 Samuel J. Rednor
Samuel J. Rednor Michael J. Ross
Michael J. Ross- 1Division of Nephrology, Albert Einstein College of Medicine/Montefiore Medical Center, Bronx, NY, United States
- 2Department of Development and Molecular Biology, Albert Einstein College of Medicine, Bronx, NY, United States
HIV-associated nephropathy (HIVAN) is an important cause of secondary focal glomerulosclerosis that occurs primarily in persons of African ancestry with advanced HIV disease. Although HIVAN is characterized by severe proteinuria and rapid progression to end stage renal disease without treatment, the phenotype is markedly attenuated by treatment with antiretroviral medications. HIV infection of glomerular and tubular epithelial cells and subsequent viral gene expression is a key contributor to HIVAN pathogenesis and the kidney can serve as reservoir for HIV strains that differ those in blood. HIV gene expression in renal epithelial cells leads to dysregulation of cellular pathways including cell cycle, inflammation, cell death, and cytoskeletal homeostasis. Polymorphisms in the APOL1 gene explain the marked predilection of HIVAN to occur in persons of African descent and HIVAN. Since HIVAN has the strongest association with APOL1 genotype of any of the APOL1-associated nephropathies, studies to determine the mechanisms by which HIV and APOL1 risk variants together promote kidney injury hold great promise to improve our understanding of the pathogenesis of APOL1-mediated kidney diseases.
Epidemiology and Clinical Presentation of HIV-Associated Nephropathy
HIV-associated nephropathy (HIVAN), was first described early in the HIV epidemic in U.S. urban centers serving large numbers of HIV-positive persons of African descent. In the early 1990's, HIVAN was the most rapidly increasing cause of ESRD in the U.S., however, the widespread use of combination antiretroviral therapy (cART), in addition to markedly reducing the incidence of mortality and progression to AIDS, has resulted in a marked reduction in the incidence of classic HIVAN (1). While the incidence of ESRD attributed to HIVAN has declined since the introduction of cART, it has not dropped as dramatically as mortality or progression to AIDS (2). Since the phenotype of HIVAN is markedly attenuated by cART (1, 3), resulting in much lower levels of proteinuria and slower progression to severe CKD/ESRD (reducing likelihood of kidney biopsy), and the United States Renal Data System no longer collects data on HIV seropositive status, it is difficult to reliably estimate the current incidence/prevalence of HIVAN.
Patients with classic HIVAN present with rapidly progressive renal failure in conjunction with severe proteinuria and most have advanced HIV disease/AIDS with of CD4 counts <200 cells/mm3. These patients typically have enlarged hyperechoic kidneys on ultrasound with a bland urine sediment (4, 5). This classic presentation is rarely encountered in patients receiving cART. There are no serologic tests that accurately predict the presence of HIVAN and since HIV-positive patients are at increased risk for comorbidities that increase the risk of kidney disease, including diabetes mellitus and hepatitis C infection, HIVAN can only be definitively diagnosed by kidney biopsy.
There have been isolated case reports of HIVAN occurring in HIV-2 positive patients but the incidence of kidney disease in the setting of HIV-2 infection is unknown (6). Though the mechanism by which HIV-2 causes kidney disease has not been studied, since the HIV-2 genome encodes most of the same genes as HIV-1, it is plausible that the pathogenesis of HIVAN occurring in HIV-1 and HIV-2-positive patients is similar (7).
Histopathology
HIVAN is characterized by the presence of FSGS and is most commonly associated with the collapsing variant (8). Proliferation and hypertrophy of overlying glomerular epithelial cells is often present, which can result in the presence of pseudocrescents [Figure 1, (9)].
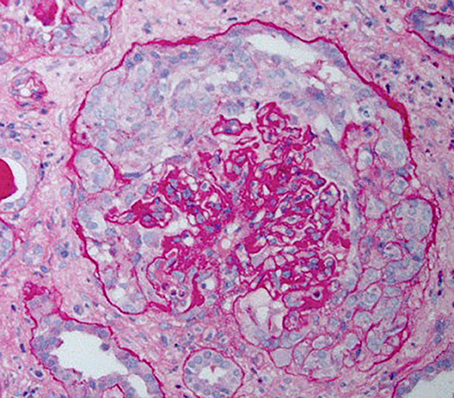
Figure 1. Periodic acid–Schiff stained kidney biopsy from a patient with HIVAN demonstrating collapsed glomerulus with focal global sclerosis and overlying pseudocrescent composed of proliferating glomerular epithelial cells. There is also prominent tubular atrophy and interstitial fibrosis.
Tubulointerstitial disease is an important component of the histopathology of HIVAN and may overshadow the severity of glomerular injury. Classic findings include dilation of tubular into “microcysts” (at least three times the diameter of normal adjacent tubules) and interstitial fibrosis and inflammation (8).
There are no specific immunofluorescence findings in HIVAN, however, as these patients often have increased plasma levels of immunoglobulins and coexisting infections, variable amounts of immunoglobulins and/or complement may be present and if prominent, may reflect the presence of a superimposed immune complex disease. Electron microscopy reveals podocyte foot process effacement, normal basement membrane thickness, absence of immune deposits, and presence of endothelial cell tubuloreticular inclusions, which likely reflect high circulating interferon levels present in patients with HIVAN (8).
Infection of Renal Epithelial Cells by HIV
HIV can infect renal tubular epithelial cells (RTEc), podocytes, and parietal epithelial cells and this infection is not specific to HIVAN but also occurs in patients with other forms of kidney disease, including HIV-positive patients with diabetic kidney disease (10, 11). Since renal epithelial cells do not normally express CD4, the primary receptor for HIV-1, or the co-receptors CCR5 and CXCR4 (12), the mechanisms by which infection occurs remain incompletely understood. Ray et al. reported that cell-free clinical HIV viral isolates were able to infect human RTEc via a CD4-independent mechanism and that HIV-infected mononuclear cells were able to mediate direct cell-cell transfer of HIV to RTEc (11).
More recently, investigators demonstrated that transfer of HIV directly from infected lymphocytes to RTEc was markedly more efficient than infection by cell free virus and that cell-cell viral transfer did not require expression of CD4 on target cells or the HIV envelope protein (Env) and was mediated in part, by heparan sulfate proteoglycans (13). Importantly, further studies showed that infected RTEc can also transfer HIV to uninfected primary T lymphocytes, suggesting that the infected RTEc may serve as a viral reservoir capable of infecting lymphocytes (14). These observations have important implications beyond the pathogenesis of HIVAN as they demonstrate that therapies aimed at fully curing HIV infection will need to include strategies to eliminate HIV infection in the kidneys.
Infection of renal epithelial cells is also an important issue in the context of kidney transplantation as a recent study involving 19 HIV-positive patients who received HIV-negative kidney allografts, found that 13 allografts had detectable HIV RNA in podocytes and/or RTEc, despite having undetectable plasma viral RNA. Moreover, those with infected kidneys had greater podocyte and RTEc injury and more rapid loss of kidney function (15).
