 Lylia Ouboussad
Lylia Ouboussad Agata N. Burska
Agata N. Burska Andrew Melville
Andrew Melville Maya H. Buch
Maya H. Buch- 1Leeds Institute of Rheumatic and Musculoskeletal Medicine, University of Leeds, Leeds, United Kingdom
- 2NIHR Leeds Biomedical Research Centre, Leeds Teaching Hospitals NHS Trust, Leeds, United Kingdom
The treatment of rheumatoid arthritis (RA) has been transformed with the introduction of biologic disease modifying anti-rheumatic drugs (bDMARD) and more recently, targeted synthetic DMARD (tsDMARD) therapies in the form of janus-kinase inhibitors. Nevertheless, response to these agents varies such that a trial and error approach is adopted; leading to poor patient quality of life, and long-term outcomes. There is thus an urgent need to identify effective biomarkers to guide treatment selection. A wealth of research has been invested in this field but with minimal progress. Increasingly recognized is the importance of evaluating synovial tissue, the primary site of RA, as opposed to peripheral blood-based investigation. In this mini-review, we summarize the literature supporting synovial tissue heterogeneity, the conceptual basis for stratified therapy. This includes recognition of distinct synovial pathobiological subtypes and associated molecular pathways. We also review synovial tissue studies that have been conducted to evaluate the effect of individual bDMARD and tsDMARD on the cellular and molecular characteristics, with a view to identifying tissue predictors of response. Initial observations are being brought into the clinical trial landscape with stratified biopsy trials to validate toward implementation. Furthermore, development of tissue based omics technology holds still more promise in advancing our understanding of disease processes and guiding future drug selection.
Introduction
Rheumatoid arthritis (RA) is a complex, genetically and biologically heterogeneous autoimmune disease. It is characterized by a systemic inflammatory arthritis. The treatment of patients with RA has evolved considerably in recent years owing to the successful development and widespread use of biologic disease modifying anti-rheumatic drug (bDMARD) therapy, with more recent introduction of targeted synthetic DMARDs (tsDMARD) in the form of small molecules inhibitors. However, up to 40% patients in clinical trials fail to respond, also reflected in real-world practice; and a sizeable proportion fail to achieve the target of therapy, mainly clinical remission where appropriate or low disease activity (1, 2). Personalized medicine, i.e., tailoring therapy to individual patient (or, put simply, “choosing the right drug for the right patient”), has the potential to improve response rates, but has proven challenging to implement. If it is to be successful, the identification of reliable biomarkers will be of prime importance.
In this mini-review, we summarize the evidence for synovial tissue heterogeneity, and tissue studies that have evaluated change in cellular and molecular markers following currently available bDMARD and tsDMARD specifically that could aid treatment selection.
The Synovium, Principal Target of Inflammation
The synovium is the principal target of inflammation in RA, undergoing marked pathological changes compared to healthy tissue. The study of RA synovial tissue has offered insights at a cellular level into multiple aspects of the disease, from identifying pathogenic processes and pathways (3, 4); to explaining clinical manifestations. Furthermore, changes in synovial tissue following successful treatment allow better understanding of mechanism of drug action (5–7).
Synovial tissue samples can be obtained via arthroscopic or ultrasound (US)-guided biopsies. The US-guided approach has been shown to be safe, with reproducible tissue quality/RNA yield (8), and has the advantage of enabling joint assessment for synovial thickness (gray-scale score) and vascularity (Power Doppler-PD), associated with active synovial inflammation (9).
Healthy Synovium
In health, the synovial membrane contains relatively few cells, consisting of an intimal lining layer of 1–2 cell thickness and a distinct synovial sublining layer (10). The intima comprises fibroblast-like synoviocytes (FLS, also known as synovial fibroblasts or type B synoviocytes) intercalated with macrophage-like synoviocytes (MLS, also called type A synoviocytes) (11). The sub-lining layer is a well-vascularized connective tissue, containing collagen fibers and evenly dispersed FLS and MLS (11).
The synovial membrane is key to the structure and function of the healthy synovial joint. The synovial membrane controls transport to and from the synovial cavity, thus maintaining the composition of synovial fluid as well as overall joint homeostasis and integrity. The intimal lining is particularly important, as its lack of tight junctions or a true basement membrane allows the ingress and egress of various cells and proteins (12). Intimal FLS orchestrate proceedings, controlling the synovial fluid volume, secreting hyaluronan for lubrication, clearing intra-articular debris, regulating various immunological processes, and maintaining the extracellular matrix (ECM) of the sublining (13).
RA Synovium
In RA, the synovial tissue becomes markedly expanded, with a striking increase in cellular infiltration. This leads to hallmark “pannus” formation at cartilage-bone interfaces; pannus can be composed of macrophages, FLS, leucocytes, plasma cells, and mast cells (14), and behaves like a locally invasive tumor, mediating damage and erosion formation in later disease (15). The intimal lining can expand to 10–20 cells in thickness, partly due to an increase in FLS, but mostly due to infiltration by bone marrow-derived MLS recruited from the circulation (15). Highly activated macrophages send pro-inflammatory signals to intimal FLS, inducing invasiveness, and to B cells, which in turn produce various pro-inflammatory mediators. Paracrine and autocrine signaling networks develop in this way, further propagating synovitis (16). Sub-lining MLS have been associated with disease activity (17) and synovial inflammation measured on magnetic resonance imaging (MRI) (18), and therefore appear of paramount importance to the inflammatory joint reaction (19). Proliferation of FLS are a prime cause of synovial hyperplasia, and major mediators of damage to cartilage and bone, via both direct and indirect interactions, including production of inflammatory mediators, adhesion molecules, proteolytic enzymes and pro-osteoclastogenic factors (13). T cells are able to establish important crosstalk with antibody-producing plasma cells (15, 20, 21). When present, CD3+ T cells in the RA synovium are mostly found in deeper sub-lining layers, where they may be homogeneously or randomly distributed, or clustered in follicle-like structures (19). Similarly, B cells, when present, are mostly organized in follicular structures, which can act as pro-inflammatory, immunological niches (19).
