 Yun Freudenberg-Hua
Yun Freudenberg-Hua Wentian Li
Wentian Li Peter Davies1
Peter Davies1
- 1Litwin-Zucker Center for the study of Alzheimer’s Disease, The Feinstein Institute for Medical Research, Northwell Health, Manhasset, NY, United States
- 2Division of Geriatric Psychiatry, Zucker Hillside Hospital, Northwell Health, Glen Oaks, NY, United States
- 3Robert S Boas Center for Genomics and Human Genetics, The Feinstein Institute for Medical Research, Northwell Health, Manhasset, NY, United States
Alzheimer’s disease (AD) is the most common type of dementia, which has a substantial genetic component. AD affects predominantly older people. Accordingly, the prevalence of dementia has been rising as the population ages. To date, there are no effective interventions that can cure or halt the progression of AD. The only available treatments are the management of certain symptoms and consequences of dementia. The current state-of-the-art medical care for AD comprises three simple principles: prevent the preventable, achieve early diagnosis, and manage the manageable symptoms. This review provides a summary of the current state of knowledge of risk factors for AD, biological diagnostic testing, and prospects for treatment. Special emphasis is given to recent advances in genetics of AD and the way genomic data may support prevention, early intervention, and development of effective pharmacological treatments. Mutations in the APP, PSEN1, and PSEN2 genes cause early onset Alzheimer’s disease (EOAD) that follows a Mendelian inheritance pattern. For late onset Alzheimer’s disease (LOAD), APOE4 was identified as a major risk allele more than two decades ago. Population-based genome-wide association studies of late onset AD have now additionally identified common variants at roughly 30 genetic loci. Furthermore, rare variants (allele frequency <1%) that influence the risk for LOAD have been identified in several genes. These genetic advances have broadened our insights into the biological underpinnings of AD. Moreover, the known genetic risk variants could be used to identify presymptomatic individuals at risk for AD and support diagnostic assessment of symptomatic subjects. Genetic knowledge may also facilitate precision medicine. The goal of precision medicine is to use biological knowledge and other health information to predict individual disease risk, understand disease etiology, identify disease subcategories, improve diagnosis, and provide personalized treatment strategies. We discuss the potential role of genetics in advancing precision medicine for AD along with its ethical challenges. We outline strategies to implement genomics into translational clinical research that will not only improve accuracy of dementia diagnosis, thus enabling more personalized treatment strategies, but may also speed up the discovery of novel drugs and interventions.
Introduction
Alzheimer’s disease (AD) is the most common form of dementia (1) accounting for 60–80% of dementia diagnosis and affects nearly 50 million people worldwide (2). The worldwide number of affected individuals is expected to reach 66 million by 2030, and 131 million by 2050 (3) as the number of older adults increases. One in 10 people over age 65 and every third person over age 85 in the US has a diagnosis of AD (4). The global financial toll of dementia was estimated to be 818 billion US dollars in 2015, an increase of 35% since 2010 and this cost is expected to further rise together with the prevalence of AD (2). The majority of the costs are related to family and social care of patients, rather than medical care. About 5% of all AD patients show cognitive symptoms before age 65 and are classified as early onset Alzheimer’s disease (EOAD) (5). Patients showing clinical symptoms after age 65 are classified as having late onset Alzheimer’s disease (LOAD). Here, we provide a summary of the clinical, neuropathological, fluid, and imaging biomarkers of AD along with a more comprehensive review of genetic findings in both Mendelian and sporadic forms of AD. We discuss how genetic analysis as applied in Mendelian randomization (MR) may be helpful in validating causality of modifiable risk factors that could advance preventive measures. Moreover, genetic data may be useful to facilitate precision medicine. The goal of precision medicine is to integrate clinical, genetic, and life style data to enable clinicians to efficiently and accurately predict the most appropriate course of action for a patient (6). We emphasize the ways genetics may facilitate precision medicine in AD: (1) identifying at risk individuals through risk prediction, (2) improving diagnostic precision, and (3) expediting the discovery of targetable disease mechanisms for drug development. Due to the large number of published articles in biomedical research of AD, we refer to more recent comprehensive reviews written by domain experts and supplement these with other findings.
Literature Selection
Our goal of writing this narrative review (7) is to discuss how genetics may not only advance basic research on disease mechanisms but also play a role in facilitating precision medicine in AD. We provide summaries about clinical and neuropathological features, research on imaging and fluid biomarkers, as well as modifiable risk factors of AD by referring to high-quality recent systematic reviews and meta-analyses. Unpublished or original data, submitted manuscripts, or personal communications are excluded. More recent scientifically rigorous and high-impact studies on these topics that were found in the PubMed database, but not previously reviewed and those having a historical impact were also included. Over the past 20 years, our understanding about genetic research has expanded together with the rapidly advancing technology. The quality requirement for genetic studies has also evolved from candidate gene approaches, which were often criticized for producing inconsistent and non-replicable results (8), to more thoroughly conducted and well-powered genome-wide studies (9). We included publications of the Mendelian AD genes as well as publications that were referred and curated by the National Human Genome Research Institute-European Bioinformatics Institute (NHGRI-EBI) Catalog of published genome-wide association studies (GWAS Catalog) (10). In addition, we included high-quality association studies reporting rare variants that meet the “analytically rigorous” criteria for GWAS (9) or are otherwise statistically thorough.
Clinical Features of AD
In 1906, the German psychiatrist Alois Alzheimer first described the clinical features of an early-onset case of AD with its pathognomonic hallmarks—extracellular amyloid (neuritic) plaques and intracellular neurofibrillary tangles (NFT) (11). Patients typically show an insidious onset and continuous cognitive decline, which typically starts with an amnestic presentation with impaired ability to remember new information. The cognitive decline may further affect language, reasoning, executive function, visuospatial abilities, and the illness is often accompanied by personality and behavioral changes that affect the social function of the patient. In an advanced disease stage, patients are completely dependent on their caregivers for daily functioning such as getting dressed, toileting, mobility, and eating. The NINCDS-ADRDA criteria for diagnosing possible and probable AD are being widely used (12) and have a sensitivity and specificity of ~70% for distinguishing between AD patients and people without dementia. However, they were less accurate distinguishing between different types of dementias (13, 14). The median survival time of patients from the symptom onset is reported to be 9 years (15).
Neuropathology of AD
Over many years, definitive diagnosis of AD could only be made by the “gold standard” of postmortem neuropathological examination, using a combination of CERAD score for neuritic plaques containing amyloid beta (Aβ) (16) together with Braak staging of NFT consisting of abnormally hyperphosphorylated tau (17). This had been defined in the National Institute on Aging (NIA)-Reagan Criteria (18). However, only half of the brains of patients with the clinical diagnosis of probable AD showed “pure” AD pathology (19). In 2011, the NIA and the Alzheimer’s Association (AA) revised the diagnostic criteria aimed at integrating the advances of imaging and cerebrospinal fluid (CSF) biomarkers to model the three stages of AD that include preclinical stage, mild cognitive impairment, and dementia (12, 20–22). The updated criteria are now used in AD research and ongoing efforts exist to refine these criteria (23). It is important to emphasize that Aβ deposits have not been proven to be causal for late onset AD. In addition to Aβ and NFT, other neuropathological features such as TDP-43 immunoreactive inclusions and Lewy bodies may coexist, along with findings like cerebral amyloid angiopathy, cerebrovascular disease, and hippocampal sclerosis (19, 24–27). It is important to note that AD pathologies were also found in nearly all brain autopsies of cognitively normal individuals above age 80, even among those considered as high-cognitive performers (28, 29). Although some cognitively normal elderly had severe AD pathologies, as a group, they showed less severe AD pathologies than dementia patients. Signs of vascular injuries ranged from 32% among high cognitive performers to 64% in late dementia subjects.
Imaging and Biomarkers
To provide early and accurate diagnosis of AD, extensive efforts have been made into developing sophisticated methods to assess pathology in the living human brain. However, to date, no test or combination of tests that could accurately diagnose AD is available for broad clinical use outside of AD research centers (4). CSF levels of Aβ42, tau, and hyperphosphorylated tau (ptau) as markers for amyloid, neuronal injury, and tangles, respectively, have been the main fluid biomarkers used in AD research (30, 31). In CSF of AD patients, a decreased level of Aβ42 has been consistently found (32), whereas the concentrations of tau and ptau are increased (31). Levels of CSF tau and ptau, but not Aβ42, were found to correlate with brain atrophy in AD (33). Interestingly, a reduction of CSF Aβ42 had been shown to correlate with brain atrophy in non-demented elderly indicating a potential preclinical stage (33).
Unaddressed problems preventing broad clinical utility of biomarkers include incomplete clinical validity, inconsistent predictive value, and assay variability (34). The consensus from experts in the field of biomarkers concludes that CSF AD biomarkers may be used alongside clinical measures to identify or exclude AD as an underlying cause particularly in uncertain and atypical clinical presentations (35).
