 Paola Valdivieso
Paola Valdivieso Martino V. Franchi
Martino V. Franchi Christian Gerber3
Christian Gerber3 Martin Flück
Martin Flück
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Med., 20 March 2018
Sec. Pathology
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00077
Non-specific chronic low back pain (nsCLBP) is a multifactorial condition of unknown etiology and pathogenesis. Physical and genetic factors may influence the predisposition of individuals to CLBP, which in many instances share a musculoskeletal origin. A reduced pain level in low back pain patients that participate in exercise therapy highlights that disuse-related muscle deconditioning may predispose individuals to nsCLBP. In this context, musculoskeletal pain may be the consequence of capillary rarefaction in inactive muscle as this would lower local tissue drainage and washing out of toxic waste. Muscle activity is translated into an angio-adaptative process, which implicates angiogenic-gene expression and individual response differences due to heritable modifications of such genes (gene polymorphisms). The pathophysiologic mechanism underlying nsCLBP is still largely unaddressed. We hypothesize that capillary rarefaction due to a deconditioning of dorsal muscle groups exacerbates nsCLBP by increasing noxious sensation, reducing muscle strength and fatigue resistance by initiating a downward spiral of local deconditioning of back muscles which diminishes their load-bearing capacity. We address the idea that specific factors such as angiotensin-converting enzyme and Tenascin-C might play an important role in altering susceptibility to nsCLBP via their effects on microvascular perfusion and vascular remodeling of skeletal muscle, inflammation, and pain sensation. The genetic profile may help to explain the individual predisposition to nsCLBP, thus identifying subgroups of patients, which could benefit from ad hoc treatment types. Future therapeutic approaches aimed at relieving the pain associated with nsCLBP should be based on the verification of mechanistic processes of activity-induced angio-adaptation and muscle-perfusion.
Low back pain (LBP) is the most common cause for years lived with a disability (1) with 60–80% of the adult population reporting LBP for some time along their lives (2). Problematic is the fact that only 5–15% of all LBP cases can be associated with a specific cause. The majority of patients with LBP present non-specific form, in which no common anatomical origin of the pathology can be identified and matched to the patient’s complaints (3). Non-specific LBP accounts for 70–90% of all LBP cases (3–5). Most LBP cases recover within days or weeks without further complications after first signs of pain manifest (6). However, a minority of about 10% does not recover and develop chronic LBP (CLBP), identified as a condition lasting longer than three months (3, 7). Although CLBP is recognized to present a multifactorial origin, the occurring pain in this group of non-specific chronic low back pain (nsCLBP) is often associated with musculoskeletal disorders (8).
The muscular system is of pivotal importance for the stability and movement of the spine (9). Inappropriate use of local musculoskeletal system blunts the impact of physiological cues which are required to maintain the functional capacity of muscle through instructing homeostatic processes and adaptation (10). As a consequence, the musculoskeletal system is deconditioned and the capacity of stabilize posture and move is compromised (11, 12). On one hand, muscle deconditioning, due to a congenital or acquired lowering of muscle activity can be seen as a cause of nsCLBP. This may cause constant overloading of the lumbar muscles and subsequent increase in muscular fatigue (13, 14) which ultimately may lead to nsCLBP. On the other hand, muscle deconditioning can be regarded as a consequence of CLBP due to a vicious cycle consisting of an initial injury, which may lead to pain and subsequent avoidance behavior regarding physical activities due to the fear of re-injury (15). Such vicious cycle may lead to muscle deconditioning, leaving the patient unable to sustain “normal” everyday loading (16–18). Whether muscle deconditioning represents a possible cause or, instead, consequence of nsCLBP, is still a point of debate (17, 19).
In a recent review, Steele et al. (15) have pointed out that “deconditioning” (i.e., defined by the authors as a “decrease in function”) may not be only caused by “disuse” [defined by Verbunt et al. as a decrease in physical activity levels (17)]. In LBP, trunk muscles concomitantly undergo a specific acute-to-chronic phase remodeling which usually leads to a reduced cross-sectional area (CSA) of involved muscles (20). This phenomenon is well understood to reflect a specific local response of muscle tissue to the reduced impact of use-related metabolic and mechanical cues, or muscle disuse (21).
Muscular disuse is defined as the response to decreased usage, leading to a reductive remodeling of the tissue (22), accompanied with a shift from slow to fast myosin isoforms muscle phenotype (23), markedly decreased force output and fatigue resistance, lower neural activation, reduced metabolic supply due to capillary rarefaction, and a diminished local oxidative capacity (24). This physiological definition of disuse distinguished from the one describing disuse as a “general lack or reduction of physical activity level.” Lower physical activity levels can indeed be associated with local muscle disuse and deconditioning (25), but some authors have found inconclusive relationships between physical activity and CLBP (17), as whole body activity levels seem not to differ between healthy controls and CLBP patients (26). However, in the formed instances physical activity was assessed by accelerometer and average daily metabolic rate to resting metabolic rate ratio (26). As no information on localized (i.e., trunk) muscle activation or energy expenditure was provided, the presence of concomitant local muscle disuse cannot be excluded. Thus, muscle disuse may still be regarded as a cause of nsCLBP pathology. To date, as it is unsolved whether muscle deconditioning is cause or consequence of disuse; there is an urge for longitudinal investigations with clearly defined and standardized measures for conditioning levels (strength and endurance) and strict control of influencing factors (17).
Morphological indicators of muscle deconditioning are provided by imaging and histochemical studies of lumbar musculature. During deconditioning, skeletal muscles go through remodeling processes that lead subsequently to a significant loss in mass (15). After prolonged physical inactivity, paraspinal muscles undergo atrophic processes (27, 28): morphometric estimates based on magnetic resonance imaging (MRI) presented a decrease of CSA in paraspinal muscle, especially the multifidus (20, 29). Similar patterns of atrophy have been demonstrated using ultrasound imaging (30, 31) and computed tomography (CT) (32–34). Although the significance of paraspinal muscle atrophy role in predicting long term of LBP has been challenged (35, 36), there is evidence for a time-dependent selective ipsilateral atrophy of distinct muscle groups specific for the symptomatic side (37, 38). This remodeling process varies between the acute and CLBP phase, which affects psoas major/erector spinae and multifidus/erector spinae, respectively (37). Goubert et al. stated in a recent review that, in nsCLBP patients, multifidus and paraspinal muscle, but not erector spinae, present the most significant reduction in CSA (39). Nonetheless, transverse relaxation time (T2) MRI analyses revealed abnormalities and asymmetries of low back muscles in patients with acute LBP (40) and such alterations in MRI-derived T2 can be associated with muscle (i.e., multifidus) disuse/altered use of multifidus muscle even after cessation of LBP (41).
