 Daniel C. Zank
Daniel C. Zank Marta Bueno
Marta Bueno Ana L. Mora1,2*
Ana L. Mora1,2* Mauricio Rojas
Mauricio Rojas- 1Division of Pulmonary, Allergy and Critical Care Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 2Vascular Medicine Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 3McGowan Institute of Regenerative Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 4Dorothy P. & Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
At present, the etiology of idiopathic pulmonary fibrosis (IPF) remains elusive. Over the past two decades, however, researchers have identified and described the underlying processes that result in metabolic dysregulation, metabolic reprogramming, and mitochondrial dysfunction observed in the cells of IPF lungs. Metabolic changes and mitochondrial dysfunction in IPF include decreased efficiency of electron transport chain function with increasing production of reactive oxygen species, decreased mitochondrial biogenesis, and impaired mitochondrial macroautophagy, a key pathway for the removal of dysfunctional mitochondria. Metabolic changes in IPF have potential impact on lung cell function, differentiation, and activation of fibrotic responses. These alterations result in activation of TGF-β and predispose to the development of pulmonary fibrosis. IPF is a disease of the aged, and many of these same bioenergetic changes are present to a lesser extent with normal aging, raising the possibility that these anticipated alterations in metabolic processes play important roles in creating susceptibility to the development of IPF. This review explores what is known regarding the cellular metabolic and mitochondrial changes that are found in IPF, and examines this body of literature to identify future research direction and potential points of intervention in the pathogenesis of IPF.
Introduction
Aging is life’s natural destiny. It is biologically defined by a progressive impairment in vital functions coupled with diminished fitness to adapt to environmental stimuli and respond to stress (homeostenosis) (1). Among the predictable cellular alterations that underlie homeostenosis, metabolic dysregulation and alterations in mitochondrial function have been shown to contribute to the aging phenotype. Aging is also associated with increased susceptibility to a wide range of chronic diseases. Lung pathologies are no exception, and the prevalence of several interstitial lung diseases, most notably idiopathic pulmonary fibrosis (IPF), has been found to increase considerably with age. IPF is a progressive and irreversible lung disease, generally diagnosed in the sixth decade of life, whose etiology remains unknown and therapeutic options remain limited (2). Many of the changes in bioenergetics and mitochondrial function seen with aging are also seen in the fibrotic lung and may contribute to IPF (Table 1).
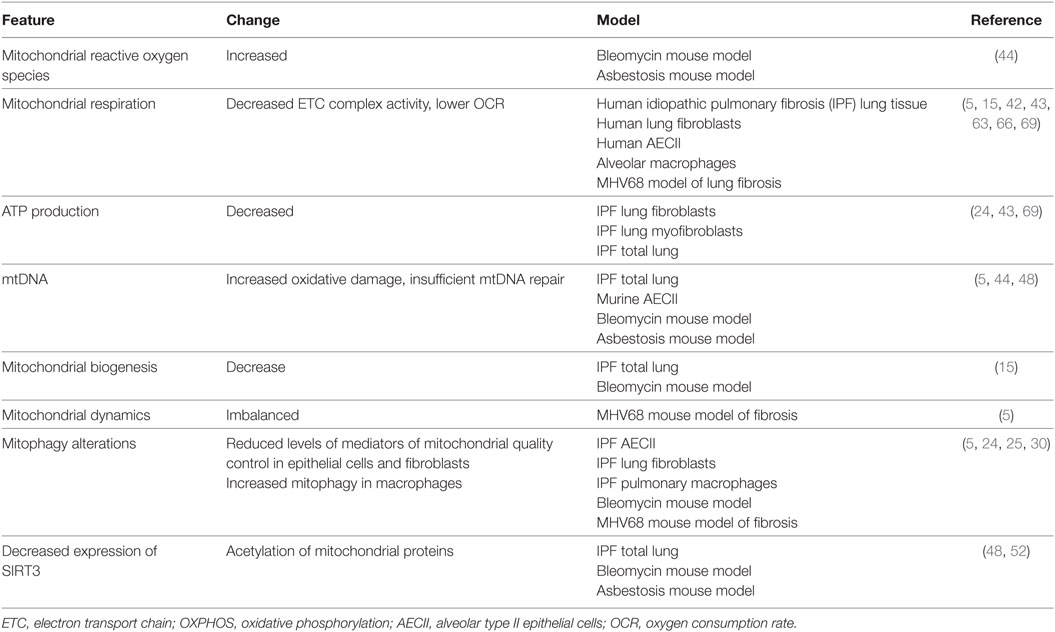
Table 1. Mitochondrial changes in the fibrotic lung.
