 Monika Pathak
Monika Pathak Bubacarr Gibril Kaira
Bubacarr Gibril Kaira Alexandre Slater
Alexandre Slater Jonas Emsley
Jonas Emsley- Centre for Biomolecular Sciences, School of Pharmacy, University of Nottingham, Nottingham, United Kingdom
The contact activation system (CAS) or contact pathway is central to the crosstalk between coagulation and inflammation and contributes to diverse disorders affecting the cardiovascular system. CAS initiation contributes to thrombosis but is not required for hemostasis and can trigger plasma coagulation via the intrinsic pathway [through factor XI (FXI)] and inflammation via bradykinin release. Activation of factor XII (FXII) is the principal starting point for the cascade of proteolytic cleavages involving FXI, prekallikrein (PK), and cofactor high molecular weight kininogen (HK) but the precise location and cell receptor interactions controlling these reactions remains unclear. FXII, PK, FXI, and HK utilize key protein domains to mediate binding interactions to cognate cell receptors and diverse ligands, which regulates protease activation. The assembly of contact factors has been demonstrated on the cell membranes of a variety of cell types and microorganisms. The cooperation between the contact factors and endothelial cells, platelets, and leukocytes contributes to pathways driving thrombosis yet the basis of these interactions and the relationship with activation of the contact factors remains undefined. This review focuses on cell receptor interactions of contact proteins and FXI to develop a cell-based model for the regulation of contact activation.
Introduction
The contact activation system (CAS) includes serine proteases factor XII (FXII), plasma prekallikrein (PK), coagulation factor XI (FXI), and high molecular weight kininogen (HK) which is the non-enzymatic cofactor of FXI and PK (1, 2). The CAS is thought to be central to crosstalk between coagulation and inflammation and the underlying cause for various disorders affecting the cardiovascular system (1, 3). Two branches of the CAS have been identified as (i) the inflammatory branch activates contact factors FXII and PK on the surface of endothelial cells resulting in release of the peptide bradykinin (BK) and (ii) plasma coagulation branch activates FXII and FXI on the surface of platelets (Figure 1) (4–6). Contact of FXII with diverse negatively charged activators leads to a change in the conformation of FXII that subsequently generates activated FXII (FXIIa) in trivial amounts (7–10). FXIIa then activates PK to form active kallikrein (PKa) (11). Reciprocal activation of FXII by PKa and PK by FXIIa occurs and subsequently PKa proteolytically liberates BK via cleavage of its precursor HK. Binding of BK to its receptor on endothelial cells results in activation of several pro-inflammatory signaling pathways leading to vasodilation, pain, and neutrophil chemotaxis (12, 13). CAS factors also participate in fibrinolytic and angiogenic pathways (14, 15).
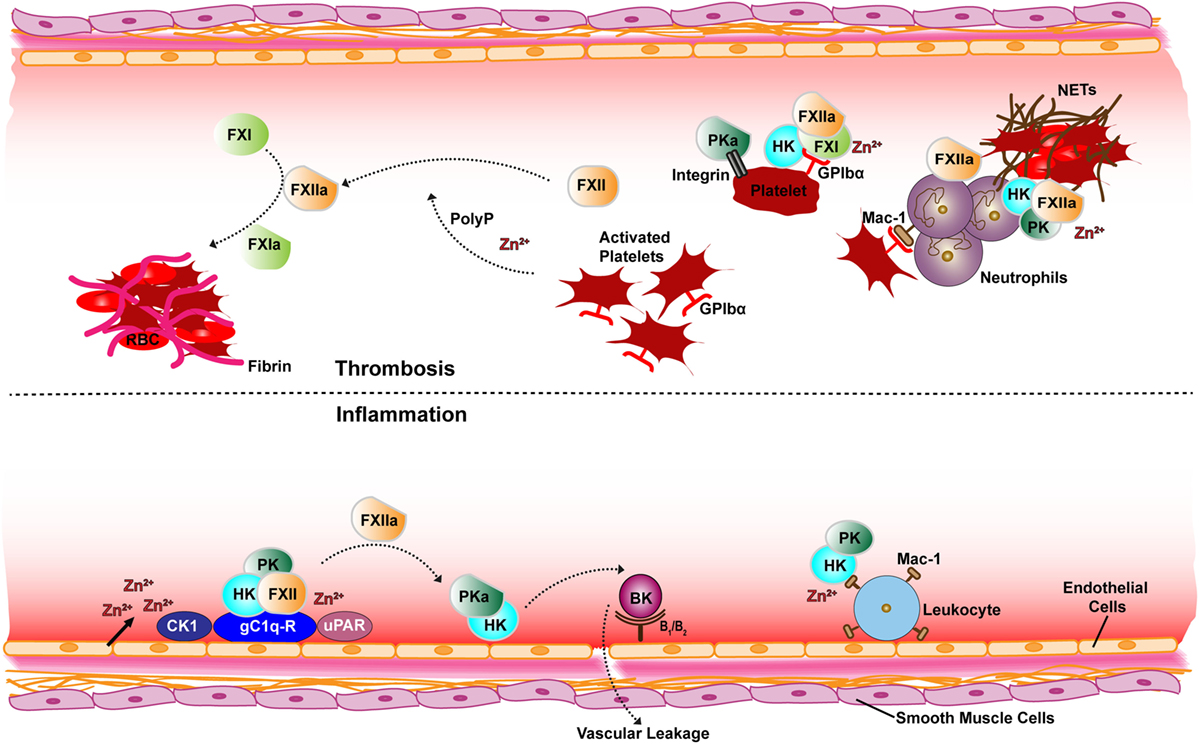
Figure 1. Schematic overview of receptor and cofactor interactions of the contact factors for thrombotic (top) and inflammatory (bottom) pathways. Assembly of the contact system via gC1q-R (with elevated Zn2+) generates PKa, FXIIa, and BK is produced on the surface of endothelial cells (bottom). Also shown is the FXII and HK interactions with the uPAR receptor and CK1. FXIIa activates plasma coagulation cascades via FXI on the surface of platelets (top). The platelet GP1b-IX receptor GP1bα chain interaction with FXI–HK and PKa binding to the activated platelet integrin αIIbβ3 is also depicted. Neutrophils are also depicted releasing NETs known to associate with contact proteins. Abbreviations: FXII, factor XII; FXIIa, activated FXII; PK, prekallikrein; PKa, activated PK; FXI, factor XI; FXIa, activated FXI; HK, high molecular-weight kininogen; BK, bradykinin; uPAR, urokinase receptor; GPIbα, platelet glycoprotein Ib; CK1, cytokeratin 1; gC1q-R, receptor for complement protein C1q; Mac-1, macrophage-1 antigen receptor; PolyP, polyphosphate; RBC, red blood cells; NETs, neutrophil extracellular traps.
Contact activation is best known as the coagulation mechanism that is activated by artificial surfaces and is the basis of the widely used aPTT hemostatic assay (2). Negatively charged polymers including nucleic acids (DNA and RNA) and polyphosphate (PolyP) are activators of the contact pathway via FXII auto-activation (16–19). Platelet-derived PolyP is mainly secreted as short chain polymers following platelet activation and has been linked to FXII activation and thrombus formation (20). Activated platelets also retain PolyP on their cell surface (membrane-associated) assembled into nanoparticles that can potently activate FXII (20). Purified DNA and RNA have been shown to bind and activate contact factors and enhance thrombin generation and clot formation in plasma based studies (21, 22). Genetic knockout studies in murine models of cardiovascular disease and genetic linkage studies in humans have implicated the contact factors in contributing to diverse cardiovascular disease processes, including thrombosis (23–29), hypertension (30), atherosclerosis (31, 32), and stroke (33, 34).
CAS factors are not considered vital for normal hemostasis in vivo as evidenced by patients with FXII deficiency exhibiting no bleeding tendencies (35, 36). However, some cases of FXI-deficiency in humans manifest with a strong bleeding phenotype (37). The first clinical trial revealed prevention of venous thrombosis by targeting FXI without compromising normal hemostasis (38, 39). In humans identification of a gain-of-function mutation in the F12 gene (encodes FXII) was shown to be linked to aggressive attacks of tissue swelling in hereditary angioedema, a rare life threatening inherited edema disorder in which excessive formation of BK leads to recurrent episodes of acute swelling and increased vascular permeability (40, 41). In this context the presence of a hyperactive FXII mutant does not seem to cause thrombosis in these patients which is consistent with previous observations that the FXII–PK–HK branch and BK production can operate separately from FXI activation and plasma coagulation (40). What leads to this mechanistic uncoupling of FXIIa driven thrombosis and inflammation is unknown raising the question as to how the proteases are regulated and highlights the importance of understanding the precise cell receptors utilized for CAS regulation.