Magnetic resonance imaging data have been used in order to measure an alteration of imaging signal intensity, which can be regarded as an index of muscle quality and increase in fat content, which in turn has been found associated with LBP (42, 43).
In addition, histological studies have pointed out changes in the fiber type distribution of back muscles, presenting a shift (in paraspinal muscle) toward the fast-fatiguable type IIX fibers (14, 44, 45).
Functional indicators of muscle deconditioning in patients with nsCLBP are represented by weakness of trunk muscles, measured either by isometric or isokinetic contractions (46) and reduced endurance of lumbar extensor muscles (47). There is evidence that individuals with CLBP have a delayed activation of back muscle activity when performing prone hip extension (48). Lower activation patterns (EMG) of multifidus and other lumbar muscles have been showed during sitting in non-specific CLBP patients compared with healthy controls (49).
It is well established that muscle strength and endurance are compromised in consequence of muscle atrophy and deconditioning (50–53). Therapeutic interventions should be identified and applied to mitigate the atrophic process. Previous training interventions were performed in patients with CLBP, in which lumbar multifidus muscle CSA was found to increase after 10 weeks of stabilization training combined with dynamic-static resistance training (32).
However, there are controversial opinions concerning the relationship between improving trunk muscle performance (i.e., strength/endurance) and relief of painful symptoms and disability (54). We identify that this criticism is related to the lack of a specific exercise treatment to halt LBP and evidence for casual relationship between changes in strength or CSA and pain and disability level, respectively (55). The note of a relationship between pain and muscle’s force producing capacity is provided by the fact that a failure of paraspinal muscles in protecting passive spinal and pelvic structures from excessive loading in daily living maneuvers or sport activities may result in damage to these pain-sensitive structures thus elevating noxious influences. For this reason, specific forms of resistance exercise are often recommended for back pain patients. The conditioning of musculature by progressive resistance exercise improves muscle hypertrophy, strength/endurance performance, and cardiovascular capacity (56). In addition, therapeutic training that improves both of the former parameters was found to relieve symptoms of CLBP and disability (54, 57) related to musculoskeletal disorders. There is also evidence for a combination of electrical stimulation or ultrasound therapy and exercises in increasing quality of life by improving pain, muscle strength/endurance and spine mobility (58).
In the late 1700s, a Scottish surgeon (John Hunter) observed that “blood goes to where it is needed” (59). Along this original line of observations it is now understood that the primary effect of mechanical-stimuli such as repeated muscle contractions will produce a significant increase in cardiac output followed by an increase in blood volume. Thus, the vast majority of the increased metabolic and mechanical demand is allocated to the contracting skeletal muscle and adequate muscle perfusion is crucial (60).
Muscle tissue is dependent on sufficient blood perfusion in terms of maintaining oxygen- and nutrient levels according to its demands (61). Insufficiency of both, oxygen- and nutrient levels, as well as insufficient removal of toxic products of the muscle metabolism, causes pain, and deteriorate physiological functioning (62). Thus, capillary regression or rarefaction, caused by physical deconditioning, chronic vasoconstriction, maladaptive alterations/diseases of the microvascular bed, or endothelial dysfunction in muscle tissue (63–65), may lead to muscle ischemia in consequence of an insufficient blood flow and decreased oxygen extraction within the muscle tissue due to an associated reduction in aerobic capacity of muscle fibers (66).
Previous literature highlighted the evidence for a potential connection between impaired perfusion (i.e., atherosclerosis and stenosis of lumbar arteries) and CLBP (67, 68). Other studies showed that an insufficient lower limb perfusion may induce a similar symptom as neuropathic pain (69, 70). Interestingly, prolonged retraction of the paraspinal muscle as induced during spinal surgery may reduce muscle perfusion and trigger back pain and disability (71). Paraspinal compartment syndrome is associated with reduced tissue perfusion that causes ischemic pain (72) as well as disk degeneration (67).
Taken together, these data point to insufficient blood supply, with evidence for hypoxic or ischemic conditions within the lumbar compartment, which may affect the metabolism of vertebral bone, nerve roots, intervertebral disks, and lumbar muscles. Interestingly, some study suggests that CLBP patients present an impairment to deliver oxygen in erector spinae muscle, which can be partly restored during exercise (73–75).
Aberrant governance of back muscle strength and endurance through use dependent vascular perfusion may represent a critical event in the etiology and progression of CLBP. In this context, a critical lack of dynamic back muscle work is a possible causal factor leading to muscle deconditioning through rarefaction of capillaries and reduced aerobic capacity (76). Regression of muscle capillaries is a common consequence in physical deconditioning (i.e., hind limb unloading) and chronic diseases such as diabetes, obesity, chronic heart failure, or chronic obstructive pulmonary disease (76, 77). The consequent microcirculatory dysfunction exacerbates the compounds underlying the pathology through further inflammation and impaired delivery of oxygen and substrate to tissues (78, 79). Although the pathophysiologic mechanisms remain unclear, it is known that capillary supply supports the homeostatic processes in muscle tissue: a qualitative and quantitative improvement in muscle perfusion, following arteriolar vasodilatation and growth of the capillary tree, enhances muscle metabolic capacity to augment resistance to fatigue (80, 81). Moreover, a better perfusion could explain analgesia produced by washing out of toxic wastes and reducing muscle and joint stiffness (82).