Heterogeneity of RA Synovitis
RA synovitis is highly heterogeneous, with diverse cellular and molecular signatures (22, 23). In recent years distinct patterns have been recognized, primarily according to the composition, organization and localization of cellular infiltrates. Studies have revealed RA synovial ‘pathotypes’ (7, 24), namely, lymphoid, myeloid, pauci-immune, and fibroid variants (other patterns, such as granulomatous synovitis, have also been described). The lymphoid pathotype is characterized by lymphoid infiltrates, which may be diffuse (small, loosely arranged lymphocyte clusters) or follicular (large aggregates of lymphocytes organized in ectopic lymphoid structures). The latter may develop germinal centers containing T follicular helper (Tfh) cells highly expressing of programmed cell-death (PD-1), C-X-C chemokine receptor 5 (CXCR5), B-cell lymphoma (Bcl6), and Inducible T cell costimulator (ICOS) (7, 24, 25). Cellular composition of tissue defined as myeloid pathotype shows a less abundant B and T cells aggregates compared to the lymphoid subgroup, and presence of sublining macrophages. By contrast, the ‘pauci-immune’ (7) (or ‘low inflammatory’) pathotype shows minimal infiltrating immune cells (24). The fibroid pathotype has complete absence of aggregates and little immune infiltration comprising hyperplastic tissues.
FLS are also not a uniform population but segregate into different phenotypes based, in part, on their cytokine profiles (26). Additionally, functionally distinct disease-associated subsets of fibroblasts are recognized in RA synovium (27) including a study based on surface expression of CD34, THY1, and CDH11 (28). T and B cells infiltrating the inflamed synovium in RA show the highest degree of qualitative and quantitative heterogeneity. Whilst the relation of fibroblast subsets to clinical outcomes remains to be elucidated, these may prove to be instructive biomarkers.
Synovial Tissue Gene Expression Profiles
Early gene expression of RA synovial tissue studies identified distinct profiles and revealed the presence of multiple activated signaling pathways (29–31). Perhaps unsurprisingly given its clinical heterogeneity, expression of molecular signatures in RA is likewise heterogeneous. Gene expression profiles can be modulated by disease activity and the burden of inflammation in synovial tissue (32). Gene expression in RA synovial (intimal) lining cells specifically has been analyzed using a laser mediated micro-dissection (LIMM) approach (33). Data analysis using clustering revealed two distinct RA subgroups associated with increased expression levels of inflammation-related genes [compared with osteoarthritis (OA) control tissue] involved in the tumor necrosis factor TNF-activated interferon regulatory factor (IRF1)- interferon (IFN)- signal transducer and activator of transcription 1 (STAT1)- pathway (34). Three molecularly distinct forms of RA tissues have also been identified by the same group; the first characterized by genes involved in inflammation and the adaptive immune response [matrix metalloproteinase (MMP) 1 and 3 genes, STAT-encoding and -induced genes and antigen-presenting-cell–related genes], the second characterized by genes involved in extracellular matrix remodeling (genes involved in degradation of cartilage and subchondral bone), and the third with a low-inflammation gene signature similar to that of osteoarthritis (30, 31). Increased receptor activator of nuclear factor kappa-B ligand (RANKL) (35) and decreased osteoprotegerin expression (36) have also been detected in actively inflamed RA synovial tissue. These findings, along with the lack of tissue repair signatures, support the hypothesis of inflammation-driven joint remodeling in RA, characterized by uncoupling of destructive and reparative processes (37). A number of transcription factor families, such as nuclear factor kB (NF-kB) and the activator protein 1 (AP-1), were established early on as chief regulators of gene expression in the inflamed synovium (38). Gene expression analysis of FLS indicates the presence of 2 subtypes, with high-inflammatory FLS expressing transforming growth factor (TGF)-β/activin A–inducible genes and FLS from low inflammatory synovial tissue predominantly expressing growth factor genes (39). Distinct molecular signatures indicating pathways relating to T cell-mediated immunity and major histocompatibility complex (MHC) class II mediated immunity (amongst others) upregulated in early RA, and pathways relating to the cell cycle upregulated in later disease (40) have been reported. Similarly differential gene expression between high and low inflammatory subsets of RA patients in relation to disease duration has been observed (29).
Gene Expression Analysis Across Synovial Pathotypes
Differential gene expression has also been confirmed across the RA synovial pathotypes described earlier, providing further evidence for different molecular mechanisms underlying these variants. The lymphoid type is characterized by increased expression of genes associated with B cell and plasmablast activation and differentiation [including CD19, CD20, X-box binding protein XBP1, immunoglobulin heavy and light chains, CD38 and C-X-C motif chemokine ligand 13 (CXCL13)], as well as the Janus kinase JAK/STAT pathway and interleukin 17 (IL-17) signaling (24). In another study, patients with lymphoid aggregates again displayed activation of the JAK/STAT pathway, but also the IL-7 pathway, as well as genes associated with lymphoid neogenesis [such as CXCL13, C-C chemokine ligand 21 (CCL21), and receptor CCR7 and Lymphotoxin alpha (LTα)] and B-cell receptor activation, supporting the existence of a link between tertiary lymphoid structures and the local humoral response (41). In the myeloid pathotype, activation of NF-κB pathway genes (including TNFα, IL-1β, IL-1RA, intracellular adhesion molecule ICAM1, and MyD88), the inflammatory chemokines CCL2 and IL-8, and granulocyte and inflammatory macrophage lineage genes (such as S100A12, CD14, and OSCAR) were identified. In the fibroid pathotype, genes associated with fibroblast and osteoclast/osteoblast regulation were found to be involved, including fibroblast growth factor FGF2, FGF9, BMP6, and osteoprotegerin. Higher expression of Wnt and TGFβ signaling pathway components, as well as “angiogenesis module” genes, were also identified (24). The pauci-immune variant shares characteristics with the aforementioned pathotypes in terms of inflammatory response gene expression, with “M2 monocyte module” genes particularly activated (24, 42). Expression of IL-6, IL-6 receptor components (IL-6R and IL-6ST/gp130), and its associated signaling component STAT3 was broadly observed across all phenotypes, consistent with the multiple roles of the IL-6 pathway in both lymphocyte and fibroblast biology (24, 43). The existence of different gene expression profiles according to RA histological pathotype was also confirmed by Klimiuk et al. who demonstrated increased transcriptional activity of TNFα, IL-1, IFNγ, IL-10, and TGFβ in follicular synovitis, compared with diffuse synovitis (44).
Recently, a machine learning algorithm was able to predict RA synovial gene expression subtype according to 20 histological features. Three subtypes were pre-identified based on RNA-seq clustering: high inflammatory, low inflammatory, and mixed. The high inflammatory subtype showed enrichment of pathways of immunity, immune cell signaling (including SH2, SH3, JAK/STAT, and TNF-mediated signaling), immunoglobulins, chemokines, and cytokines. The low inflammatory subtype was defined by enrichment of transforming growth factor β pathways, glycoprotein synthesis, and cell adhesion genes (45). Distinct myeloid and lymphoid synovial histological subtypes were not identified, in contrast to previous studies (24), but the high inflammatory subtype displayed elevated expression of genes previously attributed to these in the literature.