A recent study elucidated a novel mechanism by which HIV-1 infects human podocytes (16). Podocytes cultured from children with HIVAN supported low level productive infection when exposed to cell free virus. The ability of cell-free HIV-1 to infect these podocytes was dependent upon the presence of the HIV envelope (env) gene and cell surface proteoglycans. Expression of transmembrane TNFα promoted HIV infection and subsequent integration into genomic DNA. The role of transfer of HIV-1 from infected mononuclear cells to podocytes was not investigated in this study.
The renal epithelium may also harbor viral strains that differ from those in blood from the same patient. In one study, investigators used laser capture microdissection to isolate DNA from RTEc in kidney biopsies from patients with HIVAN. Analysis of HIV sequences amplified from this DNA demonstrated diversity in the viral env sequences, suggesting ongoing replication and evolution of the virus. Further, comparison of kidney-derived sequences to those amplified from blood revealed that kidney and blood-derived viral sequences clustered separately, suggesting that the renal epithelium is a separate viral compartment that may harbor unique viral quasispecies (17). Recent work from Blasi, et al demonstrated similar findings using urine specimens. They found that 12 of 24 patients with HIV RNA detectable in plasma also had virus in their urine. Analysis of viral env sequences from blood and urine revealed that urine-derived sequences clustered separately from blood-derived sequences (18).
Studies in macaques demonstrated that the ability of chimeric simian-human immunodeficiency (SHIV) viral clones to cause glomerular and tubular injury varied significantly, strongly suggesting that viral sequence variation is an important determinant of kidney disease (19). It is not known whether patients' kidneys harbor quasispecies with distinct variations in HIV genes that mediate renal injury or alter response to cART. Whether glomerular epithelial cells can serve as viral reservoirs also remains to be determined.
Pathomechanisms of HIVAN
Role for HIV Genes in Causing HIVAN
The HIV-1 genome encodes 9 genes (Figure 2). Numerous in vitro and animal models have been used to study the mechanisms by which viral infection of renal epithelial cells can lead to HIVAN. The most commonly used model (“Tg26”) is transgenic for an HIV provirus lacking the gag and pol genes. These mice develop severe proteinuria, progressive kidney failure, and histologic kidney injury that closely models HIVAN (20). Since gag and pol encode the major structural and enzymatic viral proteins, these mice do not produce virus, thereby demonstrating that viral replication is not necessary for the HIVAN phenotype. Numerous transgenic rodent models have been generated, expressing various HIV genes using different promoters and together, these studies demonstrate that expression of nef and/or vpr is sufficient to generate the full HIVAN phenotype and the remaining genes are not necessary for the HIVAN phenotype in rodents (21).
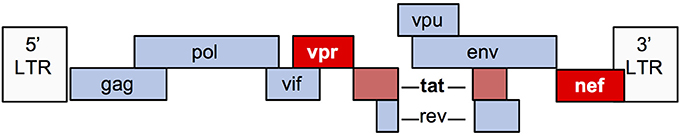
Figure 2. Schematic diagram of the HIV-1 genome.
Nef is a 27–34 kD myristoylated protein with important roles in the HIV lifecycle. Nef promotes viral transcription and activation of T cells, while helping infected cells to avoid immune surveillance by decreasing cell surface expression of several receptors including CD4, CXCR4, CCR5, and major histocompatibility complex class I (MHC-I) (22). Nef also has myriad effects upon cellular signaling, including activation of Src family kinases (23).
Vpr is a 14 kD protein that is important for nuclear import of HIV preintegration complexes. Vpr also has several other important effects upon infected cells, including inducing cell cycle arrest in G2/M phase and is an important mediator of HIV-induced injury and death (24).
Tat is critical for transactivation of HIV transcription in human cells, but is less active in murine cells due to lack of cyclin T1 in the mouse genome (25), which may explain why Tat does not have an important role in murine HIVAN models (26). However, in vitro studies using human cells suggest that Tat may contribute to glomerular injury in HIVAN, in part, via its ability to upregulate proinflammatory cytokines (27, 28).
Mechanisms of Glomerular Injury
Cell Cycle Dysregulation and Dedifferentiation
During the course of glomerular development, podocytes undergo proliferation and maturation through exquisitely controlled developmental programs, resulting in mature podocytes, which are terminally differentiated and unable to proliferate (29). Cell cycle dysregulation and aberrant podocyte cell cycle reentry is a hallmark of HIVAN pathogenesis. It has long been appreciated that in HIVAN, the proliferation marker Ki67 is expressed in podocytes overlying glomerular capillaries as well as in cells comprising the pseudocrescents surrounding the glomerular tufts in HIVAN biopsies and HIV-transgenic mice (9, 30). Though most early work in the field identified these cells as podocytes, more recent studies suggest that some or all cells comprising pseudocrescents in HIVAN and non-HIVAN collapsing FSGS may be parietal epithelial cells (PECs) (31). These discrepant findings may be explained by subsequent studies demonstrating that parietal epithelial cells may express podocyte genes at low levels (32) and studies in mice showing that PECs can be recruited onto glomerular tufts in the setting of glomerular injury (33, 34).
The cyclin dependent kinase (CDK) inhibitors p27 and p57 are highly expressed in podocytes and help to maintain them in a quiescent state by inhibiting activation of CDKs (35). p27 and p57 are down regulated in podocytes in HIVAN biopsies, thereby permitting CDK activation and entry into the cell cycle. In addition, Nef induces activation of Src family kinases in podocytes, which subsequently leads to activation of Stat3 and MAPK-induced proliferation and dedifferentiation (36). Blocking these pathways prevents HIV-induced podocyte proliferation and/or dedifferentiation and restores expression of key podocyte proteins including synaptopodin and WT-1 (36–38). Notch signaling pathways are also activated in HIVAN and inhibition of Notch signaling using a gamma secretase inhibitor prevents podocyte proliferation induced by Nef and Tat in vitro (39). Other signaling pathways that have been implicated in the pathogenesis of podocyte proliferation and/or dedifferentiation in HIVAN include retinoic acid receptor signaling (40) mammalian target of rapamycin (mTOR) (41) and Kruppel-like factors (42, 43).
Podocyte function is highly dependent upon maintenance of a complex and dynamic cytoskeleton. In HIVAN, there is widespread foot process effacement, which is due, in part to cytoskeletal dysregulation. Activation of Src kinase by HIV gene expression leads to activation of Rac1 and inhibition of RhoA, which results in loss of actin stress fiber formation (44). HIV Tat has recently been shown to promote fibroblast growth factor-2 mediated dysregulation of MAPK and RhoA in human podocytes in vitro (28). Moreover, HIV gene expression also reduces expression of podocyte genes, such as synaptopodin, that are important mediators of cytoskeletal integrity (45, 46).
Cellular Injury and Death
Several studies have delineated mechanisms of HIV-induced podocyte death. Since podocytes are terminally differentiated cells, aberrant reentry of podocytes into the cell cycle can lead to mitotic catastrophe (47). Nef expression in HIV infected glomerular epithelial cells may induce death of podocytes via this mechanism while inducing proliferation of parietal epithelial cells in an attempt to replace podocytes. Many podocyte genes are dysregulated by HIV infection in vitro and in animal models and genes with likely roles in HIV-induced podocyte death include APOL1 (discussed below) (48–50), Fas (51), KLF6 (52), p53 (53), and p66ShcA (54), the NLRP3 inflammasome (55).