In normal adult skeletal muscle, the genesis and turnover of capillaries and muscle fibers are inter-related (80, 83, 84). Being skeletal muscle a plastic tissue, under repeated mechanical stimuli, angiogenesis is requested to compensate perfusion distance between capillaries and muscle fibers. Several potential stimuli have been postulated to underlie angiogenesis in exercised muscle. An increase in blood flow within a vessel causes increases in shear stress and lowered oxygenation, which is a major stimulus in the remodeling and capillary growth (85). In fact, abluminal mechanical stress (i.e., stretch) of muscle tissue with static or repeated cycles of muscle contraction represents the physiological growth stimulus for the embedded capillaries which is equally effective as liminal sheer stress to induce growth of the capillary endothelium through the sprouting of existing blood vessels (84, 86). In this process, vasoactive factors initiate and control the remodeling of the basement membrane that surrounds the capillary endothelium by regulating the proliferative activity and motility of endothelial cells (ECs). Capillary remodeling encompasses changes in the connections between the ECs and the extracellular matrix (ECM) proteins through the activation and enhanced synthesis of adhesive and cytoskeletal proteins and extracellular proteases. Molecular control of the angiogenic process is then orchestrated by the interplay between the activation of ECs and turnover of the ECM. There is growing evidence suggesting that matrix-derived factors play a critical role in regulating muscle angiogenesis (87). The endothelium and myogenic control collaborate in vascular adaptation in response to several stimuli during vasodilatation/vasoconstriction responses (88).
On the basis of the aforementioned evidence, we develop the idea that individuals with nsCLBP might present an attenuation of muscle-perfusion, due to muscle deconditioning and concomitant disuse. In this scenario, an altered perfusion and rarefaction of capillaries may be the result of a complex interplay between genetic predisposition and environmental cues (such as muscle deconditioning).
Several studies have investigated a possible linkage between the musculoskeletal system and genetic polymorphisms affecting pro- and anti-angiogenic factors, in particular those being associated with the remodeling of the ECM and the proliferation of ECs (89). Perfusion-related angio-adaptation may implicate the regulation of muscle’s vascular bed by two potentially angiogenic modulators, angiotensin-converting enzyme (ACE) and Tenascin-C (TNC).
In a recent investigation, we have illustrated how ACE and (TNC) cooperate during muscular and vascular-remodeling (90). In cultured smooth muscle cells, TNC expression is induced by the vasoconstrictor peptide angiotensin II and is found increased in vascular structures in patients with hypertension (91–93). TNC is highly expressed in migrating ECs and branching sites of blood vessels (91, 94, 95). Suspicion is high that vasoconstriction and remodeling of resistance vessels are somehow connected and that, while TNC mediates the effect of angiotensin II, ACE in turn catalyzes the conversion of angiotensin I to angiotensin II.
We then propose that the aforementioned gene polymorphisms, being associated with muscular hypoperfusion due to an enhanced vasoconstriction, may importantly modify the angio-adaptation process. Consequently, we advocate investigating the association between single-nucleotide polymorphisms (SNPs) within the ACE and TNC genes together with changes in morphologic and functional characteristics of skeletal muscle, and clinical outcomes (pain, disability) in patients with nsCLBP in response to therapeutic exercise training (Figure 1, for a synopsis). The proposed mechanism may then operate in two distinct ways: on one hand, vasoconstriction will be regulated by ACE-I/D polymorphism, with the presence (I-allele) or the absence (D-allele) in intron 16 of the ACE gene. Presence of the I-allele may then lower ACE activity in skeletal muscle and the production of Angiotensin II (96, 97). On the other hand, remodeling will also be associated with polymorphism in the TNC gene (98). In particular, rs2104772 polymorphism describes a thymidine (T)-to-adenosine (A) exchange at nucleotide position 44513 in the TNC gene that instructs the substitution of leucine by isoleucine at amino acid position 1677 and it may alter the molecular elasticity of the TNC Fn-III-D domain (99, 100). Speculatively, via this molecular effect, the TNC T-allele cooperates with the ACE D/D modulated vasoconstriction to blunt vascular remodeling and increased perfusion in back muscles after exercise training compared with other polymorphisms. There is evidence for a direct modulatory connection between the ACE enzyme and pain sensation through modification of the balance between the vasoconstrictive angiotensin peptide and pain-modulatory and vasodilatatory peptides of the kinin family, bradykinin and substance P (101, 102). For instance, ACE degrades substance P into its inactive form and ACE-regulated vasoconstriction exacerbates muscle pain by an acidosis-related process (103). There is in fact mounting evidence that acidosis could activate muscle pain sensation mediated by activation of acid-sensing ion channels (104) and there is mounting evidence that proton-sensing receptors are involved in the Transition from Acute to Chronic Pain (105).
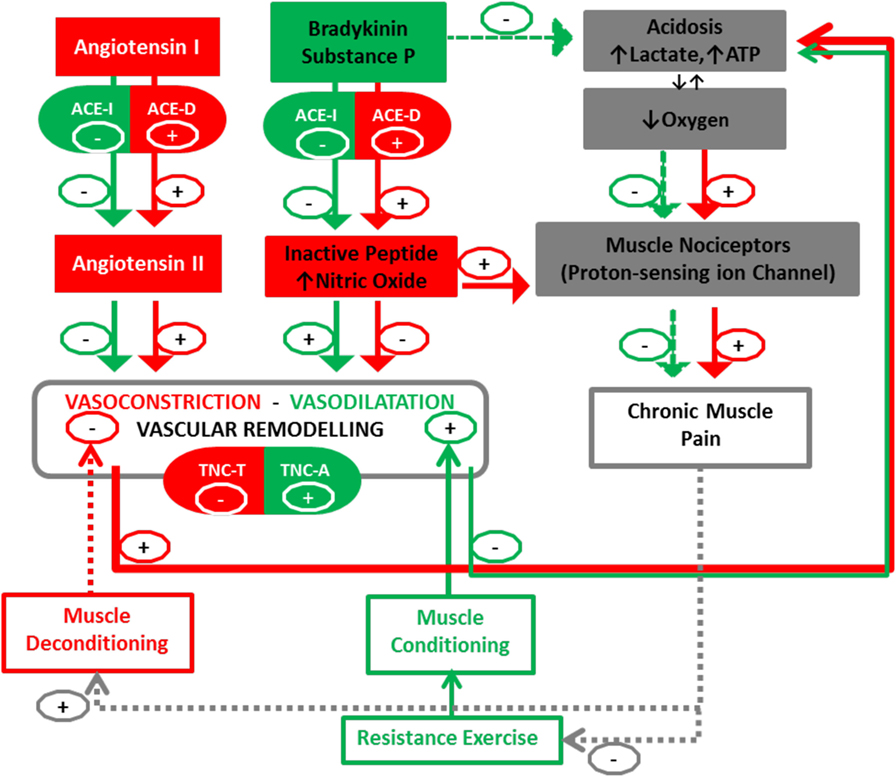
Figure 1. Peripheral blood perfusion in relationship with angiogenic factors (ACE and TNC) and chronic muscle pain. Interactions between muscle deconditioning/conditioning responses and the Angio-adaptation process. Arrows (+) indicate promotion and Arrows (−) indicate reduction. Documented interactions are fully colored in green and red, while dotted lines represent our postulated interactions in this pathway. ACE I, angiotensin-converting enzyme insertion; ACE D, angiotensin-converting enzyme deletion; TNC, Tenascin-C (rs2104772); A, adenine; T, thymine. The TNC-T and ACE-D polymorphism may result in a less-efficient vascular remodeling-perfusion and exacerbation of muscle pain by an increase of nitric oxide (NO) and acidosis-related process.