In parallel to CSF biomarkers, major advances were made to measure Aβ and tau deposits in vivo with help of brain imaging. Using a combination of 18-fluoro-deoxyglucose-positron emission tomography (PET), which measures cerebral glucose metabolism, and Pittsburgh compound B (PIB) PET measuring the Aβ deposition along with CSF biomarkers, it was demonstrated that subjects with known Mendelian AD mutations have CSF Aβ changes, brain amyloidosis, tauopathy, brain atrophy, and decreased glucose metabolism in that same temporal order starting 20 years before the clinical onset of AD (36). More recently, voxel-based hierarchical clustering was applied to cross-sectional flortaucipir PET imaging for ptau and PIB–PET for Aβ in 88 elderly cognitively normal individuals (37). The study identified four tau clusters and four Aβ clusters based on spatial features. It shows that tau clusters map to the temporal lobe and orbitofrontal cortex and expand to parietal and frontal lobes roughly corresponding to Braak tau stages (38), whereas Aβ deposits are dispersed in widespread heteromodal cortex. The finding that tau and Aβ deposits displayed distinguishable locations with some overlap, particularly in the association cortex, suggested that AD is a tau-centered disease with amyloid effects.
Risk Factors for AD
Currently known risk factors for AD include age, sex, cardiovascular risk factors, metabolic risk factors, sleep apnea, family history, and certain genetic variants (2, 4). Thus, both modifiable and non-modifiable risk factors have been associated with LOAD risk. The non-modifiable factors include sex, aging, and the genetic risk.
Genetics of Autosomal Dominant AD
A recent systematic review of studies from the US, Europe, India, and China shows that the worldwide proportion of EOAD is around 5% of all AD cases (39). Of note, only 30–60% of EOAD patients have a positive family history for dementia, and about 10–14% have a family history that is consistent with autosomal dominant inheritance (40–42). Thus, in addition to the Mendelian disease presentation of EOAD, a substantial proportion of EOAD cases fall into the category of sporadic and genetically complex disease. For the Mendelian cases, three genes that carry mutations causal for autosomal dominant AD were identified in the 1990s, namely APP (43), PSEN1 (44), and PSEN2 (45, 46). The APP gene encodes amyloid precursor protein which is proteolytically processed into Aβ peptides by β- and γ-secretase. Most pathogenic mutations in APP have been reported to either increase Aβ production or influence the ratio of Aβ peptides of different length (e.g., the Aβ42/Aβ40 ratio) resulting in increased self-aggregation (47). Notably, at the same site of a disease causing APP mutation that increases APP cleavage, a protective variant leading to a different amino acid change was found that decreases APP cleavage (48). PSEN1 and PSEN2 genes encode part of the γ-secretase complex and PSEN1 accounts for most of the known mutations for autosomal dominant AD. The majority of pathogenic PSEN1 mutations impair γ-secretase-dependent cleavage of APP and decrease the production of both Aβ42 and Aβ40 (49). These genetic findings in autosomal dominantly inherited EOAD (48, 50) provide strong support for the amyloid hypothesis implicating that Aβ plays an initiating role in AD. A recent review presented a large body of evidence from over 25 years of research supporting the generalizability of amyloid hypothesis (51). However, there are also findings that contradict amyloid being the main driving cause for the more common sporadic manifestations of AD (52). For example, elevated amyloid deposition is frequently found in cognitively normal subjects (28, 53–55) and CSF level of Aβ and Aβ imaging with PIB–PET do not correlate with cognitive decline (56). Furthermore, Aβ production is reduced by most PSEN1 mutations (49). The anatomic and temporal discordance between Aβ pathology, tau aggregation, and neurodegeneration has led to the postulation of Aβ being an initiator of a complex cascade that ends in tau-medicated neurodegeneration (57).
Genetics of LOAD
For the majority of AD patients, no known causal genetic mutations have been identified. LOAD as well as many cases of EOAD are genetically complex and have multifactorial causes, which is similar to other chronic common diseases. A large population-based twin study estimated that genetic factors contribute 58–79% of etiologic role for LOAD (58). More than 20 years ago, APOE4 (also called APOE ε4) allele of the APOE gene has been identified as a major genetic risk factor for LOAD (59, 60). The APOE gene has two missense variants at amino acid residues 112 and 158 leading to three common haplotypes, which are typically referred to as APOE alleles ε2 (Cys and Cys), ε3 (Cys and Arg), and ε4 (Arg and Arg). Among Caucasians, homozygous ε4 carriers show the highest life time risk for AD (68–91%) (61–64) with an odds ratio (OR) of 11–12.9 compared with homozygous ε3 carriers. Individuals carrying one copy of ε4 have a threefold risk increase for AD compared with people having no ε4 allele, and the ε2 allele is protective against AD (Figure 1). In African-Americans and Hispanic populations the OR of APOE4 is found to be less pronounced compared to Caucasians. It is important to note that unlike the mutations in autosomal dominant forms of AD, APOE4 is not a sufficient determinant of AD even in old aged individuals. We have previously reported a homozygous APOE4 carrier who reached the age of 95 years without overt signs of dementia (65).
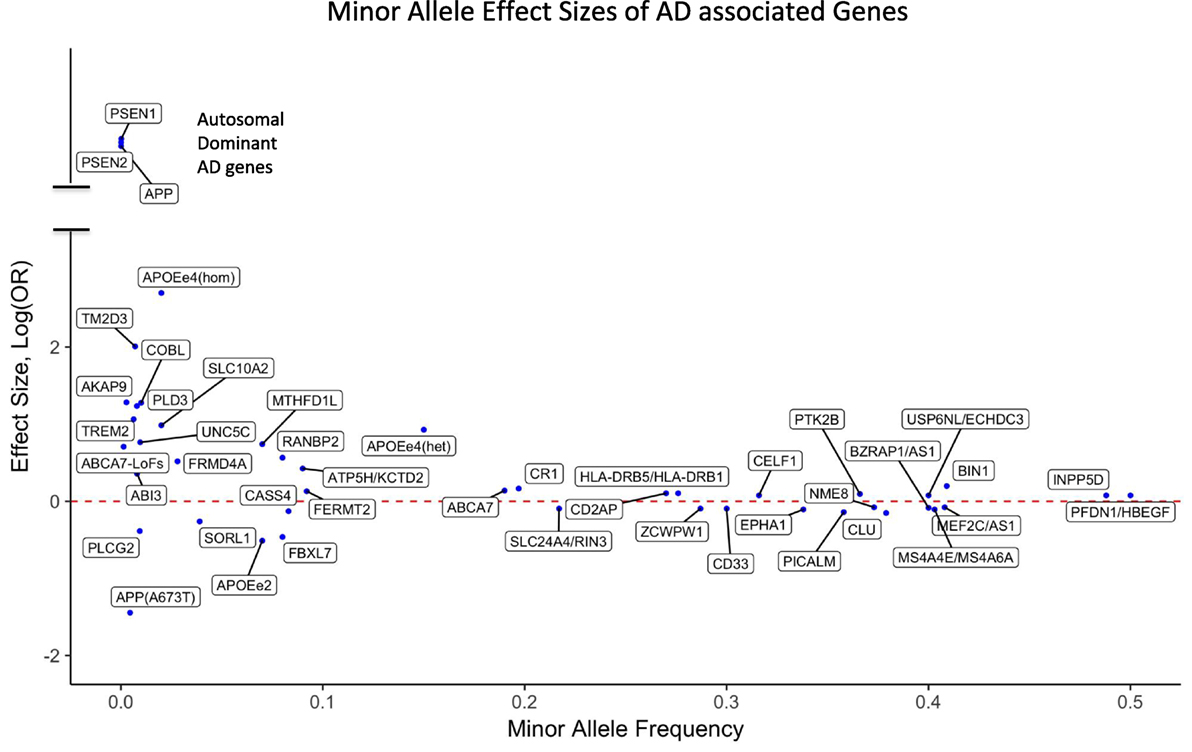
Figure 1. Effect sizes of AD associated variants for the respective minor alleles. The red dotted line indicates OR = 1 [log(OR) = 0]. Minor alleles with log(OR) above the line are risk alleles and below the line are protective. Abbreviations: APOEe4(hom), homozygosity for the APOE4 allele; APOEe4(het), heterozygosity for the APOE4 allele; ABCA7-LoFs, aggregated effects of loss-of-function variants in ABCA7; OR, odds ratio; AD, Alzheimer’s disease.
APOE encodes a lipid carrier Apolipoprotein E (ApoE) that is found both in the periphery and the central nervous system (66). The risk effects of APOE4 in AD were linked to ApoE’s pleiotropic functions that lead to reduced cholesterol transport, less efficient Aβ clearance and more aggregation, triggering neurotoxicity through Tau phosphorylation, increased brain neuronal activity and atrophy, reduced synaptic plasticity, and greater neuroinflammation. The large body of literature investigating the functional mechanism of ApoE in AD has been recently summarized (67–69). Most recently, ApoE has been shown to affect tau pathogenesis, neuroinflammation, and tau-mediated neurodegeneration independently of amyloid-β pathology in transgenic mice (70).