Synovial Tissue Studies to Predict Response to Biologic and Targeted Therapies
General Synovial Tissue Biomarkers of Response To Therapy
CD68 Macrophage
Effective treatment can modify synovial histology, cytokine and gene expression, with ineffective treatment having little impact, thus providing a means to assess for pathological response (46). Synovial sublining (CD68) macrophage numbers and macrophage expressed cytokines have been shown to correlate with disease activity, and change in sublining macrophage to be the optimal indicator of effective therapy, thus providing a potential early predictive biomarker of drug response (6, 47, 48). A recent study demonstrated that the transcriptional profile of isolated RA synovial macrophages highlighted different subpopulations of patients and identified 6 novel transcriptional modules that were associated with disease activity and therapy (49). The authors suggest that transcriptional signatures in macrophages regardless of location (sublining vs. synovial lining) predict responsiveness to specific non-biologic and/or biologic therapies.
Synovial Pathotypes and Response
A study by Dennis et al. suggested myeloid and lymphoid pathoypes may predict therapeutic sucess with TNF inhibitors (TNFi) and IL-6-targeted tocilizumab, respectively (24). Analysis of serum chemokines further suggested these two pathotypes correlate with raised serum suloble intercellular adhesion molecule 1 (sICAM) and CXCL13 (sICAM/CXCL13) compared to high CXCL13/sICAM, respectively. These initial observations however have not been validated in other cohorts using the serum correlates (50) indicating the need for additional such synovial tissue studies. Nevertheless, stratifying patients by synovial pathotype may inform choice of targeted therapy.
Multiple types of therapies will be discussed in detail below, these are summarized in Table 1 together with key findings which indicate response to biologic and synthetic targeted DMARDs.
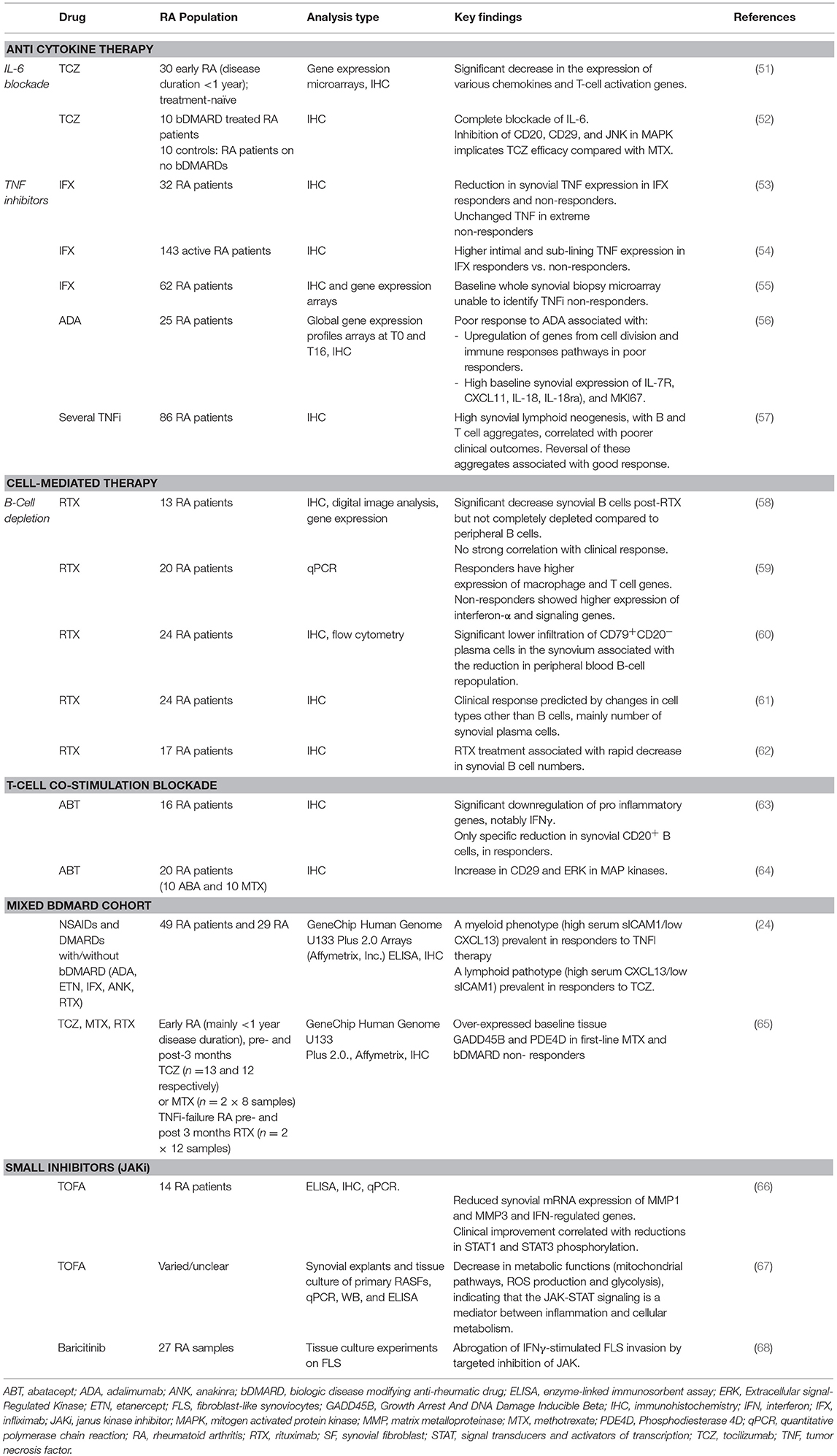
Table 1. Rheumatoid synovial tissue studies of biologic and targeted synthetic DMARDs.
Anti-cytokine Therapies
Tumor-Necrosis Factor-Inhibitors
Synovial studies have offered useful insights into the mechanism of action of TNFi. TNFi have been shown to regulate chemokine and leukocyte trafficking (69) likely explaining the reduction in the synovial cellular infiltrate observed; with reductions in synovial tissue expression of IL-6, IL-8, granulocyte macrophage colony stimulating factor (GM-CSF), macrophage chemoattractant protein-1 (MCP-1), IL-1β, TNF, and vascular endothelial growth factor (VEGF) (70).