Intriguingly, substance P signaling was recently pointed out to exert an anti-nociceptive role through modulation of activation potential threshold in certain muscle nociceptors as well (103). While the underlying patho-physiological process remains to be disentangled, ACE-mediated inhibition of substance P emerges as a potentially important factor affecting pain sensation in nsCLBP.
However, there is evidence that repeated nociceptive stimuli can alter peripheral and/or central sensitization, resulting in the chronicity of pain with both nociceptive and neuropathic pain components (106). Combination pharmacotherapy is often necessary in patients with CLBP, in order to manage both nociceptive and neuropathic pain (107).
It is noteworthy to highlight that the first line therapy in CLBP recommend the use of vasoactive drugs. Acetaminophen (paracetamol) and non-steroidal anti-inflammatory drugs “NSAIDs” or opioids are common drugs and have widespread use for acute and chronic musculoskeletal disorders. The advantage is a peripheral effect in alleviating ischemic pain by independent mechanisms of vasodilation that increase blood flow in nerve and in muscle tissue.
The postulated connection of CLBP and lower muscle perfusion provides a conceptual framework to develop a treatment that tackles the pain generating mechanism with a personalized approach.
Toward this end, studies should first investigate the association between CLBP and polymorphisms for use-dependent angiogenic factor expression that may predict the CLBP pathology. Successively, investigations should gain insights into such polymorphisms and their significant role in vascular muscle remodeling in response to exercise training. From here, potential tailored exercise programs should be identified, aimed at improving strength and fatigue resistance of the back muscles in order to likely protect or minimize the effects of injurious events and reduce sensitivity to pain.
PV conceived the manuscript. PV, MFr, and MFl contributed to the writing of the manuscript. PV, MFr, CG, and MFl edited and produced the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LT and the handling editor declared their shared affiliation.
1. GBD 2013 DALYs and HALE Collaborators, Murray CJ, Barber RM, Foreman KJ, Abbasoglu Ozgoren A, Abd-Allah F, et al. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990–2013: quantifying the epidemiological transition. Lancet (2015) 386(10009):2145–91. doi:10.1016/S0140-6736(15)61340-X
2. Conway R, Behennah J, Fisher J, Osborne N, Steele J. Associations between trunk extension endurance and isolated lumbar extension strength in both asymptomatic participants and those with chronic low back pain. Healthcare (Basel) (2016) 4(3):E70. doi:10.3390/healthcare4030070
3. Deyo RA, Weinstein JN. Low back pain. N Engl J Med (2001) 344(5):363–70. doi:10.1056/NEJM200102013440508
4. Maher C, Underwood M, Buchbinder R. Non-specific low back pain. Lancet (2017) 389(10070):736–47. doi:10.1016/S0140-6736(16)30970-9
5. Balague F, Mannion AF, Pellise F, Cedraschi C. Non-specific low back pain. Lancet (2012) 379(9814):482–91. doi:10.1016/S0140-6736(11)60610-7
6. Coste J, Schiano PJ, Leparc JM, Paolaggi JB. Clinical and psychological classification of non-specific low-back pain. A new study in primary care practice. Rev Epidemiol Sante Publique (1995) 43(2):127–38.
7. Andersson GB. Epidemiological features of chronic low-back pain. Lancet (1999) 354(9178):581–5. doi:10.1016/S0140-6736(99)01312-4
8. Musculoskeletal Disorders and the Workplace: Low Back and Upper Extremities. Washington, DC: National Academies Press (2001). Avaliable from https://www.ncbi.nlm.nih.gov/books/NBK222440/
9. Cholewicki J, McGill SM. Mechanical stability of the in vivo lumbar spine: implications for injury and chronic low back pain. Clin Biomech (Bristol, Avon) (1996) 11(1):1–15. doi:10.1016/0268-0033(95)00035-6
11. Mayer TG, Barnes D, Nichols G, Kishino ND, Coval K, Piel B, et al. Progressive isoinertial lifting evaluation. II. A comparison with isokinetic lifting in a disabled chronic low-back pain industrial population. Spine (Phila Pa 1976) (1988) 13(9):998–1002. doi:10.1097/00007632-198809000-00006
12. Bousema EJ, Verbunt JA, Seelen HA, Vlaeyen JW, Knottnerus JA. Disuse and physical deconditioning in the first year after the onset of back pain. Pain (2007) 130(3):279–86. doi:10.1016/j.pain.2007.03.024
13. Kravitz E, Moore ME, Glaros A. Paralumbar muscle activity in chronic low back pain. Arch Phys Med Rehabil (1981) 62(4):172–6.
