 Eugenio D. Hottz
Eugenio D. Hottz Fernando A. Bozza
Fernando A. Bozza Patrícia T. Bozza
Patrícia T. Bozza
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 30 April 2018
Sec. Hematology
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00121
This article is part of the Research TopicMolecular Bridges Between Hematology and InflammationView all 5 articles
Platelets are essential effector cells in hemostasis. Aside from their role in coagulation, platelets are now recognized as major inflammatory cells with key roles in the innate and adaptive arms of the immune system. Activated platelets have key thromboinflammatory functions linking coagulation to immune responses in various infections, including in response to virus. Recent studies have revealed that platelets exhibit several pattern recognition receptors (PRR) including those from the toll-like receptor, NOD-like receptor, and C-type lectin receptor family and are first-line sentinels in detecting and responding to pathogens in the vasculature. Here, we review the main mechanisms of platelets interaction with viruses, including their ability to sustain viral infection and replication, their expression of specialized PRR, and activation of thromboinflammatory responses against viruses. Finally, we discuss the role of platelet-derived mediators and platelet interaction with vascular and immune cells in protective and pathophysiologic responses to dengue, influenza, and human immunodeficiency virus 1 infections.
Platelets are highly specialized effectors of hemostasis with essential functions in vascular thrombosis and wound healing. For a long time, cellular activities attributed to platelets were restricted to rapid procoagulant responses mediated by G-protein-coupled receptors leading to platelet aggregation, enzymatic activation of eicosanoid synthesis and granule secretion (1, 2). Aside from this traditional view, it is now known that platelets express various pattern recognition receptors (PRR) and respond to infecting microorganisms, initiating cellular activities that participate in the immune and inflammatory network against diverse pathogens, including viruses (1–3). While traditional platelet activation by G-protein-coupled receptors is usually rapid, platelet PRR activation in responses to infectious and immune stimuli can be delayed and sustained, lasting hours after initial aggregation and secretion (1, 4). For example, platelets have stored RNA molecules and diverse mechanisms for posttranscriptionaly process intronic RNA using specialized pathways to synthesize immunoregulatory proteins such as cytokines and antimicrobial peptides (5–7). Through this new view, platelets are now recognized as first-line sentinels in detecting and responding to pathogens and damage signals in the vasculature and also in the extravascular space.
Pattern recognition receptors are cellular sensors that recognize molecular structures broadly shared among pathogens, the so called pathogen-associated molecular patterns (PAMPs); or cellular molecules modified and/or released during tissue damage called damage-associated molecular patterns (DAMPs). PRR from different classes have been shown to be expressed and functional in platelets, including those from C-type lectin receptors (CLR) and toll-like receptors (TLRs) families (2, 8, 9). CLR are a large family of surface proteins containing at least one carbohydrate-binding domain which are specialized in the recognition of bacterial, fungi, or viral glycans (10–12). Some viruses can exploit certain CLR for viral attachment and entry in host cells, including in platelets (13, 14). The TLRs, a family of transmembrane cellular sensors, are the best described class of PRR in platelets. While virtually all TLRs (1–10 in human) are detected at some level (mRNA or protein) in platelets (1–3, 8, 15–18), most of functional characterization of TLR-mediated platelet responses were reported for those specialized in bacterial molecules, especially regarding platelet TLR-4 activation by lipopolysaccharide (LPS) from Gram-negative bacteria (4, 6, 19–22). Recently, the expression and functionality of endosomal TLR-3, -7, and -9, related to recognition and response to viral genome in nucleated cells, were also reported in platelets (23–25). The presence of cytoplasmic PRR in platelets is far less explored, and it remains a subject of controversy whether platelets are able to sustain the replication of viral genome allowing its recognition by cytoplasmic sensors.
Two major cytoplasmic PRR from the NOD-like receptor (NLR) family were recently reported to be expressed and functional in platelets: the nucleotide-binding domain leucine rich repeat containing pyrin 3 (NLRP3), a major sensor for the activation of inflammasome that recognizes various bacterial, viral, and tissue damage signals; and the nucleotide-binding oligomerization domain 2 that recognizes the bacterial cell wall peptidoglycan component muramyl dipeptide (26–28). Other intracellular sensors including retinoic acid-inducible gene I and melanoma differentiation-associated gene 5, which are highly specialized in viral RNA recognition, are expressed in human megakaryocytes in response to type I interferon (IFN-α and -β) (29), but their expression in platelets, as well the ability of platelets to express other IFN-stimulated genes (ISGs) and to perform IFN-induced antiviral response are not known.
Megakaryocytes, platelets’ mother cells, transfer to platelets all cellular components responsible for their hemostatic and immune functions including granule-stored chemokines, immune receptors, RNA molecules, and spliceosomes (5, 23, 30, 31). Megakaryocytes have been shown to be susceptible to various viruses (29, 32–37). In addition, megakaryocytes express PRR and cytokine receptors, and there is evidence that TLR agonists or cytokine engagement affects megakaryocytic maturation and thrombopoiesis (25, 34, 38, 39). Megakaryocytes and megakaryocytic cell lines respond to viral infections or viral PAMPs by secreting high levels of α and β IFN (25, 29, 34, 40, 41), which reduce platelet production in vitro through an autocrine IFNAR signaling (25, 40–42). Besides their consequences reducing the numbers of new platelets, megakaryocyte infection, PRR engagement, and/or cytokine signaling are expected to change the phenotype of platelet progeny during infections, influencing platelet-mediated immune and inflammatory processes at the periphery. However, participation of megakaryocytes in immune response still deserves more in-depth investigation. In this review, we discuss the molecular mechanisms involved in platelet interactions with viruses including PRR and intracellular pathways related to platelet inflammatory activities during viral infections. We focused our discussion on the contributions of human platelets to pathophysiologic and protective responses during viral infections of major concern in human health globally including acquired immunodeficiency syndrome (AIDS), dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS), and influenza pneumonia.
Viral infection of a susceptible cell initiates with virus binding to a surface receptor that mediate its internalization through the endocytic pathway. Successful infection relies on the ability of the virus to escape endosomal acidification and lysosomal fusion through diverse mechanisms, delivering its genome to the cytoplasm or nucleus depending on the virus type (43–45). For each step of viral replication, the host cell has evolved mechanisms for recognizing and fighting viral infection, mainly by inducing the expression of antiviral restriction factors in bystander cells through type I IFN signaling (46–48). Platelets have been reported to express surface receptors able to mediate binding and entry of various viruses [reviewed in Ref. (49–51)]. Even though platelets do not have nucleus, they have all the molecular machinery to synthesize proteins from stored mRNA (5, 17, 52, 53), which may also implicate in the ability to translate proteins from RNA viruses. In addition, some of the PRR related to viral recognition were recently identified to be present and functional in platelets, and platelet participation in immune response to virus has been investigated in experimental viral infections and in human patients naturally infected with viruses (23–26, 54–56). Recent studies of these types have increased our understanding of how platelet responses triggered by interactions with viruses may both limit viral proliferation and pathogen burden, or complicate inflammation and disease pathogenesis. The main mechanisms for platelet interaction with viruses and the PRR involved in viral recognition by platelets known so far are discussed in this chapter and summarized in Figure 1.
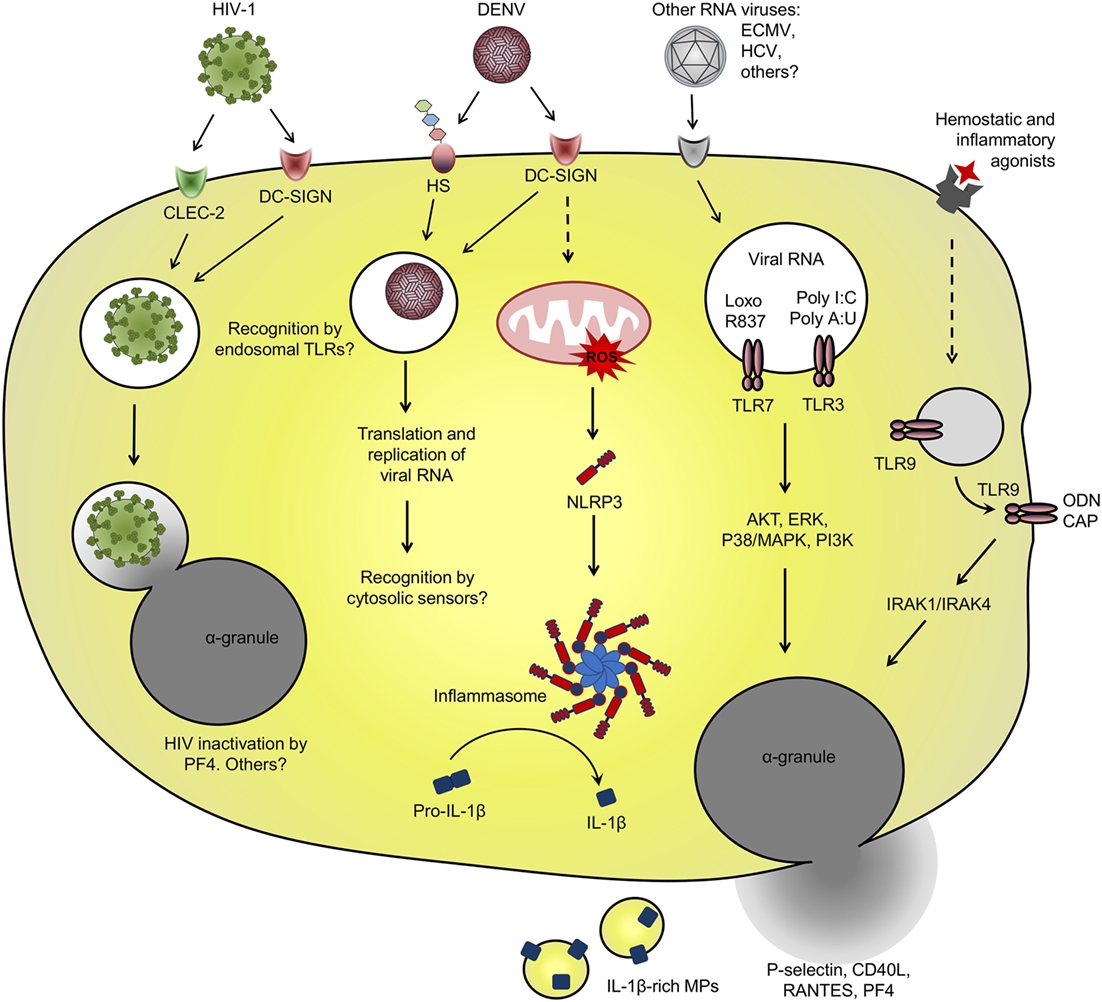
Figure 1. Platelet interaction with viruses and virus-related pathogen-associated molecular patterns (PAMPs): schematic representation of the main receptors and pathways involved in virus binding and internalization, and pattern recognition receptor involved in recognition of viral PAMP by platelets. See text for details and references.
Among diverse surface receptors involved in platelet interactions with viruses (49–51), the CLR dendritic cell-specific ICAM-3-grabbing non-integrin (DC-SIGN) is involved in dengue virus (DENV) and human immunodeficiency virus 1 (HIV-1) binding and entry in platelets (57, 58). DC-SIGN is a PRR that recognizes mannose-terminal-containing pathogen-associated carbohydrates. DC-SIGN also binds glycosylated domains on the envelope (E) protein of DENV and glycoprotein 120 (GP120) of HIV-1, a recognized mechanism for viral entry in dendritic cells (13, 45, 50, 59, 60). HIV-1 entry in dendritic cells through DC-SIGN or other C-type lectins occurs without viral membrane fusion and independently on the receptor CD4 or the coreceptors CXCR4 or CCR5 (13, 45, 50). Through this non-canonical pathway, endocytosed HIV particles sustain their infectivity for several days allowing trans-infection to CD4+ T cells (13, 45). Even through platelets internalize HIV-1 viral particles though similar mechanisms involving DC-SIGN or C-type lectin-like receptor 2 (CLEC-2) for viral attachment (14, 57), it is still controversial if platelets can perform HIV-1 trans-infection to T cells, and opposite results have been found on this regard (14, 61), as will be discussed in Section “Platelet Activation in HIV Infection.”
Platelet ability to endocytose viral particles is not an HIV-specific feature. The viral genome of DENV and hepatitis C virus (HCV), both from Flaviviridae family, have been detected in circulating platelets from infected patients (62–67). HCV genome was also detected in platelets from healthy volunteers that were exposed to HCV in vitro (65, 68). HCV capture by platelets depended on the binding domain of HCV envelope protein E2 but was not dependent on the surface receptor CD81 (65). In another in vitro study, HCV viral particles were able to bind to recombinant human glycoprotein VI (GPVI) (69), which may be a potential receptor for HCV attachment on platelets. However, the role played by GPVI on HCV binding and entry in platelets has not been addressed (69). Endocytosed DENV-like particles have been also evidenced by ultrastructural analysis of platelets from patients with dengue (62). In vitro studies identified the mechanisms of DENV binding and internalization by platelets requiring DC-SIGN and heparan sulfate proteoglycans for viral attachment (58). When isolated platelets are infected with DENV in vitro, positive- and negative-sense viral RNA as well DENV non-structural protein 1 accumulate in platelets, indicating replication and translation of viral genome (58, 70). However, infective viral particles do not accumulate in platelet pellets or culture supernatants over time (58, 70). These data indicate that even though platelets support DENV replication, they do not assemble or release mature viral particles, strongly suggesting an abortive DENV infection in platelets.
We and others have shown that in vitro infection of platelets with DENV induces platelet activation (26, 64, 71, 72), which is inhibited by anti-DC-SIGN neutralizing antibodies (72). Beyond DENV binding to DC-SIGN, heparan sulfate or other surface receptors, DENV immunocomplexes formed with cross-reactive antibodies may enhance the infection of Fcγ receptor (FcγR)-bearing phagocytes in secondary DENV infections (73). Similarly, DENV capture by platelets is increased in the presence of antibodies (74), but whether this interaction leads to DENV replication and/or FcγR-mediated platelet activation remains unknown. Regarding this issue, immunocomplexes of influenza A (H1N1) virus with specific or cross-reactive (H3N2) antibodies activate platelets through FcγRIIA, increasing platelet degranulation and shedding of microparticles (MPs) (75). It remains unknown, however, whether virus attachment to platelet surface receptors is sufficient to initiate downstream activation of platelets or if virus internalization and replication are required for subsequent activation of intracellular PRR in platelets (Figure 1).