In addition to the well-established effects of APOE, GWAS have identified more than 30 genomic loci that are associated with AD risk. Unlike the APOE variants, the majorities of GWAS identified risk variants do not alter the protein sequence and are not necessarily the actual causal variants. Instead, an associated variant may be in linkage disequilibrium with an unidentified causal variant that may alter protein sequence, splicing patterns, or gene expression. In GWAS for LOAD, genes that are located near the associated variants are considered potential risk genes, but further evidences are necessary to support their actual etiological role. As of September 1, 2017, the NHGRI-EBI GWAS Catalog (10) listed 74 published GWAS studies on LOAD. We manually curated this list by merging multiple reports for the same locus into one row (Table 1). It is clear that some gene loci have been replicated by two or more GWAS or meta-analysis. These genes are BIN1, CD2AP, CLU, CR1, EPHA1, MS4A4E/MS4A6A, PICALM, and TREM2. The confidence for these genes to be actual AD genes is higher compared with those genes supported by a distant variant in one single study. For example, one association signal on Chromosome 2 was supported by an intergenic variant rs17034806 that is located 200 kb from the gene RANBP2 (71). In Table 1, if a locus is implicated in more than one association study or is supported by meta-analysis, we show the strongest association signal.
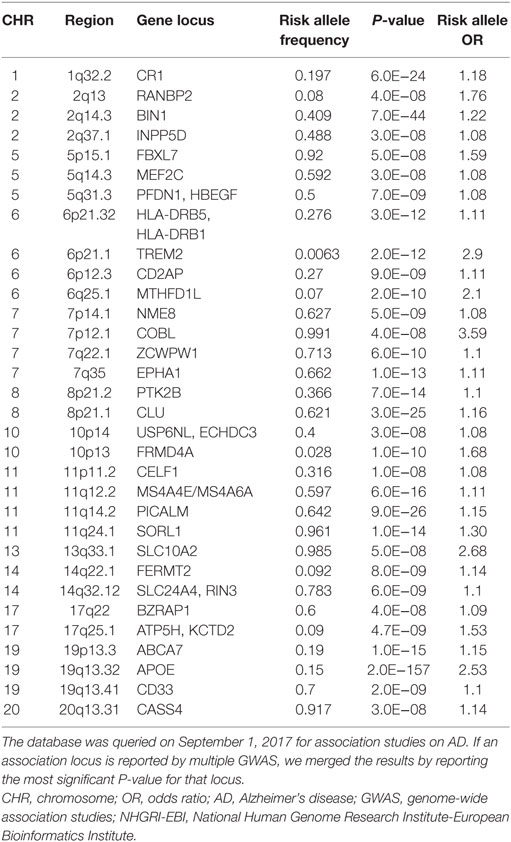
Table 1. AD associated loci from the NHGRI-EBI GWAS Catalog.
Although GWAS have been a powerful method to uncover risk loci in AD, they are less suitable to discover infrequent or rare variants. A recent estimate indicates that only 30.6% of the genetic variance can be explained by known AD single-nucleotide polymorphisms (SNPs), but a sizeable fraction of the unidentified risk variants may be located close to the known risk SNPs, potentially as rare variants (72). Consistent with an important role of rare variants, our investigation using whole genome sequencing (WGS) showed an increased burden of rare loss-of-function variants in immune genes in AD compared with cognitively healthy centenarians (73). Large-scale sequencing, such as whole exome sequencing (WES) and WGS, has already identified new genes that harbor rare variants typically missed by GWAS. Rare variants that increase the risk for AD have been identified in TREM2 (74, 75), PLD3 (76, 77), UNC5C (78), AKAP9 (79), ADAM10 (80), and ABI3 (81). Moreover, the burden of rare coding variants in risk genes identified by GWAS such as ABCA7 (82–84) as well as in Mendelian genes for AD had been found to be increased among LOAD patients compared with unaffected general population (85, 86). The potential impact of rare variants in AD is further underscored by rare and low-frequency protective variants such as APOE2 allele (61, 67), APP A673T (48), and PLGC2 P522R (81). The effect sizes of both GWAS loci and genes harboring reported rare AD-associated variants are presented in Figure 1.
Undoubtedly, the search for rare risk variants with high-effect sizes for LOAD faces many obstacles. First, many studies are underpowered to separate true signals from false-positive associations as tens of thousands of cases and controls are usually required to achieve genome-wide significance level of P < 5E−8. Second, allele frequencies of rare variants are more likely to vary between population cohorts of different ethnic backgrounds due to founder effects, making replication studies difficult to conduct. For example, risk allele frequencies in PLD3 in controls of one cohort may be higher than that of cases in another cohort, while combined result may be nominally significant (77) or not significant at all (87–89). Third, the necessarily small number of carriers of rare variants makes the respective association studies particularly prone to be impacted by factors such as age, APOE4 carrier status, and different genotyping and sequencing platforms.
Pathways Implicated by Risk Genes for AD
The established AD associated genes exert pleiotropic functions across many molecular pathways. Several of these pathways stand out by providing insights for the disease mechanisms that may play a role in the etiology of AD (90–92). Major pathways include inflammatory response (ABCA7, CD33, CLU, CR1, MS4A, INPP5D, TREM2, PLCG2, PTK2B, and ABI3), lipid metabolism (APOE, CLU, ABCA7, and PLCG2), as well as endocytosis/vesicle-mediated transport (BIN1, PICALM, CD2AP, EPHA1, and SORL1). Other functional categories include regulation of cell cycle (RANBP2), oxidative stress response (MEF2C), and axon guidance (UNC5C).
A role of innate immunity and inflammation in AD etiology is independently supported by a large body of functional evidence (93–95). Among the risk genes from the immune pathways, TREM2 stands out with its high effect-size of AD risk (74, 75). TREM2 stands for triggering receptor expressed on myeloid cells 2, a single-transmembrane protein expressed by monocytic myeloid cells. Both ApoE and Clusterin (encoded by CLU) are extracellular chaperons that prevent protein aggregation. In addition, both bind to the microglial receptor TREM2 and thus may promote uptake of Aβ by microglia (96). Studies on animal and human brains indicated that the TREM2 risk variant p.R47H impairs TREM2 detection of lipid ligands leading to microglia dysfunction (97, 98). In addition to TREM2, the two newly identified AD risk genes ABI3 and PLCG2 are highly expressed in microglia as well (81).
The abundance of genomics data in the public domain can be utilized not only to confirm the known connections among AD genes but also to reveal potentially new genes involved in the disease. Figure 2 shows an example of a network representation of AD genes by the GeneMANIA software tool (99). AD genes, as well as other genes deemed to be appropriate by the program, can be linked by criteria such as coexpression, physical interaction (PI) studies, or being part of the same pathway. Figure 2 shows an example of visualization of PI and pathways of a subset of AD genes reviewed in this article. The known high impact AD genes (APP, APOE, PSEN2, and PSEN1) are also highly connected genes. New genes introduced by this program may be further investigated as potential candidate genes for AD. As the computational methods to integrate larger biological data sets continue to improve and be refined, known risk genes may predict gene sets (100) and pathways that can be targeted by drugs.
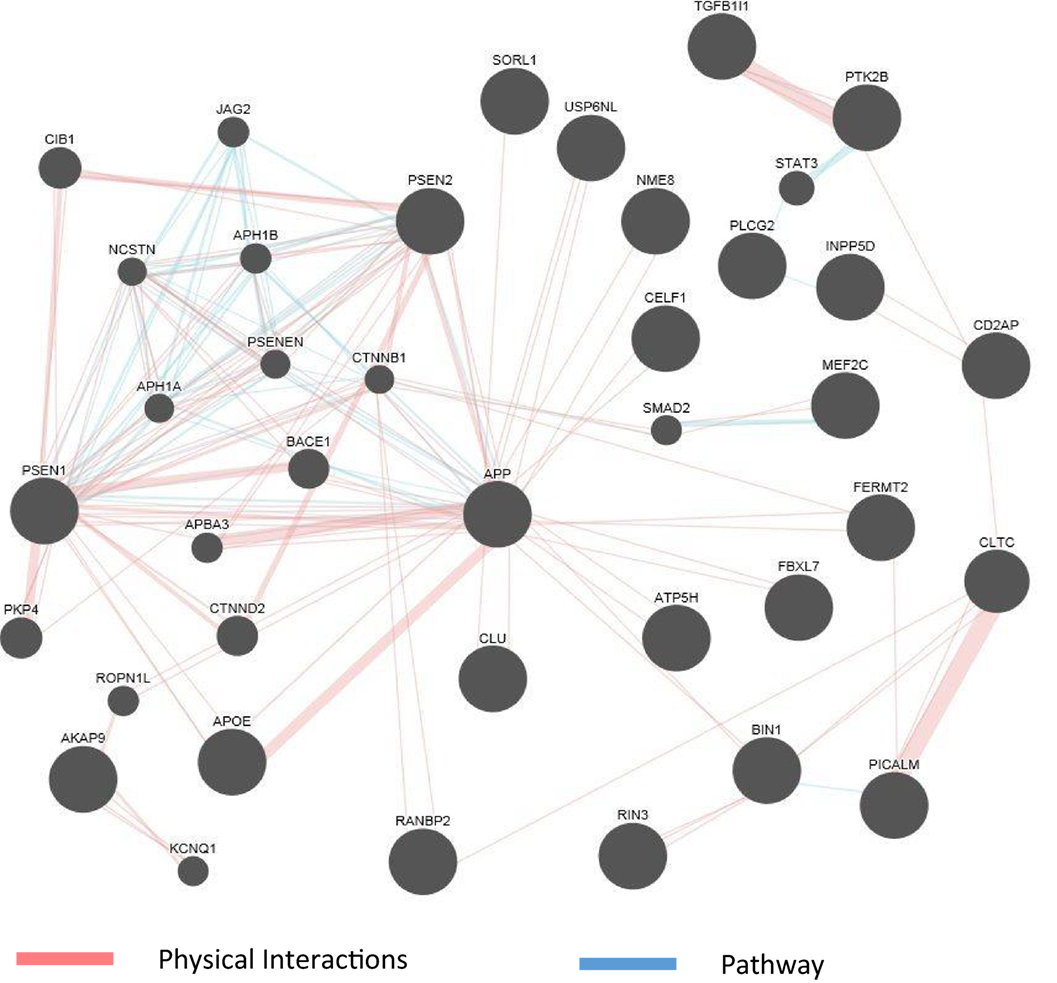
Figure 2. GeneMania network for physical interaction (PI) and pathway. An example of GeneMANIA network when only the PI and pathway links are used. Alzheimer’s disease genes from input list are presented as large black circles, and other genes deemed to be associated with the cluster are small black circles. Genes not linked to the main cluster are discarded.