14. Mannion AF, Kaser L, Weber E, Rhyner A, Dvorak J, Muntener M. Influence of age and duration of symptoms on fibre type distribution and size of the back muscles in chronic low back pain patients. Eur Spine J (2000) 9(4):273–81. doi:10.1007/s005860000189
15. Steele J, Bruce-Low S, Smith D. A reappraisal of the deconditioning hypothesis in low back pain: review of evidence from a triumvirate of research methods on specific lumbar extensor deconditioning. Curr Med Res Opin (2014) 30(5):865–911. doi:10.1185/03007995.2013.875465
16. Verbunt JA, Seelen HA, Vlaeyen JW, van de Heijden GJ, Heuts PH, Pons K, et al. Disuse and deconditioning in chronic low back pain: concepts and hypotheses on contributing mechanisms. Eur J Pain (2003) 7(1):9–21. doi:10.1016/S1090-3801(02)00071-X
17. Verbunt JA, Smeets RJ, Wittink HM. Cause or effect? Deconditioning and chronic low back pain. Pain (2010) 149(3):428–30. doi:10.1016/j.pain.2010.01.020
18. Smeets RJ, Vlaeyen JW, Kester AD, Knottnerus JA. Reduction of pain catastrophizing mediates the outcome of both physical and cognitive-behavioral treatment in chronic low back pain. J Pain (2006) 7(4):261–71. doi:10.1016/j.jpain.2005.10.011
19. Brumagne S, Cordo P, Lysens R, Verschueren S, Swinnen S. The role of paraspinal muscle spindles in lumbosacral position sense in individuals with and without low back pain. Spine (Phila Pa 1976) (2000) 25(8):989–94. doi:10.1097/00007632-200004150-00015
20. Fortin M, Macedo LG. Multifidus and paraspinal muscle group cross-sectional areas of patients with low back pain and control patients: a systematic review with a focus on blinding. Phys Ther (2013) 93(7):873–88. doi:10.2522/ptj.20120457
21. Thomason DB, Booth FW. Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol (1985) (1990) 68(1):1–12. doi:10.1152/jappl.1990.68.1.1
22. Stein TP, Wade CE. Metabolic consequences of muscle disuse atrophy. J Nutr (2005) 135(7):1824S–8S. doi:10.1093/jn/135.7.1824S
23. Harridge SD. Plasticity of human skeletal muscle: gene expression to in vivo function. Exp Physiol (2007) 92(5):783–97. doi:10.1113/expphysiol.2006.036525
24. Bogdanis GC. Effects of physical activity and inactivity on muscle fatigue. Front Physiol (2012) 3:142. doi:10.3389/fphys.2012.00142
25. Bell KE, von Allmen MT, Devries MC, Phillips SM. Muscle disuse as a pivotal problem in sarcopenia-related muscle loss and dysfunction. J Frailty Aging (2016) 5(1):33–41. doi:10.14283/jfa.2016.78
26. Verbunt JA, Westerterp KR, van der Heijden GJ, Seelen HA, Vlaeyen JW, Knottnerus JA. Physical activity in daily life in patients with chronic low back pain. Arch Phys Med Rehabil (2001) 82(6):726–30. doi:10.1053/apmr.2001.23182
27. Kader DF, Wardlaw D, Smith FW. Correlation between the MRI changes in the lumbar multifidus muscles and leg pain. Clin Radiol (2000) 55(2):145–9. doi:10.1053/crad.1999.0340
28. Kamaz M, Kiresi D, Oguz H, Emlik D, Levendoglu F. CT measurement of trunk muscle areas in patients with chronic low back pain. Diagn Interv Radiol (2007) 13(3):144–8.
29. Barker KL, Shamley DR, Jackson D. Changes in the cross-sectional area of multifidus and psoas in patients with unilateral back pain: the relationship to pain and disability. Spine (Phila Pa 1976) (2004) 29(22):E515–9. doi:10.1097/01.brs.0000144405.11661.eb
30. Hides J, Gilmore C, Stanton W, Bohlscheid E. Multifidus size and symmetry among chronic LBP and healthy asymptomatic subjects. Man Ther (2008) 13(1):43–9. doi:10.1016/j.math.2006.07.017
31. Wallwork TL, Stanton WR, Freke M, Hides JA. The effect of chronic low back pain on size and contraction of the lumbar multifidus muscle. Man Ther (2009) 14(5):496–500. doi:10.1016/j.math.2008.09.006
32. Danneels LA, Cools AM, Vanderstraeten GG, Cambier DC, Witvrouw EE, Bourgois J, et al. The effects of three different training modalities on the cross-sectional area of the paravertebral muscles. Scand J Med Sci Sports (2001) 11(6):335–41. doi:10.1034/j.1600-0838.2001.110604.x
33. Danneels LA, Vanderstraeten GG, Cambier DC, Witvrouw EE, Bourgois J, Dankaerts W, et al. Effects of three different training modalities on the cross sectional area of the lumbar multifidus muscle in patients with chronic low back pain. Br J Sports Med (2001) 35(3):186–91. doi:10.1136/bjsm.35.3.186
34. Danneels LA, Vanderstraeten GG, Cambier DC, Witvrouw EE, De Cuyper HJ. CT imaging of trunk muscles in chronic low back pain patients and healthy control subjects. Eur Spine J (2000) 9(4):266–72. doi:10.1007/s005860000190
35. Fortin M, Gibbons LE, Videman T, Battie MC. Do variations in paraspinal muscle morphology and composition predict low back pain in men? Scand J Med Sci Sports (2015) 25(6):880–7. doi:10.1111/sms.12301
36. Kalichman L, Carmeli E, Been E. The association between imaging parameters of the paraspinal muscles, spinal degeneration, and low back pain. Biomed Res Int (2017) 2017:2562957. doi:10.1155/2017/2562957
37. Wan Q, Lin C, Li X, Zeng W, Ma C. MRI assessment of paraspinal muscles in patients with acute and chronic unilateral low back pain. Br J Radiol (2015) 88(1053):20140546. doi:10.1259/bjr.20140546
38. Huang Q, Zhang Y, Li D, Yang D, Huo M, Maruyama H. The evaluation of chronic low back pain by determining the ratio of the lumbar multifidus muscle cross-sectional areas of the unaffected and affected sides. J Phys Ther Sci (2014) 26(10):1613–4. doi:10.1589/jpts.26.1613
39. Goubert D, Oosterwijck JV, Meeus M, Danneels L. Structural changes of lumbar muscles in non-specific low back pain: a systematic review. Pain Physician (2016) 19(7):E985–1000.