Among PRR involved in recognition and response against viruses, at least the endosomal TLR-3, -7, and -9 have already been demonstrated in megakaryocytes and platelets (23–25). Intracellular TLR-3 and -9 are highly expressed by megakaryocytes and there is evidence for at least TLR-3 functional responses in megakaryocytic cell lines (23, 25). Megakaryocytes infected with RNA viruses or stimulated with TLR-3 synthetic agonists poly I:C or poly A:U respond with increased type I IFN secretion and expression of ISGs (25, 29, 41). Similarly, isolated platelets stimulated with TLR-3 agonists translocate α-granule-stored factors (P-selectin and CD40L) to surface and have enhanced procoagulant responses to thrombin or other traditional agonists (25, 76). However, it is not clear if TLR-3 activation in platelets and megakaryocytes participates in immune response against viral infections.
While in nucleated cells the endosomal TLRs are restricted to intracellular compartment (77), TLR-3 and -9 in platelets have been shown to translocate to surface after platelet activation (8, 19, 23). In proplatelet-forming megakaryocytes, TLR-9 in electron-dense tubular system-related granules are transferred to platelet progeny. TLR-9-containing granules in platelets do not colocalize with α- or dense-granules, neither with TLR-7 or -8 or other endosomal proteins (23). Even though platelet TLR-9 is not expressed in endosomes, platelet stimulation promotes TLR-9 translocation to surface and pre-activated platelets are able to bind and respond to extracellular unmethylated CpG-containing oligodeoxynucleotide (ODN) (23). Besides ODN from viral or bacterial origin, TLR-9 also recognizes the endogenous DAMP carboxyalkylpyrrole (CAP) protein adducts formed during vascular oxidative stress. CAP adducts activate TLR-9–MyD88 signaling in platelets leading to platelet degranulation and aggregation in vitro and to in vivo thrombosis in atherosclerotic mice (78). However, whether and how surface-translocated TLR-9 in platelets are activated during viral infections deserves further investigation.
Toll-like receptor-7 signaling in platelets appears to be more similar to that of nucleated cells. During experimental encephalomyocarditis virus (ECMV) infection in mice, platelets are able to internalize and recognize viral RNA through TLR-7 (24). In this model, infected mice developed profound thrombocytopenia while platelets formed aggregates with neutrophils. Platelet TLR-7 also recognizes the synthetic agonist loxoribine which similarly causes platelet–neutrophil aggregates and thrombocytopenia in mice. Additional in vitro experiments with murine and human cells demonstrated that platelet–neutrophil aggregation in response to RNA virus or synthetic agonist require TLR-7 expression and endosomal-dependent signaling in platelets, but not in neutrophils. Importantly, transfusion of TLR-7-expressing platelets delayed the mortality of TLR-7-deficient mice after experimental ECMV infection, indicating that platelet TLR-7 may have important roles in immune response against RNA viruses (24).
Regardless of virus attachment to surface receptors or recognition by PRR in platelets, platelet activation can contribute to immune responses against viruses through a diversity of mechanisms including the release of chemokines that promote endothelial signaling and leukocyte migration or by physically interacting with leukocytes changing their responses (56, 79, 80). Recently, platelet factor 4 (PF4/CXCL4), a chemokine secreted exclusively by platelets and megakaryocytes, was identified as an essential mediator for neutrophil recruitment and virus clearance in influenza A infection (also see Platelet Activation in Influenza) (56). Another mechanism of protection involving platelet and neutrophils is platelet-mediated extrusion of neutrophil extracellular traps (NET). The ability of platelets to induce NET release has been extensively reported in bacterial infections, including in response to LPS (22, 81, 82). In an experimental model of myxoma virus infection, infected mice presented platelet–neutrophil aggregates and NET extrusion in the liver vasculature (79). In this model, platelet-mediated NET release protected liver cells from subsequent viral challenge (79). Platelets have been also identified as central players for preservation of spleen architecture and effective CD8+ T cell response against lymphocytic choriomeningitis arenavirus (LCMV), a murine model of viral hemorrhagic fever (80). In this model, partial platelet depletion to a degree that did not increase viral-induced hemorrhage significantly impaired viral clearance through defective assembly of virus-specific CD8+ T cell response (80). In a genetic model of hepatitis B virus infection, however, platelet-induced CD8+ T cell intrahepatic accumulation culminated in liver damage as consequence of cytotoxic activity of recruited lymphocytes toward elimination of infected cells (83). Even though the mechanisms of platelet activation and platelet–leukocyte signaling were not explored in these models, these data convincingly show that platelets play major roles in innate and adaptive immune responses against viruses.
Because of combined antiretroviral therapy (ART), in the last decades the epidemiology of HIV has changed from high mortality by opportunistic infections in AIDS to long-term non-infectious complication of HIV infection (84, 85). However, even though sustained virologic control is achieved by ART, people living with HIV still experience increased mortality associated with higher rates of long-term comorbidities including cardiovascular diseases, depression, HIV-associated neurocognitive disorders, and non-AIDS cancers (86–90). Many of long-term complications in HIV-infected individuals are related to continuing immune suppression (87, 91, 92) and/or sustained inflammation (93–95). Ischemic thrombotic and cardiovascular events represent some of the most frequent long-term complications and leading cause of death among virologically suppressed HIV-infected subjects (86, 89, 91, 92, 96, 97). Considering the potential role for platelets in the cardiovascular risk of people living with HIV, much attention has been given on the implications of platelet activation in the pathogenesis of HIV infection.
Increased platelet activation in HIV-infected subjects and AIDS patients has been extensively reported in the last two decades (55, 98–104). In AIDS patients, markers of platelet activation correlate with patient viral loads and the nadir of CD4+ T cell counts (98–100, 102). P-selectin surface expression on platelets and plasma markers of platelet activation as soluble P-selectin and CD40L (sP-selectin and sCD40L) are increased in ART naïve patients and decrease during the first weeks after antiretroviral treatment (98, 102). However, there is evidence for sustained platelet activation after months to years of virologic suppression by ART (55, 103, 105, 106). Increased platelet activation in people living with HIV is associated with measures of inflammation and coagulopathy including elevated levels of TNF-α, tissue factor, and D-dimers (98, 99, 101). Consistently, antiplatelet therapy with aspirin has been shown to reduce not only platelet activation but also activation of monocytes and T cells from CD4+ and CD8+ subsets in HIV-infected subjects undergoing virologic suppression by ART (55).
Ultrastructural analysis of platelets from HIV-1-infected subjects or in vitro-infected platelets identify HIV-1 internalization in endosome-like vesicles (57, 107, 108). These ultrastructural studies show activation-associated morphology in HIV-containing platelets, indicating that platelets become activated after direct binding and internalization of HIV-1 viral particles (57, 108). In addition, HIV-1 transactivator of transcription (Tat), a viral protein that is released in the circulation of infected subjects, is able to activate platelets increasing the translocation and secretion of CD40L and P-selectin (109). Tat-mediated platelet activation occurs through its binding to β3 integrin and CCR3 chemokine receptor on platelets (109). Injection of recombinant Tat or Tat-expressing retrovirus increases platelet P-selectin expression and plasma levels of PF4 and sCD40L in mice (109–111). Nevertheless, other mechanisms beyond platelet interaction with HIV-1 or Tat may account for platelet activation in people living with HIV since platelet activation persists in virologically suppressed individuals (55, 103). One possible mechanism is platelet TLR-4 activation by LPS from microbial translocation, which is considered a central feature of HIV pathogenesis and has been documented to persist for years after initiation of ART (112). Other possibility is the effects of ART itself. Regarding this, abacavir-containing antiretroviral regimens have been associated with platelet hyperreactivity while raltegravir-based therapy was associated with reduced platelet activation (103, 113, 114). Ritonavir-containing protease inhibitor-based ART has been also associated with increased platelet activation when compared with samples obtained before ART initiation in a small cohort of protease-inhibitor naïve patients (104). In in vitro experiments, abacavir and its metabolite carbovir triphosphate have been shown to potentiate platelet responsiveness to prothrombotic agonists by competitively inhibiting the activity of guanylyl cyclase when compared with non-guanosine nucleotide analogs (115, 116). New studies are still necessary to investigate the impact of ART in platelet activation and long-term comorbidities in people living with HIV.
Activated platelets in HIV-infected individuals express molecules including P-selectin, CD40L, and TF that participate in the thromboinflammatory state related to long-term comorbidities of HIV infection (98, 99, 103, 104, 235). In ex vivo aggregometry assay, platelets from HIV-infected subjects are more sensitive to aggregation under suboptimal prothrombotic stimulation (55, 113). In addition to prothrombotic responses, platelet activation participates in the immune and inflammatory network of HIV infection through diverse mechanisms involving secretion of stored factors and interactions with leukocytes as summarized in Figure 2 and discussed below.
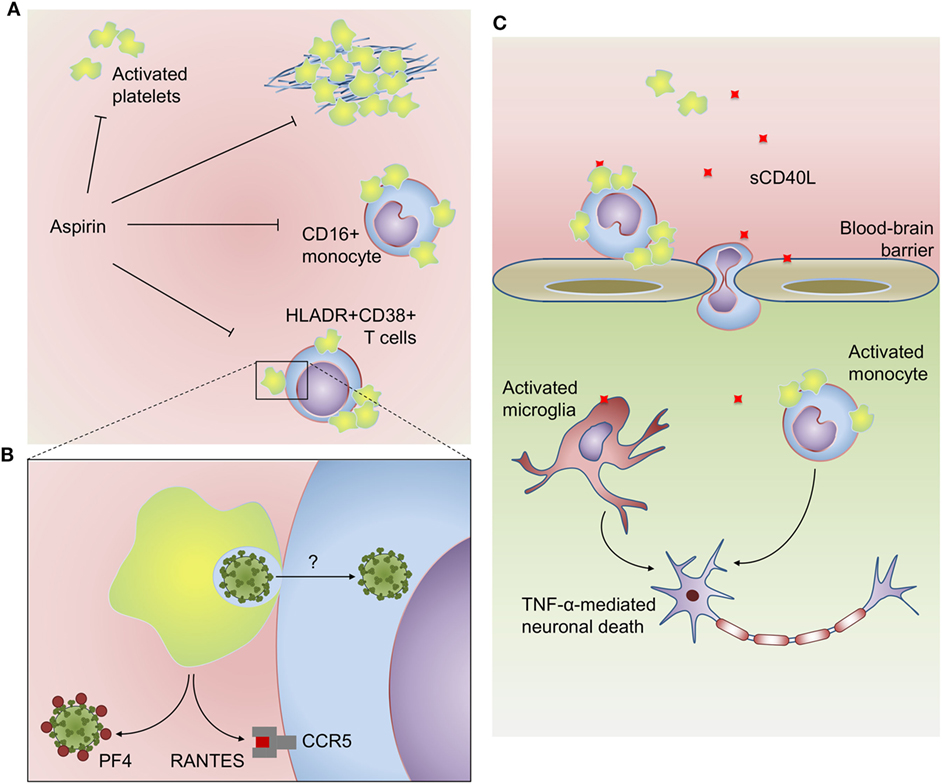
Figure 2. Platelets participate in inflammatory, virologic, and prothrombotic responses in HIV infection: (A) platelets from HIV-infected subjects have increased activation at baseline and hyperreactive aggregation under suboptimal prothrombotic stimuli. Activated platelets also form aggregates with CD16+ inflammatory monocytes and HLADR+CD38+ memory T cells in people living with HIV. HIV-infected subjects undertaken antiplatelet therapy with aspirin had reduced platelet activation, platelet hyperreactivity, and platelet–monocyte aggregates, as well as reduced activation of monocytes (sCD14) and T cells (HLADR+CD38+). (B) Platelets infected with high inoculum of reporter lentivirus are able to trans-infect susceptible T cells in vitro. Activated platelets can also inhibit human immunodeficiency virus 1 infection through the secretion of the HIV suppressive chemokines PF4/CXCL4 and RANTES/CCL5. (C) Platelet-derived CD40L plays a major role in HIV-associated neurocognitive disorders. Platelet-derived CD40L increases blood–brain barrier permeability and adhesion of GR1+CCR2+ leukocytes on brain microvasculature. CD40L-activated platelets induce monocyte migration though brain microvascular endothelial cells, and platelet–monocyte aggregates accumulate in brain microvasculature and parenchyma. Activated monocytes and microglia are more sensitive to CD40L-induced TNF-α secretion, which contributes to neuronal apoptosis. See the text for details and references.
Activated platelets interact with several classes of leukocytes including monocytes, neutrophils, and lymphocytes. These heterotypic aggregates are formed by the binding of platelet P-selectin to leukocyte P-selectin glycoprotein ligand 1 (PSGL-1), a molecular interaction that not only tethers the cells together but also signals gene expression and functional activating pathways in the leukocyte (1, 2, 117–119). Circulating platelet–monocyte and platelet–lymphocyte aggregates have been reported in increased numbers in HIV-infected subjects even after virologic suppression by ART (103, 120). Platelet aggregates formation with monocytes or with CD4+ and CD8+ T cells in people living with HIV correlate with P-selectin expression on platelets (120–122). In HIV-infected subjects under virologic suppression, aggregates of activated platelets with CD4+ and CD8+ T cells form preferentially with lymphocytes of memory phenotype (120). Interestingly, memory lymphocytes isolated from HIV-infected individuals have increased avidity for recombinant P-selectin ex vivo, suggesting that besides platelet activation, T cell activation state also contributes to platelet–lymphocyte adhesion in HIV infection (120). Platelet–monocyte aggregates in HIV-infected people are more frequent among CD16+ inflammatory monocytes and associates to increased levels of sCD163, a marker of monocyte activation (121, 122). While, similarly to lymphocytes, inflammatory monocytes may be more avid targets for activated platelets, it is tempting to speculate whether platelet adhesion contributes to monocyte or lymphocyte inflammatory activation in HIV infection. However, new studies are still necessary to determine the effects of platelet–leukocyte aggregates on monocyte- and T cell-mediated responses during HIV infection.