Several studies have sought to identify predictors of response to TNF blockade through examination of synovial tissue cytokine expression. Baseline synovial TNF levels (intimal and sub-lining) predicted response to infliximab in one study (54), although another similar study did not reproduce this finding (53). Decreased sub-lining TNF expression was, however, seen in responders. A prospective study of 86 patients found higher proportions of synovial lymphoid aggregates in poor responders to treatment, despite higher rates of TNFi use. Baseline lymphoid aggregates were an independent predictor of poor response in multivariate analysis, and reversal of these histological changes was seen in over half of treatment responders (57). Addition of lymphocyte aggregates to sub-lining TNF expression (54) improved infliximab response prediction, but still only accounting for 29% variance (71); thus insufficient for clinical application. An early RA synovial gene expression study found that mRNA levels pertaining to several inflammatory pathways were associated with response to TNFi therapy, suggesting a role for synovial gene expression profiles as response predictors (72). Another study identified a number of negative predictors of response to adalimumab, another TNFi biologic, including baseline synovial expression of IL-7 receptor alpha chain (IL-7R), CXCL11, IL-18, IL-18 receptor accessory (IL-18rap), and MKI67 (63). However, a larger gene expression study using whole synovial tissue samples pre- and post-infliximab did not identify any predictors, perhaps because of the confounding presence of lymphoid aggregates (55).
Tocilizumab
Tocilizumab is a clinically effective humanized anti–IL-6R monoclonal antibody that inhibits membrane IL-6R– and soluble IL-6R (sIL-6R)–mediated signaling. The aforementioned study by Dennis et al. (24), suggested lymphoid pathotype as predictive of response. In another study, paired synovial tissue biopsies taken at baseline and post-treatment with tocilizumab showed a significant decrease in the expression of various chemokines and T-cell activation genes (51). When compared with gene expression data following other treatments, results showed strong correlation with methotrexate and B-cell depleting agent rituximab, but notable differences with adalimumab (51). A further study of synovial histology post-tocilizumab demonstrated a complete block of synovial IL-6 and a significant reduction of B-cells, CD29 and phospho-JNK. ERK was increased in the tocilizumab group compared to a methotrexate-treated control group, whilst TNF, MMP-3, and CD68 were similarly expressed in both groups. Therefore, inhibition of IL-6/CD20/CD29 may be differentially involved in tocilizumab efficacy compared with methotrexate (52). A more recent study in 33 early RA patients suggested higher expression of TNF-induced transcripts in early RA synovitis was associated with higher disease activity, and predicted poor response to first-line therapy (that comprised either methotrexate, tocilizumab or rituximab therapy) (65). Finally, an exploratory study by Das et al. suggested persistent synovial IL-6 mRNA expression (following rituximab inefficacy) associated with subsequent tocilizumab response (73).
Cell Mediated Therapies
B-Cell Depletion: Rituximab
Treatment with the anti-CD20 monoclonal antibody rituximab significantly decreases synovial B cells, but, unlike in the periphery, does not completely eradicate them. In addition, synovial B cell depletion does not correlate strongly with clinical response in RA, suggesting the effects of rituximab on synovial B cells may be necessary but not sufficient for inducing clinical efficacy (58). A separate study of RA synovial histology pre- and post-rituximab confirmed these findings, but also examined changes in other cell populations at 4 and 16 weeks. A reduction in short-lived CD138+ plasma cells, possibly generated locally within the synovial membrane, was found to predict clinical response, whilst delayed reductions in T cell, intimal macrophages and lymphoid aggregates were also seen, highlighting the role of B cells in sustaining inflammation and cell recruitment (74). Another study suggested that clinical response to rituximab is associated with higher residual levels of CD79+CD20− plasma cells in the synovium (together with persistence of circultaing ACPA+ IgM plasmablasts) (60). In addition, there is evidence that baseline synovial gene expression may be able to predict response to rituximab (and lack of response), as composite “gene scores” were found to correlate with changes in disease activity (DAS-28 score) in one study (59). Genes relating to macrophage and T cell function were activated in responders.
At a more fundamental level, B cells have shown to be central to T-cell mediated synovial inflammation. This was elegantly demonstrated by a study showing that synovial T-cell clones adoptively transferred into human leukocyte antigen (HLA)-DR-matched synovial tissues xenotransplanted into severe combine immunodeficient (SCID) mice are able to enhance local production of IFNγ, TNF, and IL-1β, but only when transplanted tissues contain B-cell follicles (75). Furthermore, treatment of synovial grafts with anti-CD20 depleting agents induces not only a decrease in B-cell density but also a disruption of the overall lymphoid architecture and reduction of cytokine expression, as well as a dramatic depletion of T cells and macrophages, in keeping with the existence of an active cell network supported by B cells.
T-Cell Co-stimulation Blockade (Abatacept (CTLA4-Fc))
Abatacept, a recombinant fusion protein approved for the treatment of RA, blocks T cell co-stimulation by competing with CD28 for CD80/86 on antigen presenting cells. Synovial studies of the effect of and mechanism of abatacept are relatively lacking. A study of 16 RA patients compared synovial tissue pre- and 16 weeks post-abatacept in terms of gene expression and immunohistochemistry. Amongst responders, there was notable downregulation of several pro-inflammatory mediators, particularly the T-cell-related cytokine IFNγ. However, only a specific reduction in synovial CD20+ B cells without significant disruption in other cell populations was observed (contrasting with the observations following anti-cytokine therapies, perhaps in keeping with the more immunomodulatory role of CTLA4) (63). Whilst effects on tertiary lymphoid structures were not analyzed, these observations suggest that disruption of T-/B-cell interactions may be critical to abatacept's mode of action. In contrast to this study, a smaller study on 5 patients treated with abatacept indicated inhibition of cell proliferation, with decreases in the expression of MMP-3, CD68, CD4, CD8, CD20, CD80, and CD86 in the synovium (64).
Small Molecule Janus-Kinase (JAK) Inhibitors
Multiple inflammatory cytokines signal via JAK-STAT pathway. Thus, JAK/STAT signaling plays a key role in several immune mediated inflammatory diseases, including RA (76). As small molecules with intracellular targets (i.e., JAK family members), JAK inhibitors represent a novel targeted therapeutic approach in RA (77).
Tofacitinib is an oral JAK inhibitor effective for the treatment of RA (78). It is a pan-selective JAKi, blocking signaling mediated via JAK1, JAK3 and, to a lesser extent, JAK2 (79). A comparison of RA synovial tissue at pre- and 4 weeks post-treatment with tofacitinib showed no change in an overall inflammation score or levels of T cells, B cells or macrophages, but reduced expression of MMPs (MMP1 and MMP3) and interferon-regulated genes, notably CXCL10. Furthermore, clinical improvement at 4 months was found to correlate with reductions in STAT1 and STAT3 phosphorylation, indicating the importance of IFNγ and IL-6 inhibition, respectively (66). In addition, a recent metabolomics study showed that adding tofacitinib to RA synovial explants and synovial fibroblasts in vitro led to decreased mitochondrial pathway activity, reactive oxygen species (ROS) production and glycolysis, suggesting modulation of cellular metabolism may contribute to its therapeutic effect (67).