Mitochondrial Alterations with Aging and IPF
Over the past few decades, our understanding of the function and role of mitochondria has moved beyond the initial view of mitochondria as nothing more than a cellular energy generator producing ATP through oxidative phosphorylation (OXPHOS). Mitochondria are now understood to have their own life cycle consisting of biogenesis, fission/fusion dynamics, and recycling through macroautophagy (mitophagy), and to act as an important bidirectional signaling platform, communicating with the nucleus as well as other organelles. Multiple signaling pathways converge and interact to regulate the linked processes of mitochondrial energetics, biogenesis, production of reactive oxygen species (ROS), mitochondrial DNA (mtDNA) preservation and repair, and mitophagy. Dysregulation of many of these regulatory mechanisms that control mitochondrial function have recently been identified in the epithelial cells, fibroblasts, and macrophages in IPF lungs (3). Mitochondrial dysfunction in IPF lung cells contributes to maladaptation to cellular stress, creating vulnerability to injury and promoting the development of pulmonary fibrosis (3–6) (Figure 1).
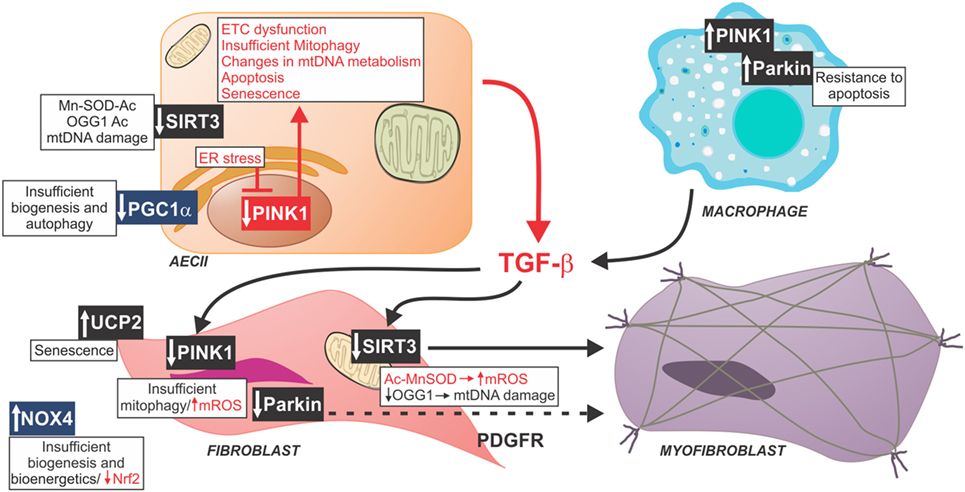
Figure 1. Schematic of profibrotic pathways mediated by mitochondrial dysfunction. Reduced PGC1α in fibrotic lungs has been associated with insufficient biogenesis and diminished autophagy. Similarly, decreased PINK1, a key modulator of mitophagy, and decreased SIRT3 have been found in AECII from fibrotic lungs associated with mitochondrial dysfunction, and increased activation of TGF-β. Profibrotic macrophages exhibit increased rates of mitophagy and resistance to apoptosis resulting in greater release of TGF-β. TGF-β affects mitochondrial function in fibroblasts through decreasing PINK1, Parkin, and SIRT3. Idiopathic pulmonary fibrosis fibroblasts have increased mtDNA damage, mitochondrial dysfunction, impaired mitochondrial biogenesis, and increased rate of senescence. All of these factors contribute to in fibroblast-to-myofibroblast differentiation. Abbreviations: Ac, acetyl group; ETC, type II alveolar epithelial cell electron transport chain, Mn-SOD, manganese super oxide dismutase; mtDNA, mitochondrial DNA, mtROS, mitochondrial reactive oxygen species; NOX4, NADPH oxidase 4; Nrf2, nuclear factor (erythroid-derived 2)-like-2 factor; OGG1, 8-oxoguanine DNA glycosylase 1; PDGFR, platelet-derived growth factor receptor; PINK1, PTEN-induced putative kinase 1; UCP2, uncoupling protein 2; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha.
