic Alzheimer model. Mol Imaging. (2016) 15:1536012115624919. doi: 10.1177/1536012115624919
30. Waldron AM, Wyffels L, Verhaeghe J, Richardson JC, Schmidt M, Stroobants S, et al. Longitudinal characterization of [18F]-FDG and [18F]-AV45 uptake in the double transgenic TASTPM mouse model. J Alzheimers Dis. (2017) 55:1537–48. doi: 10.3233/JAD-160760
31. Li XY, Men WW, Zhu H, Lei JF, Zuo FX, Wang ZJ, et al. Age- and brain region-specific changes of glucose metabolic disorder, learning, and memory dysfunction in early Alzheimer's disease assessed in APP/PS1 transgenic mice using (18)F-FDG-PET. Int J Mol Sci. (2016) 17:E1707. doi: 10.3390/ijms17101707
32. Poisnel G, Herard AS, El Tannir El Tayara N, Bourrin E, Volk A, Kober F, et al. Increased regional cerebral glucose uptake in an APP/PS1 model of Alzheimer's disease. Neurobiol Aging. (2012) 33:1995–2005. doi: 10.1016/j.neurobiolaging.2011.09.026
33. Waldron AM, Wintmolders C, Bottelbergs A, Kelley JB, Schmidt ME, Stroobants S, et al. In vivo molecular neuroimaging of glucose utilization and its association with fibrillar amyloid-beta load in aged APPPS1-21 mice. Alzheimers Res Ther. (2015) 7:76. doi: 10.1186/s13195-015-0158-6
34. Takkinen JS, Lopez-Picon FR, Al Majidi R, Eskola O, Krzyczmonik A, Keller T, et al. Brain energy metabolism and neuroinflammation in ageing APP/PS1-21 mice using longitudinal (18)F-FDG and (18)F-DPA-714 PET imaging. J Cereb Blood Flow Metab. (2017) 37:2870–82. doi: 10.1177/0271678X16677990
35. Brendel M, Probst F, Jaworska A, Overhoff F, Korzhova V, Albert NL, et al. Glial activation and glucose metabolism in a transgenic amyloid mouse model: a triple-tracer PET study. J Nucl Med. (2016) 57:954–60. doi: 10.2967/jnumed.115.167858
36. Rojas S, Herance JR, Gispert JD, Abad S, Torrent E, Jimenez X, et al. In vivo evaluation of amyloid deposition and brain glucose metabolism of 5XFAD mice using positron emission tomography. Neurobiol Aging. (2013) 34:1790–8. doi: 10.1016/j.neurobiolaging.2012.12.027
37. Macdonald IR, Debay DR, Reid GA, O'leary TP, Jollymore CT, Mawko G, et al. Early detection of cerebral glucose uptake changes in the 5XFAD mouse. Curr Alzheimer Res. (2014) 11:450–60. doi: 10.2174/1567205011666140505111354
38. Sancheti H, Akopian G, Yin F, Brinton RD, Walsh JP, Cadenas E. Age-dependent modulation of synaptic plasticity and insulin mimetic effect of lipoic acid on a mouse model of Alzheimer's disease. PLoS ONE. (2013) 8:e69830. doi: 10.1371/journal.pone.0069830
39. Bouter C, Henniges P, Franke TN, Irwin C, Sahlmann CO, Sichler ME, et al. 18F-FDG-PET detects drastic changes in brain metabolism in the Tg4-42 model of Alzheimer's disease. Front Aging Neurosci. (2019) 10:425. doi: 10.3389/fnagi.2018.00425
40. Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. (1996) 274:99–102. doi: 10.1126/science.274.5284.99
41. Holcomb L, Gordon MN, Mcgowan E, Yu X, Benkovic S, Jantzen P, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. (1998) 4:97–100. doi: 10.1038/nm0198-097
42. Elder GA, Gama Sosa MA, De Gasperi R. Transgenic mouse models of Alzheimer's disease. Mt Sinai J Med. (2010) 77:69–81. doi: 10.1002/msj.20159
43. Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, Van Der Kolk N, et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. (2004) 165:1289–300. doi: 10.1016/S0002-9440(10)63388-3