The contact factors each have individual properties including recognizing foreign substances and interacting with different cell types (29, 32, 42, 43) and bacteria (44–46). CAS has been implicated in viral pathogenesis (47) and as a component of the innate immune response (48). The assembly of contact factors has been demonstrated on the membranes of a variety of cell types and in this review we summarize the known cell receptor and cofactor interactions of contact factors and FXI.
Contact Factor Domain Structure
It is well established that the vitamin K-dependant coagulation factors, such as pro-thrombin, factor X, and factor IX (FIX), utilize Gla domains and calcium ions (Ca2+) to recognize the phosphatidylserine exposed on activated cell membranes and these interactions control efficient proteolytic cleavage and activation of these factors on a specific surface (49, 50). Contact factors FXII, PK, and FXI do not have Gla domains but instead utilize key protein modules of apple and fibronectin-like domains to mediate binding interactions to cognate cell receptors and diverse ligands, which appropriately regulate their substrate recognition and enzyme activation. Another key difference is that zinc (Zn2+) ions are implicated as critical to the function of the contact factors as opposed to Ca2+ ions, which are required for Gla domain structure and binding to cell membranes.
Factor XII
Factor XII is a 80 kDa glycosylated protein consisting of a single polypeptide chain and circulates in plasma as a zymogen with a concentration of 40 μg/ml (375 nM). Upon contact with anionic surfaces, in the presence of Zn2+ ions, FXII undergoes a conformational rearrangement leading to auto-activation or cleavage in trans by kallikrein to generate FXIIa. FXIIa consists of two chains; an N-terminal 52-kDa heavy chain and a C-terminal 28-kDa serine protease domain (51), linked together by a disulfide bond. The domain structure of the FXII heavy chain is composed of an N-terminal fibronectin type-II domain (FnII), an epidermal growth factor like domain (EGFI), a fibronectin type-I domain (FnI), a second EGF-like domain (EGFII), a kringle domain, and a distinctive proline-rich domain (52). The FXII heavy chain mediates binding to Zn2+ ions and negatively charged surfaces (53–55). FXII has been shown to bind to the urokinase receptor (uPAR) and platelet glycoprotein Ib (GPIb-IX) receptor (56, 57).
Prekallikrein
Prekallikrein is a glycoprotein of molecular weight 88 kDa consisting of a single polypeptide chain that circulates in plasma as a zymogen at a concentration of 50 μg/ml (490 nM), with an estimated 75% bound non covalently to HK (58). FXIIa or β-FXIIa (the isolated protease domain fragment) cleaves PK resulting in a two-chain enzyme kallikrein (PKa), consisting of a 52-kDa heavy chain and 33–36 kDa light chain corresponding to the serine protease domain and both chains are linked together by a disulfide bond. PK shares 58% amino acid sequence identity with FXI and both proteins have the characteristic feature of four apple domains (A1–A4) (59, 60). The HK binding region on PK is localized in the central portion of the A2 domain with possible binding sites in other apple domains (61). PK binds to endothelial cells, platelets, and granulocytes in a Zn2+-dependent interaction via the PK–HK complex (2).
High Molecular Weight Kininogen
High molecular weight kininogen is a 120 kDa non-enzymatic glycoprotein with a plasma concentration of 80 μg/ml (670 nM). Granulocytes, platelets, and endothelial cells contain HK, but plasma HK is most likely synthesized in the liver. HK is made up of six domains (D1–D6) and cleavage of HK by kallikrein PKa results in HKa, a two-chain protein consisting of a heavy chain (D1–D3; 64 kDa) and a light chain (D5–D6; 56 kDa) releasing the short BK peptide (D4) (62). The D6 domain has binding sites for PK and FXI (63). HK binds to cell surfaces in a Zn2+-dependent manner (64, 65). Both D5 and D3 domain mediate cell receptor binding of HK to endothelial cells, platelets, and neutrophils (62, 64, 66, 67). HK is constitutively bound to cell surfaces and mediates not only recruitment of PK and FXI to cell membranes but also functions to enhance interactions between different cell types (68, 69).
Factor XI
Factor XI is a dimer of 80 kDa subunits that circulates in plasma at a concentration of 5 μg/ml (30 nM) tightly bound in a non-covalent complex with HK (70). Activation of FXI by thrombin or α-FXIIa yields FXIa that consists of a heavy chain of four apple domains (A1-A4) and a light chain of the catalytic serine protease domain covalently linked together via a disulfide bond. Apple domains A1 and A2 contain a binding site for thrombin and HK, respectively (50, 59). A3 has a FIX, heparin, and GPIb-IX binding site and A4 contains a cysteine residue that forms the disulfide bond needed for FXI dimer formation (42). FXI–HK complex binding to platelets has been reported to occur via the GPIb-IX receptor (71).
Cell Interactions of Contact Factors
The interaction of contact factors with different cell types has been shown to cooperatively contribute to thrombotic pathways in animal models (29). Despite decades of research investigating in vitro characterization of CAS activation by polymers (15, 52), a detailed understanding of the receptors and cofactors that regulate CAS protease activation on cellular surfaces remains elusive. Figure 1 summarizes the key cell interactions of contact factors and below we consider each cell specific interaction in turn.
Endothelial Cells
The binding of the contact factors to endothelial cells is important to localize the production of the vasoactive peptide BK to the correct surface where its receptors are located. HK is bound tightly to PK and FXI but there is evidence that PK and FXI can also bind endothelial cells in the absence of HK (72, 73). One of the major endothelial cell receptors that binds HK and FXII is gC1q-R (receptor for the complement protein C1q) (74–76), also known as p32 or hyaluronic acid binding protein 1. Most human cell types, including lymphocytes, endothelial cells, and dendritic cells express gC1q-R. gC1q-R is a multi-compartmental, multi-ligand binding cellular protein localized predominantly in the mitochondria as well as the plasma membrane, cytosol, Golgi, endoplasmic reticulum, and the nucleus (77–79). gC1q-R is present in the mitochondrial matrix where it binds and inhibits cellular splicing factors (80) and is also observed to inhibit splicing in HIV transcripts (81). In Drosophila embryos a fly ortholog of gC1q-R was identified as a histone chaperone that exchanges protamines for histones (82). Human gC1q-R binds with high affinity to all subclasses of histones (H1, H2A, H2B, H3.1, and H4) and blocks the pathophysiological activities of histones in a murine model for histone-induced shock (83). gC1q-R also binds host-defense peptides (84). In endothelial cells, inflammatory mediators and lipopolysaccharides (LPS) upregulate expression of gC1q-R in a time and concentration-dependent manner (85). gC1q-R was first discovered as binding the globular heads of the complement protein C1q (74) and is implicated in diverse biological pathways, including adipogenesis and insulin signaling (86), regulation of RNA splicing (80), the proliferation of tumor cells (87) and atherosclerosis (88).
The interaction of gC1q-R (89) with both PK–HK and FXII on the surface of endothelial cells promotes the production of PKa and FXIIa as gC1q-R is an endogenous activator of CAS (90). Biochemically, the properties of gC1q-R are consistent with this as it is a highly anionic multimeric protein capable of binding to both FXII and HK (78, 91) in a Zn2+-dependent manner (74, 76). Binding studies calculated a KD of 0.7–0.8 nM for the interaction of HK with gC1q-R, and no difference in binding affinity was observed between HK and HKa (92). Previous studies suggest that FXII and HK compete with one another for binding to gC1q-R and for the same site on endothelial cells (43, 93, 94).
CAS can be regulated by gC1q-R as in the resting state gC1q-R is predominantly found in the cytoplasm and mitochondria and is only released to the cell surface upon activation of endothelial cells (95, 96). However, gC1q-R alone is not sufficient for full CAS activation as elevated Zn2+ ions are required for FXII recruitment to the complex. The origin of the Zn2+ has been shown to come from endothelial cells or activated platelets (43). Additional endothelial cell receptors for the contact factors are uPAR and cytokeratin 1 (CK1) (43, 69). These two proteins have been reported to form a multiprotein complex with gC1q-R on the surface of endothelial cells (Figure 2A), which is able to bind FXII (97). Additionally, the complex of uPAR with CK1 was shown to bind HK resulting in PK activation at the endothelial cell surface (98).