Mitochondrial Biogenesis
Mitochondrial biogenesis, the process of producing additional mitochondria and by extension cellular energy production capacity, is under the control of master regulators PPARγ coactivator-1α (PGC-1α) and PGC-1β, which are nutritional sensors able to induce expression of nuclear respiration factors 1 and 2 (NRF). NRFs upregulate expression of the nuclear and mitochondrial mechanisms necessary for mitochondrial biogenesis (7). With aging, the capacity for mitochondrial biogenesis declines through age-related reduction in upstream activators of PGC-1α and PGC-1β such as AMP-activated protein kinase (AMPK) as well as p53-mediated, senescence-associated repression of these key regulators of mitochondrial biogenesis (8, 9). Moderate exercise, calorie restriction, and resveratrol supplementation have been shown to increase PGC-1α activity and ameliorates aging-related decline in mitochondrial biogenesis (10–12). Expression of PGC1a is reduced in lungs from IPF patients and in fibrotic mouse lungs after bleomycin treatment. Highlighting the critical role of mitochondrial biogenesis in the susceptibility to lung fibrosis, mice deficient in PGC1a are more susceptible to bleomycin-induced lung fibrosis. Thyroid hormone (T4) signaling is known to restore mitochondrial health and function through a PGC-1α-dependent pathway (13, 14). Concordantly, experimental provision of aerosolized T3 and a T4 mimetic attenuated bleomycin- and TGF-β-induced fibrosis in mouse models. T3 supplementation was also associated with reduced alveolar epithelial cell apoptosis, improvement in mitochondrial electron transport chain function, and normalization of swollen mitochondrial morphology. This improvement was not seen in PGC-1α knockout mice. These investigations were conducted in response to the finding of increased level and activity of iodothyronine deiodinase 2 in IPF lungs, which is the enzyme responsible for conversion of inactive T4 to the physiologically active T3 form. This is presumed to be an adaptive response to increase the availability of active T3 in fibrotic IPF lung (15).
Idiopathic pulmonary fibrosis and experimental models of pulmonary fibrosis are associated with telomere shortening and DNA damage, which activates the DNA damage sensor poly[ADP-ribose] polymerase 1 (PARP-1) and the checkpoint inhibitor p53. These signaling mechanisms feedback to reduce activation of PPARγ coactivators and reduce mitochondrial biogenesis (9, 16). Impaired mitochondrial biogenesis in IPF potentially creates a mismatch in cellular energy demand and production capacity with resulting mitochondrial dysfunction. Additionally, the ROS-producing enzyme NADPH oxidase-4 (Nox4), which is upregulated in IPF lung fibroblasts, represses mitochondrial biogenesis through direct effect on NRF2 and mitochondrial transcription factor A (TFAM) independent of PGC-1α (17, 18). While these pathways have been demonstrated to play a role in age and disease-related alterations in cellular bioenergetics, and the downstream consequences, such as ROS production, DNA damage, and induction of senescence, are features of lung fibrosis, additional translational investigations in IPF lungs are needed to firmly establish the role of these perturbations in the development of IPF.
Mitophagy
Mitophagy is a selective and adaptive response that targets mitochondria for turnover, regulates the number of mitochondria to match cellular energy needs, and removes damaged and dysfunctional mitochondria that can cause cellular stress. Mitophagy and mitochondrial turnover are important processes in maintaining cellular integrity and health. Mitochondrial damage, whether from metabolic alterations, increased ROS production, or an accumulation of mtDNA damage and mutations affecting transcription, results in a reduction of mitochondrial transmembrane potential, reduced ATP production, further increases in ROS, and leakage of mitochondrial contents into the cytosol that, if left unchecked, would eventually lead to cellular injury and apoptosis (19–23).