44. Cavanaugh SE, Pippin JJ, Barnard ND. Animal models of Alzheimer disease: historical pitfalls and a path forward. ALTEX. (2014) 31:279–302. doi: 10.14573/altex.1310071
45. Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horre K, Van Houtvin T, et al. Gamma-secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science. (2009) 324:639–42. doi: 10.1126/science.1171176
46. Jackson HM, Onos KD, Pepper KW, Graham LC, Akeson EC, Byers C, et al. DBA/2J genetic background exacerbates spontaneous lethal seizures but lessens amyloid deposition in a mouse model of Alzheimer's disease. PLoS ONE. (2015) 10:e0125897. doi: 10.1371/journal.pone.0125897
47. Waldron AM, Wyffels L, Verhaeghe J, Bottelbergs A, Richardson J, Kelley J, et al. Quantitative muPET imaging of cerebral glucose metabolism and amyloidosis in the TASTPM double transgenic mouse model of Alzheimer's disease. Curr Alzheimer Res. (2015) 12:694–703. doi: 10.2174/1567205012666150710104713
48. Herholz K, Carter SF, Jones M. Positron emission tomography imaging in dementia. Br J Radiol. (2007) 80:S160–7. doi: 10.1259/bjr/97295129
49. Mosconi L. Glucose metabolism in normal aging and Alzheimer's disease: methodological and physiological considerations for PET studies. Clin Transl Imaging. (2013) 1. doi: 10.1007/s40336-013-0026-y
50. Marcus C, Mena E, Subramaniam RM. Brain PET in the diagnosis of Alzheimer's disease. Clin Nucl Med. (2014) 39:e413–22; quiz e423-416. doi: 10.1097/RLU.0000000000000547
51. Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. (2003) 10:463–70. doi: 10.1038/sj.mn.7800212
52. Ishii K, Willoch F, Minoshima S, Drzezga A, Ficaro EP, Cross DJ, et al. Statistical brain mapping of 18F-FDG PET in Alzheimer's disease: validation of anatomic standardization for atrophied brains. J Nucl Med. (2001) 42:548–57.
53. Dukart J, Mueller K, Horstmann A, Vogt B, Frisch S, Barthel H, et al. Differential effects of global and cerebellar normalization on detection and differentiation of dementia in FDG-PET studies. Neuroimage. (2010) 49:1490–5. doi: 10.1016/j.neuroimage.2009.09.017
54. Dukart J, Perneczky R, Forster S, Barthel H, Diehl-Schmid J, Draganski B, et al. Reference cluster normalization improves detection of frontotemporal lobar degeneration by means of FDG-PET. PLoS ONE. (2013) 8:e55415. doi: 10.1371/journal.pone.0055415
55. Wong KP, Sha W, Zhang X, Huang SC. Effects of administration route, dietary condition, and blood glucose level on kinetics and uptake of 18F-FDG in mice. J Nucl Med. (2011) 52:800–7. doi: 10.2967/jnumed.110.085092
Keywords: 18F-FDG-PET, Alzheimer's disease, mouse model, PET, APP, presenilin
Citation: Bouter C and Bouter Y (2019) 18F-FDG-PET in Mouse Models of Alzheimer's Disease. Front. Med. 6:71. doi: 10.3389/fmed.2019.00071
Received: 16 January 2019; Accepted: 21 March 2019;
Published: 18 April 2019.
Edited by:
Francesco Cicone, Lausanne University Hospital (CHUV), SwitzerlandReviewed by:
Vanessa Cossu, University of Genoa, ItalyCopyright © 2019 Bouter and Bouter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yvonne Bouter, eXZvbm5lLmJvdXRlckBtZWQudW5pLWdvZXR0aW5nZW4uZGU=; eXZvbm5lX2JvdXRAZ214LmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.