Among stored factors secreted upon platelet activation, two major chemokines from α-granules, RANTES/CCL5 and PF4/CXCL4, are considered endogenous inhibitors of HIV-1 replication regarding their ability to bind HIV-1 coreceptor CCR5 and HIV-1 GP120, respectively (123, 124). Together with other chemokine ligands of HIV-1 coreceptors MIP-1α/CCL3 and MIP-1β/CCL4 for CCR5 and SDF-1/CXCL12 for CXCR4 (123), platelet-derived RANTES and PF4 may be central mediators in the control of HIV-1 replication in vivo [for more details on HIV-1 suppressive chemokines see Ref. (125)]. Accordingly, genetic polymorphisms involved with the expression of these chemokines and their receptors have been reported as key determinants of the outcome in HIV-1 infection (126–129). These epidemiologic studies are supported by ultrastructural analysis of platelets from HIV-1-infected subjects or in vitro-infected platelets that show evidence for α-granules fusion with virus-containing compartments indicating HIV inactivation and/or degradation by α-granule proteins (57, 108). In addition, coculture with platelets is able to prevent T cell infection through PF4-dependent mechanisms regardless of CCR5 or CXCR4 coreceptor tropism (61, 124). In another study, however, when platelets were infected with HIV-1 in a high viral load and exposed to T cells few hours postinfection, platelets were able to trans-infect T cells instead of preventing their infection (14). These results suggest that platelets may have a dual role in HIV-1 spreading, being able to inactivate endocytosed viruses or shelter them depending on viral load and platelets α-granule content (14, 61) (Figures 1 and 2B). Of note, platelet exhaustion of α-granule chemokines has been reported in association with high viral load in HIV/AIDS patients (98).
Other platelet-derived mediators as TGF-β and CD40L have been implicated in pathologic mechanisms of HIV-associated long-term comorbidities as consequence of infection or ART (111, 130–132). TGF-β from activated platelets has been identified as a critical factor for heart tissue fibrosis and cardiac dysfunction in mice chronically treated with the antiretroviral ritonavir (132); and CD40L, besides being a key mediator for B cell germinative center formation and isotype switching, plays immunopathogenic roles in HIV-associated neurocognitive disorders (Figure 2C) as discussed below (109, 111, 121).
The levels of sCD40L are increased in plasma and cerebrospinal fluid from patients with HIV-associated cognitive impairment compared with HIV-infected subjects without cognitive damage (130). In mice experimentally infected with transgenic viruses (retrovirus expressing HIV Tat or HIV expressing murine leukemia virus GP80 in replacement of GP120) and in Tat-injected mice, platelet-derived CD40L is a crucial component for increased blood–brain barrier permeability and leukocyte recruitment to brain microvasculature (109, 111, 131). CD40L reciprocally increases platelet activation promoting the formation of platelet–monocyte aggregates (121). Adhesion of CD40L-activated platelets to monocytes increase their ability to transmigrate brain microvascular endothelial cells in vitro; and increased numbers of platelet–monocyte aggregates have been observed on microvascular bed and in vascular lumen of brain sections from patients who died by HIV-associated encephalopathy (121). Recruited monocytes and activated resident microglia secrete pro-inflammatory cytokines in response to CD40L, including TNF-α, amplifying local inflammation, and neuronal death, as suggested by in vitro experiments with Tat-activated monocytes and microglia (130). These evidences from infected patients together with in vivo and in vitro studies suggest that platelet-derived CD40L plays a central role in HIV-associated neurocognitive disorder.
Dengue is an arthropod-born viral disease caused by one of four antigenically related DENV serotypes (DENV-1 to -4). It is the most frequent hemorrhagic viral disease and re-emergent infection in the world (133, 134). Recently, it was estimated that over 2.5 billion people live in high-risk transmission areas with more than 90 million symptomatic infections occurring annually (134). DENV infection induces a spectrum of clinical manifestations that range from mild self-limited dengue fever to life-threatening severe dengue. While mild dengue presents as undifferentiated febrile illness, severe dengue syndrome progress with hemodynamic dysfunction including coagulopathy and vasculopathy associated with hypovolemia, hypotension, shock, organ dysfunction, and eventually death (133, 135–137). Thrombocytopenia is a common feature in dengue syndromes, and the drop of platelet counts is temporally coincident with the hemodynamic instability and progression to severity, while its recovery associates with clinical improvement and hospital discharge (136, 138–142). The pathophysiologic mechanisms underlying severe dengue cases are not completely understood. Overwhelming immune activation with increased levels of cytokines and other pro-inflammatory mediators that target the vascular endothelium is considered to favor DENV pathology and severity (140, 143–146). Activation of various immune cells including B and T cells, monocytes, macrophages, and dendritic cells has been shown to participate in this process (147–152). Even through thrombocytopenia is a hallmark of dengue infection, the role played by platelets in dengue immunopathology was only recently addressed.
Platelet activation has been demonstrated in patients with dengue and increased platelet activation associated with disease severity (26, 54, 64, 72, 153, 154). Surface markers of activation including P-selectin and CD63 expression (translocated from α- and dense-granules, respectively), phosphatidylserine exposure, and inside-out activation of αIIbβ3 integrin all correlate with the decline of platelet counts during dengue infection (64, 72, 155). Increased platelet activation may contribute to platelet loss by mediating platelet deposition in the microvascular bed at the periphery. In agreement, platelet aggregates and fibrinogen deposition have been detected in microvessels of postmortem histology and skin biopsies from severe dengue cases (156, 157). In a flow model, perfusion of DENV-infected whole blood on histamine-activated endothelial cells formed increased area of adhered platelets and platelet-von Willebrand factor strings compared with uninfected blood (64). Platelets have been also shown to adhere on DENV-infected endothelial cells in vitro, which reciprocally increased platelet activation (158). Aggregation of activated platelets with leukocytes may also contribute to thrombocytopenia in dengue, and circulating platelet–leukocyte aggregates have been shown among monocytes, lymphocytes, and granulocytes from dengue patients (54, 159). Accordingly, the levels of platelet–monocyte aggregates in the circulation of dengue patients negatively correlate with platelet counts (54). The participation of platelet–leukocyte aggregates (as well other platelet-mediated responses) in inflammatory amplification and dengue pathogenesis will be discussed further in this section and is highlighted in Figure 3.
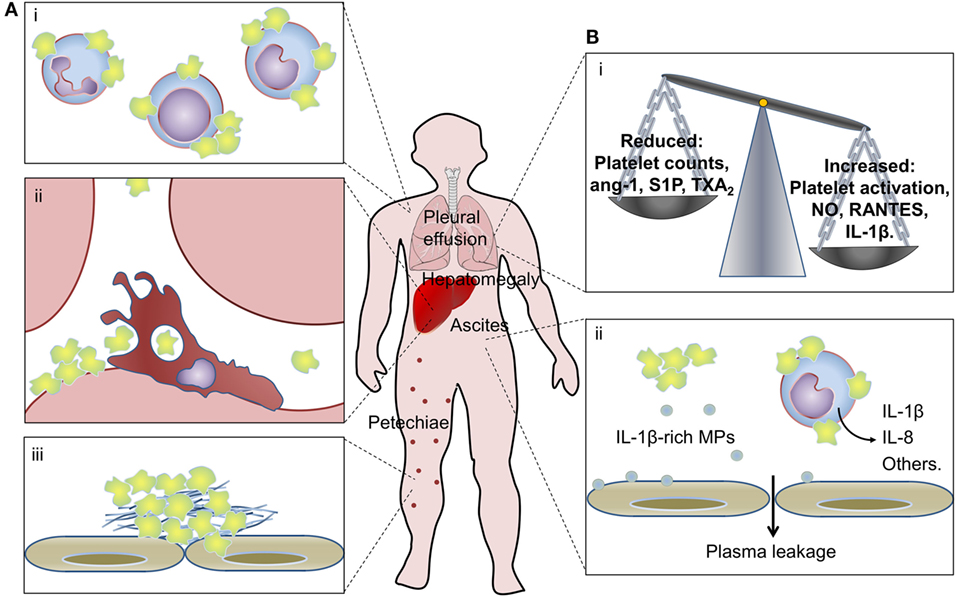
Figure 3. Platelets in dengue pathogenesis. (A) Peripheral mechanisms of thrombocytopenia in dengue: (i) platelet–leukocyte aggregates are formed with monocytes, lymphocytes, and neutrophils in the circulation of dengue patients; (ii) increased platelet sequestration in the liver of patients with dengue hemorrhagic fever/dengue shock syndrome and clearance of apoptotic platelets by resident macrophages through phosphatidylserine-mediated phagocytosis; and (iii) platelet adhesion and platelet clot formation on microvascular bed at the periphery may all contribute to thrombocytopenia in dengue. (B) Contributions of platelet activation and thrombocytopenia to inflammatory amplification and increased vascular permeability in dengue: (i) thrombocytopenia and reduced levels of platelet-derived endothelium-stabilizing factors alongside increased production of pro-inflammatory vasoactive factors by activated platelets that remain in circulation may contribute to dengue-associated vasculopathy; (ii) platelet synthesis of IL-1β and release of IL-1β-containing microparticles (MPs) associate with increased vascular permeability in dengue patients, and IL-1-β-rich MPs shed from dengue virus-infected platelets increase endothelial permeability in vitro; activated platelets from dengue-infected patients also induce pro-inflammatory cytokine secretion by monocytes. See the text for details and references.
The exact triggers of platelet activation in dengue infection are not completely clear. As discussed earlier, the mechanisms of DENV binding and replication in platelets and those involved in activation of DENV-infected platelets have been investigated (58, 64, 72). Consistently, DENV genome copies in platelets positively correlate with platelet activation in acute dengue infection (64). However, platelet activation in patients with dengue is maximal at the critical phase of infection, when DENV particles are no longer circulating (72, 153), indicating the existence of additional mechanisms governing platelet activation in non-viremic phases of infection. Mechanisms possibly involved in platelet activation in dengue include platelet adhesion to activated endothelium (158), increased thrombin generation (138, 160–162), and increased production of pro-inflammatory mediators as platelet-activating factor (PAF) (163–165). Regarding the latter, experimental DENV infection in mice lacking PAF receptor results in mild disease with reduced thrombocytopenia, inflammation, and mortality (166). Recently, a quantitative proteome approach of platelets from patients with dengue revealed novel mechanisms and pathways of platelet activation involving the binding of cell-free histone H2A (154). Cell-free histones are recognized DAMP signals that activate platelets through TLR-2 and -4 in vitro and in vivo (167–169). Injection of histones in mice leads to histone accumulation in sites of thrombosis and to thrombocytopenia (168–170). In dengue patients, increased levels of circulating cell-free histone H2A and histone sequestration by circulating platelets associate with disease severity (154). When platelets from healthy volunteers are exposed to plasma from dengue patients, cell-free histone H2A binds to and activates platelets ex vivo, indicating that circulating cell-free histones may contribute to platelet activation in dengue (154).
Aside from platelet activation and intravascular thrombosis, increased platelet apoptosis may contribute to thrombocytopenia in dengue infection. Increased rates of apoptotic platelets circulate in dengue-infected patients (54, 72, 155). In vitro infection with any serotype of DENV also culminates in the activation of an intrinsic cell death program in platelets that involves mitochondrial dysfunction and activation of apoptotic caspases (64, 72, 155). Patients with dengue hemorrhagic fever (DHF) present higher frequencies of platelet apoptosis compared with mild dengue (155). Accordingly, platelet life-span is reduced in DHF, and patients with DHF/DSS present increased platelet sequestration in the liver (171). In parallel ex vivo experiments, cultured macrophages phagocytized apoptotic platelets from dengue patients chiefly depending on phosphatidylserine recognition by the phagocyte (155). These studies suggest that platelet retention in the reticuloendothelial system with phagocytosis of apoptotic platelets by resident macrophages may represent a key mechanism of thrombocytopenia in dengue.
Besides platelet apoptosis and aggregation at the periphery, central mechanisms of bone marrow suppression may also contribute to thrombocytopenia in dengue (172, 173). DENV propagation in bone marrow (33, 174, 175) may contribute to suppression of hematopoiesis in all lineages, including the megakaryocytic (174, 176, 177). Bone marrow aspirates from dengue patients show moderate hypocellularity and arrest of maturation in granulocyte, megakaryocyte, and erythroid/myeloid precursors (178–181). In agreement, experimental DENV infection of humanized mice shows transient thrombocytopenia with reduced numbers of human megakaryocytic progenitors and megakaryocytes in the marrow (182). In in vitro experiments, DENV infection of megakaryocyte progenitor cells culminates in loss of their proliferative capacity and cell death by apoptosis (177). Regarding mature megakaryocytes, it has been demonstrated in non-human primates and ex vivo infection of human marrow cells that megakaryocytes are the main target for DENV in the marrow (32, 33), but whether DENV infection of mature megakaryocytes impacts platelet production, quantitative or qualitatively, remains to be explored.
Thrombocytopenia may be involved in the increased vascular permeability of dengue patients. Platelets participate in the regulation of endothelial barrier function and are required for the maintenance of basal endothelial permeability in physiological conditions (2, 183, 184). In in vitro experiments, isolated platelets or supernatants from rested platelets are able to reduce the permeability of endothelial monolayers in a concentration-dependent fashion (185–188). Platelet-rich plasma is also able to revert TNF-α-induced endothelial permeability in vitro and LPS-induced alveolar-capillary leak in experimental endotoxemia in mice (189). Various platelet-derived proteins and lipid mediators have been identified as vascular endothelium-stabilizing factors including lysophosphatidic acid, sphingosine-1-phosphate (S1P), thromboxane A2 (TXA2), and angiopoietin-1 (188–193). In agreement, reduced plasma levels of angiopoietin-1 alongside increased levels of its antagonist angiopoietin-2 are associated with shock presentation in children with DHF (194). Similarly, plasma levels of TXA2 and S1P are reduced in patients presenting ultrasound-evidence of plasma leakage or that progress to shock (195–197). These results together with the association between thrombocytopenia and plasma leakage in dengue patients (136, 138–142) suggest that platelets at steady state are required for preservation of vascular barrier integrity, and reductions in platelet counts and platelet-derived endothelium-stabilizing factors might increase vascular permeability in dengue. Paradoxically, activated platelets participate in inflammatory processes that trigger increased vascular permeability as well.