Selective mitophagy of damaged mitochondria occurs through PTEN-induced putative kinase 1 (PINK1)-Parkin signaling with PINK1 acting as a sensor of mitochondrial membrane depolarization and subsequently activating Parkin, which labels the dysfunctional mitochondrion for trafficking to the autophagosome (19, 24). With aging, a decrease in PINK1 has been observed coupled with a decrease in markers of autophagy and an increase in the size of mitochondria (5, 25). These findings demonstrate that capacity for mitophagy declines as we age.
Deficient mitophagy has been associated with IPF and development of pulmonary fibrosis in response to injury (5, 16, 25–27). Deficiency or dysfunction of multiple mediators of mitophagy has been implicated in the pathobiology of IPF making maintenance or augmentation of mitophagy an attractive avenue of potential intervention. Our work has shown that type II alveolar epithelial cells from IPF lungs are deficient in PINK1 resulting in an accumulation of dysmorphic mitochondria with reduced transmembrane potential, reduced electron transport chain (ETC) activity, increased ROS production, and increased opening of the mitochondrial permeability transition pore. PINK1 knockout was also found to be sufficient to recapitulate this mitochondrial phenotype and confer vulnerability to fibrosis in response to lung injury (5). PINK1 expression has been found to diminish with age and persistent endoplasmic reticulum stress. ER stress has been shown to induce expression of ATF3, a transcriptional repressor of PINK1, in alveolar epithelial cells (28). Interestingly, deficiency of PINK1 in lung epithelial cells was also associated with upregulation of markers of senescence (p16 and p21) and increased levels of TGF-β expression, a key mediator of fibrogenic processes in IPF (5, 28).
As adaptive responses, autophagy and mitophagy are essential to maintaining normal fibroblast function and preventing apoptosis. Impairment of mitophagy, mediated by Parkin deficiency, in IPF lung fibroblasts has been associated with increased deposition of extracellular matrix under profibrotic conditions such as exposure to TGF-β. Defects in mitophagy and autophagy result in increased production of ROS and activation of platelet-derived growth factor receptor (PDGFR)/mammalian target of rapamycin (mTOR) signaling pathways that enhance fibroblast to myofibroblast transformation (24). Supporting these data, Parkin-deficient mice develop more severe lung fibrosis. Induction of autophagy and treatment with antioxidants reduced fibroblast to myofibroblast differentiation, myofibroblast proliferation, and fibrogenesis suggesting that this process is driven by increased mitochondrial ROS (24, 25). Indeed, we now recognize that a subset of surfactant protein C mutations, which causes a familial form of pulmonary fibrosis, acts through mistrafficking of surfactant protein C, accumulation within endosomes, and a late block of macroautophagy and mitophagy within type II alveolar epithelial cells. This results in proteostasis and an accumulation of dysfunctional mitochondria with increased mitochondrial mass but decreased transmembrane potential, which may increase susceptibility to pulmonary fibrosis when an additional “second hit” stressor is present (29).
TGF-β has been shown to variably affect key mediators of mitophagy and promote fibrogenesis. Treatment of alveolar epithelial cells with TGF-β results in initial stabilization of PINK1 on the surface of mitochondria and induction of mitophagy, which appears to be an adaptive response to increased ROS production due to TGF-β’s effect on the ETC (30). Longer term exposure to TGF-β results in impaired mitophagy through downregulation of phosphatase and tensin homolog (PTEN), and a subsequent reduction in PINK1 expression (25). Another potential activator of autophagy is T4, which is a well-known activator of AMPK and inhibitor of mTOR (15). Supporting this role of T4 signaling in PINK1-mediated mitochondrial homeostasis and mitophagy, the therapeutic effect of inhaled T3 in mouse models of lung fibrosis required an active PINK1 signaling pathway (15).
On the contrary, pulmonary macrophages isolated from IPF lungs demonstrate increased mitophagy. IPF macrophages are a source of activated TGF-β, and macrophage resistance to apoptosis is necessary for disease progression. IPF macrophages activate TGF-β by producing mitochondrial H2O2 through activation of the pro-survival kinase, protein kinase B (Akt1). Increased Akt1 activation and ROS production induce mitophagy as a protective measure and prevent macrophage apoptosis, which stabilizes macrophages to release additional TGF-β and promote local fibroblast activation and proliferation. Blocking mitophagy in alveolar macrophages is protective against bleomycin-induced fibrosis (27). This finding exemplifies the role of mitophagy as an adaptive survival response that, in this case, serves to preserve and promote macrophage signaling.