In the pre-cART era, small clinical studies suggested that treatment with ACE inhibitors improved renal outcomes in HIVAN (56, 57). Several studies have also demonstrated that ACE inhibitors and angiotensin receptor blockers (ARBs) also prevent renal injury in HIV transgenic mice (58, 59). Interestingly, the protective effects of ARBs in HIV-transgenic mice appear to be independent of blockade of angiotensin II receptor type 1 on podocytes (60).
Dysregulated cell-cell and cell-matrix adhesion also contribute to glomerular injury in HIVAN. Sidekick-1 (Sdk-1) is a transmembrane immunoglobulin family protein that mediates cell-cell adhesion. HIV expression increases Sdk-1 expression which is primarily detected in glomerular pseudocrescents HIV-transgenic mice. Sdk-1 mediated dysregulation of cell-cell adhesion may contribute to the clustering of glomerular epithelial cells that is characteristic of collapsing glomerulopathy in HIVAN (61, 62). HIV expression also suppresses activation of the small GTPase RAP1 by increasing expression of RAP1GAP, which is a negative regulator of RAP1. RAP1 is an important regulator of cell-cell and cell-matrix adhesion. Reduced RAP1 expression reduces β1-integrin abundance, leading to podocyte detachment and glomerulosclerosis in mice (63). Major mechanisms of HIV-induced glomerular injury are summarized in Figure 3.
Figure 3. Schematic diagram demonstrating major mechanisms by which HIV-1 infection promotes glomerular and tubular injury.
Tubulointerstitial Injury in HIVAN
Cell Cycle Dysregulation
Though Ki-67 is also upregulated in RTEc in HIVAN biopsies, it is not clear whether this is a primary pathogenic process or is an adaptive response to replace RTEc undergoing apoptosis. Also, though Ki-67 is commonly used as a marker of proliferation, it is expressed during all phases of cell cycle other than G0 (64). Since Vpr causes G2/M phase arrest and dysregulated cytokinesis in RTEc in vitro and in mice (65, 66), it is possible that the increased Ki-67 in RTEc in HIVAN reflects increased proportion of cells that arrested in cell cycle phases other than G0 and not ongoing proliferation per se. Moreover, since G2/M phase arrest of RTEc has recently been demonstrated to be an important mechanism contributing to tubulointerstitial injury and fibrosis in non-HIV kidney injury in mice (67), it is likely that mechanisms of cell cycle dysregulation in HIVAN may have direct relevance to our understanding of the tubulointerstitial injury in other proteinuric kidney diseases.
Cellular Injury and Death
Apoptosis is increased in HIVAN biopsies, with the greatest levels of apoptosis found in RTEc (68, 69). Vpr expression induces apoptosis in RTEc in vitro, which is mediated via persistent unregulated activation of ERK, resulting in caspase 8 activation, which cleaves BID to tBID, leading to mitochondrial membrane permeablization and apoptosis (65). Mitochondrial injury and Vpr-induced apoptosis are dependent upon expression of the ubiquitin-like protein FAT10 (70). Interestingly, suppression of FAT10 expression prevents apoptosis primarily in cells with Vpr-induced hyperploidy in vitro.
Homeo-domain interacting protein kinase 2 (HIPK2) has also recently been implicated in the pathogenesis of HIV-induced RTEc apoptosis and tubulointerstitial fibrosis. HIPK2 expression is increased, primarily in tubular cells in HIV-transgenic mice and in HIVAN biopsy specimens and promotes RTEc apoptosis via activation of p53, TGF-β-SMAD3, and Wnt/Notch pathways which together promote tubulointerstitial fibrosis (71).
Inflammation
The predominant transcriptional response of RTEc after HIV infection is increased expression of proinflammatory mediators, many of which are NF-κB and interferon-inducible genes (72, 73). A primary role for NF-κB-induced inflammation in the pathogenesis of HIVAN is supported by studies demonstrating that pharmacologic NF-κB inhibition prevents renal disease in two different murine models of HIV-induced kidney disease (74, 75). Further, clinical studies (primarily from the pre-cART era) demonstrated that glucocorticoid treatment is associated with improved renal outcomes in HIVAN (76–78) and that the primary histologic change observed after treatment is reduced tubulointerstitial inflammation (79).
Treatment of HIV-transgenic mice with the mTOR inhibitor sirolimus is also protective against the HIVAN phenotype in HIV-transgenic mice. Sirolimus was shown to reduce HIV transcription in podocytes in vitro and in mice (80) and prevent expression of mediators of epithelial mesenchymal transition (81) in these models. Since mTOR inhibition can have myriad cellular effects, including immunosuppression and activation of autophagy (a cytoprotective and immunomodulatory pathway), it is likely that many pathways are involved in mTOR-induced protection in murine models of HIVAN.
Major mechanisms of HIV-induced tubular injury are summarized in Figure 3.
Role of Apoliproprotein L1 Variants in HIVAN
Epidemiology
The risk of HIVAN in persons of African ancestry is much higher than in other racial/ethnic groups and black race is associated with a 12.2-fold increased risk of HIVAN (82). The genetic basis of this disparity was elucidated in a seminal study in 2010 in which the authors reported that variants of the APOL1 gene, which encodes the Apolipoprotein L1 (ApoL1) protein, are strongly associated with risk of kidney disease. Two variants (G1 and G2) are associated with increased risk of kidney disease compared to wild type (G0). The G1 allele encodes two missense mutations in the APOL1 protein whereas G2 encodes a two-amino acid deletion (Figure 4). Kidney risk attributed to the G1 and G2 alleles generally occurs in an autosomal recessive pattern and persons who are homozygous for either allele or compound heterozygous for G1 and G2 are at approximately equally increased risk of kidney disease (83).
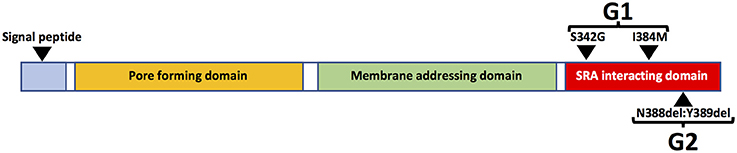
Figure 4. Schematic diagram of major APOL1 protein domains and G1 and G2 sequence variants.
Persons with APOL1 high risk genotypes are at 10.5- and 7.3-fold increased risk of developing non-HIV related FSGS and hypertension-attributed ESRD, respectively (83, 84). Remarkably, subsequent studies found that APOL1 high risk genotypes are associated with 29-fold increased risk of HIVAN in African Americans and an 89-fold increase risk in South Africans and HIV-positive patients are at 50% lifetime risk of developing HIVAN without antiretroviral treatment (85, 86). Importantly, though nearly all studies have demonstrated that heterozygosity for APOL1 risk alleles does not increase the risk of kidney disease, Kasembeli et al reported that in HIV positive South Africans, heterozygosity for the G1, but not the G2 allele, was associated with increased risk of HIVAN (86). Though this observation requires replication in another cohort, if true, it may be due to differences in genetic admixture of the South African population or environmental factors.