Baricitinib, a JAK inhibitor targeting JAK1/JAK2, is another licensed treatment for RA (80). A study specifically examining FLS activity in RA showed that baricitinib abrogates IFNγ-induced invasiveness of FLS (68), which is of importance given their key contribution to pannus formation (aggressive cell masses that destroy articular cartilage and bone), one of the hallmarks of RA synovial pathobiology (81).
Conclusion
It is well-accepted that the considerable advances in the treatment of RA need to be accompanied by a stratified approach that mitigates against the current trial and error approach of treatment decision-making, and the associated individual patient and health-economic consequences. Significant investment in biomarker studies has failed to deliver clinically meaningful tools, with the vast majority focusing on peripheral blood-based evaluation. The emphasis on synovial tissue, the primary site of RA is intuitive, from which tissue and thus disease subtypes are emerging.
The need to pull through benchside investigation of tissue biomarkers to the bedside demands more refined and innovative stratified trial design (82). We will soon see the outcomes of such initiatives [including STRAP—Stratification of Biologic Therapies for RA by Pathobiology (ISRCTN10618686) and R4-RA—A Randomized, open labeled study in anti-TNFa inadequate responders to investigate the mechanisms for Response—Resistance to Rituximab vs. Tocilizumab in RA (ISRCTN97443826)] that will inform future tissue driven trial design. These trials and other tissue-based programmes such as the recently established NIH Accelerating Medicines Partnership (AMP) RA/SLE network will also exploit high-dimensional analyses including mass cytometry, RNA-seq of selected cell populations, and single cell RNA-seq (83). Whilst the sheer volume of data in itself presents massive challenges in the clinically meaningful interpretation, the richness of data matched with improved sophisticated analytical techniques holds the promise of being able to join the field of personalized RA targeted therapy use.
Author Contributions
LO, AB, and AM: literature Search, write up, final approval of the manuscript; MB: conception and design of the work, write up, final approval of the manuscript.
Funding
MB research grants Pfizer Ltd., Roche, UCB. Consulting fees, Abbvie, Lilly Merck-Serono, Pfizer Ltd., Sandoz, Sanofi.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Nam J, Winthrop K, van Vollenhoven RF, Pavelka K, Valesini G, Hensor E, et al. Current evidence for the management of rheumatoid arthritis with biological disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of RA. Ann Rheum Dis. (2010) 69:976–86. doi: 10.1136/ard.2009.126573
2. Sokka T, Hetland ML, Mäkinen H, Kautiainen H, Hørslev-Petersen K, Luukkainen RK, et al. Remission and rheumatoid arthritis: data on patients receiving usual care in twenty-four countries. Arthritis Rheumat. (2008) 58:2642–51. doi: 10.1002/art.23794
3. Devauchelle V, Marion S, Cagnard N, Mistou S, Falgarone G, Breban M, et al. DNA microarray allows molecular profiling of rheumatoid arthritis and identification of pathophysiological targets. Genes Immun. (2004) 5:597–608. doi: 10.1038/sj.gene.6364132
4. Gerlag DM, Tak PP. How to perform and analyse synovial biopsies. Best Pract Res Clin Rheumatol. (2009) 23:221–32. doi: 10.1016/j.berh.2013.03.006
5. Tak P. Lessons learnt from the synovial tissue response to anti-rheumatic treatment. Rheumatology. (2000) 39:817–20. doi: 10.1093/rheumatology/39.8.817
6. Tak PP, Smeets TJ, Daha MR, Kluin PM, Meijers KA, Brand R, et al. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheumat. (1997) 40:217–25. doi: 10.1002/art.1780400206
7. Pitzalis C, Kelly S, Humby F. New learnings on the pathophysiology of RA from synovial biopsies. Curr Opin Rheumatol. (2013) 25:334–44. doi: 10.1097/BOR.0b013e32835fd8eb
8. Kelly S, Humby F, Filer A, Ng N, Di Cicco M, Hands R, et al. Ultrasound-guided synovial biopsy: a safe, well-tolerated and reliable technique for obtaining high-quality synovial tissue from both large and small joints in early arthritis patients. Ann Rheuma Dis. (2015) 74:611–7. doi: 10.1136/annrheumdis-2013-204603
9. Koski JM, Saarakkala S, Helle M, Hakulinen U, Heikkinen JO, Hermunen H. Power Doppler ultrasonography and synovitis: correlating ultrasound imaging with histopathological findings and evaluating the performance of ultrasound equipments. Ann Rheuma Dis. (2006) 65:1590–5. doi: 10.1136/ard.2005.051235
10. Smith MD. The normal synovium. Open Rheumatol J. (2011) 5:100–6. doi: 10.2174/1874312901105010100
11. Lindblad S, Hedfors E. The synovial membrane of healthy individuals–immunohistochemical overlap with synovitis. Clin Exp Immunol. (1987) 69:41–7.
12. Steenvoorden MMC, Tolboom TCA, van der Pluijm G, Löwik C, Visser CPJ, DeGroot J, et al. Transition of healthy to diseased synovial tissue in rheumatoid arthritis is associated with gain of mesenchymal/fibrotic characteristics. Arthritis Res Therapy. (2006) 8:R165. doi: 10.1186/ar2073
13. Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. (2013) 9:24. doi: 10.1038/nrrheum.2012.190
14. Bromley M, Woolley DE. Histopathology of the rheumatoid lesion. Arthritis Rheumat. (1984) 27:857–63. doi: 10.1002/art.1780270804
15. Firestein GS. Etiology and Pathogenesis of Rheumatoid Arthritis. Kelley's Textbook of Rheumatology. Philadelphia, PA: Elsevier (2012), 921–66.
16. Tak PP, Bresnihan B. The pathogenesis and prevention of joint damage in rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis Rheumat. (2000) 43:2619–33. doi: 10.1002/1529-0131(200012)43:12<2619::AID-ANR1>3.0.CO;2-V
17. Vordenbäumen S, Sewerin P, Lögters T, Miese F, Schleich C, Bleck E, et al. Inflammation and vascularisation markers of arthroscopically-guided finger joint synovial biospies reflect global disease activity in rheumatoid arthritis. Clin Exp Rheumatol. (2014) 32:117–20.