Polygenic Risk Scores
Because many AD risk SNPs are common variants, every individual necessarily inherits multiple such risk alleles. A polygenic risks score (PRS) (101) can be calculated based on the number of common genetic risk factors present in an individual’s genome, which may be used as predictor for AD risk (102, 103). Using the area under the curve receiver operator characteristic method, PRS may capture nearly all of the genetic liability from common risk variants for AD. However, the efficacy of a genetic predictor is dependent on prevalence and heritability of a disease (104). In AD, the prevalence is highly dependent on age. For the younger age group (65–74) PRS profile captured 90% of the phenotypic variance that can be attributed to common SNPs, which was estimated to be about 24%. Even though it is still controversial whether PRS is a good enough predictor for clinical use (105, 106), it may be useful to identify high-risk subjects where disease prevention studies can focus.
Modifiable Risk Factors for AD
Observational studies have suggested that diabetes, mid-life obesity, mid-life hypertension, high cholesterol, and smoking are modifiable risk factors for AD (107). In terms of modifiable protective factors, education has been robustly shown to reduce AD risk (108). However, for many modifiable factors, no consistent pattern was found across studies (109). A recent comprehensive meta-analysis of 93 modifiable risk factors was conducted from 323 retrospective case/control and prospective cohort studies, which were selected after a systematic review of 16,906 publications (110). This study analyzed associations between AD risk and medical, dietary and occupational exposures as well as serum biochemistry, preexisting diseases, lifestyle, and psychological factors. The identified potentially protective factors include medical exposures of estrogen, statin, antihypertensive medications, and non-steroidal anti-inflammatory drugs, along with dietary exposures to folate, vitamin E/C, and coffee. Other potentially beneficial factors include a history of arthritis, heart disease, and cancer, cognitive activity, current smoking (in Western population), light-to-moderate drinking, and stress. Factors associated with increased risk were hyperhomocysteinemia, depression, frailty, carotid atherosclerosis, hypertension, low diastolic blood pressure, and low education. Evidence for metabolic factors appeared to be inconsistent. Notably, type 2 diabetes mellitus was associated with increased risk in an Asian population, but metabolic syndrome was associated with decreased risk. Moreover, both high body mass index (BMI) in mid-life and low BMI in late-life were associated with increased risk. Most recently, the Lancet Commissions estimated the population attributable fraction of the following modifiable risk factors: hearing loss (9.1%), “less education” (7.5%), followed by smoking, depression, physical inactivity, social isolation, hypertension, diabetes, and mid-life obesity in a declining order (2). The authors estimated that about 35% of total dementia risk may be attributable to a combination of these risk factors. Any preventive interventions addressing these factors can be applied independently of the presence of other factors like genetic risk. However, identifying individuals who would benefit most from a certain intervention due to their genetic risks remains an open question.
It has been widely hypothesized that factors such as physical activities that protect cardiovascular health would also protect the brain from AD and other dementias. A prospective interventional trial (111) along with observational studies (112–117) supports the notion that physical activity may reduce dementia risk. However, a recent meta-analysis of several randomized controlled trials (118) does not support the beneficial effects of long-term exercise on dementia or cognitive decline. A recent large trial with random assignment of intensive lifestyle intervention over 10 years showed that sustained relative weight loss and increases in physical activity did not alter the subsequent prevalence of cognitive impairment in diabetic and obese patients (119). It is currently uncertain whether life style intervention would prevent AD.
Another method to address the causal relationship of a potential modifiable risk factor (exposure) with an outcome such AD is MR. MR infers causation between the exposure and the outcome if the genetic variants associated with the exposure are also associated with the outcome. In other words, if a clinical risk factor P1 is causal for a disease P2, then genetic risk variants G associated with P1 would also be associated with P2 (G → P1 → P2) (120, 121). In principle, MR is expected to avoid bias from reverse causation and generally reduce confounding from other modifiable environmental exposures as it is a common problem in observational studies. Thus, it may provide relatively unbiased estimates of the effect of the modifiable risk factor being studied (122). A limitation of the MR approach is that at least one genetic variant that can reliably predict the exposure is required.
Larsson et al. (123) applied MR on genetic data from over 17,000 AD cases and over 37,000 controls to analyze the effect of 24 potentially modifiable risk factors. Assuming linear association and absence of any alternative causal pathways, genetically predicted higher educational attainment was found to significantly lower odds for AD. This finding is consistent with observational studies. Surprisingly, suggestive evidence was also found for genetically predicted higher quantity of cigarette smoking and lower odds of AD, which is inconsistent with results from cohort studies (124). In addition, genetically predicted higher 25-hydroxyvitamin D concentrations were associated with decreased AD odds, whereas higher coffee consumption with increased odds. Genetically predicted alcohol consumption, serum folate, serum vitamin B12, homocysteine, cardiometabolic factors, and C reactive proteins were not predicted to influence AD risk. One limitation of this study is that the authors used summary of association results rather than actual genotypes. Another MR study using different intermediate factors on the same set of GWAS data found that genetically predicted higher systolic blood pressure may be protective for AD (125), which is compatible with the reported protective effect of higher diastolic blood pressure (110). This result is nonetheless counterintuitive, given the known detrimental health effects of hypertension. This study also found a protective effect of genetically predicted higher smoking quantity. In addition, findings on cholesterol were not consistent with a causal effect on AD risk, after controlling for the confounding effect of APOE. Clearly, more research on larger datasets that include recorded clinical and lifestyle factors are needed to confirm or reject causal implications of some modifiable risk factors of AD.
In addition to the MR approach, there are other attempts to find interplay between genetic and environmental factors. An example is to study gene–environment interactions (126) and one study have shown that estrogen use may be associated with less cognitive decline among APOE4 negative women (127).
Current State of Development of Treatment for AD and Future Outlook
Currently, no disease modifying treatment is available for AD. The only treatments available are treating symptoms, but not the causes of the disease and its progression (128). This statement holds despite the stunning fact that between 2002 and 2014, more than 400 drug trials for AD have been performed but subsequently failed (129). More recently, several large drug trials aiming at reducing the amyloid burden had failed to show efficacy. Attempts to reduce Aβ production (130) as well as immunotherapeutic approaches to clear amyloid plaques from the brain did not show efficacy in slowing down or halting the course of AD (131, 132). Biogen’s immunotherapeutic drug Aducanumab reported positive Phase 1 results on removing brain Aβ plaques and clinical benefits (133). The result of a larger phase 3 trial is still pending.
Explanations of the failure of so many drug trials targeting Aβ argue for possible flaws in the amyloid hypothesis, or the possibility that the disease being too advanced at the time of intervention (131, 134). Drug trials in presymptomatic mutation carriers of autosomal dominant AD may shed light on whether targeting amyloid will yield any therapeutic effect (135). Ongoing drug trials include targeting anti-amyloid, anti-tau, anti-inflammatory, neuroprotection, stem cell therapy, and metabolism (136).
Advances of information technology have enabled health care providers to collect, store, and analyze large quantities of individual health data ranging from clinical information such as diagnostic test results and medication history to lifestyle factors such as smoking. At the same time, scientific community is equipped with methods to generate, process, and analyze large datasets from genomics, imaging, transcriptomics, and many other data-intensive researches. The current concept of precision medicine (137) considers clinical, behavioral, and molecular data to predict personalized disease risk, implement preventive measures, make more accurate diagnosis, and recommend treatments that maximize therapeutic effects and minimize adverse effects. To facilitate precision medicine the National Institute of Health (NIH) launched the All of Us research program, which plans to enroll one million participants (https://allofus.nih.gov/about/about-all-us-research-program).