40. Clark BC, Walkowski S, Conatser RR, Eland DC, Howell JN. Muscle functional magnetic resonance imaging and acute low back pain: a pilot study to characterize lumbar muscle activity asymmetries and examine the effects of osteopathic manipulative treatment. Osteopath Med Prim Care (2009) 3:7. doi:10.1186/1750-4732-3-7
41. D’Hooge R, Cagnie B, Crombez G, Vanderstraeten G, Achten E, Danneels L. Lumbar muscle dysfunction during remission of unilateral recurrent nonspecific low-back pain: evaluation with muscle functional MRI. Clin J Pain (2013) 29(3):187–94. doi:10.1097/AJP.0b013e31824ed170
42. Teichtahl AJ, Urquhart DM, Wang Y, Wluka AE, O’Sullivan R, Jones G, et al. Physical inactivity is associated with narrower lumbar intervertebral discs, high fat content of paraspinal muscles and low back pain and disability. Arthritis Res Ther (2015) 17:114. doi:10.1186/s13075-015-0629-y
43. D’Hooge R, Cagnie B, Crombez G, Vanderstraeten G, Dolphens M, Danneels L. Increased intramuscular fatty infiltration without differences in lumbar muscle cross-sectional area during remission of unilateral recurrent low back pain. Man Ther (2012) 17(6):584–8. doi:10.1016/j.math.2012.06.007
44. Mannion AF. Fibre type characteristics and function of the human paraspinal muscles: normal values and changes in association with low back pain. J Electromyogr Kinesiol (1999) 9(6):363–77. doi:10.1016/S1050-6411(99)00010-3
45. Mannion AF, Dumas GA, Stevenson JM, Cooper RG. The influence of muscle fiber size and type distribution on electromyographic measures of back muscle fatigability. Spine (Phila Pa 1976) (1998) 23(5):576–84. doi:10.1097/00007632-199803010-00010
46. Cho KH, Beom JW, Lee TS, Lim JH, Lee TH, Yuk JH. Trunk muscles strength as a risk factor for nonspecific low back pain: a pilot study. Ann Rehabil Med (2014) 38(2):234–40. doi:10.5535/arm.2014.38.2.234
47. Payne N, Gledhill N, Katzmarzyk PT, Jamnik V. Health-related fitness, physical activity, and history of back pain. Can J Appl Physiol (2000) 25(4):236–49. doi:10.1139/h00-018
48. Suehiro T, Mizutani M, Ishida H, Kobara K, Osaka H, Watanabe S. Individuals with chronic low back pain demonstrate delayed onset of the back muscle activity during prone hip extension. J Electromyogr Kinesiol (2015) 25(4):675–80. doi:10.1016/j.jelekin.2015.04.013
49. Dankaerts W, O’Sullivan P, Burnett A, Straker L. Differences in sitting postures are associated with nonspecific chronic low back pain disorders when patients are subclassified. Spine (Phila Pa 1976) (2006) 31(6):698–704. doi:10.1097/01.brs.0000202532.76925.d2
50. Teasell R, Dittmer DK. Complications of immobilization and bed rest. Part 2: other complications. Can Fam Physician (1993) 39:1440–2, 1445–6.
51. Dittmer DK, Teasell R. Complications of immobilization and bed rest. Part 1: musculoskeletal and cardiovascular complications. Can Fam Physician (1993) 39:1428–32, 1435–7.
52. Gogia P, Schneider VS, LeBlanc AD, Krebs J, Kasson C, Pientok C. Bed rest effect on extremity muscle torque in healthy men. Arch Phys Med Rehabil (1988) 69(12):1030–2.
53. Cassisi JE, Robinson ME, O’Conner P, MacMillan M. Trunk strength and lumbar paraspinal muscle activity during isometric exercise in chronic low-back pain patients and controls. Spine (Phila Pa 1976) (1993) 18(2):245–51. doi:10.1097/00007632-199302000-00012
54. Steele J, Bruce-Low S. Steiger et al. 2011: relationships and specificity in CLBP rehabilitation through exercise. Eur Spine J (2012) 21(9):1887; author reply 8–9. doi:10.1007/s00586-012-2449-y
55. Steele J, Bruce-Low S, Smith D. A review of the clinical value of isolated lumbar extension resistance training for chronic low back pain. PM R (2015) 7(2):169–87. doi:10.1016/j.pmrj.2014.10.009
56. Carpenter DM, Nelson BW. Low back strengthening for the prevention and treatment of low back pain. Med Sci Sports Exerc (1999) 31(1):18–24. doi:10.1097/00005768-199901000-00005
57. Rainville J, Hartigan C, Martinez E, Limke J, Jouve C, Finno M. Exercise as a treatment for chronic low back pain. Spine J (2004) 4(1):106–15. doi:10.1016/S1529-9430(03)00174-8
58. Durmus D, Durmaz Y, Canturk F. Effects of therapeutic ultrasound and electrical stimulation program on pain, trunk muscle strength, disability, walking performance, quality of life, and depression in patients with low back pain: a randomized-controlled trial. Rheumatol Int (2010) 30(7):901–10. doi:10.1007/s00296-009-1072-7
59. Rowell LB. Ideas about control of skeletal and cardiac muscle blood flow (1876–2003): cycles of revision and new vision. J Appl Physiol (1985) (2004) 97(1):384–92. doi:10.1152/japplphysiol.01220.2003