18. Vordenbäumen S, Schleich C, Lögters T, Sewerin P, Bleck E, Pauly T, et al. Dynamic contrast-enhanced magnetic resonance imaging of metacarpophalangeal joints reflects histological signs of synovitis in rheumatoid arthritis. Arthritis Res Therapy. (2014) 16:452. doi: 10.1186/s13075-014-0452-
19. Mucke J, Hoyer A, Brinks R, Bleck E, Pauly T, Schneider M, et al. Inhomogeneity of immune cell composition in the synovial sublining: linear mixed modelling indicates differences in distribution and spatial decline of CD68+ macrophages in osteoarthritis and rheumatoid arthritis. Arthritis Res Therapy. (2016) 18:170. doi: 10.1186/s13075-016-1057-3
20. Buckley CD, McGettrick HM. Leukocyte trafficking between stromal compartments: lessons from rheumatoid arthritis. Nat Rev Rheumatol. (2018) 14:476–87. doi: 10.1038/s41584-018-0042-4
21. Fonseca J, Canhao H, Resende C, Saraiva F, da Costa JT, Pimentão JB, et al. Histology of the synovial tissue: value of semiquantitative analysis for the prediction of joint erosions in rheumatoid arthritis. Clin Exp Rheumatol. (2000) 18:559–64.
22. Klimiuk PA, Goronzy JJ. Tissue cytokine patterns distinguish variants of rheumatoid synovitis. Am J Pathol. (1997) 151:1311–9.
23. Lauwerys BR, Hernández-Lobato D, Gramme P, Ducreux J, Dessy A, Focant I, et al. Heterogeneity of synovial molecular patterns in patients with arthritis. PLoS ONE. (2015) 10:e0122104. doi: 10.1371/journal.pone.0122104
24. Dennis G, Holweg CT, Kummerfeld SK, Choy DF, Setiadi AF, Hackney JA, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Therapy. (2014) 16:R90. doi: 10.1186/ar4555
25. Orr C, Najm A, Biniecka M, McGarry T, Ng Ct, Young F, et al. Synovial immunophenotype and anti–citrullinated peptide antibodies in rheumatoid arthritis patients: relationship to treatment response and radiologic prognosis. Arthritis Rheumatol. (2017) 69:2114–23. doi: 10.1002/art.40218
26. Croft AP, Naylor AJ, Marshall JL, Hardie DL, Zimmermann B, Turner J, et al. Rheumatoid synovial fibroblasts differentiate into distinct subsets in the presence of cytokines and cartilage. Arthritis Res Therapy. (2016) 18:270. doi: 10.1186/s13075-016-1156-1
27. Stephenson W, Donlin LT, Butler A, Rozo C, Bracken B, Rashidfarrokhi A, et al. Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation. Nat Commun. (2018) 9:791. doi: 10.1038/s41467-017-02659-x
28. Mizoguchi F, Slowikowski K, Wei K, Marshall JL, Rao DA, Chang SK, et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat Commun. (2018) 9:789. doi: 10.1038/s41467-018-02892-y
29. van Baarsen LG, Wijbrandts CA, Timmer TC, van der Pouw Kraan TC, Tak PP, Verweij CL. Synovial tissue heterogeneity in rheumatoid arthritis in relation to disease activity and biomarkers in peripheral blood. Arthritis Rheumatism. (2010) 62:1602–7. doi: 10.1002/art.27415
30. Van der Pouw Kraan T, Van Gaalen F, Huizinga T, Pieterman E, Breedveld F, Verweij C. Discovery of distinctive gene expression profiles in rheumatoid synovium using cDNA microarray technology: evidence for the existence of multiple pathways of tissue destruction and repair. Genes Immunity. (2003) 4:187–96. doi: 10.1038/sj.gene.6363975
31. van der Pouw Kraan TC, van Gaalen FA, Kasperkovitz PV, Verbeet NL, Smeets TJ, Kraan MC, et al. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheumat. (2003) 48:2132–45. doi: 10.1002/art.11096
32. Townsend MJ. Molecular and cellular heterogeneity in the Rheumatoid Arthritis synovium: clinical correlates of synovitis. Best Pract Res Clin Rheumatol. (2014) 28:539–49. doi: 10.1016/j.berh.2014.10.024
33. Yoshida S, Arakawa F, Higuchi F, Ishibashi Y, Goto M, Sugita Y, et al. Gene expression analysis of rheumatoid arthritis synovial lining regions by cDNA microarray combined with laser microdissection: up-regulation of inflammation-associated STAT1, IRF1, CXCL9, CXCL10, and CCL5. Scand J Rheumatol. (2012) 41:170–9. doi: 10.3109/03009742.2011.623137
34. Hashimoto A, Tarner IH, Bohle RM, Gaumann A, Manetti M, Distler O, et al. Analysis of vascular gene expression in arthritic synovium by laser-mediated microdissection. Arthritis Rheumat. (2007) 56:1094–105. doi: 10.1002/art.22450
35. Crotti T, Smith M, Weedon H, Ahern M, Findlay D, Kraan M, et al. Receptor activator NF-κB ligand (RANKL) expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathy, osteoarthritis, and from normal patients: semiquantitative and quantitative analysis. Anna Rheumat Dis. (2002) 61:1047–54. doi: 10.1136/ard.61.12.1047
36. Haynes D, Barg E, Crotti T, Holding C, Weedon H, Atkins G, et al. Osteoprotegerin expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathies and osteoarthritis and normal controls. Rheumatology. (2003) 42:123–34. doi: 10.1093/rheumatology/keg047
37. Bugatti S, Manzo A, Bombardieri M, Vitolo B, Humby F, Kelly S, et al. Synovial tissue heterogeneity and peripheral blood biomarkers. Curr Rheumatol Rep. (2011) 13:440. doi: 10.1007/s11926-011-0201-y
38. Tak PP, Firestein GS. NF-κB: a key role in inflammatory diseases. J Clin Invest. (2001) 107:7–11. doi: 10.1172/JCI11830
39. Kasperkovitz P, Verbeet N, Smeets T, van Rietschoten J, Kraan M, van der Pouw Kraan T, et al. Activation of the STAT1 pathway in rheumatoid arthritis. Ann Rheumat Dis. (2004) 63:233–9. doi: 10.1136/ard.2003.013276
40. Lequerre T, Bansard C, Vittecoq O, Derambure C, Hiron M, Daveau M, et al. Early and long-standing rheumatoid arthritis: distinct molecular signatures identified by gene-expression profiling in synovia. Arthritis Res Therapy. (2009) 11:R99. doi: 10.1186/ar2744
41. Timmer TCG, Baltus B, Vondenhoff M, Huizinga TWJ, Tak PP, Verweij CL, et al. Inflammation and ectopic lymphoid structures in rheumatoid arthritis synovial tissues dissected by genomics technology: identification of the interleukin-7 signaling pathway in tissues with lymphoid neogenesis. Arthritis Rheumat. (2007) 56:2492–502. doi: 10.1002/art.22748
42. Manzo A, Bugatti S, Caporali R, Montecucco C. Histopathology of the synovial tissue: perspectives for biomarker development in chronic inflammatory arthritides. Reumatismo. (2018) 70:121–32. doi: 10.4081/reumatismo.2018.1057
43. Srirangan S, Choy EH. The role of interleukin 6 in the pathophysiology of rheumatoid arthritis. Therape Adv Musculoskeletal Dis. (2010) 2:247–56. doi: 10.1177/1759720X10378372
44. Klimiuk PA, Sierakowski S, Latosiewicz R, Skowronski J, Cylwik JP, Cylwik B, et al. Histological patterns of synovitis and serum chemokines in patients with rheumatoid arthritis. J Rheumatol. (2005) 32:1666–72.