Under the assumption that the treatment success of a potentially effective pharmacological intervention depends on its initiation in the presymptomatic stage, the identification of at risk subjects will be crucial to maximize treatment effect. Currently, a prevention trial as part of the Dominantly Inherited Alzheimer Network (DIAN) is under way (138). However, results from DIAN may not be representative for the majority of at risk subjects, as most AD patients do not carry Mendelian mutations. Independently, imaging amyloid and tau was shown to identify such at risk subjects (139). In reality, however, large-scale application of imaging biomarkers as a broad population screening method is difficult to implement, due to its invasiveness, high cost, and limited availability of equipment. Other fluid biomarkers have been useful in research studies (21), but their broad use in clinical settings was limited due to lack of established reproducible assays and the reluctance of patients to agree to lumbar puncture procedure (140). Most recently, reports on high-performance plasma amyloid-β biomarkers showed promising accuracy in predicting brain amyloid-β burden (141). Unlike these biomarkers, known genetic risks of a subject remain stable over time and are not influenced by any confounding factors. Currently, genetic risk factors can be assessed at a very low cost starting at around $50 per sample for array-based genotyping data. These arrays cover common variants that may include disease risk variants, which can be further used to impute additional disease risk variants. Moreover, the cost for more comprehensive WES and WGS is down trending toward several hundred dollars. Thus, it is feasible that genetic risk profiles may be used alone or combined with other biomarkers to select at risk subjects in preclinical stage for closer follow-ups and enrollment into preventive studies.
Genetic testing may also increase diagnostic precision in patients with dementia. A recent study showed that known pathogenic mutations for AD and frontotemporal dementia were found with similar proportion in familial LOAD and sporadic LOAD patients. Mutations for Parkinson’s diseases (PD) and amyotrophic lateral sclerosis were also found in clinically diagnosed AD subjects (86). Therefore, genetic testing may prevent other neurodegenerative diseases, which may even have some treatment options, from being misdiagnosed as AD. Combined with fluid and imaging biomarkers, genetics may further increase diagnostic accuracy to ensure clinical trials are done in truly AD patients. Furthermore, instead of treating AD as a homogeneous disease, genetics and other diagnostic methods hold the potential to identify functional disease subtypes that could be specifically targeted.
Another advantage of genetic screening, especially in subjects with family history of dementia, would be the improved risk assessment. An accurate risk assessment could lead to specific consultation for preventive measures addressing modifiable risk factors, such as early use of hearing aids and managing metabolic symptoms. Linking genomic data and electronic health record (EHR) may further help researchers to identify how genetic factors interact with other health conditions such as the impact of medication use on disease risk. For example, an EHR-based analysis found that salbutamol, a β2-adrenoreceptor agonist often prescribed for asthma, is associated with a 34% lower risk of PD and propranolol, a drug frequently prescribed for hypertension, with increased risk (142). Similar approaches of EHR mining may discover medications that alter AD risk. Genetic risk factors had strongly supported a role of immune pathways in AD. Analysis of large EHR data could find out whether drugs that target the immune system had an impact on risk for AD.
Large-scale genetic testing may come from consumer genetic services as they become more broadly available. More than three million people already had their DNA tested at 23&Me and received their carrier status of APOE4 among other risk variants affecting health. Currently, there are hundreds of companies offering similar services and the list is growing (143). The number of people equipped with personal genetic data will likely continue to increase in the general population. Such consumer genetic data may be integrated into EHR to assist diagnostic assessments and choice of treatment. For example, clinicians may consider avoiding propranolol and other β-blockers for patients with genetic predisposition for PD.
In addition to risk variants, genetic studies will identify more protective variants against AD. As the sample size becomes larger, researchers may identify potentially protective factors in subjects who carry strong risk factors such as homozygosity of APOE4, but do not develop AD at an advanced age (65). Identification of protective variants in such a population may lead to possible new drugs that act through a similar mechanism. A recent example for protective genetic variants fueling new effective therapeutics was the development of PCSK9 inhibitor for hypercholesterolemia (144, 145). The newly identified gene PLCG2 that harbors rare protective variants is highly expressed in microglia and may be a target to be exploited for drug discovery in AD (81). Certainly, a hope is that ongoing sequencing efforts (146) would identify more protective variants that can be targeted by drugs.
A workflow for clinical translational research implementing clinical assessments, genetics, and biomarkers into clinical research (as discussed above) is graphically described in Figure 3. Of course, large-scale population level genetic testing also brings ethical challenges. Clinicians and researchers need to take into account the respective guidelines for genetic testing (147). Current studies indicate that the majority of individuals tested for autosomal dominant forms of AD under a standardized counseling protocol demonstrated effective coping skills. Negative psychological reactions were absent after several months and the testing was perceived to be beneficial. The potential benefits, harms, and dilemmas of genetic testing and impacts on family members were detailed in a case report (148). If results of risk factors are returned to the participants, counseling needs to be provided and psychosocial support should be made available. It is important that patients and customers of consumer genetics services understand that typical risk factors are not deterministic for AD. The ethical, legal, and social implications of genetic testing such as testing-induced harm and discrimination are an active area of research at NIH (149).
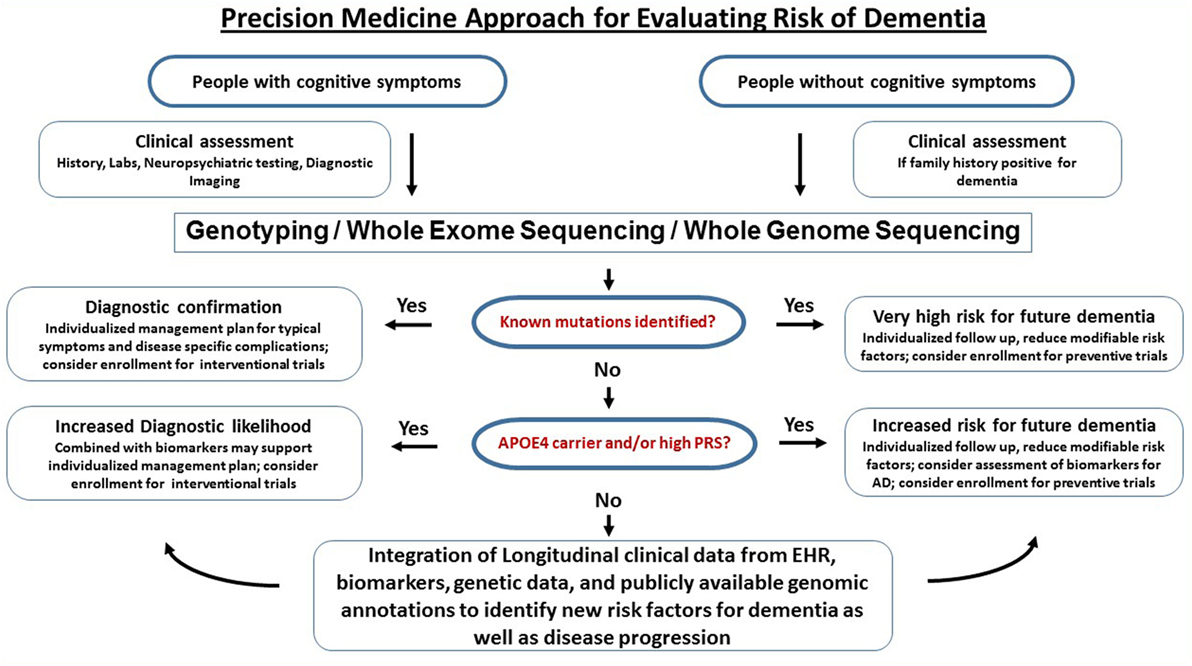
Figure 3. Precision medicine approach for dementia. This is a graphical outline of how genetic and genomic information could be combined and integrated with electronic health records (EHRs) to improve the accuracy of dementia diagnosis and facilitate drug discovery. Middle-aged and older people (e.g., age > 50) are enrolled in an ongoing protocol that includes medical and family history, diagnostic assessment, and access to EHR. For those who have signs of cognitive impairment, genetic testing using either mutation-panels, genotyping arrays, whole exome sequencing or whole genome sequencing depending on the clinical question is performed alongside biomarkers. If a dementia diagnosis is confirmed through genetics and biomarkers, the patients are referred to specialized behavioral and pharmacological intervention and have the option to participate in drug trials. For the majority of subjects who do not have definitive biological findings, a likelihood risk score may be estimated based on the genetic and biomarker profiles. These risk scores may provide support for clinical diagnosis and identify subjects at risk for dementia. The presymptomatic at risk subject may be enrolled in longitudinal studies on prevention and those who never develop dementia despite having high risk may be studied to identify protective factors.
In summary, the current approach for AD consists of optimizing modifiable risk factors to reduce and delay symptom onset as well as symptomatic treatment after disease onset. The dawn of the big data era may make it feasible to advance precision medicine by systematically integrating massive biological data generated by next-generation genomic sequencing, biomarker testing, and EHRs. This development is likely to shed more light to the complex biology of AD and accelerate development of better prevention, diagnosis, and treatments.