The APOL1 G1 and G2 alleles are unique to persons of African ancestry, which is explained by the fact that these alleles arose relatively recently in Africa. Even within Africa, the prevalence of these alleles varies widely—the prevalence of the G1 allele is highest in western African countries including Ghana and Nigeria where the prevalence of the allele is >40%, but the prevalence of the G2 allele is more variable at 6–24% (87). In the United States, the prevalence of the G1 and G2 alleles in African Americans is 20–22 and 13–15% respectively, and 10–15% have high risk APOL1 genotypes. Due to the transatlantic slave trade that occurred from Western Africa the sixteenth to the nineteenth centuries, the APOL1 risk alleles are also found among persons living in the Caribbean and Latin America at highly variable frequencies (88).
Putative Mechanisms of APOL1-Mediated Kidney Injury
The mechanisms by which APOL1 variants promote killing of trypanosomes may provide important insights into how they promote renal injury. APOL1 functions in part, as an innate immune response protein, and is induced by interferons and TNF-α (89). APOL1 is unique to a few primate species and is not encoded in the genomes of non-primate species. Trypanosoma brucei rhodesiense causes African Sleeping Sickness in humans who are homozygous for the APOL1 G0 allele because it produces Serum Resistance Associated (SRA) protein, which binds APOL1, preventing it from killing the parasite. The changes in APOL1 encoded by the G1 and G2 alleles prevent the SRA protein from binding/inactivating APOL1, thereby protecting persons harboring one or both of these alleles from African Sleeping Sickness induced by Trypanosoma brucei rhodesiense (90).
APOL1 can be expressed in numerous cell types and tissues, especially in the presence of interferons and/or TNFα (89). There are also several isoforms of APOL1, most of which contain an N-terminal signal peptide, which is likely necessary for extracellular secretion. The liver is the source of most circulating APOL1 protein, which is complexed in HDL3 particles, which are also synthesized in liver (91). There are also APOL1 isoforms that lack the N-terminal signal peptide sequence (Figure 4) and are expressed as intracellular proteins (49). Plasma APOL1 levels do not correlate with risk of kidney disease (92, 93). Moreover, in the setting of kidney transplantation, the presence of high risk APOL1 genotypes in the kidney donor, but not the recipient, is strongly associated with adverse allograft outcomes (94–96). These data strongly suggest that local production of APOL1 in kidney cells is more important than circulating APOL1 in promoting kidney injury.
Nearly 15% of African Americans have high risk genotypes, the majority of whom never develop kidney disease. Additional factors or “second hits” are therefore necessary to unmask the deleterious effects of APOL1 upon the kidney. Several of these second hits have been identified, including exogenous interferon administration (89), lupus nephritis (97, 98), and sickle cell disease (99), but HIV is the most potent “second hit” to promote kidney disease in persons with high risk APOL1 genotypes (85, 86). HIVAN is therefore an important model disease, the study of which may hold important insights mechanisms by which APOL1 promotes kidney disease. Putative mechanisms by which APOL1 and HIV-1 may together promote kidney injury are depicted in Figure 5.
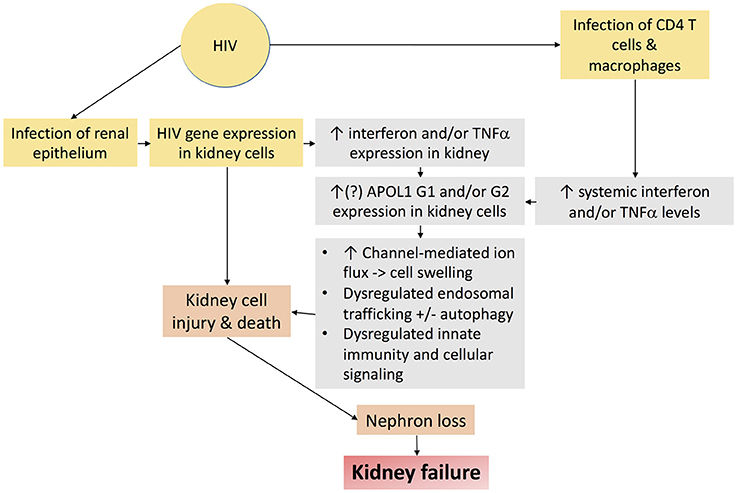
Figure 5. Schematic diagram demonstrating putative mechanisms by which HIV-1 infection and APOL1 together promote kidney injury.
Though several types of kidney cells have been reported to produce APOL1 in vivo, including podocytes, tubular epithelial cells, endothelial cells, and vascular smooth muscle cells (100, 101), it remains unclear which cells are the most important targets of APOL1-induced injury. Moreover, while cytokines strongly induce APOL1 expression in vitro and many of these same cytokines are commonly increased in the context of chronic kidney injury, APOL1 expression in biopsy specimens from patients with HIVAN has been reported to be lower than control specimens. Though it is possible, if not likely that APOL1 expression is increased at some point in the course of the disease process, it is not clear whether increased expression of APOL1 is an important contributor to disease pathogenesis. This point also has important implications with regard to relevance of current in vitro and animal models of APOL1-mediated injury, most of which rely on overexpression of APOL1 at non-physiologic levels or expression of APOL1 transgenes in animals that normally lack APOL1 in their genomes.
Numerous publications have elucidated potential mechanisms by which APOL1 risk alleles may promote kidney injury but relatively few have focused upon APOL1-mediated injury in the context of HIV infection. APOL1 kills trypanosomes by inserting into acidified endosomes, which when recycled to the plasma membrane at neutral pH become high conductance cation channels, resulting in rapid swelling and lysis of the parasite (102). Forced overexpression of APOL1 G1 and G2 alleles induce greater cytotoxicity than the G0 allele in vitro (49, 50, 89) but it remains unclear whether APOL1 promotes kidney injury via its function as a channel. Transgenic mice expressing the G0 or G2 alleles of APOL1 in podocytes under control of the nephrin promoter do not develop kidney disease (103) but mice expressing the G2 but not the G0 allele in podocytes under control of the podocin promoter developed FSGS lesions (48). Podocyte injury induced by the G2 allele in the latter study was associated with impaired endosomal trafficking and autophagic flux, leading to pyroptosis (48). In vitro studies with human podocytes found that HIV infection markedly increased the toxicity of APOL1 overexpression (50).
The results of a recent provocative study suggest that APOL1-induced cytotoxicity and channel function may be artifacts of in vitro overexpression and that these effects may not occur when APOL1 is expressed at physiologic levels (104). Since this study used inducible APOL1 in immortalized cell lines, additional studies are needed to determine if this observation is relevant in primary kidney cells. However, it is clear that since nearly all previous studies on the role of APOL1 in inducing cellular injury used overexpression models, further studies are needed in which APOL1 is expressed at physiologically relevant levels in physiologically relevant cells.