Activated platelets in patients with dengue or platelets infected with DENV in vitro secrete various factors with vasorelaxing or endothelial activating functions, including small molecules (serotonin and nitric oxide) (153, 198, 199), granule-stored chemokine (RANTES/CCL5 and PF4/CXCL4) (154), and newly-synthesized IL-1β (26). Among these factors, at least nitric oxide, RANTES, and IL-1β have been linked to disease severity and vascular instability in dengue-infected patients (140, 200–204). Similarly, platelet activation markers have been associated with measures of plasma leakage in patients with dengue (26, 54, 153). We recently reported increased expression of IL-1β in platelets from patients with dengue and in DENV-infected platelets in vitro. Newly synthesized pro-IL-1β was processed by inflammasome-dependent caspase-1 activity, as demonstrated in platelets from infected patients and in functional assays using inflammasome and caspase-1 inhibitors in vitro (26). In this model, IL-1β processing and secretion required the generation of reactive oxygen species in mitochondria as an endogenous signal for NLRP3 activation (Figure 1). Biologically active mature IL-1β was released from platelets in suspension and in IL-1β-rich MPs. In patients with dengue, IL-1β expression in platelets and platelet-derived MPs and caspase-1 activity in platelets correlated with clinical signs of increased vascular permeability; and MPs recovered from DENV-infected platelets increased endothelial cell permeability in vitro, which was blocked by IL-1 receptor antagonist. These observations in platelets from patients with dengue and from in vitro infection models and functional assays provided evidence for NLRP3 inflammasome activation in platelets culminating in the release of IL-1β-containing MPs as an important pathogenic mechanism for vasculopathy in dengue syndrome (26).
Platelet–leukocyte aggregates also participate in leukocyte immunoregulation and inflammatory response in dengue. As mentioned before, platelets form aggregates with lymphocytes, monocytes, and granulocytes during dengue infection (54, 159). Circulating platelet–monocyte and platelet–neutrophil aggregates were also demonstrated in a model of dengue-induced hemorrhage in rhesus macaques (205). In this model, microscopic evidence of platelet–monocyte aggregates and platelet phagocytosis by monocytes in peripheral blood was also provided (205). In recent experiments from our group, ex vivo heterologous interactions of platelets from dengue-infected patients with monocytes from healthy volunteers demonstrated the ability of activated and apoptotic platelets from infected patients to modulate the synthesis and secretion of IL-1β, IL-8, and IL-10 by monocytes. The same responses were not achieved by platelets from healthy volunteers interacted with monocytes from heterologous healthy subjects or dengue patients (54). Complementary in vitro experiments demonstrated that modulation of cytokine secretion by monocytes required P-selectin-mediated adhesion and recognition of phosphatidylserine on apoptotic platelets, which provided a previously unrecognized signal for IL-10 secretion in platelet–monocyte complexes (54). Beyond cytokines, activated platelets have been shown to trigger the synthesis of chemokines, adhesion molecules, metalloproteinase-9, and cyclooxygenase-2 by monocytes (118, 119, 206, 207), which were all implicated in dengue immunopathology in other studies (195, 208–213).
The consequences of platelet–neutrophil and platelet–lymphocyte interactions in dengue pathogenesis is far less explored. Regarding platelet interaction with lymphocytes, the ability of platelets to cross-present exogenous protein antigens to CD8+ T cells through major histocompatibility complex (MHC) class I was recently demonstrated in vitro and in experimental cerebral malaria in vivo (214). In this study, platelets effectively activated antigen-specific CD8+ T cells through presentation of pathogen-derived antigen in MHC class I (214). Very recent experiments investigating the ability of mature megakaryocytes to process exogenous proteins and present their peptides in MHC class I indicate that megakaryocytes also trigger CD8+ T cell activation and proliferation in vitro and in vivo. More importantly, megakaryocytes are able to transfer antigen-loaded MHC class I complexes to platelets during thrombopoiesis (31). Platelets isolated from patients with dengue have increased expression of MHC class I as evidenced by mass spectrometry-based proteome and western blot (154). This increased MHC class I content may derive from megakaryocytes, which have been identified as the main target cells for DENV infection in marrow (32, 33); or from DENV infection of platelets at circulation (62–64). In in vitro experiments, platelets infected with DENV increased MHC class I expression and surface display through mechanisms depending on proteasome activity (154). Whether peptides processed by platelet proteasome/immunoproteasome and presented in MHC class I on platelets derived from viral antigens or self-proteins requires further investigation. In experimental models or patients with idiopathic thrombocytopenic purpura and in platelet transfusion-refractory individuals, platelet MHC class I-mediated CD8+ T cell cytotoxicity leads to platelet clearance and inflammatory cytokine secretion (31, 214–219), both important pathogenic mechanisms in dengue. Nevertheless, new studies are still necessary to investigate the role played by platelet MHC class I expression and platelet–lymphocyte interactions in dengue pathogenesis, specially thrombocytopenia and cytokine storm.
Influenza pneumonia, caused by virulent strains of influenza A virus (IAV), remains a major global health problem with seasonal influenza epidemics and unexpected pandemics that have occurred for more than a century. For example, the virulent H1N1 strain of influenza identified in the 2009 flu pandemic caused increased morbidity and mortality worldwide (220–222). Patients admitted in intensive care unit (ICU) with severe influenza pneumonia usually presented acute lung injury and acute respiratory distress syndrome (ALI/ARDS) which have exacerbated immune response and increased systemic and airway inflammation as central features of pathogenesis (221, 223, 224). Pathophysiologic mechanisms of severe influenza pneumonia include overwhelming immune activation at the airways resulting in alveolar-capillary barrier damage, edema, pulmonary microvascular thrombosis, and progressive loss of respiratory capacity (224–226). Because of disrupted alveolar-capillary barrier integrity in influenza pneumonia, platelets may interact with influenza virus or influenza-IgG immunocomplexes even if the virus is restricted to alveoli and airways compartment (75, 227). In addition, there has been evidence for platelet activation by locally generated agonists and damage signals (227, 228). The participation of platelets in local immune and inflammatory responses during influenza infection (Figure 4) makes a perfect example of platelet activities at the extravascular space.
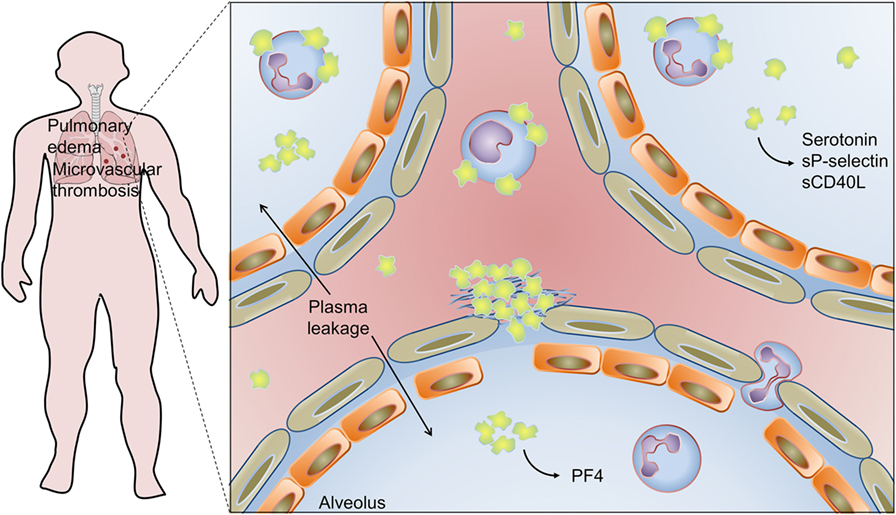
Figure 4. Platelet activation and infiltration to airways in influenza pneumonia: pulmonary edema and microvascular thrombosis with platelet–fibrin clot deposition in areas of disrupted alveolar-capillary barrier integrity are histopathological features of influenza pneumonia. Platelets, platelet–platelet aggregates, and platelet–neutrophil aggregates have been also detected in extravascular space in the lungs, and platelet–monocyte aggregates have been demonstrated in circulation. Activated platelets secrete serotonin, sCD40L, sP-selectin, and PF4 in the airways. Platelet activation is involved in pathophysiologic mechanisms of lung injury and pulmonary edema in influenza. On the other hand, platelet secretion of PF4 is an essential mechanism for neutrophil recruitment to the lungs and innate immunity against influenza virus. See the text for details and references.
Increased platelet activation and increased formation of platelet–monocyte aggregates have been reported in the blood of critically ill H1N1 influenza patients presenting ALI/ARDS (229). In this study, platelet activation and platelet–monocyte aggregates’ formation were higher in H1N1 influenza patients compared with patients with bacterial pneumonia at ICU (229). Activated platelets and platelet–monocyte aggregates have been also shown in influenza vaccinated subjects (230–232). Increased platelet activation and platelet–monocyte aggregation after influenza vaccination correlated with the expansion of inflammatory CD14highCD16+ and reduction of classical CD14highCD16− monocyte subsets, which was prevented by antiplatelet therapy with aspirin or clopidogrel (230, 232). Complementary in vitro experiments indicated that concurrent signaling from P-selectin-PSGL-1 binding and secreted prostaglandin E2 expanded the subset of CD16+ inflammatory monocytes in platelet–monocyte cocultures (230). However, whether and how platelet–monocyte aggregates contribute to the expansion of inflammatory monocytes in influenza or other infections in humans remains to be demonstrated.
Histopathological studies of lungs in experimental influenza A infection in mice and autopsies from patients who expired from H1N1 influenza documented frequent microvascular thrombosis in areas of disrupted integrity of alveolar-capillary barrier (56, 226–228). In influenza A-infected mice, platelet infiltration to lung parenchyma was evidenced by CD41 and PF4 immunohistochemistry, transmission electron microscopy, and platelet counting in bronchoalveolar lavage (BAL) (56, 75, 227, 228). Ultrastructural analysis of infiltrated platelets shows evidence of platelet activation and formation of platelet–neutrophil aggregates, which are in agreement with increased levels of serotonin, sP-selectin, and sCD40L in BAL (227, 228). In these models, platelet activation has been observed in both airways and blood from infected mice, advocating in favor of multiple (parallel or sequential) mechanisms for platelet activation (56, 227, 228). These mechanisms may involve, in addition to direct contact with the virus or virus-IgG immunocomplexes, the generation of agonists and damage signals that activate infiltrated and circulating platelets in severe influenza pneumonia (75, 227, 228). Recent in vitro experiments investigating platelet responses to H1N1 in the presence of influenza immune serum demonstrated that complete thromboinflammatory phenotype of activated platelets depends on the synergistic activation of FcγRIIA by immunocomplexes and protease activating receptor (PAR) by thrombin (75).
Studies of experimental influenza A infection plus pharmacological and genetic models have supported the participation of platelets in the pathogenesis of airway inflammation and lung injury. Influenza A infection together with PAR-4 agonist or antagonist were able to, respectively, exacerbate or recover lung inflammation, plasma leakage and mortality with no change on influenza viral load (227). Genetic deficiency of integrin αIIbβ3 or treatment with the integrin αIIbβ3 inhibitor eptifibatide reduced influenza- or influenza plus PAR-4 agonist-induced platelet infiltration and activation in the lungs, and rescued infected mice from lung injury and mortality (227). Other antiplatelet drugs including aspirin and P2Y purinergic receptor inhibitors also protected influenza-infected mice from platelet accumulation in lungs and mortality (227, 233). In complementary in vitro experiments, it was demonstrated that platelet attachment to infected pulmonary microvascular endothelial cells depended on integrin-mediated adhesion and was also blocked by pharmacological inhibitors of platelet activation (233). Very recent studies investigating the role of extracellular histones in influenza-mediated lung injury and vascular thrombosis has evidenced the deposition of cell-free histones in association with activated platelets and neutrophils in areas of alveolar damage in lungs of infected mice (228). Increased levels of cell-free histones have been observed in BAL from influenza-infected mice and in nasal wash from influenza A, but not influenza B, infected patients (228, 234). Even though this study has demonstrated activated platelets in BAL of infected mice, the particular role of cell-free histones in platelet activation and platelet infiltration in the airways has not been addressed (228). Altogether, these experiments indicate that pathogenic mechanisms involving activation-dependent platelet adhesion, infiltration, and inflammatory response in the lungs contribute to influenza pneumonia and ARDS. These evidences from experimental infection models shed light on novel targets for therapeutic intervention in influenza-associated ALI/ARDS. Accordingly, supplementation of antiviral therapy with antiplatelet drugs or antihistone antibodies has improved histological index of lung injury and increased the survival of infected mice when compared with antiviral therapy alone (228, 233).
Another recent work indicates that platelets have protective activities in innate immune response against IAV. Increased levels of PF4 accumulate in blood and lung of mice experimentally infected with H1N1 influenza. Based on genetic models, PF4 in blood and lungs from infected mice have essential roles in innate mechanisms of viral clearance and survival. Experimental infection of PF4-deficient mice resulted in exacerbated lung inflammation, alveolar damage, and increased mortality (56). Mice lacking PF4 were unable to provide effective clearance of the virus even though specific T and B cell-mediated adaptive immunity were responsive (56). Deficient viral clearance was related to insufficient production of neutrophil chemoattractant chemokines and, consequently, to lower neutrophil migration to the circulation and lungs. PF4 intravenous injection or instillation in the lungs recovered the levels of chemokines, infiltration of neutrophils, and survival of PF4-deficient mice (56). These experimental observations together with the ones aforementioned indicate that platelets accumulate at intra- and extravascular space in the lungs during influenza A infection, and that they are effectors of both host protection and, in severe influenza pneumonia, lung injury (56, 227).
Emerging evidences identify platelets as dynamic cells that participate in inflammation and prothrombotic responses in many vascular and inflammatory processes, including viral infections. New platelet functions have emerged over time and platelets are getting increasingly recognized as immune cells. Pathophysiological mechanisms involving platelet responses have been reported in HIV infection, dengue fever, and influenza pneumonia in naturally infected patients and in experimental infection models as discussed here. Thrombocytopenia, platelet secretion of stored and newly synthesized factors, and complex interactions with leukocytes comprise platelet features that may have both injurious and protective immune consequences in viral infections. Increasing our understanding on immunoregulatory functions of platelets in viral infections will undoubtedly improve our knowledge on diseases pathogenesis, clinical management, and therapeutic options.
EH, FB and PB contributed to development of the concepts and design of the review article. EH wrote the manuscript and prepared the figures. PB and FB reviewed the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The work of the authors is supported by CNPq, CAPES, FAPERJ, and PAPES/FIOCRUZ.
1. Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol (2012) 34:5–30. doi:10.1007/s00281-011-0286-4
2. Middleton EA, Weyrich AS, Zimmerman GA. Platelets in pulmonary immune responses and inflammatory lung diseases. Physiol Rev (2016) 96:1211–59. doi:10.1152/physrev.00038.2015
3. Semple JW, Italiano JE, Freedman J. Platelets and the immune continuum. Nat Rev Immunol (2011) 11:264–74. doi:10.1038/nri2956
4. Shashkin PN, Brown GT, Ghosh A, Marathe GK, McIntyre TM. Lipopolysaccharide is a direct agonist for platelet RNA splicing. J Immunol (2008) 181:3495–502. doi:10.4049/jimmunol.181.5.3495
5. Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell (2005) 122:379–91. doi:10.1016/j.cell.2005.06.015
6. Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1-rich microparticles. J Immunol (2011) 186:5489–96. doi:10.4049/jimmunol.1001623
7. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of β-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog (2011) 7(11):e1006333. doi:10.1371/journal.ppat.1002355
8. Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of toll-like receptor molecules on human platelets. Immunol Cell Biol (2005) 83:196–8. doi:10.1111/j.1440-1711.2005.01314.x
9. D’Atri LP, Schattner M. Platelet toll-like receptors in thromboinflammation. Front Biosci (Landmark Ed) (2017) 22:1867–83. doi:10.2741/4576
10. Shiokawa M, Yamasaki S, Saijo S. C-type lectin receptors in anti-fungal immunity. Curr Opin Microbiol (2017) 40:123–30. doi:10.1016/j.mib.2017.11.004
11. Marakalala MJ, Ndlovu H. Signaling C-type lectin receptors in antimycobacterial immunity. PLoS Pathog (2017) 13:e1006333. doi:10.1371/journal.ppat.1006333
12. Monteiro JT, Lepenies B. Myeloid C-type lectin receptors in viral recognition and antiviral immunity. Viruses (2017) 9:59. doi:10.3390/v9030059
13. Turville SG, Cameron PU, Handley A, Lin G, Pöhlmann S, Doms RW, et al. Diversity of receptors binding HIV on dendritic cell subsets. Nat Immunol (2002) 3:975–83. doi:10.1038/ni841
14. Chaipan C, Soilleux EJ, Simpson P, Hofmann H, Gramberg T, Marzi A, et al. DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J Virol (2006) 80:8951–60. doi:10.1128/JVI.00136-06
15. Cognasse F, Nguyen KA, Damien P, McNicol A, Pozzetto B, Hamzeh-Cognasse H, et al. The inflammatory role of platelets via their TLRs and siglec receptors. Front Immunol (2015) 6:83. doi:10.3389/fimmu.2015.00083
16. Koupenova M, Mick E, Mikhalev E, Benjamin EJ, Tanriverdi K, Freedman JE. Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors. Arterioscler Thromb Vasc Biol (2015) 35:1030–7. doi:10.1161/ATVBAHA.114.304954
17. Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, et al. Genome wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood (2011) 118(14):e101–11. doi:10.1182/blood-2011-03-339705
18. Garraud O, Cognasse F. Platelet toll-like receptor expression: the link between “danger” ligands and inflammation. Inflamm Allergy Drug Targets (2010) 9(5):322–33. doi:10.2174/187152810793937991
19. Aslam R, Speck ER, Kim M, Crow AR, Bang KWA, Nestel FP, et al. Platelet toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-α production in vivo. Blood (2006) 107(2):637–41. doi:10.1182/blood-2005-06-2202
20. Andonegui G, Kerfoot S, McNagny K, Ebbert K, Patel K, Kubes P. Platelets express functional toll-like receptor-4 (TLR4). Blood (2005) 106:2417–23. doi:10.1182/blood-2005-03-0916.Supported
21. Ståhl AL, Svensson M, Mörgelin M, Svanborg C, Tarr PI, Mooney JC, et al. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood (2006) 108:167–76. doi:10.1182/blood-2005-08-3219
22. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13:463–9. doi:10.1038/nm1565
23. Thon JN, Peters CG, Machlus KR, Aslam R, Rowley J, Macleod H, et al. T granules in human platelets function in TLR9 organization and signaling. J Cell Biol (2012) 198:561–74. doi:10.1083/jcb.201111136
24. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood (2014) 124:791–802. doi:10.1182/blood-2013-11-536003
25. D’Atri LP, Etulain J, Rivadeneyra L, Lapponi MJ, Centurion M, Cheng K, et al. Expression and functionality of toll-like receptor 3 in the megakaryocytic lineage. J Thromb Haemost (2015) 13:839–50. doi:10.1111/jth.12842
26. Hottz ED, Lopes JF, Freitas C, Valls-De-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood (2013) 122:3405–14. doi:10.1182/blood-2013-05-504449
27. Zhang S, Zhang S, Hu L, Zhai L, Xue R, Ye J, et al. Nucleotide-binding oligomerization domain 2 receptor is expressed in platelets and enhances platelet activation and thrombosis. Circulation (2015) 131:1160–70. doi:10.1161/CIRCULATIONAHA.114.013743
28. Hottz ED, Monteiro APT, Bozza FA, Bozza PT. Inflammasome in platelets: allying coagulation and inflammation in infectious and sterile diseases? Mediators Inflamm (2015) 2015:12–5. doi:10.1155/2015/435783
29. Lütteke N, Raftery MJ, Lalwani P, Lee MH, Giese T, Voigt S, et al. Switch to high-level virus replication and HLA class I upregulation in differentiating megakaryocytic cells after infection with pathogenic hantavirus. Virology (2010) 405:70–80. doi:10.1016/j.virol.2010.05.028
30. Cecchetti L, Tolley ND, Michetti N, Bury L, Weyrich AS, Gresele P. Megakaryocytes differentially sort mRNAs for matrix metalloproteinases and their inhibitors into platelets: a mechanism for regulating synthetic events. Blood (2011) 118:1903–11. doi:10.1182/blood-2010-12-324517
31. Zufferey A, Speck ER, Machlus KR, Aslam R, Guo L, McVey MJ, et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv (2017) 1:1773–85. doi:10.1182/bloodadvances.2017007021
32. Clark KB, Noisakran S, Onlamoon N, Hsiao HM, Roback J, Villinger F, et al. Multiploid CD61+ cells are the pre-dominant cell lineage infected during acute dengue virus infection in bone marrow. PLoS One (2012) 7:e52902. doi:10.1371/journal.pone.0052902
33. Noisakran S, Onlamoon N, Hsiao HM, Clark KB, Villinger F, Ansari AA, et al. Infection of bone marrow cells by dengue virus in vivo. Exp Hematol (2012) 40:250–9.e4. doi:10.1016/j.exphem.2011.11.011
34. Negrotto S, J De Giusti C, Lapponi MJ, Etulain J, Rivadeneyra L, Pozner RG, et al. Expression and functionality of type I interferon receptor in the megakaryocytic lineage. J Thromb Haemost (2011) 9:2477–85. doi:10.1111/j.1538-7836.2011.04530.x
35. Voulgaropoulou F, Pontow SE, Ratner L. Productive infection of CD34+-cell-derived megakaryocytes by X4 and R5 HIV-1 isolates. Virology (2000) 269:78–85. doi:10.1006/viro.2000.0193
36. Chelucci C, Federico M, Guerriero R, Mattia G, Casella I, Pelosi E, et al. Productive human immunodeficiency virus-1 infection of purified megakaryocytic progenitors/precursors and maturing megakaryocytes. Blood (1998) 91:1225–34.
37. Li X, Jeffers LJ, Garon C, Fischer ER, Scheffel J, Moore B, et al. Persistence of hepatitis C virus in a human megakaryoblastic leukaemia cell line. J Viral Hepat (1999) 6:107–14. doi:10.1046/j.1365-2893.1999.00140.x
38. Beaulieu LM, Lin E, Mick E, Koupenova M, Weinberg EO, Kramer CD, et al. Interleukin 1 receptor 1 and interleukin 1β regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol (2014) 34:552–64. doi:10.1161/ATVBAHA.113.302700
39. Beaulieu LM, Lin E, Morin KM, Tanriverdi K, Freedman JE. Regulatory effects of TLR2 on megakaryocytic cell function. Blood (2011) 117:5963–74. doi:10.1182/blood-2010-09-304949
40. Rivadeneyra L, Pozner RG, Meiss R, Fondevila C, Gómez RM, Schattner M. Poly (I:C) downregulates platelet production and function through type I interferon. Thromb Haemost (2015) 114:982–93. doi:10.1160/TH14-11-0951
41. Pozner RG, Ure AE, Jaquenod de Giusti C, D’Atri LP, Italiano JE, Torres O, et al. Junín virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type I IFN signaling. PLoS Pathog (2010) 6:e1000847. doi:10.1371/journal.ppat.1000847
42. Yamane A, Nakamura T, Suzuki H, Ito M, Ohnishi Y, Ikeda Y, et al. Interferon-alpha 2b-induced thrombocytopenia is caused by inhibition of platelet production but not proliferation and endomitosis in human megakaryocytes. Blood (2008) 112:542–50. doi:10.1182/blood-2007-12-125906
43. Mukhopadhyay S, Kuhn RJ, Rossmann MG. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol (2005) 3:13–22. doi:10.1038/nrmicro1067
44. Blijleven JS, Boonstra S, Onck PR, van der Giessen E, van Oijen AM. Mechanisms of influenza viral membrane fusion. Semin Cell Dev Biol (2016) 60:78–88. doi:10.1016/j.semcdb.2016.07.007
45. Dutartre H, Clavière M, Journo C, Mahieux R. Cell-free versus cell-to-cell infection by human immunodeficiency virus type 1 and human T-lymphotropic virus type 1: exploring the link among viral source, viral trafficking, and viral replication. J Virol (2016) 90:7607–17. doi:10.1128/JVI.00407-16
46. Brass AL, Huang I-C, Benita Y, John SP, Krishnan MN, Feeley EM, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell (2009) 139:1243–54. doi:10.1016/j.cell.2009.12.017
47. Oshiumi H, Kouwaki T, Seya T. Accessory factors of cytoplasmic viral RNA sensors required for antiviral innate immune response. Front Immunol (2016) 7:200. doi:10.3389/fimmu.2016.00200
48. Hotter D, Kirchhoff F. Interferons and beyond: induction of antiretroviral restriction factors. J Leukoc Biol (2018) 103(3):465–477. doi:10.1002/JLB.3MR0717-307R
49. Chabert A, Hamzeh-Cognasse H, Pozzetto B, Cognasse F, Schattner M, Gomez RM, et al. Human platelets and their capacity of binding viruses: meaning and challenges? BMC Immunol (2015) 16:26. doi:10.1186/s12865-015-0092-1
50. Flaujac C, Boukour S, Cramer-Bordé E. Platelets and viruses: an ambivalent relationship. Cell Mol Life Sci (2010) 67:545–56. doi:10.1007/s00018-009-0209-x
51. Assinger A. Platelets and infection – an emerging role of platelets in viral infection. Front Immunol (2014) 5:649. doi:10.3389/fimmu.2014.00649
52. Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, et al. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost (2011) 9(4):748–58. doi:10.1111/j.1538-7836.2011.04208.x
53. Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, Spencer E, et al. mTOR-dependent synthesis of Bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood (2007) 109(5):1975–83. doi:10.1182/blood-2006-08-042192
54. Hottz ED, Medeiros-de-Moraes IM, Vieira-de-Abreu A, de Assis EF, Vals-de-Souza R, Castro-Faria-Neto HC, et al. Platelet activation and apoptosis modulate monocyte inflammatory responses in dengue. J Immunol (2014) 193:1864–72. doi:10.4049/jimmunol.1400091
55. O’Brien M, Montenont E, Hu L, Nardi MA, Valdes V, Merolla M, et al. Aspirin attenuates platelet activation and immune activation in HIV-1-infected subjects on antiretroviral therapy: a pilot study. J Acquir Immune Defic Syndr (2013) 63:280–8. doi:10.1097/QAI.0b013e31828a292c
56. Guo L, Feng K, Wang YC, Mei JJ, Ning RT, Zheng HW, et al. Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol (2017) 10:1529–41. doi:10.1038/mi.2017.1
57. Boukour S, Massé JM, Bénit L, Dubart-Kupperschmitt A, Cramer EM. Lentivirus degradation and DC-SIGN expression by human platelets and megakaryocytes. J Thromb Haemost (2006) 4:426–35. doi:10.1111/j.1538-7836.2006.01749.x
58. Simon AY, Sutherland MR, Pryzdial ELG. Dengue virus binding and replication by platelets. Blood (2015) 126(3):378–85. doi:10.1182/blood-2014-09-598029
59. Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med (2003) 197(7):823–9. doi:10.1084/jem.20021840
60. Pokidysheva E, Zhang Y, Battisti AJ, Bator-Kelly CM, Chipman PR, Xiao C, et al. Cryo-EM reconstruction of dengue virus in complex with the carbohydrate recognition domain of DC-SIGN. Cell (2006) 124(3):485–93. doi:10.1016/j.cell.2005.11.042
61. Solomon Tsegaye T, Gnirß K, Rahe-Meyer N, Kiene M, Krämer-Kühl A, Behrens G, et al. Platelet activation suppresses HIV-1 infection of T cells. Retrovirology (2013) 10:48. doi:10.1186/1742-4690-10-48
62. Noisakran S, Chokephaibulkit K, Songprakhon P, Onlamoon N, Hsiao H-M, Villinger F, et al. A re-evaluation of the mechanisms leading to dengue hemorrhagic fever. Ann N Y Acad Sci (2009) 1171(Suppl 1):E24–35. doi:10.1111/j.1749-6632.2009.05050.x
63. Noisakran S, Gibbons RV, Songprakhon P, Jairungsri A, Ajariyakhajorn C, Nisalak A, et al. Detection of dengue virus in platelets isolated from dengue patients. Southeast Asian J Trop Med Public Health (2009) 40(2):253–62.