45. Orange DE, Agius P, DiCarlo EF, Robine N, Geiger H, Szymonifka J, et al. Identification of three rheumatoid arthritis disease subtypes by machine learning integration of synovial histologic features and RNA sequencing Data. Arthritis Rheumatol. (2018) 70:690–701. doi: 10.1002/art.40428
46. Baeten D, Houbiers J, Kruithof E, Vandooren B, Van den Bosch F, Boots AM, et al. Synovial inflammation does not change in the absence of effective treatment: implications for the use of synovial histopathology as biomarker in early phase clinical trials in rheumatoid arthritis. Ann Rheumat Dis. (2006) 65:990–7. doi: 10.1136/ard.2005.047852
47. Bresnihan B, Tak PP, Emery P, Klareskog L, Breedveld F. Synovial biopsy in arthritis research: five years of concerted european collaboration. Ann Rheumat Dis. (2000) 59:506–11. doi: 10.1136/ard.59.7.506
48. Haringman JJ, Gerlag DM, Zwinderman AH, Smeets TJ, Kraan MC, Baeten D, et al. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheumat Dis. (2005) 64:834–8. doi: 10.1136/ard.2004.029751
49. Mandelin AM, Homan PJ, Shaffer AM, Cuda CM, Dominguez ST, Bacalao E, et al. Transcriptional profiling of synovial macrophages using minimally invasive ultrasound-guided synovial biopsies in rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:841–54. doi: 10.1002/art.40453
50. Sornasse T, Gabay C, Townsend M, Laubender R, Wang J, Tuckwell K. THU0131 Levels of cxcl13 and sicam1 correlate with disease activity score in rheumatoid arthritis (RA) patients treated with tocilizumab (TCZ). Ann Rheumat Dis. (2018) 77(Suppl. 2):286–7.
51. Ducreux J, Durez P, Galant C, Nzeusseu Toukap A, Van den Eynde B, Houssiau FA, et al. Global molecular effects of tocilizumab therapy in rheumatoid arthritis synovium. Arthritis Rheumat. (2014) 66:15–23. doi: 10.1002/art.38202
52. Kanbe K, Chen Q, Nakamura A, Hobo K. Inhibition of MAP kinase in synovium by treatment with tocilizumab in rheumatoid arthritis. Clin Rheumatol. (2011) 30:1407. doi: 10.1007/s10067-011-1833-z
53. Buch MH, Reece RJ, Quinn MA, English A, Cunnane G, Henshaw K, et al. The value of synovial cytokine expression in predicting the clinical response to TNF antagonist therapy (infliximab). Rheumatology. (2008) 47:1469–75. doi: 10.1093/rheumatology/ken261
54. Wijbrandts CA, Dijkgraaf MG, Kraan MC, Vinkenoog M, Smeets TJ, Dinant H, et al. The clinical response to infliximab in rheumatoid arthritis is in part dependent on pretreatment tumour necrosis factor α expression in the synovium. Ann Rheumat Dis. (2008) 67:1139–44. doi: 10.1136/ard.2007.080440
55. Lindberg J, Wijbrandts CA, Van Baarsen LG, Nader G, Klareskog L, Catrina A, et al. The gene expression profile in the synovium as a predictor of the clinical response to infliximab treatment in rheumatoid arthritis. PLoS ONE. (2010) 5:e11310. doi: 10.1371/journal.pone.0011310
56. Badot V, Galant C, Toukap AN, Theate I, Maudoux A-L, Van den Eynde BJ, et al. Gene expression profiling in the synovium identifies a predictive signature of absence of response to adalimumab therapy in rheumatoid arthritis. Arthritis Res Therapy. (2009) 11:R57. doi: 10.1186/ar2678
57. Cañete JD, Celis R, Moll C, Izquierdo E, Marsal S, Sanmartí R, et al. Clinical significance of synovial lymphoid neogenesis and its reversal after anti-tumour necrosis factor α therapy in rheumatoid arthritis. Ann Rheumat Dis. (2009) 68:751–6. doi: 10.1136/ard.2008.089284
58. Kavanaugh A, Rosengren S, Lee SJ, Hammaker D, Firestein GS, Kalunian K, et al. Assessment of rituximab's immunomodulatory synovial effects (ARISE trial). 1: clinical and synovial biomarker results. Ann Rheumat Dis. (2008) 67:402–8. doi: 10.1136/ard.2007.074229
59. Hogan VE, Holweg CT, Choy DF, Kummerfeld SK, Hackney JA, Teng YK, et al. Pretreatment synovial transcriptional profile is associated with early and late clinical response in rheumatoid arthritis patients treated with rituximab. Ann Rheum Dis. (2012) 71:1888–94. doi: 10.1136/annrheumdis-2011-201115
60. Teng YO, Levarht EN, Toes RE, Huizinga TW, van Laar JM. Residual inflammation after rituximab treatment is associated with sustained synovial plasma cell infiltration and enhanced B cell repopulation. Ann Rheumat Dis. (2009) 68:1011–6. doi: 10.1136/ard.2008.092791
61. Thurlings RM, Vos K, Wijbrandts CA, Zwinderman AH, Gerlag DM, Tak PP. Synovial tissue response to rituximab: mechanism of action and identification of biomarkers of response. Ann Rheumat Dis. (2008) 67:917–25. doi: 10.1136/ard.2007.080960
62. Vos K, Thurlings RM, Wijbrandts CA, van Schaardenburg D, Gerlag DM, Tak PP. Early effects of rituximab on the synovial cell infiltrate in patients with rheumatoid arthritis. Arthritis Rheumat. (2007) 56:772–8. doi: 10.1002/art.22400
63. Buch MH, Boyle DL, Rosengren S, Saleem B, Reece RJ, Rhodes LA, et al. Mode of action of abatacept in rheumatoid arthritis patients having failed tumour necrosis factor blockade: a histological, gene expression and dynamic magnetic resonance imaging pilot study. Ann Rheumat Dis. (2009) 68:1220–7. doi: 10.1136/ard.2008.091876
64. Kanbe K, Oh K, Chiba J, Inoue Y, Taguchi M, Yabuki A. Analysis of mitogen-activated protein kinases in bone and cartilage of patients with rheumatoid arthritis treated with abatacept. Clin Med Insights Arthritis Musculoskeletal Disord. (2016) 9:51–6. doi: 10.4137/CMAMD.S34424