Future Perspectives
Though antiretroviral medications have emerged as an effective strategy for the prevention and treatment of kidney disease in HIV-positive persons, there remains an urgent need for research to answer key questions with important implications for the care for all persons living with HIV and/or kidney disease including: (1) How do antiretroviral medications prevent/treat HIVAN without reducing HIV infection of renal epithelial cells? (2) Does the kidney harbor viral strains with unique antiretroviral resistance patterns? (3) Is it possible to eradicate HIV-1 from the kidney without worsening kidney injury by killing infected epithelial cells? (4) How do APOL1 risk variants promote renal injury and why is HIV-1 such a potent “second hit” in persons with APOL1 high risk genotypes? (5) Since HIVAN is the only “APOL1 nephropathy” with an effective treatment available (antiretrovirals), can insights into the pathogenesis of HIVAN inform treatment of APOL1-associated kidney disease in HIV-negative patients? (6) How can we develop in vitro and animal models that faithfully model the in vivo function(s) of APOL1?
Conclusions
HIVAN is caused by infection of renal epithelial cells in genetically susceptible persons. In addition to its importance in HIVAN pathogenesis, the infected renal epithelia can serve as a viral reservoir that must be addressed in future attempts to cure HIV-positive patients. HIV-infection induces dysregulation of host genes and several cellular pathways, including inflammation, cell cycle, cytoskeletal homeostasis, and cell death. Polymorphisms in the APOL1 gene account for the majority of excess risk of HIVAN attributed to African ancestry and of all APOL1-associated nephropathies, HIVAN has the strongest association with APOL1 risk genotypes. Though antiretroviral therapies have markedly reduced the burden of HIVAN in HIV-positive patients, studies to elucidate how HIV increases susceptibility to kidney disease in persons with high risk APOL1 genotypes promise to provide key insights that may help develop novel strategies for the prevention and treatment of APOL1 nephropathies in HIV-negative patients.
Author Contributions
SR prepared the initial draft of this manuscript. MR revised and expanded the scope of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the NIH/NIDDK grants R01DK108346 and R01DK101338.
References
1. Mallipattu SK, Salem F, Wyatt CM. The changing epidemiology of HIV-related chronic kidney disease in the era of antiretroviral therapy. Kidney Int. (2014) 86:259–65. doi: 10.1038/ki.2014.44
2. Centers for Disease Control Prevention (2016),. Diagnoses of HIV Infection in the United States and Dependent Areas 2016. HIV Surveillance Report 28, http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html. (Accessed Jan, 23 2018).
3. USRDS (2012). US Renal Data System. USRDS 2012 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United State. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD.
4. Wyatt CM, Klotman PE, D'Agati VD. HIV-associated nephropathy: clinical presentation, pathology, and epidemiology in the era of antiretroviral therapy. Semin Nephrol. (2008) 28:513–22. doi: 10.1016/j.semnephrol.2008.08.005
5. Atta MG. Diagnosis and natural history of HIV-associated nephropathy. Adv Chronic Kidney Dis. (2010) 17:52–8. doi: 10.1053/j.ackd.2009.08.005
6. Izzedine H, Damond F, Brocheriou I, Ghosn J, Lassal H, Deray G. HIV-2 infection and HIV-associated nephropathy. Aids (2006) 20:949–50. doi: 10.1097/01.aids.0000218566.05274.2b
7. Campbell-Yesufu OT, Gandhi RT. Update on human immunodeficiency virus (HIV)-2 infection. Clin Infect Dis. (2011) 52:780–7. doi: 10.1093/cid/ciq248
8. D'Agati V, Suh JI, Carbone L, Cheng JT, Appel G. Pathology of HIV-associated nephropathy: a detailed morphologic and comparative study. Kidney Int. (1989) 35:1358–70.
9. Barisoni L, Kriz W, Mundel P, D'Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (1999) 10:51–61.
10. Bruggeman LA, Ross MD, Tanji N, Cara A, Dikman S, Gordon RE, et al. Renal epithelium is a previously unrecognized site of HIV-1 infection. J Am Soc Nephrol. (2000) 11:2079–87.
11. Ray PE, Liu XH, Henry D, Dye LIIII, Xu L, Orenstein JM, et al. Infection of human primary renal epithelial cells with HIV-1 from children with HIV-associated nephropathy. Kidney Int. (1998) 53:1217–29. doi: 10.1046/j.1523-1755.1998.00900.x
12. Eitner F, Cui Y, Hudkins KL, Stokes MB, Segerer S, Mack M, et al. Chemokine receptor CCR5 and CXCR4 expression in HIV-associated kidney disease. J Am Soc Nephrol. (2000) 11:856–67.
13. Chen P, Chen BK, Mosoian A, Hays T, Ross MJ, Klotman PE, et al. Virological synapses allow HIV-1 uptake and gene expression in renal tubular epithelial cells. J Am Soc Nephrol. (2011) 22:496–507. doi: 10.1681/ASN.2010040379
14. Blasi M, Balakumaran B, Chen P, Negri DR, Cara A, Chen BK, et al. Renal epithelial cells produce and spread HIV-1 via T-cell contact. AIDS (2014) 28:2345–53. doi: 10.1097/QAD.0000000000000398
15. Canaud G, Dejucq-Rainsford N, Avettand-Fenoel V, Viard JP, Anglicheau D, Bienaime F, et al. The kidney as a reservoir for HIV-1 after renal transplantation. J Am Soc Nephrol. (2013) 25:407–19. doi: 10.1681/ASN.2013050564
16. Li J, Das JR, Tang P, Han Z, Jaiswal JK, Ray PE. Transmembrane TNF- facilitates HIV-1 infection of podocytes cultured from children with HIV-associated nephropathy. J Am Soc Nephrol. (2016) 28:862–75. doi: 10.1681/ASN.2016050564
17. Marras D, Bruggeman LA, Gao F, Tanji N, Mansukhani MM, Cara A, et al. Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat Med. (2002) 8:522–6. doi: 10.1038/nm0502-522
18. Blasi M, Carpenter JH, Balakumaran B, Cara A, Gao F, Klotman ME. Identification of HIV-1 genitourinary tract compartmentalization by analyzing the env gene sequences in urine. AIDS (2015) 29:1651–7. doi: 10.1097/QAD.0000000000000757
19. Stephens EB, Tian C, Dalton SB, Gattone VH. Simian-human immunodeficiency virus-associated nephropathy in macaques. AIDS Res Hum Retroviruses (2000) 16:1295–306. doi: 10.1089/08892220050117050
20. Dickie P, Felser J, Eckhaus M, Bryant J, Silver J, Marinos N, et al. HIV-associated nephropathy in transgenic mice expressing HIV-1 genes. Virology (1991) 185:109–19. doi: 10.1016/0042-6822(91)90759-5
21. Zuo Y, Matsusaka T, Zhong J, Ma J, Ma LJ, Hanna Z, et al. HIV-1 genes vpr and nef synergistically damage podocytes, leading to glomerulosclerosis. J Am Soc Nephrol. (2006) 17:2832–43. doi: 10.1681/ASN.2005080878
22. Pereira EA, daSilva LL. HIV-1 nef: taking control of protein trafficking. Traffic (2016) 17:976–96. doi: 10.1111/tra.12412