64. Ojha A, Nandi D, Batra H, Singhal R, Annarapu GK, Bhattacharyya S, et al. Platelet activation determines the severity of thrombocytopenia in dengue infection. Sci Rep (2017) 7:1–10. doi:10.1038/srep41697
65. Hamaia S, Li C, Allain JP. The dynamics of hepatitis C virus binding to platelets and 2 mononuclear cell lines. Blood (2001) 98:2293–300. doi:10.1182/blood.V98.8.2293
66. de Almeida AJ, Campos-de-Magalhães M, de Melo Marçal OP, Brandão-Mello CE, Okawa MY, de Oliveira RV, et al. Hepatitis C virus-associated thrombocytopenia: a controlled prospective, virological study. Ann Hematol (2004) 83:434–40. doi:10.1007/s00277-004-0844-0
67. de Almeida AJ, Campos-de-Magalhães M, Brandão-Mello CE, de Oliveira RV, do Espirito-Santo MP, Yoshida CFT, et al. Detection of hepatitis C virus in platelets: evaluating its relationship to antiviral therapy outcome. Hepatogastroenterology (2009) 56(90):429–36.
68. Padovani JL, Corvino SM, Drexler JF, Silva GF, Pardini MI, Grotto RM. In vitro detection of hepatitis C virus in platelets from uninfected individuals exposed to the virus. Rev Soc Bras Med Trop (2013) 46:154–5. doi:10.1590/0037-8682-1627-2013
69. Zahn A, Jennings N, Ouwehand WH, Allain J-P. Hepatitis C virus interacts with human platelet glycoprotein VI. J Gen Virol (2006) 87:2243–51. doi:10.1099/vir.0.81826-0
70. Kar M, Singla M, Chandele A, Kabra SK, Lodha R, Medigeshi GR. Dengue virus entry and replication does not lead to productive infection in platelets. Open Forum Infect Dis (2017) 4:1–5. doi:10.1093/ofid/ofx051
71. Ghosh K, Gangodkar S, Jain P, Shetty S, Ramjee S, Poddar P, et al. Imaging the interaction between dengue 2 virus and human blood platelets using atomic force and electron microscopy. J Electron Microsc (Tokyo) (2008) 57:113–8. doi:10.1093/jmicro/dfn007
72. Hottz ED, Oliveira MF, Nunes PCG, Nogueira RMR, Valls-de-Souza R, Da Poian AT, et al. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J Thromb Haemost (2013) 11:951–62. doi:10.1111/jth.12178
73. Cruz-Oliveira C, Freire JM, Conceição TM, Higa LM, Castanho MARB, Da Poian AT. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol Rev (2015) 39(2):155–70. doi:10.1093/femsre/fuu004
74. Wang S, He R, Patarapotikul J, Innis BL, Anderson R. Antibody-enhanced binding of dengue-2 virus to human platelets. Virology (1995) 213(1):254–7. doi:10.1006/viro.1995.1567
75. Boilard E, Paré G, Rousseau M, Cloutier N, Dubuc I, Lévesque T, et al. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood (2014) 123:2854–63. doi:10.1182/blood-2013-07-515536
76. Anabel AS, Eduardo PC, Pedro Antonio HC, Carlos SM, Juana NM, Honorio TA, et al. Human platelets express toll-like receptor 3 and respond to poly I:C. Hum Immunol (2014) 75:1244–51. doi:10.1016/j.humimm.2014.09.013
77. Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity (2010) 32(3):305–15. doi:10.1016/j.immuni.2010.03.012
78. Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, et al. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ Res (2013) 112:103–12. doi:10.1161/CIRCRESAHA.112.274241
79. Jenne CN, Wong CHY, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe (2013) 13:169–80. doi:10.1016/j.chom.2013.01.005
80. Loria GD, Romagnoli PA, Moseley NB, Rucavado A, Altman JD. Platelets support a protective immune response to LCMV by preventing splenic necrosis. Blood (2013) 121:940–50. doi:10.1182/blood-2011-08-376822
81. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe (2012) 12:324–33. doi:10.1016/j.chom.2012.06.011
82. Kim SJ, Jenne CN. Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. Semin Immunol (2016) 28:546–54. doi:10.1016/j.smim.2016.10.013
83. Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, et al. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med (2005) 11:1167–9. doi:10.1038/nm1317
84. Lewden C, Chene G, Morlat P, Raffi F, Dupon M, Dellamonica P, et al. HIV-infected adults with a CD4 cell count greater than 500 cells/mm3 on long-term combination antiretroviral therapy reach same mortality rates as the general population. J Acquir Immune Defic Syndr (2007) 46(1):72–7. doi:10.1097/QAI.0b013e318134257a
85. Palella FJ Jr, Baker RK, Moorman AC, Chmiel JS, Wood KC, Brooks JT, et al. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. J Acquir Immune Defic Syndr (2006) 43:27–34. doi:10.1097/01.qai.0000233310.90484.16
86. Lazar R, Kersanske L, Xia Q, Daskalakis D, Braunstein SL. Hospitalization rates among people with HIV/AIDS in New York city, 2013. Clin Infect Dis (2017) 65:469–76. doi:10.1093/cid/cix343
87. Marin B, Thiébaut R, Bucher HC, Rondeau V, Costagliola D, Dorrucci M, et al. Non-AIDS-defining deaths and immunodeficiency in the era of combination antiretroviral therapy. AIDS (2009) 23(13):1743–53. doi:10.1097/QAD.0b013e32832e9b78
88. Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet (2007) 370(9581):59–67. doi:10.1016/S0140-6736(07)61050-2
89. Goehringer F, Bonnet F, Salmon D, Cacoub P, Paye A, Chêne G, et al. Causes of death in HIV-infected individuals with immunovirologic success in a national prospective survey. AIDS Res Hum Retroviruses (2017) 33(2):187–93. doi:10.1089/aid.2016.0222
90. Hasse B, Ledergerber B, Furrer H, Battegay M, Hirschel B, Cavassini M, et al. Morbidity and aging in HIV-infected persons: the Swiss HIV cohort study. Clin Infect Dis (2011) 53(11):1130–9. doi:10.1093/cid/cir626
91. Lang S, Mary-Krause M, Simon A, Partisani M, Gilquin J, Cotte L, et al. HIV replication and immune status are independent predictors of the risk of myocardial infarction in HIV-infected individuals. Clin Infect Dis (2012) 55(4):600–7. doi:10.1093/cid/cis489
92. Lichtenstein KA, Armon C, Buchacz K, Chmiel JS, Buckner K, Tedaldi EM, et al. Low CD4+ T cell count is a risk factor for cardiovascular disease events in the HIV outpatient study. Clin Infect Dis (2010) 51(4):435–47. doi:10.1086/655144
93. Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med (2008) 5(10):e203. doi:10.1371/journal.pmed.0050203
94. McDonald B, Moyo S, Gabaitiri L, Gaseitsiwe S, Bussmann H, Koethe JR, et al. Persistently elevated serum interleukin-6 predicts mortality among adults receiving combination antiretroviral therapy in Botswana: results from a clinical trial. AIDS Res Hum Retroviruses (2013) 29(7):993–9. doi:10.1089/aid.2012.0309
95. Tien PC, Choi AI, Zolopa AR, Benson C, Tracy R, Scherzer R, et al. Inflammation and mortality in HIV-infected adults: analysis of the FRAM study cohort. J Acquir Immune Defic Syndr (2010) 55(3):316–22. doi:10.1097/QAI.0b013e3181e66216
96. D’Arminio Monforte A. Cardio- and cerebrovascular events in HIV-infected persons. AIDS (2004) 18(13):1811–7. doi:10.1097/00002030-200409030-00010
97. van Lelyveld SF, Gras L, Kesselring A, Zhang S, De Wolf F, Wensing AMJ, et al. Long-term complications in patients with poor immunological recovery despite virological successful HAART in Dutch ATHENA cohort. AIDS (2012) 26(4):465–74. doi:10.1097/QAD.0b013e32834f32f8
98. Holme PA, Müller F, Solum NO, Brosstad F, Frøland SS, Aukrust P. Enhanced activation of platelets with abnormal release of RANTES in human immunodeficiency virus type 1 infection. FASEB J (1998) 12(1):79–89. doi:10.1096/fasebj.12.1.79
99. Mayne E, Funderburg NT, Sieg SF, Asaad R, Kalinowska M, Rodriguez B, et al. Increased platelet and microparticle activation in HIV infection. J Acquir Immune Defic Syndr (2012) 59:340–6. doi:10.1097/QAI.0b013e3182439355
100. Nkambule BB, Davison GM, Ipp H. The evaluation of platelet function in HIV infected, asymptomatic treatment-naïve individuals using flow cytometry. Thromb Res (2015) 135(6):1131–9. doi:10.1016/j.thromres.2015.01.031
101. Nkambule BB, Davison G, Ipp H. The value of flow cytometry in the measurement of platelet activation and aggregation in human immunodeficiency virus infection. Platelets (2015) 26(3):250–7. doi:10.3109/09537104.2014.909021
102. O’Halloran JA, Dunne E, Gurwith MMP, Lambert JJS, Sheehan GJ, Feeney ER, et al. The effect of initiation of antiretroviral therapy on monocyte, endothelial and platelet function in HIV-1 infection. HIV Med (2015) 16:608–19. doi:10.1111/hiv.12270
103. Tunjungputri RN, Van Der Ven AJ, Schonsberg A, Mathan TS, Koopmans P, Roest M, et al. Reduced platelet hyperreactivity and platelet–monocyte aggregation in HIV-infected individuals receiving a raltegravir-based regimen. AIDS (2014) 28:2091–6. doi:10.1097/QAD.0000000000000415
104. von Hentig N, Förster A-K, Kuczka K, Klinkhardt U, Klauke S, Gute P, et al. Platelet-leucocyte adhesion markers before and after the initiation of antiretroviral therapy with HIV protease inhibitors. J Antimicrob Chemother (2008) 62:1118–21. doi:10.1093/jac/dkn333
105. Wolf K, Tsakiris DA, Weber R, Erb P, Battegay M; Swiss HIV Cohort Study. Antiretroviral therapy reduces markers of endothelial and coagulation activation in patients infected with human immunodeficiency virus type 1. J Infect Dis (2002) 185:456–62. doi:10.1086/338572
106. Corrales-Medina VF, Simkins J, Chirinos JA, Serpa JA, Horstman LL, Jy W, et al. Increased levels of platelet microparticles in HIV-infected patients with good response to highly active antiretroviral therapy. J Acquir Immune Defic Syndr (2010) 54:217–8. doi:10.1097/QAI.0b013e3181c8f4c9
107. Zucker-Franklin D, Seremetis S, Zheng ZY. Internalization of human immunodeficiency virus type I and other retroviruses by megakaryocytes and platelets. Blood (1990) 75:1920–3.
108. Youssefian T, Drouin A, Massé JM, Guichard J, Cramer EM. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus ocurs in a specific subcellular compoartmente and is enhanced by platelet activation. Blood (2002) 99:4021–9. doi:10.1182/blood-2001-12-0191.Supported
109. Wang J, Zhang W, Nardi MA, Li Z. HIV-1 tat-induced platelet activation and release of CD154 contribute to HIV-1-associated autoimmune thrombocytopenia. J Thromb Haemost (2011) 9:562–73. doi:10.1111/j.1538-7836.2010.04168.x
110. Davidson DC, Hirschman MP, Spinelli SL, Morrell CN, Schifitto G, Phipps RP, et al. Antiplatelet activity of valproic acid contributes to decreased soluble CD40 ligand production in HIV type 1-infected individuals. J Immunol (2011) 186:584–91. doi:10.4049/jimmunol.1001911
111. Davidson DC, Hirschman MP, Sun A, Singh MV, Kasischke K, Maggirwar SB. Excess soluble CD40L contributes to blood brain barrier permeability in vivo: implications for HIV-associated neurocognitive disorders. PLoS One (2012) 7:e51793. doi:10.1371/journal.pone.0051793
112. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med (2006) 12:1365–71. doi:10.1038/nm1511
113. Satchell CS, O’Halloran JA, Cotter AG, Peace AJ, O’Connor EF, Tedesco AF, et al. Increased platelet reactivity in HIV-1-infected patients receiving abacavir-containing antiretroviral therapy. J Infect Dis (2011) 204:1202–10. doi:10.1093/infdis/jir509
114. Falcinelli E, Francisci D, Belfiori B, Petito E, Guglielmini G, Malincarne L, et al. In vivo platelet activation and platelet hyperreactivity in abacavir-treated HIV-infected patients. Thromb Haemost (2013) 110:349–57. doi:10.1160/TH12-07-0504
115. Baum PD, Sullam PM, Stoddart CA, McCune JM. Abacavir increases platelet reactivity via competitive inhibition of soluble guanylyl cyclase. AIDS (2011) 25:2243–8. doi:10.1097/QAD.0b013e32834d3cc3
116. Diallo YL, Ronique Ollivier V, Ronique Joly V, Faille DE, Catalano G, Jandrot-Perrus M, et al. Abacavir has no prothrombotic effect on platelets in vitro. J Antimicrob Chemother (2016) 71(12):3506–9. doi:10.1093/jac/dkw303
117. Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, et al. PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell (1989) 59:305–12. doi:10.1016/0092-8674(89)90292-4
118. Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, et al. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest (1996) 97:1525–34. doi:10.1172/JCI118575
119. Weyrich AS, Denis MM, Kuhlmann-Eyre JR, Spencer ED, Dixon DA, Marathe GK, et al. Dipyridamole selectively inhibits inflammatory gene expression in platelet-monocyte aggregates. Circulation (2005) 111:633–42. doi:10.1161/01.CIR.0000154607.90506.45
120. Green SA, Smith M, Hasley RB, Stephany D, Harned A, Nagashima K, et al. Activated platelet-T-cell conjugates in peripheral blood of patients with HIV infection: coupling coagulation/inflammation and T cells. AIDS (2015) 29:1297–308. doi:10.1097/QAD.0000000000000701
121. Singh MV, Davidson DC, Jackson JW, Singh VB, Silva J, Ramirez SH, et al. Characterization of platelet-monocyte complexes in HIV-1-infected individuals: possible role in HIV-associated neuroinflammation. J Immunol (2014) 192:4674–84. doi:10.4049/jimmunol.1302318
122. Liang H, Duan Z, Li D, Li D, Wang Z, Ren L, et al. Higher levels of circulating monocyte-platelet aggregates are correlated with viremia and increased sCD163 levels in HIV-1 infection. Cell Mol Immunol (2015) 12:435–43. doi:10.1038/cmi.2014.66
123. Cocchi F, Devico AL, Garzino-demo A, Arya SK, Gallo RC, Lussot P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science (1995) 270:1811–5. doi:10.1126/science.270.5243.1811
124. Auerbach DJ, Lin Y, Miao H, Cimbro R, DiFiore MJ, Gianolini ME, et al. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc Natl Acad Sci U S A (2012) 109:9569–74. doi:10.1073/pnas.1207314109
125. Lusso P. HIV and the chemokine system: 10 years later. EMBO J (2006) 25:447–56. doi:10.1038/sj.emboj.7600947
126. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber M, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature (1996) 382:722–6. doi:10.1038/382722a0
127. Winkler C, Modi W, Smith MW, Nelson GW, Wu X, Carrington M, et al. Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. ALIVE Study, Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC). Science (1998) 279:389–93. doi:10.1007/s11033-009-9890-y
128. Liu H, Chao D, Nakayama EE, Taguchi H, Goto M, Xin X, et al. Polymorphism in RANTES chemokine promoter affects HIV-1 disease progression. Proc Natl Acad Sci U S A (1999) 96:4581–5. doi:10.1073/pnas.96.8.4581
129. Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science (2005) 307:1434–40. doi:10.1126/science.1101160
130. Sui Z, Sniderhan LF, Schifitto G, Phipps RP, Gelbard HA, Dewhurst S, et al. Functional synergy between CD40 ligand and HIV-1 tat contributes to inflammation: implications in HIV type 1 dementia. J Immunol (2007) 178:3226–36. doi:10.4049/jimmunol.178.5.3226
131. Jones LD, Jackson JW, Maggirwar SB. Modeling HIV-1 induced neuroinflammation in mice: role of platelets in mediating blood-brain barrier dysfunction. PLoS One (2016) 11:e0151702. doi:10.1371/journal.pone.0151702
132. Laurence J, Elhadad S, Robison T, Terry H, Varshney R, Woolington S, et al. HIV protease inhibitor-induced cardiac dysfunction and fibrosis is mediated by platelet-derived TGF-β 1 and can be suppressed by exogenous carbon monoxide. PLoS One (2017) 12:e0187185. doi:10.1371/journal.pone.0187185
133. World Health Organization. Dengue: Guidelines for Diagnosis, Treatment, Prevention, and Control. Geneva: TDR WHO/HTM/NTD/DEN (2009).
134. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature (2013) 496:504–7. doi:10.1038/nature12060
135. Muller DA, Depelsenaire ACI, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis (2017) 215:S89–95. doi:10.1093/infdis/jiw649
136. Pang J, Thein T-L, Leo Y-S, Lye DC. Early clinical and laboratory risk factors of intensive care unit requirement during 2004–2008 dengue epidemics in Singapore: a matched case-control study. BMC Infect Dis (2014) 14:649. doi:10.1186/s12879-014-0649-2
137. Tomashek KM, Lorenzi OD, Andújar-Pérez DA, Torres-Velásquez BC, Hunsperger EA, Munoz-Jordan JL, et al. Clinical and epidemiologic characteristics of dengue and other etiologic agents among patients with acute febrile illness, Puerto Rico, 2012–2015. PLoS Negl Trop Dis (2017) 11:e0005859. doi:10.1371/journal.pntd.0005859
138. Krishnamurti C, Kalayanarooj S, Cutting MA, Peat RA, Rothwell SW, Reid TJ, et al. Mechanisms of hemorrhage in dengue without circulatory collapse. Am J Trop Med Hyg (2001) 65:840–7. doi:10.4269/ajtmh.2001.65.840
139. Mourão MP, Lacerda MV, Macedo VO, Santos JB. Thrombocytopenia in patients with dengue virus infection in the Brazilian Amazon. Platelets (2007) 18:605–12. doi:10.1080/09537100701426604
140. Bozza FA, Cruz OG, Zagne SM, Azeredo EL, Nogueira RM, Assis EF, et al. Multiplex cytokine profile from dengue patients: MIP-1beta and IFN-gamma as predictive factors for severity. BMC Infect Dis (2008) 8:86. doi:10.1186/1471-2334-8-86
141. Wills B, Tran VN, Nguyen TH, Truong TT, Tran TN, Nguyen MD, et al. Hemostatic changes in Vietnamese children with mild dengue correlate with the severity of vascular leakage rather than bleeding. Am J Trop Med Hyg (2009) 81:638–44. doi:10.4269/ajtmh.2009.08-0008
142. Huy NT, Van Giang T, Thuy DHD, Kikuchi M, Hien TT, Zamora J, et al. Factors associated with dengue shock syndrome: a systematic review and meta-analysis. PLoS Negl Trop Dis (2013) 7:e2412. doi:10.1371/journal.pntd.0002412
143. Pang T, Cardosa MJ, Guzman MG. Of cascades and perfect storms: the immunopathogenesis of dengue haemorrhagic fever-dengue shock syndrome (DHF/DSS). Immunol Cell Biol (2007) 85:43–5. doi:10.1038/sj.icb.7100008
144. Tramontini Gomes de Sousa Cardozo F, Baimukanova G, Lanteri MC, Keating SM, Moraes Ferreira F, Heitman J, et al. Serum from dengue virus-infected patients with and without plasma leakage differentially affects endothelial cells barrier function in vitro. PLoS One (2017) 12:e0178820. doi:10.1371/journal.pone.0178820
145. Malavige GN, Ogg GS. Pathogenesis of vascular leak in dengue virus infection. Immunology (2017) 151:261–9. doi:10.1111/imm.12748
146. Srikiatkhachorn A, Mathew A, Rothman AL. Immune-mediated cytokine storm and its role in severe dengue. Semin Immunopathol (2017) 39:563–74. doi:10.1007/s00281-017-0625-1
147. Mongkolsapaya J, Dejnirattisai W, Xu X, Vasanawathana S, Tangthawornchaikul N, Chairunsri A, et al. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat Med (2003) 9:921–7. doi:10.1038/nm887
148. Mathew A, Rothman AL. Understanding the contribution of cellular immunity to dengue disease pathogenesis. Immunol Rev (2008) 225:300–13. doi:10.1111/j.1600-065X.2008.00678.x
149. Puerta-Guardo H, Raya-Sandino A, Gonzalez-Mariscal L, Rosales VH, Ayala-Davila J, Chavez-Mungia B, et al. The cytokine response of U937-derived macrophages infected through antibody-dependent enhancement of dengue virus disrupts cell apical-junction complexes and increases vascular permeability. J Virol (2013) 87:7486–501. doi:10.1128/JVI.00085-13
150. Duyen HTL, Cerny D, Trung DT, Pang J, Velumani S, Toh YX, et al. Skin dendritic cell and T cell activation associated with dengue shock syndrome. Sci Rep (2017) 7:14224. doi:10.1038/s41598-017-14640-1
151. Schmid MA, Diamond MS, Harris E. Dendritic cells in dengue virus infection: targets of virus replication and mediators of immunity. Front Immunol (2014) 5:647. doi:10.3389/fimmu.2014.00647
152. Yong YK, Tan HY, Jen SH, Shankar EM, Natkunam SK, Sathar J, et al. Aberrant monocyte responses predict and characterize dengue virus infection in individuals with severe disease. J Transl Med (2017) 15:121. doi:10.1186/s12967-017-1226-4
153. Michels M, Alisjahbana B, de Groot PG, Indrati AR, Fijnheer R, Puspita M, et al. Platelet function alterations in dengue are associated with plasma leakage. Thromb Haemost (2014) 112:352–62. doi:10.1160/TH14-01-0056
154. Trugilho MRO, Hottz ED, Brunoro GVF, Teixeira-Ferreira A, Carvalho PC, Salazar GA, et al. Platelet proteome reveals novel pathways of platelet activation and platelet-mediated immunoregulation in dengue. PLoS Pathog (2017) 13:e1006385. doi:10.1371/journal.ppat.1006385
155. Alonzo MTG, Lacuesta TLV, Dimaano EM, Kurosu T, Suarez LAC, Mapua CA, et al. Platelet apoptosis and apoptotic platelet clearance by macrophages in secondary dengue virus infections. J Infect Dis (2012) 205:1321–9. doi:10.1093/infdis/jis180
156. Póvoa TF, Alves AMB, Oliveira CAB, Nuovo GJ, Chagas VLA, Paes MV. The pathology of severe dengue in multiple organs of human fatal cases: histopathology, ultrastructure and virus replication. PLoS One (2014) 9:e83386. doi:10.1371/journal.pone.0083386
157. Boonpucknavig S, Boonpucknavig V, Bhamarapravati N, Nimmannitya S. Immunofluorescence study of skin rash in patients with dengue hemorrhagic fever. Arch Pathol Lab Med (1979) 103:463–6.
158. Krishnamurti C, Peat RA, Cutting MA, Rothwell SW. Platelet adhesion to dengue-2 virus-infected endothelial cells. Am J Trop Med Hyg (2002) 66:435–41. doi:10.4269/ajtmh.2002.66.435
159. Tsai JJ, Jen YH, Chang JS, Hsiao HM, Noisakran S, Perng GC. Frequency alterations in key innate immune cell components in the peripheral blood of dengue patients detected by FACS analysis. J Innate Immun (2011) 3:530–40. doi:10.1159/000322904
160. Suharti C, van Gorp ECM, Setiati TE, Dolmans WMV, Djokomoeljanto RJ, Hack CE, et al. The role of cytokines in activation of coagulation and fibrinolysis in dengue shock syndrome. Thromb Haemost (2002) 87:42–6. doi:10.1055/s-0037-1612941
161. Chagan-Yasutan H, Lacuesta TL, Ndhlovu LC, Oguma S, Leano PSA, Telan EFO, et al. Elevated levels of full-length and thrombin-cleaved osteopontin during acute dengue virus infection are associated with coagulation abnormalities. Thromb Res (2014) 134:449–54. doi:10.1016/j.thromres.2014.05.003
162. Sosothikul D, Seksarn P, Pongsewalak S, Thisyakorn U, Lusher J. Activation of endothelial cells, coagulation and fibrinolysis in children with dengue virus infection. Thromb Haemost (2007) 97:627–34. doi:10.1160/TH06-02-0094
163. Yang KD, Wang CL, Shaio MF. Production of cytokines and platelet activating factor in secondary dengue virus infections. J Infect Dis (1995) 172:604–5. doi:10.1093/infdis/172.2.604
164. Jeewandara C, Gomes L, Wickramasinghe N, Gutowska-Owsiak D, Waithe D, Paranavitane SA, et al. Platelet activating factor contributes to vascular leak in acute dengue infection. PLoS Negl Trop Dis (2015) 9:e0003459. doi:10.1371/journal.pntd.0003459
165. Kamaladasa A, Gomes L, Jeewandara C, Shyamali NLA, Ogg GS, Malavige GN. Lipopolysaccharide acts synergistically with the dengue virus to induce monocyte production of platelet activating factor and other inflammatory mediators. Antiviral Res (2016) 133:183–90. doi:10.1016/j.antiviral.2016.07.016
166. Souza DG, Fagundes CT, Sousa LP, Amaral FA, Souza RS, Souza AL, et al. Essential role of platelet-activating factor receptor in the pathogenesis of dengue virus infection. Proc Natl Acad Sci U S A (2009) 106:14138–43. doi:10.1073/pnas.0906467106
167. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi:10.1182/blood-2011-03-343061
168. Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood (2011) 118:3708–14. doi:10.1182/blood-2011-01-332676
169. Nakahara M, Ito T, Kawahara KI, Yamamoto M, Nagasato T, Shrestha B, et al. Recombinant thrombomodulin protects mice against histone-induced lethal thromboembolism. PLoS One (2013) 8:e75961. doi:10.1371/journal.pone.0075961
170. Michels A, Albánez S, Mewburn J, Nesbitt K, Gould TJ, Liaw PC, et al. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J Thromb Haemost (2016) 14:2274–86. doi:10.1111/jth.13493
171. Mitrakul C, Poshyachinda M, Futrakul P, Sangkawibha N, Ahandrik S. Hemostatic and platelet kinetic studies in dengue hemorrhagic fever. Am J Trop Med Hyg (1977) 26:975–84. doi:10.4269/ajtmh.1977.26.975
172. Hottz E, Tolley ND, Zimmerman GA, Weyrich AS, Bozza FA. Platelets in dengue infection. Drug Discov Today Dis Mech (2011) 8:e33–8. doi:10.1016/j.ddmec.2011.09.001
173. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol (1995) 8:249–70. doi:10.1016/S0950-3536(05)80240-9
174. Nakao S, Lai CJ, Young NS. Dengue virus, a flavivirus, propagates in human bone marrow progenitors and hematopoietic cell lines. Blood (1989) 74:1235–40.
175. de Araújo JMG, Schatzmayr HG, de Filippis AMB, Dos Santos FB, Cardoso MA, Britto C, et al. A retrospective survey of dengue virus infection in fatal cases from an epidemic in Brazil. J Virol Methods (2009) 155:34–8. doi:10.1016/j.jviromet.2008.09.023
176. Murgue B, Cassar O, Guigon M, Chungue E. Dengue virus inhibits human hematopoietic progenitor growth in vitro. J Infect Dis (1997) 175:1497–501. doi:10.1086/516486
177. Basu A, Jain P, Gangodkar SV, Shetty S, Ghosh K. Dengue 2 virus inhibits in vitro megakaryocytic colony formation and induces apoptosis in thrombopoietin-inducible megakaryocytic differentiation from cord blood CD34+ cells. FEMS Immunol Med Microbiol (2008) 53:46–51. doi:10.1111/j.1574-695X.2008.00399.x
178. Young NS. Flaviviruses and bone marrow failure. JAMA (1990) 263:3065–8. doi:10.1001/jama.263.22.3065
179. Srichaikul T, Punyagupta S, Kanchanapoom T, Chanokovat C, Likittanasombat K, Leelasiri A. Hemophagocytic syndrome in dengue hemorrhagic fever with severe multiorgan complications. J Med Assoc Thai (2008) 91(1):104–9.
180. Bhamarapravati N, Tuchinda P, Boonyapaknavik V. Pathology of Thailand haemorrhagic fever: a study of 100 autopsy cases. Ann Trop Med Parasitol (1967) 61:500–10. doi:10.1080/00034983.1967.11686519
181. Na-Nakorn S, Suingdumrong A, Pootrakul S, Bhamarapravati N. Bone-marrow studies in Thai haemorrhagic fever. Bull World Health Organ (1966) 35:54–5.