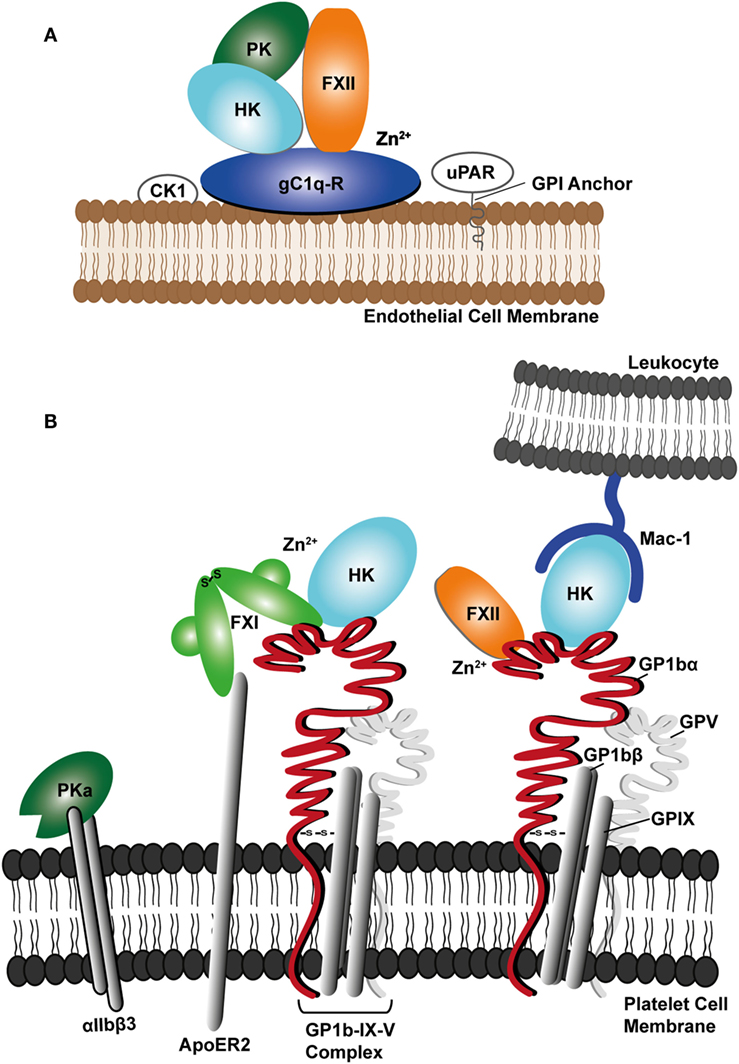
Figure 2. Schematic diagram showing (A) assembly of contact factors factor XII (FXII), prekallikrein (PK), and cofactor high molecular weight kininogen (HK) with the endothelial cell receptor complex involving the receptor for the complement protein C1q (gC1q-R), urokinase receptor (uPAR), and cytokeratin 1 (CK1). (B) Platelet receptor glycoprotein Ib complex (GPIb-IX-V) is shown forming a network of interactions with FXII, FXI, and HK and the Mac-1 receptor on leukocytes, where HK may bridge the two cell types. PKa is shown binding to the activated αIIbβ3 platelet integrin.
uPAR is a well characterized protease receptor and regulates the amount of active plasmin generated at the cell surface, through the interaction with urokinase (uPA) (99, 100), resulting in the degradation of fibrin fibers (101). Through this mechanism, uPAR has been linked with a number of biological processes, including cell migration, angiogenesis (102), tumor metastasis (103), and leukocyte migration (104). HK binding with uPAR inhibits endothelial cell migration and proliferation, and angiogenesis by disrupting the interaction of uPAR with uPA (69, 105, 106). While it is well established that HK/HKa binds to uPAR, Betapudi et al., (107) showed that the antiangiogenic effects of HK are mediated equally well in wild-type and uPAR-deficient mice, concluding that uPAR is not essential for inhibition of angiogenesis by HKa in vivo or for HKa-induced endothelial cell apoptosis in vitro. This study also failed to demonstrate an essential role for any of the previously known endothelial cell receptors for HK or HKa including the uPAR, gC1q-R, and CK1. Also, HKa inhibits angiogenesis via induction of apoptosis in proliferating endothelial cells and these effects were mediated mostly by HKa domain D5. A separate study showed an interaction between HKa domain D5 and endothelial tropomyosin underlies the antiangiogenic activity of HKa (108), indicating there is likely redundancy in the mechanisms whereby HK binds to cell surfaces.
A study using surface plasmon resonance has measured the binding of HK to these endothelial receptors revealing that HK binds with the greatest affinity to gC1q-R (0.8 nM), followed by CK1 (15 nM) and then uPAR (2.3 µM), each in a Zn2+-dependent manner (92). Both gC1q-R and CK1 showed no significant differences in binding affinity for HKa or HK, whereas uPAR bound 50-fold tighter to active HKa. It was, therefore, proposed that gC1q-R and CK1 are involved in the initial binding of HK to the cell surface where it is then cleaved by PKa. HKa is then able to selectively bind uPAR and mediate cell migration. As gC1q-R does not have a membrane anchor, additional interactions are likely critical to position the protein in the correct location on the endothelial cell surface to bind HK and FXII.
Platelets
GPIb-IX is the receptor on platelets for FXII (57), FXI (109) and HK (110, 111). GP1bα is a subunit of the GPIb–IX complex, which plays a prominent role in the initial steps of platelet adhesion (112). The GPIbα–FXI interaction has been demonstrated biochemically to be mediated by the FXI A3 domain in a Zn2+-dependent fashion (Kd ~ 52 nM). The Interaction is localized to the GPIbα N-terminal leucine-rich repeats at a site distinct to the GPIbα anionic region, and FXI binding was shown to compete with VWFA1 but not thrombin binding (71, 109, 113). Recent studies on the FXI-GPIbα receptor interactions describe a vascular coagulation and inflammatory circuit that overlaps with arterial hypertension pathways (30). The apolipoprotein E receptor 2 (ApoER2, LRP8) has also been identified as a platelet receptor for FXI (114). ApoER2 is a member of the low-density lipoprotein family of receptors and initiates platelet cell signaling through the disabled-1 adaptor protein (115). Due to the dimeric nature of FXI, it may simultaneously bind both GPIbα and ApoER2.
GPIbα is also the primary receptor on platelets for thrombin and thus plays a well characterized role in platelet activation (116). HK binding to platelets is mediated predominantly through GPIbα (117) and this binding has been shown to compete with the thrombin-binding site, which is localized to the GPIbα anionic region (118). HK has been shown to interact with GPIbα through the D3 domain and the D5 domain although the precise determinants of the interaction are not clear (117). A monoclonal antibody binding to the GPIX subunit also inhibited HK binding to platelets, indicating there may be a more extensive interaction beyond the GPIbα chain (117).
FXII binds to GPIbα and in a similar manner to HK and FXII has also been shown to inhibit the binding of thrombin (57). As both FXII and HK have domains with concentrated regions of positive charge, it is possible both recognize the GPIbα N-terminal domain anionic region in a similar fashion to the way thrombin utilizes its positively charged exosites to bind GPIbα (57, 117, 118). The platelet membrane is well known as a pro-coagulant surface and a component of this response is the feedback loop of thrombin to cleave and activate FXI (119). PK binds platelets and recently the activated enzyme PKa has been described binding to platelet integrin αIIbβ3 through its KGD and KGE motifs (Figure 2) (120). PKa (but not the zymogen) enhances ADP induced platelet activation by PAR-1 hydrolysis (120).
Leukocytes
HK, PK, FXII, and FXI have been shown to bind to the surface of neutrophils (121). Thus, release of BK on the neutrophil surface could enhance the passage of neutrophils out of the vasculature to mediate inflammatory responses (121). HK inhibits calpain (122), is involved in neutrophil aggregation (123), chemotaxis (124), and the release of elastase through degranulation (124, 125).
The major leukocyte cell receptor for HK is the activated integrin macrophage-1 antigen receptor (Mac-1) (66). HK binds to Mac-1 via the D3 and D5 domains (66, 126). Mac-1 is a multifunctional receptor expressed primarily on monocytes, macrophages, neutrophils, and natural killer cells. A variety of different ligands bind to this receptor including fibrinogen (66), intercellular adhesion molecule-1 (ICAM-1) (127) as well as HK (66). Mac-1 also mediates a variety of cell to cell interactions, including the neutrophil-platelet association involving interaction with GPIbα (128). HK mediates the adhesion of neutrophils to sites of fibrin formation and endothelial cells by inhibiting the interaction of Mac-1 with fibrinogen and ICAM-1 (129). Surfaces preadsorbed with HK are anti-adhesive to neutrophil binding (130).