23. Saksela K, Cheng G, Baltimore D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. (1995) 14:484–91.
24. Le Rouzic E, Benichou S. The Vpr protein from HIV-1: distinct roles along the viral life cycle. Retrovirology (2005) 2:11. doi: 10.1186/1742-4690-2-11
25. Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell (1998) 92:451–62. doi: 10.1016/S0092-8674(00)80939-3
26. Snyder A, Ross MJ. Murine models of Vpr-mediated pathogenesis. Curr HIV Res. (2009) 7:129–35. doi: 10.2174/157016209787581526
27. Conaldi PG, Bottelli A, Baj A, Serra C, Fiore L, Federico G, et al. Human immunodeficiency virus-1 tat induces hyperproliferation and dysregulation of renal glomerular epithelial cells. Am J Pathol. (2002) 161:53–61. doi: 10.1016/S0002-9440(10)64156-9
28. Xie X, Colberg-Poley AM, Das JR, Li J, Zhang A, Tang P, et al. The basic domain of HIV-tat transactivating protein is essential for its targeting to lipid rafts and regulating fibroblast growth factor-2 signaling in podocytes isolated from children with HIV-1-associated nephropathy. J Am Soc Nephrol. (2014) 25:1800–13. doi: 10.1681/ASN.2013070710
29. Hagen M, Pfister E, Kosel A, Shankland S, Pippin J, Amann K, et al. Cell cycle re-entry sensitizes podocytes to injury induced death. Cell Cycle (2016) 15:1929–37. doi: 10.1080/15384101.2016.1191710
30. Barisoni L, Bruggeman LA, Mundel P, D'Agati VD, Klotman PE. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int. (2000) 58:173–81. doi: 10.1046/j.1523-1755.2000.00152.x
31. Dijkman HB, Weening JJ, Smeets B, Verrijp KC, van Kuppevelt TH, Assmann KK, et al. Proliferating cells in HIV and pamidronate-associated collapsing focal segmental glomerulosclerosis are parietal epithelial cells. Kidney Int. (2006) 70:338–44. doi: 10.1038/sj.ki.5001574
32. Guhr SS, Sachs M, Wegner A, Becker JU, Meyer TN, Kietzmann L, et al. The expression of podocyte-specific proteins in parietal epithelial cells is regulated by protein degradation. Kidney Int. (2013) 84:532–44. doi: 10.1038/ki.2013.115
33. Lazzeri E, Romagnani P. Podocyte biology: differentiation of parietal epithelial cells into podocytes. Nat Rev Nephrol. (2015) 11:7–8. doi: 10.1038/nrneph.2014.218
34. Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, et al. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol. (2009) 20:333–43. doi: 10.1681/ASN.2008070795
35. Shankland SJ, Eitner F, Hudkins KL, Goodpaster T, D'Agati V, Alpers CE. Differential expression of cyclin-dependent kinase inhibitors in human glomerular disease: role in podocyte proliferation and maturation. Kidney Int (2000) 58:674–83. doi: 10.1046/j.1523-1755.2000.00213.x
36. He JC, Husain M, Sunamoto M, D'Agati VD, Klotman ME, Iyengar R, et al. Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1,2 pathways. J Clin Invest. (2004) 114:643–51. doi: 10.1172/JCI200421004
37. Feng X, Lu TC, Chuang PY, Fang W, Ratnam K, Xiong H, et al. Reduction of stat3 activity attenuates HIV-induced kidney injury. J Am Soc Nephrol. (2009) 20:2138–46. doi: 10.1681/ASN.2008080879
38. Lu TC, Wang Z, Feng X, Chuang P, Fang W, Chen Y, et al. Retinoic acid utilizes CREB and USF1 in a transcriptional feed-forward loop in order to stimulate MKP1 expression in human immunodeficiency virus-infected podocytes. Mol Cell Biol. (2008b) 28:5785–94. doi: 10.1128/MCB.00245-08
39. Sharma M, Magenheimer LK, Home T, Tamano KN, Singhal PC, Hyink DP, et al. Inhibition of Notch pathway attenuates the progression of human immunodeficiency virus-associated nephropathy. Am J Physiol Renal Physiol. (2013) 304:F1127–36. doi: 10.1152/ajprenal.00475.2012
40. Ratnam KK, Feng X, Chuang PY, Verma V, Lu TC, Wang J, et al. Role of the retinoic acid receptor-alpha in HIV-associated nephropathy. Kidney Int. (2011) 79:624–34. doi: 10.1038/ki.2010.470
41. Kumar D, Konkimalla S, Yadav A, Sataranatarajan K, Kasinath BS, Chander PN, et al. HIV-associated nephropathy: role of mammalian target of rapamycin pathway. Am J Pathol. (2010) 177:813–21. doi: 10.2353/ajpath.2010.100131
42. Mallipattu SK, Liu R, Zheng F, Narla G, Ma'ayan A, Dikman S, et al. Kruppel-like factor 15 (KLF15) is a key regulator of podocyte differentiation. J Biol Chem. (2012) 287:19122–35. doi: 10.1074/jbc.M112.345983
43. Mallipattu SK, Estrada CC, He JC. The critical role of Krüppel-like factors in kidney disease. Am J Physiol Renal Physiol. (2017) 312:F259–65. doi: 10.1152/ajprenal.00550.2016
44. Lu TC, He JC, Wang ZH, Feng X, Fukumi-Tominaga T, Chen N, et al. HIV-1 Nef disrupts the podocyte actin cytoskeleton by interacting with diaphanous interacting protein. J Biol Chem. (2008a) 283:8173–82. doi: 10.1074/jbc.M708920200
45. Husain M, D'Agati VD, He JC, Klotman ME, Klotman PE. HIV-1 Nef induces dedifferentiation of podocytes in vivo: a characteristic feature of HIVAN. Aids (2005) 19:1975–80. doi: 10.1097/01.aids.0000191918.42110.27
46. Sunamoto M, Husain M, He JC, Schwartz EJ, Klotman PE. Critical role for Nef in HIV-1-induced podocyte dedifferentiation. Kidney Int. (2003) 64:1695–701. doi: 10.1046/j.1523-1755.2003.00283.x
47. Liapis H, Romagnani P, Anders HJ. New insights into the pathology of podocyte loss: mitotic catastrophe. Am J Pathol. (2013) 183:1364–74. doi: 10.1016/j.ajpath.2013.06.033
48. Beckerman P, Bi-Karchin J, Park AS, Qiu C, Dummer PD, Soomro I, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med. (2017) 23:429–38. doi: 10.1038/nm.4287
49. Khatua AK, Cheatham AM, Kruzel ED, Singhal PC, Skorecki K, Popik W. Exon 4-encoded sequence is a major determinant of cytotoxicity of apolipoprotein L1. Am J Physiol Cell Physiol. (2015) 309:C22–37. doi: 10.1152/ajpcell.00384.2014
50. Lan X, Jhaveri A, Cheng K, Wen H, Saleem MA, Mathieson PW, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. (2014) 307:F326–36. doi: 10.1152/ajprenal.00647.2013
51. Ross MJ, Martinka S, D'Agati VD, Bruggeman LA. NF-κB regulates Fas-mediated apoptosis in HIV-associated nephropathy. J Am Soc Nephrol. (2005) 16:2403–11. doi: 10.1681/ASN.2004121101
52. Mallipattu SK, Horne SJ, D'Agati V, Narla G, Liu R, Frohman MA, et al. Krüppel-like factor 6 regulates mitochondrial function in the kidney. J Clin Invest. (2015) 125:1347–61. doi: 10.1172/JCI77084
53. Rai P, Plagov A, Lan X, Chandel N, Singh T, Lederman R, et al. mTOR plays a critical role in p53-induced oxidative kidney cell injury in HIVAN. Am J Physiol Renal Physiol. (2013) 305:F343–54. doi: 10.1152/ajprenal.00135.2013
54. Husain M, Meggs LG, Vashishtha H, Simoes S, Griffiths KO, Kumar D, et al. Inhibtion of P66SHCA longevity gene rescues podocytes from HIV-1-induced oxidative stress and apoptosis. J Biol Chem. (2009) 284:16648–58. doi: 10.1074/jbc.M109.008482
55. Haque S, Lan X, Wen H, Lederman R, Chawla A, Attia M, et al. HIV promotes NLRP3 inflammasome complex activation in murine HIV-associated nephropathy. Am J Pathol. (2016) 186:347–58. doi: 10.1016/j.ajpath.2015.10.002
56. Burns GC, Visintainer P, Mohammed NB. Effect of angiotensin converting-enzyme inhibition on progression of renal disease and mortality in HIV-associated nephropathy. J Amer Soc Nephrol. (1999) 10:155.