60. Joyner MJ, Casey DP. Regulation of increased blood flow (hyperemia) to muscles during exercise: a hierarchy of competing physiological needs. Physiol Rev (2015) 95(2):549–601. doi:10.1152/physrev.00035.2013
61. Hellsten Y. Limitations of skeletal muscle oxygen supply in ageing. J Physiol (2016) 594(8):2259–60. doi:10.1113/JP272062
62. Mense S. Muscle pain: mechanisms and clinical significance. Dtsch Arztebl Int (2008) 105(12):214–9. doi:10.3238/artzebl.2008.0214
63. Snijders T, Nederveen JP, Joanisse S, Leenders M, Verdijk LB, van Loon LJ, et al. Muscle fibre capillarization is a critical factor in muscle fibre hypertrophy during resistance exercise training in older men. J Cachexia Sarcopenia Muscle (2017) 8(2):267–76. doi:10.1002/jcsm.12137
64. Vincent MA, Clerk LH, Rattigan S, Clark MG, Barrett EJ. Active role for the vasculature in the delivery of insulin to skeletal muscle. Clin Exp Pharmacol Physiol (2005) 32(4):302–7. doi:10.1111/j.1440-1681.2005.04188.x
65. Dinenno FA, Seals DR, DeSouza CA, Tanaka H. Age-related decreases in basal limb blood flow in humans: time course, determinants and habitual exercise effects. J Physiol (2001) 531(Pt 2):573–9. doi:10.1111/j.1469-7793.2001.0573i.x
66. Fluck M, Hoppeler H. Molecular basis of skeletal muscle plasticity – from gene to form and function. Rev Physiol Biochem Pharmacol (2003) 146:159–216. doi:10.1007/s10254-002-0004-7
67. Kauppila LI. Atherosclerosis and disc degeneration/low-back pain – a systematic review. Eur J Vasc Endovasc Surg (2009) 37(6):661–70. doi:10.1016/j.ejvs.2009.02.006
68. Kurunlahti M, Karppinen J, Haapea M, Niinimaki J, Autio R, Vanharanta H, et al. Three-year follow-up of lumbar artery occlusion with magnetic resonance angiography in patients with sciatica: associations between occlusion and patient-reported symptoms. Spine (Phila Pa 1976) (2004) 29(16):1804–8; discussion 9. doi:10.1097/01.BRS.0000134576.77709.64
69. Song J, Lee JB, Suh JK. Clinicopathological considerations in patients with lumbosacral extraforaminal stenosis. J Clin Neurosci (2009) 16(5):650–4. doi:10.1016/j.jocn.2008.06.003
70. Liu JT, Liao WJ, Chang CS, Chen YH. Lower limb pain caused by insufficient muscular microcirculation. Indian J Surg (2014) 76(1):70–5. doi:10.1007/s12262-012-0628-z
71. Datta G, Gnanalingham KK, Peterson D, Mendoza N, O’Neill K, Van Dellen J, et al. Back pain and disability after lumbar laminectomy: is there a relationship to muscle retraction? Neurosurgery (2004) 54(6):1413–20; discussion 20. doi:10.1227/01.NEU.0000124751.57121.A6
72. Paryavi E, Jobin CM, Ludwig SC, Zahiri H, Cushman J. Acute exertional lumbar paraspinal compartment syndrome. Spine (Phila Pa 1976) (2010) 35(25):E1529–33. doi:10.1097/BRS.0b013e3181ec4023
73. Olivier N, Thevenon A, Berthoin S, Prieur F. An exercise therapy program can increase oxygenation and blood volume of the erector spinae muscle during exercise in chronic low back pain patients. Arch Phys Med Rehabil (2013) 94(3):536–42. doi:10.1016/j.apmr.2012.10.028
74. Kell RT, Bhambhani Y. In vivo erector spinae muscle blood volume and oxygenation measures during repetitive incremental lifting and lowering in chronic low back pain participants. Spine (Phila Pa 1976) (2006) 31(22):2630–7. doi:10.1097/01.brs.0000240647.57959.72
75. Kovacs KM, Marras WS, Litsky AS, Gupta P, Ferguson SA. Localized oxygen use of healthy and low back pain individuals during controlled trunk movements. J Spinal Disord (2001) 14(2):150–8. doi:10.1097/00002517-200104000-00010
76. Olfert IM, Baum O, Hellsten Y, Egginton S. Advances and challenges in skeletal muscle angiogenesis. Am J Physiol Heart Circ Physiol (2016) 310(3):H326–36. doi:10.1152/ajpheart.00635.2015
77. Dapp C, Schmutz S, Hoppeler H, Fluck M. Transcriptional reprogramming and ultrastructure during atrophy and recovery of mouse soleus muscle. Physiol Genomics (2004) 20(1):97–107. doi:10.1152/physiolgenomics.00100.2004
78. Fraser GM, Morton JS, Schmidt SM, Bourque S, Davidge ST, Davenport MH, et al. Reduced uterine perfusion pressure decreases functional capillary density in skeletal muscle. Am J Physiol Heart Circ Physiol (2015) 309(12):H2002–7. doi:10.1152/ajpheart.00641.2015
79. Pelletier-Fleury N, Lanoe JL, Fleury B, Fardeau M. The cost of treating COPD patients with long-term oxygen therapy in a French population. Chest (1996) 110(2):411–6. doi:10.1378/chest.110.2.411
80. Hudlicka O. Development and adaptability of microvasculature in skeletal muscle. J Exp Biol (1985) 115:215–28.
81. Korthuis RJ. Skeletal Muscle Circulation. San Rafael, CA: Integrated Systems Physiology: From Molecule to Function to Disease (2011).