Platelets and leukocytes interact and coordinate innate immune responses (131). The relationship between these two cell types and the CAS has been described as being involved in a pathway termed immuno-thrombosis that can contribute to thrombus formation in animal models of disease (132). Thus, it is of interest that HK binds both cell types and has been described as a molecular bridge between GPIbα and Mac-1 enhancing the interaction between cell types through these receptors (Figure 2B) (68). Mac-1 has also been linked to neutrophil extracellular traps (NETs) formation (133, 134). This promotes microbe entrapment by fibrin clots thus facilitating microbial clearance through the engagement of phagocytic cells and leukocytes by stimulating inflammation. Thus, HK may be a key coordinating cofactor for drawing together the CAS plasma proteins and different cell types in an innate immune pathway that overlaps with thrombosis (29).
Contact Factor Interactions with Bacteria and Viruses
Negatively charged LPS or surface associated negatively charged teichoic acids (S. aureus) (135, 136) and long chain PolyP (137) from various bacteria can induce CAS activation and BK release (138). HK binding to LPS from K. pneumoniae, P. aeruginosa, S. Minnesota, and E. coli strains converts single-chain HK to two-chain HKa and releases BK (139). High levels of BK have been reported in animal models of sepsis (140).
Contact factors bind to the surface of Gram-negative bacteria as well as Gram-positive bacteria and FXII and HK dependent contact activation on fibrous structures, including curli and fimbriae in E. coli and S. typhimurium, respectively, activates CAS which is not found in mutant strains lacking either curli or fimbriae (141, 142). The streptococcal M1 protein together with human fibrinogen, initiates polymorphonuclear neutrophils to form NETs, providing a surface for binding and activation of the contact system (143).
FXII mediated contact activation (144) and coagulation during viral infections has been reported (48). Upon hantavirus infection increased FXII binding and auto-activation is observed on the surface of infected endothelial cells (47). In Herpes Simplex Virus 1 an anionic phospholipid was identified as being responsible for activating FXII leading to enhanced coagulation through CAS activation (145).
Conclusion
The assembly of contact factors on cell surfaces is mediated via a number of structurally unrelated cell receptors (uPAR, GPIbα, Mac-1) and cofactors (HK, gC1q-R, CK1). On the surface of endothelial cells, gC1q-R is primarily responsible for assembly and activation of the FXII/HK/PK. gC1q-R has structural features of a cofactor rather than a cell receptor as it lacks a means to independently anchor to the plasma membrane and it is also present as a soluble factor in plasma (146). It has been shown by several groups independently that gC1q-R binds both HK and FXII and that it is capable of activating FXII and PK only when the three proteins, FXII/HK/PK, and elevated Zn2+ ions are present (90). Thus, it is capable of acting in a similar way to polyanions such as PolyP (90). As both gC1q-R and HK have been characterized as being capable of forming interactions with cell receptors and cell surface proteins (97, 147), there may be several redundancies in the mechanism whereby the FXII/HK/PK/gC1q-R complex locates to the surface of endothelial cells.
A key question is the molecular mechanism and pathophysiological significance of the Zn2+ dependence for both CAS receptor binding and FXII enzyme activation and whether this cation is a global regulator of CAS. The plasma concentration of Zn2+ ions ranges between 10 and 20 µM (148), most of which is bound to serum albumin, resulting in free Zn2+ concentrations of approximately 0.5 µM (149). FXII binding to endothelial cells increases in the presence of Zn2+ and binding plateaued at Zn2+ concentrations of 50 µM (93). This Zn2+ concentration is 100-fold more than the free Zn2+ concentration found in the plasma, which supports Zn2+ concentration as a potential control mechanism for contact activation. Platelets are known to release Zn2+ upon activation (43, 150) but the cellular process that controls this remains unknown. Different cell types cooperate in pathways leading to thrombosis (29, 30, 151) and thus studying each cell type in isolation may have limited utility when trying to understand contact factor cell localization and contact activation in vivo.
On the surface of platelets the assembly of the FXII/HK/FXI complex may occur via GPIbα, which can also act as the receptor for thrombin, thus coordinating FXI activation via both coagulation pathways (intrinsic and extrinsic). Activation of platelets and resulting secretion of PolyP plays a role both as a cofactor for thrombin cleavage of FXI (119) and FXII activation (17, 20, 152–155).
uPAR acts as a receptor for FXII on the surface of neutrophils (156) and dendritic cells (157), mediating processes that may be independent of contact activation. Contact factors are considered to be novel drug targets for thrombotic (36) and inflammatory diseases and targeting cell surface and receptor/cofactor binding domains has the potential of introducing more selectivity over targeting the contact factor enzyme active sites. Elucidating the structure of cognate cell receptors and cofactors regulating CAS activation in inflammation and FXI activation in the plasma coagulation pathway will provide a scaffold to develop novel antagonists and therapies for diverse vascular diseases.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors acknowledge the British Heart Foundation grant RG/12/9/29775 awarded to JE.
Abbreviations
CAS, contact activation system; PK, prekallikrein; HK, high molecular weight kininogen; FXII, factor XII; BK, bradykinin; uPAR, urokinase receptor; GPIb-IX, platelet glycoprotein Ib.
References
1. Renne T. The procoagulant and proinflammatory plasma contact system. Semin Immunopathol (2012) 34(1):31–41. doi:10.1007/s00281-011-0288-2
2. Colman RW, Schmaier AH. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood (1997) 90(10):3819–43.
3. Wu Y. Contact pathway of coagulation and inflammation. Thromb J (2015) 13:1–9. doi:10.1186/s12959-015-0048-y
4. Schmaier AH. Physiologic activities of the contact activation system. Thromb Res (2014) 133(Suppl 1):S41–4. doi:10.1016/j.thromres.2014.03.018
5. Nickel KF, Long AT, Fuchs TA, Butler LM, Renne T. Factor XII as a therapeutic target in thromboembolic and inflammatory diseases. Arterioscler Thromb Vasc Biol (2017) 37(1):13–20. doi:10.1161/atvbaha.116.308595
6. Weitz JI, Fredenburgh JC. Factors XI and XII as targets for new anticoagulants. Front Med (2017) 4:19. doi:10.3389/fmed.2017.00019
7. Samuel M, Pixley RA, Villanueva MA, Colman RW, Villanueva GB. Human factor XII (Hageman factor) autoactivation by dextran sulfate. Circular dichroism, fluorescence, and ultraviolet difference spectroscopic studies. J Biol Chem (1992) 267(27):19691–7.
8. Ivanov I, Matafonov A, Sun MF, Cheng Q, Dickeson SK, Verhamme IM, et al. Proteolytic properties of single-chain factor XII: a mechanism for triggering contact activation. Blood (2017) 129(11):1527–37. doi:10.1182/blood-2016-10-744110
9. Silverberg M, Dunn JT, Garen L, Kaplan AP. Autoactivation of human Hageman factor. Demonstration utilizing a synthetic substrate. J Biol Chem (1980) 255(15):7281–6.
10. Tankersley DL, Finlayson JS. Kinetics of activation and autoactivation of human factor XII. Biochemistry (1984) 23(2):273–9. doi:10.1021/bi00297a016
11. Muller F, Gailani D, Renne T. Factor XI and XII as antithrombotic targets. Curr Opin Hematol (2011) 18(5):349–55. doi:10.1097/MOH.0b013e3283497e61
12. Kaplan AP, Joseph K. Chapter two – pathogenic mechanisms of bradykinin mediated diseases: dysregulation of an innate inflammatory pathway. In: Alt FW, editor. Advances in Immunology (Vol. 121). South Carolina: Academic Press (2014). p. 41–89.