57. Kimmel PL, Mishkin GJ, Umana WO. Captopril and renal survival in patients with human immunodeficiency virus nephropathy. Am J Kidney Dis. (1996) 28:202–8. doi: 10.1016/S0272-6386(96)90302-9
58. Zhong Y, Chen EY, Liu R, Chuang PY, Mallipattu SK, Tan CM, et al. Renoprotective effect of combined inhibition of angiotensin-converting enzyme and histone deacetylase. J Am Soc Nephrol. (2013) 24:801–11. doi: 10.1681/ASN.2012060590
59. Hiramatsu N, Hiromura K, Shigehara T, Kuroiwa T, Ideura H, Sakurai N, et al. Angiotensin II. type (2013) 1 receptor blockade inhibits the development and progression of HIV-associated nephropathy in a mouse model. J Am Soc Nephrol. (2007) 18:515–27. doi: 10.1681/ASN.2006030217
60. Shimizu A, Zhong J, Miyazaki Y, Hosoya T, Ichikawa I, Matsusaka T. ARB protects podocytes from HIV-1 nephropathy independently of podocyte AT1. Nephrol Dial Transplant. (2012) 27:3169–75. doi: 10.1093/ndt/gfs033
61. Kaufman L, Yang G, Hayashi K, Ashby JR, Huang L, Ross MJ, et al. The homophilic adhesion molecule sidekick-1 contributes to augmented podocyte aggregation in HIV-associated nephropathy. FASEB J. (2007) 21:1367–75. doi: 10.1096/fj.06-7191com
62. Kaufman L, Hayashi K, Ross MJ, Ross MD, Klotman PE. Sidekick-1 is upregulated in glomeruli in HIV-associated nephropathy. J Am Soc Nephrol. (2004) 15:1721–30. doi: 10.1097/01.ASN.0000128975.28958.C2
63. Potla U, Ni J, Vadaparampil J, Yang G, Leventhal JS, Campbell KN, et al. Podocyte-specific RAP1GAP expression contributes to focal segmental glomerulosclerosis-associated glomerular injury. J Clin Invest. (2014) 124:1757–69. doi: 10.1172/JCI67846
64. Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. (2000) 182:311–22. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9
65. Snyder A, Alsauskas ZC, Leventhal JS, Rosenstiel PE, Gong P, Chan JJ, et al. HIV-1 viral protein r induces ERK and caspase-8-dependent apoptosis in renal tubular epithelial cells. AIDS (2010) 24:1107–19. doi: 10.1097/QAD.0b013e328337b0ab
66. Rosenstiel PE, Gruosso T, Letourneau AM, Chan JJ, Leblanc A, Husain M, et al. HIV-1 Vpr inhibits cytokinesis in human proximal tubule cells. Kidney Int. (2008) 74:1049–58. doi: 10.1038/ki.2008.303
67. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. (2010) 16:535–43, 1p following 143. doi: 10.1038/nm.2144
68. Yang Y, Gubler MC, Beaufils H. Dysregulation of podocyte phenotype in idiopathic collapsing glomerulopathy and HIV-associated nephropathy. Nephron (2002) 91:416–23. doi: 10.1159/000064281
69. Bodi I, Abraham AA, Kimmel PL. Apoptosis in human immunodeficiency virus-associated nephropathy. Am J Kidney Dis. (1995) 26:286–91. doi: 10.1016/0272-6386(95)90648-7
70. Snyder A, Alsauskas Z, Gong P, Rosenstiel PE, Klotman ME, Klotman PE, et al. FAT10: a novel mediator of Vpr-induced apoptosis in human immunodeficiency virus-associated nephropathy. J Virol. (2009) 83:11983–8. doi: 10.1128/JVI.00034-09
71. Jin Y, Ratnam K, Chuang PY, Fan Y, Zhong Y, Dai Y, et al. A systems approach identifies HIPK2 as a key regulator of kidney fibrosis. Nat Med. (2012) 18:580–588. doi: 10.1038/nm.2685
72. Chen P, Yi Z, Zhang W, Klotman ME, Chen BK. HIV infection-induced transcriptional program in renal tubular epithelial cells activates a CXCR2-driven CD4+ T-cell chemotactic response. AIDS (2016) 30:1877–88. doi: 10.1097/QAD.0000000000001153
73. Ross MJ, Fan C, Ross MD, Chu TH, Shi Y, Kaufman L, et al. HIV-1 infection initiates an inflammatory cascade in human renal tubular epithelial cells. J Acquir Immune Defic Syndr. (2006) 42:1–11. doi: 10.1097/01.qai.0000218353.60099.4f
74. Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L, et al. Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem. (2012) 287:28840–51. doi: 10.1074/jbc.M112.359505
75. Heckmann A, Waltzinger C, Jolicoeur P, Dreano M, Kosco-Vilbois MH, Sagot Y. IKK2 inhibitor alleviates kidney and wasting diseases in a murine model of human AIDS. Am J Pathol. (2004) 164:1253–62. doi: 10.1016/S0002-9440(10)63213-0
76. Appel RG, Neill J. A steroid-responsive nephrotic syndrome in a patient with human immunodeficiency virus (HIV) infection. Ann Intern Med. (1990) 113:892–3. doi: 10.7326/0003-4819-113-11-892
77. Laradi A, Mallet A, Beaufils H, Allouache M, Martinez F. HIV-associated nephropathy: outcome and prognosis factors. Groupe d' Etudes Nephrologiques d'Ile de France. J Am Soc Nephrol. (1998) 9:2327–35.