65. De Groof A, Ducreux J, Humby F, Nzeusseu Toukap A, Badot V, Pitzalis C, et al. Higher expression of TNFα-induced genes in the synovium of patients with early rheumatoid arthritis correlates with disease activity, and predicts absence of response to first line therapy. Arthritis Res Therapy. (2016) 18:19. doi: 10.1186/s13075-016-0919-z
66. Boyle D, Soma K, Hodge J, Kavanaugh A, Mandel D, Mease P, et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann Rheumat Dis. (2015) 74:1311–6. doi: 10.1136/annrheumdis-2014-206028
67. McGarry T, Orr C, Wade S, Biniecka M, Wade S, Gallagher L, et al. JAK-STAT blockade alters synovial bioenergetics, mitochondrial function and pro-inflammatory mediators in Rheumatoid arthritis. Arthritis Rheumat. (2018) 70:1959–70. doi: 10.1002/art.40569
68. Karonitsch T, Beckmann D, Dalwigk K, Niederreiter B, Studenic P, Byrne RA, et al. Targeted inhibition of Janus kinases abates interfon gamma-induced invasive behaviour of fibroblast-like synoviocytes. Rheumatology. (2017) 57:572–7. doi: 10.1093/rheumatology/kex426
69. Taylor PC, Peters AM, Paleolog E, Chapman PT, Elliott MJ, McCloskey R, et al. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor α blockade in patients with rheumatoid arthritis. Arthritis Rheumat. (2000) 43:38–47. doi: 10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L
70. Ulfgren AK, Andersson U, Engström M, Klareskog L, Maini RN, Taylor PC. Systemic anti–tumor necrosis factor α therapy in rheumatoid arthritis down-regulates synovial tumor necrosis factor α synthesis. Arthritis Rheumat. (2000) 43:2391–6. doi: 10.1002/1529-0131(200011)43:11<2391::AID-ANR3>3.0.CO;2-F
71. Klaasen R, Thurlings RM, Wijbrandts CA, van Kuijk AW, Baeten D, Gerlag DM, et al. The relationship between synovial lymphocyte aggregates and the clinical response to infliximab in rheumatoid arthritis: a prospective study. Arthritis Rheumat. (2009) 60:3217–24. doi: 10.1002/art.24913
72. van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, Rustenburg F, Baggen JM, Verweij CL, et al. Responsiveness to anti-tumour necrosis factor alpha therapy is related to pre-treatment tissue inflammation levels in rheumatoid arthritis patients. Ann Rheum Dis. (2008) 67:563–6. doi: 10.1136/ard.2007.081950
73. Das S, Vital EM, Horton S, Bryer D, El-Sherbiny Y, Rawstron AC, et al. Abatacept or tocilizumab after rituximab in rheumatoid arthritis? An exploratory study suggests non-response to rituximab is associated with persistently high IL-6 and better clinical response to IL-6 blocking therapy. Ann Rheum Dis. (2014) 73:909–12. doi: 10.1136/annrheumdis-2013-204417
74. Thurlings RM, Wijbrandts CA, Mebius RE, Cantaert T, Dinant HJ, van der Pouw-Kraan TC, et al. Synovial lymphoid neogenesis does not define a specific clinical rheumatoid arthritis phenotype. Arthritis Rheumat. (2008) 58:1582–9. doi: 10.1002/art.23505
75. Takemura S, Klimiuk PA, Braun A, Goronzy JJ, Weyand CM. T cell activation in rheumatoid synovium is B cell dependent. J Immunol. (2001) 167:4710–8. doi: 10.4049/jimmunol.167.8.4710
76. O'shea JJ, Park H, Pesu M, Borie D, Changelian P. New strategies for immunosuppression: interfering with cytokines by targeting the Jak/Stat pathway. Curr Opin Rheumatol. (2005) 17:305–11. doi: 10.1097/01.bor.0000160781.07174.db
77. O'sullivan LA, Liongue C, Lewis RS, Stephenson SE, Ward AC. Cytokine receptor signaling through the Jak–Stat–Socs pathway in disease. Mol Immunol. (2007) 44:2497–506. doi: 10.1016/j.molimm.2006.11.025
78. Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. (2012) 367:495–507. doi: 10.1056/NEJMoa1109071
79. Furumoto Y, Gadina M. The arrival of JAK inhibitors: advancing the treatment of immune and hematologic disorders. BioDrugs. (2013) 27:431–8. doi: 10.1007/s40259-013-0040-7
80. Smolen JS, Genovese MC, Takeuchi T, Hyslop DL, Macias WL, Rooney T, et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol. (2018) 46(1):7-18. doi: 10.3899/jrheum.171361
81. Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev. (2008) 223:252–70. doi: 10.1111/j.1600-065X.2008.00648.x
82. Buch MH, Pavitt S, Parmar M, Emery P. Creative trial design in RA: optimizing patient outcomes. Nat Rev Rheumatol. (2013) 9:183–94. doi: 10.1038/nrrheum.2013.5
Keywords: rheumatoid arthritis, biologics, JAK inhibitors, synovial tissue, histology, cytokine, gene expression, pathotypes
Citation: Ouboussad L, Burska AN, Melville A and Buch MH (2019) Synovial Tissue Heterogeneity in Rheumatoid Arthritis and Changes With Biologic and Targeted Synthetic Therapies to Inform Stratified Therapy. Front. Med. 6:45. doi: 10.3389/fmed.2019.00045
Received: 14 December 2018; Accepted: 20 February 2019;
Published: 19 March 2019.
Edited by:
João Eurico Fonseca, University of Lisbon, PortugalReviewed by:
Mihir D. Wechalekar, Flinders Medical Centre, AustraliaVasco C. Romão, University of Lisbon, Portugal
Copyright © 2019 Ouboussad, Burska, Melville and Buch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maya H. Buch, bS5idWNoQGxlZWRzLmFjLnVr
†These authors have contributed equally to this work