13. Leeb-Lundberg LMF, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev (2005) 57(1):27. doi:10.1124/pr.57.1.2
14. Smith SA, Travers RJ, Morrissey JH. How it all starts: initiation of the clotting cascade. Crit Rev Biochem Mol Biol (2015) 50(4):326–36. doi:10.3109/10409238.2015.1050550
15. Renne T, Schmaier AH, Nickel KF, Blomback M, Maas C. In vivo roles of factor XII. Blood (2012) 120(22):4296–303. doi:10.1182/blood-2012-07-292094
16. Gajsiewicz JM, Smith SA, Morrissey JH. Polyphosphate and RNA differentially modulate the contact pathway of blood clotting. J Biol Chem (2017) 292(5):1808–14. doi:10.1074/jbc.M116.754325
17. Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell (2009) 139(6):1143–56. doi:10.1016/j.cell.2009.11.001
18. Mutch NJ. Polyphosphate as a haemostatic modulator. Biochem Soc Trans (2016) 44(1):18–24. doi:10.1042/bst20150207
19. Maas C, Oschatz C, Renne T. The plasma contact system 2.0. Semin Thromb Hemost (2011) 37(4):375–81. doi:10.1055/s-0031-1276586
20. Verhoef JJ, Barendrecht AD, Nickel KF, Dijkxhoorn K, Kenne E, Labberton L, et al. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood (2017) 129(12):1707–17. doi:10.1182/blood-2016-08-734988
21. Vu TT, Leslie BA, Stafford AR, Zhou J, Fredenburgh JC, Weitz JI. Histidine-rich glycoprotein binds DNA and RNA and attenuates their capacity to activate the intrinsic coagulation pathway. Thromb Haemost (2016) 115(1):89–98. doi:10.1160/th15-04-0336
22. Bhagirath VC, Dwivedi DJ, Liaw PC. Comparison of the proinflammatory and procoagulant properties of nuclear, mitochondrial, and bacterial DNA. Shock (2015) 44(3):265–71. doi:10.1097/shk.0000000000000397
23. Nickel KF, Renne T. Crosstalk of the plasma contact system with bacteria. Thromb Res (2012) 130(Suppl 1):S78–83. doi:10.1016/j.thromres.2012.08.284
24. Larsson M, Rayzman V, Nolte MW, Nickel KF, Bjorkqvist J, Jamsa A, et al. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med (2014) 6(222):222ra17. doi:10.1126/scitranslmed.3006804
25. Nickel KF, Ronquist G, Langer F, Labberton L, Fuchs TA, Bokemeyer C, et al. The polyphosphate-factor XII pathway drives coagulation in prostate cancer-associated thrombosis. Blood (2015) 126(11):1379–89. doi:10.1182/blood-2015-01-622811
26. Fredenburgh JC, Gross PL, Weitz JI. Emerging anticoagulant strategies. Blood (2017) 129(2):147–54. doi:10.1182/blood-2016-09-692996
27. Morange PE, Oudot-Mellakh T, Cohen W, Germain M, Saut N, Antoni G, et al. KNG1 Ile581Thr and susceptibility to venous thrombosis. Blood (2011) 117(13):3692–4. doi:10.1182/blood-2010-11-319053
28. Revenko AS, Gao D, Crosby JR, Bhattacharjee G, Zhao C, May C, et al. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood (2011) 118(19):5302–11. doi:10.1182/blood-2011-05-355248
29. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209(4):819–35. doi:10.1084/jem.20112322
30. Kossmann S, Lagrange J, Jackel S, Jurk K, Ehlken M, Schonfelder T, et al. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci Transl Med (2017) 9:375. doi:10.1126/scitranslmed.aah4923
31. Kuijpers MJ, van der Meijden PE, Feijge MA, Mattheij NJ, May F, Govers-Riemslag J, et al. Factor XII regulates the pathological process of thrombus formation on ruptured plaques. Arterioscler Thromb Vasc Biol (2014) 34(8):1674–80. doi:10.1161/atvbaha.114.303315
32. Vorlova S, Koch M, Manthey HD, Cochain C, Busch M, Chaudhari SM, et al. Coagulation factor XII induces pro-inflammatory cytokine responses in macrophages and promotes atherosclerosis in mice. Thromb Haemost (2017) 117(1):176–87. doi:10.1160/th16-06-0466
33. Simao F, Ustunkaya T, Clermont AC, Feener EP. Plasma kallikrein mediates brain hemorrhage and edema caused by tissue plasminogen activator therapy in mice after stroke. Blood (2017) 129(16):2280–90. doi:10.1182/blood-2016-09-740670
34. De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, Kleinschnitz C. Thromboinflammation in stroke brain damage. Stroke (2016) 47(4):1165–72. doi:10.1161/strokeaha.115.011238
35. Zeerleder S, Schloesser M, Redondo M, Wuillemin WA, Engel W, Furlan M, et al. Reevaluation of the incidence of thromboembolic complications in congenital factor XII deficiency – a study on 73 subjects from 14 Swiss families. Thromb Haemost (1999) 82(4):1240–6.
36. Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med (2005) 202(2):271–81. doi:10.1084/jem.20050664
37. Key NS. Epidemiologic and clinical data linking factors XI and XII to thrombosis. Hematology Am Soc Hematol Educ Program (2014) 2014(1):66–70. doi:10.1182/asheducation-2014.1.66
38. Buller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med (2015) 372(3):232–40. doi:10.1056/NEJMoa1405760
39. Buller HR, Gailani D, Weitz JI. Factor XI antisense oligonucleotide for venous thrombosis. N Engl J Med (2015) 372(17):1672. doi:10.1056/NEJMc1503223
40. Cichon S, Martin L, Hennies HC, Muller F, Van Driessche K, Karpushova A, et al. Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet (2006) 79(6):1098–104. doi:10.1086/509899
41. Bjorkqvist J, de Maat S, Lewandrowski U, Di Gennaro A, Oschatz C, Schonig K, et al. Defective glycosylation of coagulation factor XII underlies hereditary angioedema type III. J Clin Invest (2015) 125(8):3132–46. doi:10.1172/jci77139
42. Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood (2010) 115(13):2569–77. doi:10.1182/blood-2009-09-199182
43. Mahdi F, Madar ZS, Figueroa CD, Schmaier AH. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood (2002) 99(10):3585–96. doi:10.1182/blood.V99.10.3585
44. Frick IM, Bjorck L, Herwald H. The dual role of the contact system in bacterial infectious disease. Thromb Haemost (2007) 98(3):497–502. doi:10.1160/TH07-01-0051
45. Loof TG, Deicke C, Medina E. The role of coagulation/fibrinolysis during Streptococcus pyogenes infection. Front Cell Infect Microbiol (2014) 4:128. doi:10.3389/fcimb.2014.00128
46. Holm K, Frick IM, Bjorck L, Rasmussen M. Activation of the contact system at the surface of Fusobacterium necrophorum represents a possible virulence mechanism in Lemierre’s syndrome. Infect Immun (2011) 79(8):3284–90. doi:10.1128/iai.05264-11
47. Taylor SL, Wahl-Jensen V, Copeland AM, Jahrling PB, Schmaljohn CS. Endothelial cell permeability during hantavirus infection involves factor XII-dependent increased activation of the kallikrein-kinin system. PLoS Pathog (2013) 9(7):e1003470. doi:10.1371/journal.ppat.1003470
48. Long AT, Kenne E, Jung R, Fuchs TA, Renne T. Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost (2016) 14(3):427–37. doi:10.1111/jth.13235
49. Stenflo J. Contributions of Gla and EGF-like domains to the function of vitamin K-dependent coagulation factors. Crit Rev Eukaryot Gene Expr (1999) 9(1):59–88.
50. Mohammed BM, Matafonov A, Ivanov I, Sun M-F, Cheng Q, Kent Dickeson S, et al. An update on factor XI structure and function. Thromb Res (2018) 161:94–105. doi:10.1016/j.thromres.2017.10.008
51. Pathak M, Wilmann P, Awford J, Li C, Hamad BK, Fischer PM, et al. Coagulation factor XII protease domain crystal structure. J Thromb Haemost (2015) 13(4):580–91. doi:10.1111/jth.12849
52. Naudin C, Burillo E, Blankenberg S, Butler L, Renne T. Factor XII contact activation. Semin Thromb Hemost (2017) 43(8):814–26. doi:10.1055/s-0036-1598003
53. Rojkaer R, Schousboe I. Partial identification of the Zn2+-binding sites in factor XII and its activation derivatives. Eur J Biochem (1997) 247(2):491–6. doi:10.1111/j.1432-1033.1997.00491.x
54. Citarella F, Ravon DM, Pascucci B, Felici A, Fantoni A, Hack CE. Structure/function analysis of human factor XII using recombinant deletion mutants. Evidence for an additional region involved in the binding to negatively charged surfaces. Eur J Biochem (1996) 238(1):240–9. doi:10.1111/j.1432-1033.1996.0240q.x
55. Citarella F, te Velthuis H, Helmer-Citterich M, Hack CE. Identification of a putative binding site for negatively charged surfaces in the fibronectin type II domain of human factor XII – an immunochemical and homology modeling approach. Thromb Haemost (2000) 84(6):1057–65.