Author Contributions
YF-H contributed to conception of the work, data collection, literature research, data interpretation, drafting of the article, critical revision, and final approval of the version to be published. WL contributed to data interpretation, drafting of the article, and critical revision. PD contributed to conception of the work, critical revision, and final approval of the version to be published.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Vanessa Cortes for her participation in literature search and Jan Freudenberg for critical comments on the manuscript.
Funding
YF-H is supported by National Institutes of Health/National Institute on Aging grant 1K08AG054727-01A1 and Mildred and Frank Feinberg Family Foundation.
Abbreviations
AA, Alzheimer’s Association; Aβ, amyloid beta; AD, Alzheimer’s disease; BMI, body mass index; CSF, cerebrospinal fluid; DIAN, Dominantly Inherited Alzheimer Network; EOAD, early onset Alzheimer’s disease; EHR, electronic health record; FDG, 18-fluoro-deoxyglucose; GWAS, genome-wide association studies; LOAD, late onset Alzheimer’s disease; MR, Mendelian randomization; NFT, neurofibrillary tangles; NHGRI-EBI, National Human Genome Research Institute-European Bioinformatics Institute; NIH, National Institute of Health; NIA, National Institute on Aging; PD, Parkinson’s diseases; PET, positron emission tomography; PI, physical interaction; PIB, Pittsburgh compound B; PRS, polygenic risks score; SNP, single-nucleotide polymorphism; WES, whole exome sequencing; WGS, whole genome sequencing.
References
1. Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, Loewenstein D, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the state of Florida brain bank. Alzheimer Dis Assoc Disord (2002) 16:203–12. doi:10.1097/00002093-200210000-00001
2. Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, et al. Dementia prevention, intervention, and care. Lancet (2017) 390:2673–734. doi:10.1016/S0140-6736(17)31363-6
3. Prince MJ, Wu F, Guo Y, Gutierrez Robledo LM, O’Donnell M, Sullivan R, et al. The burden of disease in older people and implications for health policy and practice. Lancet (2015) 385:549–62. doi:10.1016/S0140-6736(14)61347-7
4. Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement (2016) 12:459–509. doi:10.1016/j.jalz.2016.03.001
5. Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, et al. Global prevalence of dementia: a Delphi consensus study. Lancet (2005) 366:2112–7. doi:10.1016/S0140-6736(05)67889-0
6. Aronson SJ, Rehm HL. Building the foundation for genomics in precision medicine. Nature (2015) 526:336–42. doi:10.1038/nature15816
7. McGaghie WC. Varieties of integrative scholarship: why rules of evidence, criteria, and standards matter. Acad Med (2015) 90:294–302. doi:10.1097/ACM.0000000000000585
8. Tabor HK, Risch NJ, Myers RM. Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet (2002) 3:391–7. doi:10.1038/nrg796
9. Barsh GS, Copenhaver GP, Gibson G, Williams SM. Guidelines for genome-wide association studies. PLoS Genet (2012) 8:e1002812. doi:10.1371/journal.pgen.1002812
10. MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res (2017) 45:D896–901. doi:10.1093/nar/gkw1133
11. Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift für Psychiatrie und Psychisch-Gerichtliche Medizin. (1907) 64:146–8.
12. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement (2011) 7:263–9. doi:10.1016/j.jalz.2011.03.005
13. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet (2011) 377:1019–31. doi:10.1016/S0140-6736(10)61349-9
14. Scheltens P, Blennow K, Breteler MM, De Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet (2016) 388:505–17. doi:10.1016/S0140-6736(15)01124-1
15. Walsh JS, Welch HG, Larson EB. Survival of outpatients with Alzheimer-type dementia. Ann Intern Med (1990) 113:429–34. doi:10.7326/0003-4819-113-6-429
16. Mirra SS, Heyman A, Mckeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology (1991) 41:479–86. doi:10.1212/WNL.41.4.479
17. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol (2006) 112:389–404. doi:10.1007/s00401-006-0127-z
18. Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol (1997) 56:1095–7. doi:10.1097/00005072-199710000-00002
19. Jellinger KA. Criteria for the neuropathological diagnosis of dementing disorders: routes out of the swamp? Acta Neuropathol (2009) 117:101–10. doi:10.1007/s00401-008-0466-z
20. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement (2011) 7:280–92. doi:10.1016/j.jalz.2011.03.003
21. Albert MS, Dekosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement (2011) 7:270–9. doi:10.1016/j.jalz.2011.03.008
22. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement (2012) 8:1–13. doi:10.1016/j.jalz.2011.10.007
23. Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology (2016) 87:539–47. doi:10.1212/WNL.0000000000002923
24. Nag S, Yu L, Capuano AW, Wilson RS, Leurgans SE, Bennett DA, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann Neurol (2015) 77:942–52. doi:10.1002/ana.24388
25. Crystal HA, Dickson D, Davies P, Masur D, Grober E, Lipton RB. The relative frequency of “dementia of unknown etiology” increases with age and is nearly 50% in nonagenarians. Arch Neurol (2000) 57:713–9. doi:10.1001/archneur.57.5.713
26. Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, et al. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol (2014) 127:441–50. doi:10.1007/s00401-013-1211-9
27. Rosenberg CK, Cummings TJ, Saunders AM, Widico C, Mcintyre LM, Hulette CM. Dementia with Lewy bodies and Alzheimer’s disease. Acta Neuropathol (2001) 102:621–6. doi:10.1007/s004010100415
28. Cholerton B, Larson EB, Quinn JF, Zabetian CP, Mata IF, Keene CD, et al. Precision medicine: clarity for the complexity of dementia. Am J Pathol (2016) 186:500–6. doi:10.1016/j.ajpath.2015.12.001
29. Sonnen JA, Santa Cruz K, Hemmy LS, Woltjer R, Leverenz JB, Montine KS, et al. Ecology of the aging human brain. Arch Neurol (2011) 68:1049–56. doi:10.1001/archneurol.2011.157
30. Schindler SE, Fagan AM. Autosomal dominant Alzheimer disease: a unique resource to study CSF biomarker changes in preclinical AD. Front Neurol (2015) 6:142. doi:10.3389/fneur.2015.00142
31. Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, et al. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol (2009) 66:382–9. doi:10.1001/archneurol.2008.596
32. Grimmer T, Riemenschneider M, Forstl H, Henriksen G, Klunk WE, Mathis CA, et al. Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry (2009) 65:927–34. doi:10.1016/j.biopsych.2009.01.027
33. Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol (2009) 65:176–83. doi:10.1002/ana.21559
34. Frisoni GB, Boccardi M, Barkhof F, Blennow K, Cappa S, Chiotis K, et al. Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol (2017) 16:661–76. doi:10.1016/S1474-4422(17)30159-X
35. Simonsen AH, Herukka SK, Andreasen N, Baldeiras I, Bjerke M, Blennow K, et al. Recommendations for CSF AD biomarkers in the diagnostic evaluation of dementia. Alzheimers Dement (2017) 13:274–84. doi:10.1016/j.jalz.2016.09.008
36. Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med (2012) 367:795–804. doi:10.1056/NEJMoa1202753
37. Sepulcre J, Grothe MJ, Sabuncu M, Chhatwal J, Schultz AP, Hanseeuw B, et al. Hierarchical organization of Tau and amyloid deposits in the cerebral cortex. JAMA Neurol (2017) 74:813–20. doi:10.1001/jamaneurol.2017.0263
38. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (1991) 82:239–59. doi:10.1007/BF00308809
39. Zhu XC, Tan L, Wang HF, Jiang T, Cao L, Wang C, et al. Rate of early onset Alzheimer’s disease: a systematic review and meta-analysis. Ann Transl Med (2015) 3:38. doi:10.3978/j.issn.2305-5839.2015.01.19
40. Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet (1999) 65:664–70. doi:10.1086/302553
41. Jarmolowicz AI, Chen HY, Panegyres PK. The patterns of inheritance in early-onset dementia: Alzheimer’s disease and frontotemporal dementia. Am J Alzheimers Dis Other Demen (2015) 30:299–306. doi:10.1177/1533317514545825
42. An SS, Park SA, Bagyinszky E, Bae SO, Kim YJ, Im JY, et al. A genetic screen of the mutations in the Korean patients with early-onset Alzheimer’s disease. Clin Interv Aging (2016) 11:1817–22. doi:10.2147/CIA.S116724
43. Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature (1991) 349:704–6. doi:10.1038/349704a0
44. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature (1995) 375:754–60. doi:10.1038/375754a0
45. Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature (1995) 376:775–8. doi:10.1038/376775a0
46. Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science (1995) 269:973–7. doi:10.1126/science.7638621
47. Weggen S, Beher D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimers Res Ther (2012) 4:9. doi:10.1186/alzrt107
48. Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in App protects against Alzheimer’s disease and age-related cognitive decline. Nature (2012) 488:96–9. doi:10.1038/nature11283
49. Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc Natl Acad Sci U S A (2017) 114:E476–85. doi:10.1073/pnas.1618657114
50. Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, et al. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem (2014) 289:30990–1000. doi:10.1074/jbc.M114.589069
51. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med (2016) 8:595–608. doi:10.15252/emmm.201606210
52. Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun (2014) 2:135. doi:10.1186/s40478-014-0135-5
53. Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol (2004) 55:306–19. doi:10.1002/ana.20009
54. Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology (2007) 68:1718–25. doi:10.1212/01.wnl.0000261919.22630.ea
55. Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol (2008) 65:1509–17. doi:10.1001/archneur.65.11.1509
56. Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, et al. Relationships between biomarkers in aging and dementia. Neurology (2009) 73:1193–9. doi:10.1212/WNL.0b013e3181bc010c
57. Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat Neurosci (2015) 18:800–6. doi:10.1038/nn.4018
58. Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry (2006) 63:168–74. doi:10.1001/archpsyc.63.2.168
59. Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A (1993) 90:1977–81. doi:10.1073/pnas.90.5.1977
60. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science (1993) 261:921–3. doi:10.1126/science.8346443
61. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA (1997) 278:1349–56. doi:10.1001/jama.1997.03550160069041
62. Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry (2011) 16:903–7. doi:10.1038/mp.2011.52
63. Bickeboller H, Campion D, Brice A, Amouyel P, Hannequin D, Didierjean O, et al. Apolipoprotein E and Alzheimer disease: genotype-specific risks by age and sex. Am J Hum Genet (1997) 60:439–46.