78. Smith MC, Austen JL, Carey JT, Emancipator SN, Herbener T, Gripshover B, et al. Prednisone improves renal function and proteinuria in human immunodeficiency virus-associated nephropathy. Am J Med. (1996) 101:41–8. doi: 10.1016/S0002-9343(96)00065-4
79. Briggs WA, Tanawattanacharoen S, Choi MJ, Scheel PJJr, Nadasdy T, Racusen L. Clinicopathologic correlates of prednisone treatment of human immunodeficiency virus-associated nephropathy. Am J Kidney Dis. (1996) 28:618–21. doi: 10.1016/S0272-6386(96)90477-1
80. Rai P, Plagov A, Kumar D, Pathak S, Ayasolla KR, Chawla AK, et al. Rapamycin-induced modulation of HIV gene transcription attenuates progression of HIVAN. Exp Mol Pathol. (2012) 94:255–61. doi: 10.1016/j.yexmp.2012.09.009
81. Yadav A, Kumar D, Salhan D, Rattanavich R, Maheshwari S, Adabala M, et al. Sirolimus modulates HIVAN phenotype through inhibition of epithelial mesenchymal transition. Exp Mol Pathol. (2012) 93:173–81. doi: 10.1016/j.yexmp.2012.04.021
82. Abbott KC, Hypolite I, Welch PG, Agodoa LY. Human immunodeficiency virus/acquired immunodeficiency syndrome- associated nephropathy at end-stage renal disease in the United States: patient characteristics and survival in the pre highly active antiretroviral therapy era. J Nephrol. (2001) 14:377–83.
83. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science (2010) 329:841–5. doi: 10.1126/science.1193032
84. Freedman BI, Kopp JB, Langefeld CD, Genovese G, Friedman DJ, Nelson GW, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. (2010) 21:1422–6. doi: 10.1681/ASN.2010070730
85. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (2011) 22:2129–37. doi: 10.1681/ASN.2011040388
86. Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T, et al. APOL1 risk variants are strongly associated with HIV-associated nephropathy in black south africans. J Am Soc Nephrol. (2015) 26:2882–90. doi: 10.1681/ASN.2014050469
87. Limou S, Nelson GW, Kopp JB, Winkler CA. APOL1 kidney risk alleles: population genetics and disease associations. Adv Chronic Kidney Dis. (2014) 21:426–33. doi: 10.1053/j.ackd.2014.06.005
88. Rotimi CN, Tekola-Ayele F, Baker JL, Shriner D. The African diaspora: history, adaptation and health. Curr Opin Genet Dev. (2016) 41:77–84. doi: 10.1016/j.gde.2016.08.005
89. Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D'Agati V, et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. (2015) 87:332–42. doi: 10.1038/ki.2014.270
90. Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK, Carrington M, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA. (2014) 111:E2130–9. doi: 10.1073/pnas.1400699111
91. Shukha K, Mueller JL, Chung RT, Curry MP, Friedman DJ, Pollak MR, et al. Most ApoL1 is secreted by the liver. J Am Soc Nephrol. (2016) 28:1079–83. doi: 10.1681/ASN.2016040441
92. Kozlitina J, Zhou H, Brown PN, Rohm RJ, Pan Y, Ayanoglu G, et al. Plasma levels of risk-variant APOL1 do not associate with renal disease in a population-based cohort. J Am Soc Nephrol. (2016) 27:3204–19. doi: 10.1681/ASN.2015101121
93. Bruggeman LA, O'Toole JF, Ross MD, Madhavan SM, Smurzynski M, Wu K, et al. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol. (2013) 25:634–44. doi: 10.1681/ASN.2013070700
94. Doshi MD, Ortigosa-Goggins M, Garg AX, Li L, Poggio ED, Winkler CA, et al. APOL1 genotype and renal function of black living donors. J Am Soc Nephrol. (2018) 29:1309–16. doi: 10.1681/ASN.2017060658
95. Lee BT, Kumar V, Williams TA, Abdi R, Bernhardy A, Dyer C, et al. The APOL1 genotype of African American kidney transplant recipients does not. impact 5-year allograft survival. Am J Transplant. (2012) 12:1924–8. doi: 10.1111/j.1600-6143.2012.04033.x
96. Reeves-Daniel AM, DePalma JA, Bleyer AJ, Rocco MV, Murea M, Adams PL, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant. (2011) 11:1025–30. doi: 10.1111/j.1600-6143.2011.03513.x
97. Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. (2014) 66:390–6. doi: 10.1002/art.38220
98. Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol. (2013) 24:722–5. doi: 10.1681/ASN.2012121180
99. Ashley-Koch AE, Okocha EC, Garrett ME, Soldano K, De Castro LM, Jonassaint JC, et al. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br J Haematol. (2011) 155:386–94. doi: 10.1111/j.1365-2141.2011.08832.x
100. Ma L, Shelness GS, Snipes JA, Murea M, Antinozzi PA, Cheng D, et al. Localization of APOL1 protein and mRNA in the human kidney: nondiseased tissue, primary cells, and immortalized cell lines. J Am Soc Nephrol. (2014) 26:339–48. doi: 10.1681/ASN.2013091017
101. Madhavan SM, O'Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol. (2011) 22:2119–28. doi: 10.1681/ASN.2011010069
102. Thomson R., Finkelstein A. Human trypanolytic factor APOL1 forms pH-gated cation-selective channels in planar lipid bilayers: relevance to trypanosome lysis. Proc Natl Acad Sci USA. (2015) 112:2894–9. doi: 10.1073/pnas.1421953112
103. Bruggeman LA, Wu Z, Luo L, Madhavan SM, Konieczkowski M, Drawz PE, et al. APOL1-G0 or APOL1-G2 transgenic models develop preeclampsia but not kidney disease. J Am Soc Nephrol. (2016) 27:3600–10. doi: 10.1681/ASN.2015111220
Keywords: HIV, AIDS, HIV-associated nephropathy, proteinuria, chronic kidney disease, podocyte, renal tubular epithelial cell, APOL1
Citation: Rednor SJ and Ross MJ (2018) Molecular Mechanisms of Injury in HIV-Associated Nephropathy. Front. Med. 5:177. doi: 10.3389/fmed.2018.00177
Received: 13 February 2018; Accepted: 22 May 2018;
Published: 07 June 2018.
Edited by:
Sandra Merscher, University of Miami, United StatesReviewed by:
Gurvinder Kaur, All India Institute of Medical Sciences, IndiaJoanna Mikulak, Humanitas Research Hospital, Italy
Darshana Dadhania, Cornell University, United States
Copyright © 2018 Rednor and Ross. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Ross, bWljaGFlbC5yb3NzQGVpbnN0ZWluLnl1LmVkdQ==