56. LaRusch GA, Mahdi F, Shariat-Madar Z, Adams G, Sitrin RG, Zhang WM, et al. Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood (2010) 115(24):5111–20. doi:10.1182/blood-2009-08-236430
57. Bradford HN, Pixley RA, Colman RW. Human factor XII binding to the glycoprotein Ib-IX-V complex inhibits thrombin-induced platelet aggregation. J Biol Chem (2000) 275(30):22756–63. doi:10.1074/jbc.M002591200
58. Mandle RJ, Colman RW, Kaplan AP. Identification of prekallikrein and high-molecular-weight kininogen as a complex in human plasma. Proc Natl Acad Sci U S A (1976) 73(11):4179–83. doi:10.1073/pnas.73.11.4179
59. Wong SS, Ostergaard S, Hall G, Li C, Williams PM, Stennicke H, et al. A novel DFP tripeptide motif interacts with the coagulation factor XI apple 2 domain. Blood (2016) 127(23):2915–23. doi:10.1182/blood-2015-10-676122
60. Bjorkqvist J, Jamsa A, Renne T. Plasma kallikrein: the bradykinin-producing enzyme. Thromb Haemost (2013) 110(3):399–407. doi:10.1160/th13-03-0258
61. Page JD, You JL, Harris RB, Colman RW. Localization of the binding site on plasma kallikrein for high-molecular-weight kininogen to both apple 1 and apple 4 domains of the heavy chain. Arch Biochem Biophys (1994) 314(1):159–64. doi:10.1006/abbi.1994.1424
62. Weisel JW, Nagaswami C, Woodhead JL, DeLa Cadena RA, Page JD, Colman RW. The shape of high molecular weight kininogen. Organization into structural domains, changes with activation, and interactions with prekallikrein, as determined by electron microscopy. J Biol Chem (1994) 269(13):10100–6.
63. Thompson RE, Mandle R Jr, Kaplan AP. Studies of binding of prekallikrein and Factor XI to high molecular weight kininogen and its light chain. Proc Natl Acad Sci U S A (1979) 76(10):4862–6. doi:10.1073/pnas.76.10.4862
64. Greengard JS, Griffin JH. Receptors for high molecular weight kininogen on stimulated washed human platelets. Biochemistry (1984) 23(26):6863–9. doi:10.1021/bi00321a090
65. Zini JM, Schmaier AH, Cines DB. Bradykinin regulates the expression of kininogen binding sites on endothelial cells. Blood (1993) 81(11):2936–46.
66. Wachtfogel YT, DeLa Cadena RA, Kunapuli SP, Rick L, Miller M, Schultze RL, et al. High molecular weight kininogen binds to Mac-1 on neutrophils by its heavy chain (domain 3) and its light chain (domain 5). J Biol Chem (1994) 269(30):19307–12.
67. Hasan AA, Cines DB, Herwald H, Schmaier AH, Muller-Esterl W. Mapping the cell binding site on high molecular weight kininogen domain 5. J Biol Chem (1995) 270(33):19256–61. doi:10.1074/jbc.270.33.19256
68. Chavakis T, Santoso S, Clemetson KJ, Sachs UJ, Isordia-Salas I, Pixley RA, et al. High molecular weight kininogen regulates platelet-leukocyte interactions by bridging Mac-1 and glycoprotein Ib. J Biol Chem (2003) 278(46):45375–81. doi:10.1074/jbc.M304344200
69. Colman RW, Pixley RA, Najamunnisa S, Yan W, Wang J, Mazar A, et al. Binding of high molecular weight kininogen to human endothelial cells is mediated via a site within domains 2 and 3 of the urokinase receptor. J Clin Invest (1997) 100(6):1481–7. doi:10.1172/jci119669
70. Thompson RE, Mandle R Jr, Kaplan AP. Association of factor XI and high molecular weight kininogen in human plasma. J Clin Invest (1977) 60(6):1376–80. doi:10.1172/jci108898
71. Baglia FA, Gailani D, Lopez JA, Walsh PN. Identification of a binding site for glycoprotein Ibalpha in the Apple 3 domain of factor XI. J Biol Chem (2004) 279(44):45470–6. doi:10.1074/jbc.M406727200
72. Motta G, Rojkjaer R, Hasan AA, Cines DB, Schmaier AH. High molecular weight kininogen regulates prekallikrein assembly and activation on endothelial cells: a novel mechanism for contact activation. Blood (1998) 91(2):516–28.
73. Shariat-Madar Z, Mahdi F, Schmaier AH. Factor XI assembly and activation on human umbilical vein endothelial cells in culture. Thromb Haemost (2001) 85(3):544–51. doi:10.1055/s-0037-1615619
74. Joseph K, Ghebrehiwet B, Peerschke EI, Reid KB, Kaplan AP. Identification of the zinc-dependent endothelial cell binding protein for high molecular weight kininogen and factor XII: identity with the receptor that binds to the globular “heads” of C1q (gC1q-R). Proc Natl Acad Sci U S A (1996) 93(16):8552–7. doi:10.1073/pnas.93.16.8552
75. Herwald H, Hasan AA, Godovac-Zimmermann J, Schmaier AH, Muller-Esterl W. Identification of an endothelial cell binding site on kininogen domain D3. J Biol Chem (1995) 270(24):14634–42.
76. Herwald H, Dedio J, Kellner R, Loos M, Muller-Esterl W. Isolation and characterization of the kininogen-binding protein p33 from endothelial cells. Identity with the gC1q receptor. J Biol Chem (1996) 271(22):13040–7. doi:10.1074/jbc.271.22.13040
77. Ghebrehiwet B, Lim BL, Peerschke EI, Willis AC, Reid KB. Isolation, cDNA cloning, and overexpression of a 33-kD cell surface glycoprotein that binds to the globular “heads” of C1q. J Exp Med (1994) 179(6):1809–21. doi:10.1084/jem.179.6.1809
78. Ghebrehiwet B, Lim BL, Kumar R, Feng X, Peerschke EI. gC1q-R/p33, a member of a new class of multifunctional and multicompartmental cellular proteins, is involved in inflammation and infection. Immunol Rev (2001) 180:65–77. doi:10.1034/j.1600-065X.2001.1800106.x
79. Dembitzer FR, Kinoshita Y, Burstein D, Phelps RG, Beasley MB, Garcia R, et al. gC1qR expression in normal and pathologic human tissues: differential expression in tissues of epithelial and mesenchymal origin. J Histochem Cytochem (2012) 60(6):467–74. doi:10.1369/0022155412440882
80. Petersen-Mahrt SK, Estmer C, Ohrmalm C, Matthews DA, Russell WC, Akusjarvi G. The splicing factor-associated protein, p32, regulates RNA splicing by inhibiting ASF/SF2 RNA binding and phosphorylation. EMBO J (1999) 18(4):1014–24. doi:10.1093/emboj/18.4.1014
81. Berro R, Kehn K, de la Fuente C, Pumfery A, Adair R, Wade J, et al. Acetylated Tat regulates human immunodeficiency virus type 1 splicing through its interaction with the splicing regulator p32. J Virol (2006) 80(7):3189–204. doi:10.1128/jvi.80.7.3189-3204.2006
82. Emelyanov AV, Rabbani J, Mehta M, Vershilova E, Keogh MC, Fyodorov DV. Drosophila TAP/p32 is a core histone chaperone that cooperates with NAP-1, NLP, and nucleophosmin in sperm chromatin remodeling during fertilization. Genes Dev (2014) 28(18):2027–40. doi:10.1101/gad.248583.114
83. Westman J, Smeds E, Johansson L, Morgelin M, Olin AI, Malmstrom E, et al. Treatment with p33 curtails morbidity and mortality in a histone-induced murine shock model. J Innate Immun (2014) 6(6):819–30. doi:10.1159/000363348
84. Westman J, Hansen FC, Olin AI, Morgelin M, Schmidtchen A, Herwald H. p33 (gC1q receptor) prevents cell damage by blocking the cytolytic activity of antimicrobial peptides. J Immunol (2013) 191(11):5714–21. doi:10.4049/jimmunol.1300596
85. Guo WX, Ghebrehiwet B, Weksler B, Schweitzer K, Peerschke EI. Up-regulation of endothelial cell binding proteins/receptors for complement component C1q by inflammatory cytokines. J Lab Clin Med (1999) 133(6):541–50. doi:10.1016/S0022-2143(99)90183-X
86. Kim KB, Kim BW, Choo HJ, Kwon YC, Ahn BY, Choi JS, et al. Proteome analysis of adipocyte lipid rafts reveals that gC1qR plays essential roles in adipogenesis and insulin signal transduction. Proteomics (2009) 9(9):2373–82. doi:10.1002/pmic.200800811
87. Peerschke EI, Ghebrehiwet B. cC1qR/CR and gC1qR/p33: observations in cancer. Mol Immunol (2014) 61(2):100–9. doi:10.1016/j.molimm.2014.06.011
88. Peerschke EI, Minta JO, Zhou SZ, Bini A, Gotlieb A, Colman RW, et al. Expression of gC1q-R/p33 and its major ligands in human atherosclerotic lesions. Mol Immunol (2004) 41(8):759–66. doi:10.1016/j.molimm.2004.04.020
89. Peerschke EI, Ghebrehiwet B. Human blood platelet gC1qR/p33. Immunol Rev (2001) 180:56–64. doi:10.1034/j.1600-065X.2001.1800105.x
90. Joseph K, Shibayama Y, Ghebrehiwet B, Kaplan AP. Factor XII-dependent contact activation on endothelial cells and binding proteins gC1qR and cytokeratin 1. Thromb Haemost (2001) 85(1):119–24. doi:10.1055/s-0037-1612914
91. Jiang J, Zhang Y, Krainer AR, Xu RM. Crystal structure of human p32, a doughnut-shaped acidic mitochondrial matrix protein. Proc Natl Acad Sci U S A (1999) 96(7):3572–7. doi:10.1073/pnas.96.7.3572
92. Pixley RA, Espinola RG, Ghebrehiwet B, Joseph K, Kao A, Bdeir K, et al. Interaction of high-molecular-weight kininogen with endothelial cell binding proteins suPAR, gC1qR and cytokeratin 1 determined by surface plasmon resonance (BiaCore). Thromb Haemost (2011) 105(6):1053–9. doi:10.1160/th10-09-0591
93. Reddigari SR, Shibayama Y, Brunnee T, Kaplan AP. Human Hageman factor (factor XII) and high molecular weight kininogen compete for the same binding site on human umbilical vein endothelial cells. J Biol Chem (1993) 268(16):11982–7.