64. Sando SB, Melquist S, Cannon A, Hutton ML, Sletvold O, Saltvedt I, et al. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol (2008) 8:9. doi:10.1186/1471-2377-8-9
65. Freudenberg-Hua Y, Freudenberg J, Vacic V, Abhyankar A, Emde AK, Ben-Avraham D, et al. Disease variants in genomes of 44 centenarians. Mol Genet Genomic Med (2014) 2:438–50. doi:10.1002/mgg3.86
66. Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science (1988) 240:622–30. doi:10.1126/science.3283935
67. Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol (2013) 9:106–18. doi:10.1038/nrneurol.2012.263
68. Zhao N, Liu CC, Qiao W, Bu G. Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol Psychiatry (2018) 83:347–57. doi:10.1016/j.biopsych.2017.03.003
69. Frieden C, Garai K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc Natl Acad Sci U S A (2012) 109:8913–8. doi:10.1073/pnas.1207022109
70. Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature (2017) 549:523–7. doi:10.1038/nature24016
71. Naj AC, Beecham GW, Martin ER, Gallins PJ, Powell EH, Konidari I, et al. Dementia revealed: novel chromosome 6 locus for late-onset Alzheimer disease provides genetic evidence for folate-pathway abnormalities. PLoS Genet (2010) 6:e1001130. doi:10.1371/journal.pgen.1001130
72. Ridge PG, Hoyt KB, Boehme K, Mukherjee S, Crane PK, Haines JL, et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging (2016) 41:200.e13–200.e20. doi:10.1016/j.neurobiolaging.2016.02.024
73. Freudenberg-Hua Y, Li W, Abhyankar A, Vacic V, Cortes V, Ben-Avraham D, et al. Differential burden of rare protein truncating variants in Alzheimer’s disease patients compared to centenarians. Hum Mol Genet (2016) 25:3096–105. doi:10.1093/hmg/ddw150
74. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med (2013) 368:117–27. doi:10.1056/NEJMoa1211851
75. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med (2013) 368:107–16. doi:10.1056/NEJMoa1211103
76. Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature (2014) 505:550–4. doi:10.1038/nature12825
77. Van Der Lee SJ, Holstege H, Wong TH, Jakobsdottir J, Bis JC, Chouraki V, et al. PLD3 variants in population studies. Nature (2015) 520:E2–3. doi:10.1038/nature14038
78. Wetzel-Smith MK, Hunkapiller J, Bhangale TR, Srinivasan K, Maloney JA, Atwal JK, et al. A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat Med (2014) 20:1452–7. doi:10.1038/nm.3736
79. Logue MW, Schu M, Vardarajan BN, Farrell J, Bennett DA, Buxbaum JD, et al. Two rare AKAP9 variants are associated with Alzheimer’s disease in African Americans. Alzheimers Dement (2014) 10(6):609–618.e11. doi:10.1016/j.jalz.2014.06.010
80. Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum Mol Genet (2009) 18:3987–96. doi:10.1093/hmg/ddp323
81. Sims R, Van Der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet (2017) 49:1373–84. doi:10.1038/ng.3916
82. Le Guennec K, Nicolas G, Quenez O, Charbonnier C, Wallon D, Bellenguez C, et al. ABCA7 rare variants and Alzheimer disease risk. Neurology (2016) 86:2134–7. doi:10.1212/WNL.0000000000002627
83. Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet (2015) 47:445–7. doi:10.1038/ng.3246
84. Cuyvers E, De Roeck A, Van Den Bossche T, Van Cauwenberghe C, Bettens K, Vermeulen S, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: a targeted resequencing study. Lancet Neurol (2015) 14:814–22. doi:10.1016/S1474-4422(15)00133-7
85. Cruchaga C, Haller G, Chakraverty S, Mayo K, Vallania FL, Mitra RD, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for Ad in late-onset Alzheimer’s disease families. PLoS One (2012) 7:e31039. doi:10.1371/journal.pone.0031039
86. Fernandez MV, Kim JH, Budde JP, Black K, Medvedeva A, Saef B, et al. Analysis of neurodegenerative Mendelian genes in clinically diagnosed Alzheimer disease. PLoS Genet (2017) 13:e1007045. doi:10.1371/journal.pgen.1007045
87. Heilmann S, Drichel D, Clarimon J, Fernandez V, Lacour A, Wagner H, et al. PLD3 in non-familial Alzheimer’s disease. Nature (2015) 520:E3–5. doi:10.1038/nature14039
88. Hooli BV, Lill CM, Mullin K, Qiao D, Lange C, Bertram L, et al. PLD3 gene variants and Alzheimer’s disease. Nature (2015) 520:E7–8. doi:10.1038/nature14040
89. Lambert JC, Grenier-Boley B, Bellenguez C, Pasquier F, Campion D, Dartigues JF, et al. PLD3 and sporadic Alzheimer’s disease risk. Nature (2015) 520:E1. doi:10.1038/nature14036
90. Guerreiro R, Bras J, Hardy J. SnapShot: genetics of Alzheimer’s disease. Cell (2013) 155:968–968.e1. doi:10.1016/j.cell.2013.10.037
91. Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry (2015) 77:43–51. doi:10.1016/j.biopsych.2014.05.006
92. Giri M, Zhang M, Lu Y. Genes associated with Alzheimer’s disease: an overview and current status. Clin Interv Aging (2016) 11:665–81. doi:10.2147/CIA.S105769
93. Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer’s disease. Nat Immunol (2015) 16:229–36. doi:10.1038/ni.3102
94. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol (2015) 14:388–405. doi:10.1016/S1474-4422(15)70016-5
95. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci (2015) 16:358–72. doi:10.1038/nrn3880
96. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 Binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron (2016) 91:328–40. doi:10.1016/j.neuron.2016.06.015
97. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell (2015) 160:1061–71. doi:10.1016/j.cell.2015.01.049
98. Ma L, Allen M, Sakae N, Ertekin-Taner N, Graff-Radford NR, Dickson DW, et al. Expression and processing analyses of wild type and p.R47H TREM2 variant in Alzheimer’s disease brains. Mol Neurodegener (2016) 11:72. doi:10.1186/s13024-016-0137-9
99. Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res (2010) 38:W214–20. doi:10.1093/nar/gkq537
100. Li W, Freudenberg J, Oswald M. Principles for the organization of gene-sets. Comput Biol Chem (2015) 59(Pt B):139–49. doi:10.1016/j.compbiolchem.2015.04.005
101. International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature (2009) 460:748–52. doi:10.1038/nature08185
102. Escott-Price V, Shoai M, Pither R, Williams J, Hardy J. Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol Aging (2017) 49: 214.e7–214.e11. doi:10.1016/j.neurobiolaging.2016.07.018
103. Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain (2015) 138:3673–84. doi:10.1093/brain/awv268
104. Wray NR, Yang J, Goddard ME, Visscher PM. The genetic interpretation of area under the Roc curve in genomic profiling. PLoS Genet (2010) 6:e1000864. doi:10.1371/journal.pgen.1000864
105. Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet (2013) 14:507–15. doi:10.1038/nrg3457
106. Lee SH, Harold D, Nyholt DR; ANZGene Consortium, International Endogene Consortium, Genetic and Environmental Risk for Alzheimer’s Disease Consortium. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum Mol Genet (2013) 22:832–41. doi:10.1093/hmg/dds491
107. Baumgart M, Snyder HM, Carrillo MC, Fazio S, Kim H, Johns H. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population-based perspective. Alzheimers Dement (2015) 11:718–26. doi:10.1016/j.jalz.2015.05.016
108. Kukull WA, Higdon R, Bowen JD, Mccormick WC, Teri L, Schellenberg GD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol (2002) 59:1737–46. doi:10.1001/archneur.59.11.1737
109. Power MC, Weuve J, Gagne JJ, Mcqueen MB, Viswanathan A, Blacker D. The association between blood pressure and incident Alzheimer disease: a systematic review and meta-analysis. Epidemiology (2011) 22:646–59. doi:10.1097/EDE.0b013e31822708b5
110. Xu W, Tan L, Wang HF, Jiang T, Tan MS, Tan L, et al. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry (2015) 86:1299–306. doi:10.1136/jnnp-2015-310548
111. Lautenschlager NT, Cox KL, Flicker L, Foster JK, Van Bockxmeer FM, Xiao J, et al. Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA (2008) 300:1027–37. doi:10.1001/jama.300.9.1027