82. Giombini A, Giovannini V, Di Cesare A, Pacetti P, Ichinoseki-Sekine N, Shiraishi M, et al. Hyperthermia induced by microwave diathermy in the management of muscle and tendon injuries. Br Med Bull (2007) 83:379–96. doi:10.1093/bmb/ldm020
83. Hudlicka O. Growth of capillaries in skeletal and cardiac muscle. Circ Res (1982) 50(4):451–61. doi:10.1161/01.RES.50.4.451
84. Hudlicka O. What makes blood vessels grow? J Physiol (1991) 444:1–24. doi:10.1113/jphysiol.1991.sp018863
85. Hudlicka O, Price S. The role of blood flow and/or muscle hypoxia in capillary growth in chronically stimulated fast muscles. Pflugers Arch (1990) 417(1):67–72. doi:10.1007/BF00370770
86. Egginton S, Hudlicka O. Early changes in performance, blood flow and capillary fine structure in rat fast muscles induced by electrical stimulation. J Physiol (1999) 515(Pt 1):265–75. doi:10.1111/j.1469-7793.1999.265ad.x
87. Sottile J. Regulation of angiogenesis by extracellular matrix. Biochim Biophys Acta (2004) 1654(1):13–22. doi:10.1016/j.bbcan.2003.07.002
88. Dzau VJ, Gibbons GH. Endothelium and growth factors in vascular remodeling of hypertension. Hypertension (1991) 18(5 Suppl):III115–21. doi:10.1161/01.HYP.18.5_Suppl.III115
89. Rogers MS, D’Amato RJ. Common polymorphisms in angiogenesis. Cold Spring Harb Perspect Med (2012) 2(11):a006510. doi:10.1101/cshperspect.a006510
90. van Ginkel S, Ruoss S, Valdivieso P, Degens H, Waldron S, de Haan A, et al. ACE inhibition modifies exercise-induced pro-angiogenic and mitochondrial gene transcript expression. Scand J Med Sci Sports (2016) 26(10):1180–7. doi:10.1111/sms.12572
91. Mackie EJ, Scott-Burden T, Hahn AW, Kern F, Bernhardt J, Regenass S, et al. Expression of tenascin by vascular smooth muscle cells. Alterations in hypertensive rats and stimulation by angiotensin II. Am J Pathol (1992) 141(2):377–88.
92. Gorman JL, Liu ST, Slopack D, Shariati K, Hasanee A, Olenich S, et al. Angiotensin II evokes angiogenic signals within skeletal muscle through co-ordinated effects on skeletal myocytes and endothelial cells. PLoS One (2014) 9(1):e85537. doi:10.1371/journal.pone.0085537
93. Greene AS. Life and death in the microcirculation: a role for angiotensin II. Microcirculation (1998) 5(2–3):101–7. doi:10.1038/sj.mn.7300024
94. Hahn AW, Kern F, Jonas U, John M, Buhler FR, Resink TJ. Functional aspects of vascular tenascin-C expression. J Vasc Res (1995) 32(3):162–74. doi:10.1159/000159090
95. Mustafa DA, Dekker LJ, Stingl C, Kremer A, Stoop M, Sillevis Smitt PA, et al. A proteome comparison between physiological angiogenesis and angiogenesis in glioblastoma. Mol Cell Proteomics (2012) 11(6):M111008466. doi:10.1074/mcp.M111.008466
96. Vaughan D, Huber-Abel FA, Graber F, Hoppeler H, Fluck M. The angiotensin converting enzyme insertion/deletion polymorphism alters the response of muscle energy supply lines to exercise. Eur J Appl Physiol (2013) 113(7):1719–29. doi:10.1007/s00421-012-2583-6
97. Valdivieso P, Vaughan D, Laczko E, Brogioli M, Waldron S, Rittweger J, et al. The metabolic response of skeletal muscle to endurance exercise is modified by the ACE-I/D gene polymorphism and training state. Front Physiol (2017) 8:993. doi:10.3389/fphys.2017.00993
98. Valdivieso P, Toigo M, Hoppeler H, Fluck M. T/T homozygosity of the tenascin-C gene polymorphism rs2104772 negatively influences exercise-induced angiogenesis. PLoS One (2017) 12(4):e0174864. doi:10.1371/journal.pone.0174864
99. Saunders CJ, van der Merwe L, Posthumus M, Cook J, Handley CJ, Collins M, et al. Investigation of variants within the COL27A1 and TNC genes and Achilles tendinopathy in two populations. J Orthop Res (2013) 31(4):632–7. doi:10.1002/jor.22278
100. Matsuda A, Hirota T, Akahoshi M, Shimizu M, Tamari M, Miyatake A, et al. Coding SNP in tenascin-C Fn-III-D domain associates with adult asthma. Hum Mol Genet (2005) 14(19):2779–86. doi:10.1093/hmg/ddi311
101. Campbell DJ. Angiotensin converting enzyme (ACE) inhibitors and kinin metabolism: evidence that ACE inhibitors may inhibit a kininase other than ACE. Clin Exp Pharmacol Physiol (1995) 22(12):903–11. doi:10.1111/j.1440-1681.1995.tb02325.x
102. Steckelings UM, Artuc M, Wollschlager T, Wiehstutz S, Henz BM. Angiotensin-converting enzyme inhibitors as inducers of adverse cutaneous reactions. Acta Derm Venereol (2001) 81(5):321–5. doi:10.1080/000155501317140007
103. Lin CC, Chen WN, Chen CJ, Lin YW, Zimmer A, Chen CC. An antinociceptive role for substance P in acid-induced chronic muscle pain. Proc Natl Acad Sci U S A (2012) 109(2):E76–83. doi:10.1073/pnas.1108903108
104. Pollak KA, Swenson JD, Vanhaitsma TA, Hughen RW, Jo D, White AT, et al. Exogenously applied muscle metabolites synergistically evoke sensations of muscle fatigue and pain in human subjects. Exp Physiol (2014) 99(2):368–80. doi:10.1113/expphysiol.2013.075812
105. Sun WH, Chen CC. Roles of proton-sensing receptors in the transition from acute to chronic pain. J Dent Res (2016) 95(2):135–42. doi:10.1177/0022034515618382
106. Arendt-Nielsen L, Graven-Nielsen T. Central sensitization in fibromyalgia and other musculoskeletal disorders. Curr Pain Headache Rep (2003) 7(5):355–61. doi:10.1007/s11916-003-0034-0
Keywords: non-specific chronic low back pain, muscle perfusion, gene polymorphisms, pain sensation, therapeutics
Citation: Valdivieso P, Franchi MV, Gerber C and Flück M (2018) Does a Better Perfusion of Deconditioned Muscle Tissue Release Chronic Low Back Pain? Front. Med. 5:77. doi: 10.3389/fmed.2018.00077
Received: 29 November 2017; Accepted: 07 March 2018;
Published: 20 March 2018
Edited by:
Luigi M. Terracciano, Universität Basel, SwitzerlandReviewed by:
Luigi Tornillo, Universität Basel, SwitzerlandCopyright: © 2018 Valdivieso, Franchi, Gerber and Flück. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Valdivieso, cGFvbGEudmFsZGl2aWVzb0BiYWxncmlzdC5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.