182. Sridharan A, Chen Q, Tang KF, Ooi EE, Hibberd ML, Chen J. Inhibition of megakaryocyte development in the bone marrow underlies dengue virus-induced thrombocytopenia in humanized mice. J Virol (2013) 87:11648–58. doi:10.1128/JVI.01156-13
183. Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med (2008) 359:1261–70. doi:10.1056/NEJMra0800887
184. Ho-Tin-Noé B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost (2011) 9:56–65. doi:10.1111/j.1538-7836.2011.04317.x
185. Haselton FR, Alexander JS. Platelets and a platelet-released factor enhance endothelial barrier. Am J Physiol (1992) 263:L670–8. doi:10.1152/ajplung.1992.263.6.L670
186. Patil S, Kaplan JE, Minnear FL. Protein, not adenosine or adenine nucleotides, mediates platelet decrease in endothelial permeability. Am J Physiol (1997) 273:H2304–11.
187. Lo SK, Burhop KE, Kaplan JE, Malik AB. Role of platelets in maintenance of pulmonary vascular permeability to protein. Am J Physiol (1988) 254:H763–71. doi:10.1152/ajpheart.1988.254.4.H763
188. Alexander JS, Patton WF, Christman BW, Cuiper LL, Haselton FR. Platelet-derived lysophosphatidic acid decreases endothelial permeability in vitro. Am J Physiol (1998) 274:H115–22.
189. Mammoto T, Jiang A, Jiang E, Mammoto A. Platelet-rich plasma extract prevents pulmonary edema through angiopoietin-Tie2 signaling. Am J Respir Cell Mol Biol (2015) 52:56–64. doi:10.1165/rcmb.2014-0076OC
190. Ashton AW, Yokota R, John G, Zhao S, Suadicani SO, Spray DC, et al. Inhibition of endothelial cell migration, intercellular communication, and vascular tube formation by thromboxane A(2). J Biol Chem (1999) 274:35562–70. doi:10.1074/jbc.274.50.35562
191. Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med (2000) 6:460–3. doi:10.1038/74725
192. Li JJ, Huang YQ, Basch R, Karpatkin S. Thrombin induces the release of angiopoietin-1 from platelets. Thromb Haemost (2001) 85:204–6. doi:10.1055/s-0037-1615677
193. Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest (2009) 119:1871–9. doi:10.1172/JCI38575DS1
194. Michels M, Van Der Ven AJAM, Djamiatun K, Fijnheer R, De Groot PG, Griffioen AW, et al. Imbalance of angiopoietin-1 and angiopoetin-2 in severe dengue and relationship with thrombocytopenia, endothelial activation, and vascular stability. Am J Trop Med Hyg (2012) 87:943–6. doi:10.4269/ajtmh.2012.12-0020
195. Preeyasombat C, Treepongkaruna S, Sriphrapradang A, Choubtum L. The role of prostacyclin (PGI2) and thromboxane A2 (TXA2) in pathogenesis of dengue hemorrhagic fever (DHF). J Med Assoc Thai (1999) 82:S16–21.
196. Gomes L, Fernando S, Fernando RH, Wickramasinghe N, Shyamali NLA, Ogg GS, et al. Sphingosine 1-phosphate in acute dengue infection. PLoS One (2014) 9:e113394. doi:10.1371/journal.pone.0113394
197. Michels M, Japtok L, Alisjahbana B, Wisaksana R, Sumardi U, Puspita M, et al. Decreased plasma levels of the endothelial protective sphingosine-1-phosphate are associated with dengue-induced plasma leakage. J Infect (2015) 71:480–7. doi:10.1016/j.jinf.2015.06.014
198. Matsuura C, Moraes TL, Barbosa JB, Moss MB, Siqueira MAS, Mann GE, et al. Nitric oxide activity in platelets of dengue haemorrhagic fever patients: the apparent paradoxical role of ADMA and l-NMMA. Trans R Soc Trop Med Hyg (2012) 106:174–9. doi:10.1016/j.trstmh.2011.10.009
199. Mendes-Ribeiro AC, Moss MB, Siqueira MAS, Moraes TL, Ellory JC, Mann GE, et al. Dengue fever activates the L-arginine-nitric oxide pathway: an explanation for reduced aggregation of human platelets. Clin Exp Pharmacol Physiol (2008) 35:1143–6. doi:10.1111/j.1440-1681.2008.04970.x
200. Jaiyen Y, Masrinoul P, Kalayanarooj S, Pulmanausahakul R, Ubol S. Characteristics of dengue virus-infected peripheral blood mononuclear cell death that correlates with the severity of illness. Microbiol Immunol (2009) 53:442–50. doi:10.1111/j.1348-0421.2009.00148.x
201. Sa-Ngasang A, Ohashi J, Naka I, Anantapreecha S, Sawanpanyalert P, Patarapotikul J. Association of IL1B-31C/T and IL1RA variable number of an 86-bp tandem repeat with dengue shock syndrome in Thailand. J Infect Dis (2014) 210:138–45. doi:10.1093/infdis/jiu042
202. Thein TL, Wong J, Leo YS, Ooi EE, Lye D, Yeo TW. Association between increased vascular nitric oxide bioavailability and progression to dengue hemorrhagic fever in adults. J Infect Dis (2015) 212:711–4. doi:10.1093/infdis/jiv122
203. Yacoub S, Lam PK, Huynh TT, Nguyen Ho HH, Dong Thi HT, Van Thu N, et al. Endothelial nitric oxide pathways in the pathophysiology of dengue: a prospective observational study. Clin Infect Dis (2017) 65:1453–61. doi:10.1093/cid/cix567
204. van de Weg CAM, Pannuti CS, de Araújo ESA, van den Ham H-J, Andeweg AC, Boas LSV, et al. Microbial translocation is associated with extensive immune activation in dengue virus infected patients with severe disease. PLoS Negl Trop Dis (2013) 7:e2236. doi:10.1371/journal.pntd.0002236
205. Onlamoon N, Noisakran S, Hsiao H, Duncan A, Villinger F, Ansari AA. Dengue virus-induced hemorrhage in a nonhuman primate model. Blood (2010) 115:1823–34. doi:10.1182/blood-2009-09-242990.The
206. Dixon DA, Tolley ND, Bemis-Standoli K, Martinez ML, Weyrich AS, Morrow JD, et al. Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J Clin Invest (2006) 116:2727–38. doi:10.1172/JCI27209
207. Mahoney TS, Weyrich AS, Dixon DA, McIntyre T, Prescott SM, Zimmerman GA. Cell adhesion regulates gene expression at translational checkpoints in human myeloid leukocytes. Proc Natl Acad Sci U S A (2001) 98:10284–9. doi:10.1073/pnas.181201398
208. Lee Y-R, Liu M-T, Lei H-Y, Liu C-C, Wu J-M, Tung Y-C, et al. MCP-1, a highly expressed chemokine in dengue haemorrhagic fever/dengue shock syndrome patients, may cause permeability change, possibly through reduced tight junctions of vascular endothelium cells. J Gen Virol (2006) 87:3623–30. doi:10.1099/vir.0.82093-0
209. Her Z, Kam Y-W, Gan VC, Lee B, Thein T-L, Tan JJL, et al. Severity of plasma leakage is associated with high levels of interferon γ-inducible protein 10, hepatocyte growth factor, matrix metalloproteinase 2 (MMP-2), and MMP-9 during dengue virus infection. J Infect Dis (2017) 215:42–51. doi:10.1093/infdis/jiw494
210. Kubelka CF, Azeredo EL, Gandini M, Oliveira-Pinto LM, Santos Barbosa L, Damasco PV, et al. Metalloproteinases are produced during dengue fever and MMP9 is associated with severity. J Infect (2010) 61:501–5. doi:10.1016/j.jinf.2010.09.020
211. Luplerdlop N, Missé D, Bray D, Deleuze V, Gonzalez J-P, Leardkamolkarn V, et al. Dengue-virus-infected dendritic cells trigger vascular leakage through metalloproteinase overproduction. EMBO Rep (2006) 7:1176–81. doi:10.1038/sj.embor.7400814
212. Lin C-K, Tseng C-K, Wu Y-H, Liaw C-C, Lin C-Y, Huang C-H, et al. Cyclooxygenase-2 facilitates dengue virus replication and serves as a potential target for developing antiviral agents. Sci Rep (2017) 7:44701. doi:10.1038/srep44701
213. Wu W-L, Ho L-J, Chang D-M, Chen C-H, Lai J-H. Triggering of DC migration by dengue virus stimulation of COX-2-dependent signaling cascades in vitro highlights the significance of these cascades beyond inflammation. Eur J Immunol (2009) 39:3413–22. doi:10.1002/eji.200939306
214. Chapman LM, Aggrey AA, Field DJ, Srivastava K, Ture S, Yui K, et al. Platelets present antigen in the context of MHC class I. J Immunol (2012) 189:916–23. doi:10.4049/jimmunol.1200580
215. Olsson B, Andersson P-O, Jernås M, Jacobsson S, Carlsson B, Carlsson LMS, et al. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat Med (2003) 9:1123–4. doi:10.1038/nm921
216. Chow L, Aslam R, Speck ER, Kim M, Cridland N, Webster ML, et al. A murine model of severe immune thrombocytopenia is induced by antibody- and CD8+ T cell-mediated responses that are differentially sensitive to therapy. Blood (2010) 115:1247–53. doi:10.1182/blood-2009-09-244772
217. Arthur CM, Patel SR, Sullivan HC, Winkler AM, Tormey CA, Hendrickson JE, et al. CD8+ T cells mediate antibody-independent platelet clearance in mice. Blood (2016) 127:1823–7. doi:10.1182/blood-2015-10-673426
218. Zhou H, Qiu JH, Wang T, Yu YY, Liu XN, Li X, et al. Interleukin 27 inhibits cytotoxic T-lymphocyte-mediated platelet destruction in primary immune thrombocytopenia. Blood (2014) 124:3316–9. doi:10.1182/blood-2014-06-580084
219. Panitsas FP, Theodoropoulou M, Kouraklis A, Karakantza M, Theodorou GL, Zoumbos NC, et al. Adult chronic idiopathic thrombocytopenic purpura (ITP) is the manifestation of a type-1 polarized immune response. Blood (2004) 103:2645–7. doi:10.1182/blood-2003-07-2268
220. Dominguez-Cherit G, De la Torre A, Rishu A, Pinto R, Ñamendys-Silva SA, Camacho-Ortiz A, et al. Influenza A (H1N1pdm09)-related critical illness and mortality in Mexico and Canada, 2014. Crit Care Med (2016) 44:1861–70. doi:10.1097/CCM.0000000000001830
221. Bonmarin I, Belchior E, Bergounioux J, Brun-Buisson C, Mégarbane B, Chappert JL, et al. Intensive care unit surveillance of influenza infection in France: the 2009/10 pandemic and the three subsequent seasons. Euro Surveill (2015) 20:30066. doi:10.2807/1560-7917.ES.2015.20.46.30066
222. Kumar A, Zarychanski R, Pinto R, Cook DJ, Marshall J, Lacroix J, et al. Critically Ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA (2009) 302:1872–9. doi:10.1001/jama.2009.1496
223. De Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJD, Chau TNB, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med (2006) 12:1203–7. doi:10.1038/nm1477
224. Short KR, Kroeze EJBV, Fouchier RAM, Kuiken T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis (2014) 14:57–69. doi:10.1016/S1473-3099(13)70286-X
225. Kuiken T, Riteau B, Fouchier RAM, Rimmelzwaan GF. Pathogenesis of influenza virus infections: the good, the bad and the ugly. Curr Opin Virol (2012) 2:276–86. doi:10.1016/j.coviro.2012.02.013
226. Harms PW, Schmidt LA, Smith LB, Newton DW, Pletneva MA, Walters LL, et al. Autopsy findings in eight patients with fatal H1N1 influenza. Am J Clin Pathol (2010) 134:27–35. doi:10.1309/AJCP35KOZSAVNQZW
227. Lê VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med (2015) 191:804–19. doi:10.1164/rccm.201406-1031OC
228. Ashar HK, Mueller NC, Rudd JM, Snider TA, Achanta M, Prasanthi M, et al. The role of extracellular histones in influenza virus pathogenesis. Am J Pathol (2018) 188(1):135–148. doi:10.1016/j.ajpath.2017.09.014
229. Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES, et al. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1). Chest (2012) 141:1490–5. doi:10.1378/chest.11-2860
230. Passacquale G, Vamadevan P, Pereira L, Hamid C, Corrigall V, Ferro A. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS One (2011) 6:e25595. doi:10.1371/journal.pone.0025595
231. Lanza GA, Barone L, Scalone G, Pitocco D, Sgueglia GA, Mollo R, et al. Inflammation-related effects of adjuvant influenza A vaccination on platelet activation and cardiac autonomic function. J Intern Med (2011) 269:118–25. doi:10.1111/j.1365-2796.2010.02285.x
232. Layne K, Di Giosia P, Ferro A, Passacquale G. Anti-platelet drugs attenuate the expansion of circulating CD14highCD16+monocytes under pro-inflammatory conditions. Cardiovasc Res (2016) 111:26–33. doi:10.1093/cvr/cvw089
233. Sugiyama M, Gamage A, Zyla R, Armstrong SM, Advani S, Advani A, et al. Influenza infection induces platelet-endothelial adhesion which contributes to lung injury. J Virol (2015) 90:1812–23. doi:10.1128/jvi.02599-15
234. Wygrecka M, Kosanovic D, Wujak L, Reppe K, Henneke I, Frey H, et al. Antihistone properties of C1 esterase inhibitor protect against lung injury. Am J Respir Crit Care Med (2017) 196:186–99. doi:10.1164/rccm.201604-0712OC
Keywords: platelet, HIV infection, dengue, influenza, immunology and virology, viruses
Citation: Hottz ED, Bozza FA and Bozza PT (2018) Platelets in Immune Response to Virus and Immunopathology of Viral Infections. Front. Med. 5:121. doi: 10.3389/fmed.2018.00121
Received: 02 February 2018; Accepted: 12 April 2018;
Published: 30 April 2018
Edited by:
Ana Kasirer-Friede, University of California, San Diego, United StatesReviewed by:
Olivier Garraud, Institut National de la Transfusion Sanguine, FranceCopyright: © 2018 Hottz, Bozza and Bozza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eugenio D. Hottz, ZXVnZW5pb2hvdHR6QGdtYWlsLmNvbQ==;
Patrícia T. Bozza, cGJvenphQGlvYy5maW9jcnV6LmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.