112. Weuve J, Kang JH, Manson JE, Breteler MM, Ware JH, Grodstein F. Physical activity, including walking, and cognitive function in older women. JAMA (2004) 292:1454–61. doi:10.1001/jama.292.12.1454
113. Abbott RD, White LR, Ross GW, Masaki KH, Curb JD, Petrovitch H. Walking and dementia in physically capable elderly men. JAMA (2004) 292:1447–53. doi:10.1001/jama.292.12.1447
114. Larson EB, Wang L, Bowen JD, Mccormick WC, Teri L, Crane P, et al. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med (2006) 144:73–81. doi:10.7326/0003-4819-144-2-200601170-00004
115. Podewils LJ, Guallar E, Kuller LH, Fried LP, Lopez OL, Carlson M, et al. Physical activity, Apoe genotype, and dementia risk: findings from the cardiovascular health cognition study. Am J Epidemiol (2005) 161:639–51. doi:10.1093/aje/kwi092
116. Van Gelder BM, Tijhuis MA, Kalmijn S, Giampaoli S, Nissinen A, Kromhout D. Physical activity in relation to cognitive decline in elderly men: the fine study. Neurology (2004) 63:2316–21. doi:10.1212/01.WNL.0000147474.29994.35
117. Zhou Z, Fu J, Hong YA, Wang P, Fang Y. Association between exercise and the risk of dementia: results from a nationwide longitudinal study in China. BMJ Open (2017) 7:e017497. doi:10.1136/bmjopen-2017-017497
118. Barreto PS, Demougeot L, Vellas B, Rolland Y. Exercise training for preventing dementia, mild cognitive impairment, and clinically meaningful cognitive decline: a systematic review and meta-analysis. J Gerontol A Biol Sci Med Sci (2017) Series A:glx234. doi:10.1093/gerona/glx234
119. Espeland MA, Luchsinger JA, Baker LD, Neiberg R, Kahn SE, Arnold SE, et al. Effect of a long-term intensive lifestyle intervention on prevalence of cognitive impairment. Neurology (2017) 88:2026–35. doi:10.1212/WNL.0000000000003955
120. Katan MB. Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet (1986) 1:507–8. doi:10.1016/S0140-6736(86)92972-7
121. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet (2014) 23:R89–98. doi:10.1093/hmg/ddu328
122. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA (2017) 318:1925–6. doi:10.1001/jama.2017.17219
123. Larsson SC, Traylor M, Malik R, Dichgans M, Burgess S, Markus HS, et al. Modifiable pathways in Alzheimer’s disease: Mendelian randomisation analysis. BMJ (2017) 359:j5375. doi:10.1136/bmj.j5375
124. Cataldo JK, Prochaska JJ, Glantz SA. Cigarette smoking is a risk factor for Alzheimer’s disease: an analysis controlling for tobacco industry affiliation. J Alzheimers Dis (2010) 19:465–80. doi:10.3233/JAD-2010-1240
125. Ostergaard SD, Mukherjee S, Sharp SJ, Proitsi P, Lotta LA, Day F, et al. Associations between potentially modifiable risk factors and Alzheimer disease: a Mendelian randomization study. PLoS Med (2015) 12:e1001841; discussion e1001841. doi:10.1371/journal.pmed.1001841
126. Thomas D. Gene–environment-wide association studies: emerging approaches. Nat Rev Genet (2010) 11:259–72. doi:10.1038/nrg2764
127. Yaffe K, Haan M, Byers A, Tangen C, Kuller L. Estrogen use, APOE, and cognitive decline: evidence of gene-environment interaction. Neurology (2000) 54:1949–54. doi:10.1212/WNL.54.10.1949
128. Howard R, Mcshane R, Lindesay J, Ritchie C, Baldwin A, Barber R, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med (2012) 366:893–903. doi:10.1056/NEJMoa1106668
129. Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther (2014) 6:37. doi:10.1186/alzrt269
130. Carroll J. Another Alzheimer’s drug flops in pivotal clinical trial. Science (2017). doi:10.1126/science.aal0759
131. Gold M. Phase II clinical trials of anti-amyloid beta antibodies: when is enough, enough? Alzheimers Dement (N Y) (2017) 3:402–9. doi:10.1016/j.trci.2017.04.005
132. Sacks CA, Avorn J, Kesselheim AS. The failure of solanezumab – how the Fda saved taxpayers billions. N Engl J Med (2017) 376:1706–8. doi:10.1056/NEJMp1701047
133. Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature (2016) 537:50–6. doi:10.1038/nature19323
134. Giacobini E, Gold G. Alzheimer disease therapy – moving from amyloid-beta to tau. Nat Rev Neurol (2013) 9:677–86. doi:10.1038/nrneurol.2013.223
135. Reiman EM, Langbaum JB, Tariot PN, Lopera F, Bateman RJ, Morris JC, et al. CAP – advancing the evaluation of preclinical Alzheimer disease treatments. Nat Rev Neurol (2016) 12:56–61. doi:10.1038/nrneurol.2015.177
136. Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (N Y) (2017) 3:367–84. doi:10.1016/j.trci.2017.05.002
137. Reitz C. Toward precision medicine in Alzheimer’s disease. Ann Transl Med (2016) 4:107. doi:10.21037/atm.2016.03.05
138. Bateman RJ, Benzinger TL, Berry S, Clifford DB, Duggan C, Fagan AM, et al. The Dian-Tu next generation Alzheimer’s prevention trial: adaptive design and disease progression model. Alzheimers Dement (2017) 13:8–19. doi:10.1016/j.jalz.2016.07.005
139. Hanseeuw BJ, Betensky RA, Schultz AP, Papp KV, Mormino EC, Sepulcre J, et al. Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann Neurol (2017) 81:583–96. doi:10.1002/ana.24910
140. Blennow K, Dubois B, Fagan AM, Lewczuk P, De Leon MJ, Hampel H. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement (2015) 11:58–69. doi:10.1016/j.jalz.2014.02.004
141. Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Dore V, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature (2018) 554:249–54. doi:10.1038/nature25456
142. Mittal S, Bjornevik K, Im DS, Flierl A, Dong X, Locascio JJ, et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science (2017) 357:891–8. doi:10.1126/science.aaf3934
143. Phillips AM. Only a click away – DTC genetics for ancestry, health, love … and more: a view of the business and regulatory landscape. Appl Transl Genom (2016) 8:16–22. doi:10.1016/j.atg.2016.01.001
144. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low Ldl cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet (2005) 37:161–5. doi:10.1038/ng1509
145. Defesche JC, Stefanutti C, Langslet G, Hopkins PN, Seiz W, Baccara-Dinet MT, et al. Efficacy of alirocumab in 1191 patients with a wide spectrum of mutations in genes causative for familial hypercholesterolemia. J Clin Lipidol (2017) 11(6):1338–46.e7. doi:10.1016/j.jacl.2017.08.016
146. Crane PK, Foroud T, Montine TJ, Larson EB. Alzheimer’s disease sequencing project discovery and replication criteria for cases and controls: data from a community-based prospective cohort study with autopsy follow-up. Alzheimers Dement (2017) 13:1410–3. doi:10.1016/j.jalz.2017.09.010
147. Goldman JS, Hahn SE, Catania JW, Larusse-Eckert S, Butson MB, Rumbaugh M, et al. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American college of medical genetics and the national society of genetic counselors. Genet Med (2011) 13:597–605. doi:10.1097/GIM.0b013e31821d69b8
148. Mapes MV, O’Brien BM, King LP. How should clinicians counsel a woman with a strong family history of early-onset Alzheimer’s disease about her pregnancy? AMA J Ethics (2017) 19:663–74. doi:10.1001/journalofethics.2017.19.7.ecas4-1707
Keywords: Alzheimer’s disease, genetics, genomics, risk factors, risk variants, precision medicine, genome sequencing
Citation: Freudenberg-Hua Y, Li W and Davies P (2018) The Role of Genetics in Advancing Precision Medicine for Alzheimer’s Disease—A Narrative Review. Front. Med. 5:108. doi: 10.3389/fmed.2018.00108
Received: 21 January 2018; Accepted: 03 April 2018;
Published: 24 April 2018
Edited by:
Gil Atzmon, University of Haifa, IsraelReviewed by:
Antony Bayer, Cardiff University, United KingdomWee Shiong Lim, Tan Tock Seng Hospital, Singapore
Copyright: © 2018 Freudenberg-Hua, Li and Davies. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Freudenberg-Hua, yfreuden@northwell.edu