94. Ghebrehiwet B, CebadaMora C, Tantral L, Jesty J, Peerschke EI. gC1qR/p33 serves as a molecular bridge between the complement and contact activation systems and is an important catalyst in inflammation. Adv Exp Med Biol (2006) 586:95–105. doi:10.1007/0-387-34134-x_7
95. Dedio J, Jahnen-Dechent W, Bachmann M, Muller-Esterl W. The multiligand-binding protein gC1qR, putative C1q receptor, is a mitochondrial protein. J Immunol (1998) 160(7):3534–42.
96. van Leeuwen HC, O’Hare P. Retargeting of the mitochondrial protein p32/gC1Qr to a cytoplasmic compartment and the cell surface. J Cell Sci (2001) 114(Pt 11):2115–23.
97. Joseph K, Ghebrehiwet B, Kaplan AP. Cytokeratin 1 and gC1qR mediate high molecular weight kininogen binding to endothelial cells. Clin Immunol (1999) 92(3):246–55. doi:10.1006/clim.1999.4753
98. Mahdi F, Shariat-Madar Z, Todd RF III, Figueroa CD, Schmaier AH. Expression and colocalization of cytokeratin 1 and urokinase plasminogen activator receptor on endothelial cells. Blood (2001) 97(8):2342–50. doi:10.1182/blood.V97.8.2342
99. Ploug M, Ellis V. Structure-function relationships in the receptor for urokinase-type plasminogen activator. Comparison to other members of the Ly-6 family and snake venom alpha-neurotoxins. FEBS Lett (1994) 349(2):163–8. doi:10.1016/0014-5793(94)00674-1
100. Estreicher A, Muhlhauser J, Carpentier JL, Orci L, Vassalli JD. The receptor for urokinase type plasminogen activator polarizes expression of the protease to the leading edge of migrating monocytes and promotes degradation of enzyme inhibitor complexes. J Cell Biol (1990) 111(2):783–92. doi:10.1083/jcb.111.2.783
101. Ellis V, Dano K. Plasminogen activation by receptor-bound urokinase. Semin Thromb Hemost (1991) 17(3):194–200. doi:10.1055/s-2007-1002609
102. Bacharach E, Itin A, Keshet E. In vivo patterns of expression of urokinase and its inhibitor PAI-1 suggest a concerted role in regulating physiological angiogenesis. Proc Natl Acad Sci U S A (1992) 89(22):10686–90. doi:10.1073/pnas.89.22.10686
103. Pollanen J, Stephens RW, Vaheri A. Directed plasminogen activation at the surface of normal and malignant cells. Adv Cancer Res (1991) 57:273–328. doi:10.1016/S0065-230X(08)61002-7
104. Nykjaer A, Moller B, Todd RF III, Christensen T, Andreasen PA, Gliemann J, et al. Urokinase receptor. An activation antigen in human T lymphocytes. J Immunol (1994) 152(2):505–16.
105. Cao DJ, Guo YL, Colman RW. Urokinase-type plasminogen activator receptor is involved in mediating the apoptotic effect of cleaved high molecular weight kininogen in human endothelial cells. Circ Res (2004) 94(9):1227–34. doi:10.1161/01.res.0000126567.75232.46
106. Liu Y, Sainz IM, Wu Y, Pixley R, Espinola RG, Hassan S, et al. The inhibition of tube formation in a collagen-fibrinogen, three-dimensional gel by cleaved kininogen (HKa) and HK domain 5 (D5) is dependent on Src family kinases. Exp Cell Res (2008) 314(4):774–88. doi:10.1016/j.yexcr.2007.10.008
107. Betapudi V, Shukla M, Alluri R, Merkulov S, McCrae KR. Novel role for p56/Lck in regulation of endothelial cell survival and angiogenesis. FASEB J (2016) 30(10):3515–26. doi:10.1096/fj.201500040
108. Zhang JC, Donate F, Qi X, Ziats NP, Juarez JC, Mazar AP, et al. The antiangiogenic activity of cleaved high molecular weight kininogen is mediated through binding to endothelial cell tropomyosin. Proc Natl Acad Sci U S A (2002) 99(19):12224–9. doi:10.1073/pnas.192668299
109. Baglia FA, Shrimpton CN, Emsley J, Kitagawa K, Ruggeri ZM, Lopez JA, et al. Factor XI interacts with the leucine-rich repeats of glycoprotein Ibalpha on the activated platelet. J Biol Chem (2004) 279(47):49323–9. doi:10.1074/jbc.M407889200
110. Puri RN, Zhou F, Hu CJ, Colman RF, Colman RW. High molecular weight kininogen inhibits thrombin-induced platelet aggregation and cleavage of aggregin by inhibiting binding of thrombin to platelets. Blood (1991) 77(3):500–7.
111. Joseph K, Nakazawa Y, Bahou WF, Ghebrehiwet B, Kaplan AP. Platelet glycoprotein Ib: a zinc-dependent binding protein for the heavy chain of high-molecular-weight kininogen. Mol Med (1999) 5(8):555–63.
112. Li R, Emsley J. The organizing principle of the platelet glycoprotein Ib-IX-V complex. J Thromb Haemost (2013) 11(4):605–14. doi:10.1111/jth.12144
113. Baglia FA, Shrimpton CN, Lopez JA, Walsh PN. The glycoprotein Ib-IX-V complex mediates localization of factor XI to lipid rafts on the platelet membrane. J Biol Chem (2003) 278(24):21744–50. doi:10.1074/jbc.M212991200
114. White-Adams TC, Berny MA, Tucker EI, Gertz JM, Gailani D, Urbanus RT, et al. Identification of coagulation factor XI as a ligand for platelet apolipoprotein E receptor 2 (ApoER2). Arterioscler Thromb Vasc Biol (2009) 29(10):1602–7. doi:10.1161/atvbaha.109.187393
115. Urbanus RT, Pennings MT, Derksen RH, de Groot PG. Platelet activation by dimeric beta2-glycoprotein I requires signaling via both glycoprotein Ibalpha and apolipoprotein E receptor 2’. J Thromb Haemost (2008) 6(8):1405–12. doi:10.1111/j.1538-7836.2008.03021.x
116. Harmon JT, Jamieson GA. Platelet activation by thrombin in the absence of the high-affinity thrombin receptor. Biochemistry (1988) 27(6):2151–7. doi:10.1021/bi00406a050
117. Bradford HN, Dela Cadena RA, Kunapuli SP, Dong JF, Lopez JA, Colman RW. Human kininogens regulate thrombin binding to platelets through the glycoprotein Ib-IX-V complex. Blood (1997) 90(4):1508–15.
118. Celikel R, McClintock RA, Roberts JR, Mendolicchio GL, Ware J, Varughese KI, et al. Modulation of alpha-thrombin function by distinct interactions with platelet glycoprotein Ibalpha. Science (2003) 301(5630):218–21. doi:10.1126/science.1084183
119. Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood (2011) 118(26):6963–70. doi:10.1182/blood-2011-07-368811
120. Ottaiano TF, Andrade SS, de Oliveira C, Silva MC, Buri MV, Juliano MA, et al. Plasma kallikrein enhances platelet aggregation response by subthreshold doses of ADP. Biochimie (2017) 135:72–81. doi:10.1016/j.biochi.2017.01.010
121. Henderson LM, Figueroa CD, Muller-Esterl W, Bhoola KD. Assembly of contact-phase factors on the surface of the human neutrophil membrane. Blood (1994) 84(2):474–82.
122. Bradford HN, Jameson BA, Adam AA, Wassell RP, Colman RW. Contiguous binding and inhibitory sites on kininogens required for the inhibition of platelet calpain. J Biol Chem (1993) 268(35):26546–51.
123. Schapira M, Despland E, Scott CF, Boxer LA, Colman RW. Purified human plasma kallikrein aggregates human blood neutrophils. J Clin Invest (1982) 69(5):1199–202. doi:10.1172/JCI110557
124. Kaplan AP, Kay AB, Austen KF. A prealbumin activator of prekallikrein. 3. Appearance of chemotactic activity for human neutrophils by the conversion of human prekallikrein to kallikrein. J Exp Med (1972) 135(1):81–97. doi:10.1084/jem.135.1.81
125. Wachtfogel YT, Kucich U, James HL, Scott CF, Schapira M, Zimmerman M, et al. Human plasma kallikrein releases neutrophil elastase during blood coagulation. J Clin Invest (1983) 72(5):1672–7. doi:10.1172/jci111126
126. Khan MM, Kunapuli SP, Lin Y, Majluf-Cruz A, Cadena RA, Cooper SL, et al. Three noncontiguous peptides comprise binding sites on high-molecular-weight kininogen to neutrophils. Am J Physiol (1998) 275(1 Pt 2):H145–50.
127. Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell (1991) 65(6):961–71. doi:10.1016/0092-8674(91)90548-D
128. Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med (2000) 192(2):193–204. doi:10.1084/jem.192.2.193
129. Sheng N, Fairbanks MB, Heinrikson RL, Canziani G, Chaiken IM, Mosser DM, et al. Cleaved high molecular weight kininogen binds directly to the integrin CD11b/CD18 (Mac-1) and blocks adhesion to fibrinogen and ICAM-1. Blood (2000) 95(12):3788–95.
130. Yung LY, Lim F, Khan MM, Kunapuli SP, Rick L, Colman RW, et al. Neutrophil adhesion on surfaces preadsorbed with high molecular weight kininogen under well-defined flow conditions. Immunopharmacology (1996) 32(1–3):19–23. doi:10.1016/0162-3109(96)00003-3
131. Deppermann C, Kubes P. Platelets and infection. Semin Immunol (2016) 28(6):536–45. doi:10.1016/j.smim.2016.10.005
132. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol (2013) 13(1):34–45. doi:10.1038/nri3345
133. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science (2014) 346(6214):1234–8. doi:10.1126/science.1256478
134. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol (2016) 99(1):153–62. doi:10.1189/jlb.3A0415-161R
135. Mattsson E, Herwald H, Cramer H, Persson K, Sjobring U, Bjorck L. Staphylococcus aureus induces release of bradykinin in human plasma. Infect Immun (2001) 69(6):3877–82. doi:10.1128/iai.69.6.3877-3882.2001
136. Kalter ES, van Dijk WC, Timmerman A, Verhoef J, Bouma BN. Activation of purified human plasma prekallikrein triggered by cell wall fractions of Escherichia coli and Staphylococcus aureus. J Infect Dis (1983) 148(4):682–91. doi:10.1093/infdis/148.4.682
137. Azevedo C, Saiardi A. Functions of inorganic polyphosphates in eukaryotic cells: a coat of many colours. Biochem Soc Trans (2014) 42(1):98–102. doi:10.1042/bst20130111
138. Morrissey JH, Choi SH, Smith SA. Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation. Blood (2012) 119(25):5972–9. doi:10.1182/blood-2012-03-306605
139. Yang A, Xie Z, Wang B, Colman RW, Dai J, Wu Y. An essential role of high-molecular-weight kininogen in endotoxemia. J Exp Med (2017) 214(9):2649–70. doi:10.1084/jem.20161900
140. Oehmcke S, Herwald H. Contact system activation in severe infectious diseases. J Mol Med (Berl) (2010) 88(2):121–6. doi:10.1007/s00109-009-0564-y
141. Ben Nasr A, Olsen A, Sjobring U, Muller-Esterl W, Bjorck L. Assembly of human contact phase proteins and release of bradykinin at the surface of curli-expressing Escherichia coli. Mol Microbiol (1996) 20(5):927–35. doi:10.1111/j.1365-2958.1996.tb02534.x
142. Herwald H, Morgelin M, Olsen A, Rhen M, Dahlback B, Muller-Esterl W, et al. Activation of the contact-phase system on bacterial surfaces – a clue to serious complications in infectious diseases. Nat Med (1998) 4(3):298–302. doi:10.1038/nm0398-298
143. Oehmcke S, Morgelin M, Herwald H. Activation of the human contact system on neutrophil extracellular traps. J Innate Immun (2009) 1(3):225–30. doi:10.1159/000203700
144. Pixley RA, De La Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A, et al. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J Clin Invest (1993) 91(1):61–8. doi:10.1172/jci116201
145. Gershom ES, Sutherland MR, Lollar P, Pryzdial EL. Involvement of the contact phase and intrinsic pathway in herpes simplex virus-initiated plasma coagulation. J Thromb Haemost (2010) 8(5):1037–43. doi:10.1111/j.1538-7836.2010.03789.x
146. Peerschke EI, Brandwijk RJ, Dembitzer FR, Kinoshita Y, Ghebrehiwet B. Soluble gC1qR in blood and body fluids: examination in a pancreatic cancer patient cohort. Int J Cancer Res Mol Mech (2015) 1(3):1–12. doi:10.16966/ijcrmm.110
147. Lu PD, Galanakis DK, Ghebrehiwet B, Peerschke EI. The receptor for the globular “heads” of C1q, gC1q-R, binds to fibrinogen/fibrin and impairs its polymerization. Clin Immunol (1999) 90(3):360–7. doi:10.1006/clim.1998.4660
148. Rukgauer M, Klein J, Kruse-Jarres JD. Reference values for the trace elements copper, manganese, selenium, and zinc in the serum/plasma of children, adolescents, and adults. J Trace Elem Med Biol (1997) 11(2):92–8. doi:10.1016/s0946-672x(97)80032-6
149. Scott BJ, Bradwell AR. Identification of the serum binding proteins for iron, zinc, cadmium, nickel, and calcium. Clin Chem (1983) 29(4):629–33.
150. Foley B, Johnson SA, Hackley B, Smith JC Jr, Halsted JA. Zinc content of human platelets. Proc Soc Exp Biol Med (1968) 128(1):265–9. doi:10.3181/00379727-128-32993
151. Payne H, Ponomaryov T, Watson SP, Brill A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood (2017) 129(14):2013–20. doi:10.1182/blood-2016-09-742999
152. Castaldi PA, Larrieu MJ, Caen J. Availability of platelet Factor 3 and activation of factor XII in thrombasthenia. Nature (1965) 207(995):422–4. doi:10.1038/207422a0
153. Walsh PN, Griffin JH. Platelet-coagulant protein interactions in contact activation. Ann N Y Acad Sci (1981) 370:241–52. doi:10.1111/j.1749-6632.1981.tb29737.x
154. Johne J, Blume C, Benz PM, Pozgajova M, Ullrich M, Schuh K, et al. Platelets promote coagulation factor XII-mediated proteolytic cascade systems in plasma. Biol Chem (2006) 387(2):173–8. doi:10.1515/bc.2006.023
155. Back J, Sanchez J, Elgue G, Ekdahl KN, Nilsson B. Activated human platelets induce factor XIIa-mediated contact activation. Biochem Biophys Res Commun (2010) 391(1):11–7. doi:10.1016/j.bbrc.2009.10.123
156. Stavrou EX, Fang C, Bane KL, Long AT, Naudin C, Kucukal E, et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest (2018) 128(3):944–59. doi:10.1172/jci92880
Keywords: contact activation system, factor XII, factor XI, plasma kallikrein, high molecular weight kininogen, endothelial cell, Platelet, Leukocyte
Citation: Pathak M, Kaira BG, Slater A and Emsley J (2018) Cell Receptor and Cofactor Interactions of the Contact Activation System and Factor XI. Front. Med. 5:66. doi: 10.3389/fmed.2018.00066
Received: 05 December 2017; Accepted: 26 February 2018;
Published: 21 March 2018
Edited by:
Alvin H. Schmaier, Case Western Reserve University, United StatesReviewed by:
James H. Morrissey, University of Michigan, United StatesKeith McCrae, Cleveland Clinic, United States
Copyright: © 2018 Pathak, Kaira, Slater and Emsley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonas Emsley, am9uYXMuZW1zbGV5QG5vdHRpbmdoYW0uYWMudWs=