Mitochondrial OXPHOS
The processes of mitochondrial respiration and OXPHOS are the highly efficient and preferable means of ATP production, but a natural byproduct of these processes is superoxide and hydrogen peroxide production, which is produced in increasing quantities by dysfunctional mitochondria (31). Mitochondrial ROS (mtROS) serve as a signaling mechanism acting at multiple points throughout the cell that serves to inactivate phosphatases, activate select kinases, and facilitate hypoxia-inducible factor 1-α (HIF-1α), p53, and NF-κB signaling pathways (32–36). Interestingly, mtROS levels have a biphasic effect. At low levels, mtROS stimulate an increase in antioxidant capacity, referred to as mitohormesis, which has a protective effect and has been associated with longevity and resistance to cellular injury. In higher concentrations or with sustained production, ROS propagates free radicals that cause oxidative damage to lipids, proteins, and DNA (37).
Control of OXPHOS and energy production occurs through cooperative signaling mechanisms at mitochondrial, cytosolic, and nuclear levels in order to balance OXPHOS, energetic demands of the cell, and nutrient supply. This complex process limits production of ROS. Additionally, mitochondria have multiple mechanisms for reducing mtROS, but effective regulation of ROS production and mitigation declines with aging and potentiates susceptibility to lung injury and fibrosis (18, 38, 39). Aging is associated with lower ATP production and increased ROS (40). Moreover, older animals have been shown to accumulate higher levels of oxidized proteins in response to lung injury suggesting waning cellular antioxidant defense systems (41).
A profibrotic environment promotes mitochondrial dysfunction in pulmonary epithelial cells. Studies in Mv1Lu cells demonstrate that treatment with TGF-β1 downregulates mitochondrial ETC function, particularly at complex IV, resulting in loss of mitochondrial transmembrane potential and increased mtROS production (42). Increased ROS serves to oxidize and activate latent TGF-β1 creating a self-reinforcing cycle with potential to recruit fibroblasts and promote fibrogenesis (30). This correlates with the findings that type II alveolar epithelial cells from IPF lungs have reduced ETC complex I and IV activity (5), and IPF fibroblasts have reduced ATP content and reduced rate of oxygen consumption indicating poor mitochondrial function (43).
While the effectiveness of antioxidant supplementation in preventing diseases of aging such as IPF has been disappointing and, in some cases, detrimental, overexpression of an antioxidant protective mechanism in mitochondria has shown effectiveness in abrogating the fibrotic response to bleomycin-induced lung injury. Mice that overexpress mitochondria-targeted human catalase predictably show lower levels of mtROS, but also display reduced mtDNA damage and fragmentation with decreased pulmonary fibrosis in response to oxidative injury from asbestos or bleomycin (44). This raises the possibility that mitochondrial-targeted antioxidants may prevent or abrogate the development of IPF by scavenging mtROS at the source.
mtDNA Damage
Mitochondrial DNA is significantly more susceptible to oxidative injury than nuclear DNA, likely due to proximity to the source of ROS, lack of protective histones, and relative paucity of mtDNA repair mechanisms (45). Indeed, oxidative DNA damage is a key mechanism of injury in the commonly used model of bleomycin-induced pulmonary fibrosis (46). In comparison with the nucleus, mitochondria contain fewer and less efficient mechanisms for correcting mutations and repairing DNA damage. Mitochondrial transcription factor A (TFAM) is a key regulator of mtDNA transcription and replication, which also functions to induce U-shaped bends in mtDNA. These protective nucleoids can act to sequester damaged DNA and prevent transcription until base excision repair enzymes can correct the damage (47). The base excision repair enzyme, 8-oxoguanine DNA glycosylase 1 (OGG1), plays a critical role in repair of mtDNA oxidative damage, OGG1 deficiency or inactivation predisposes to pulmonary fibrosis (26, 48). The processes of mtROS production and mtDNA damage are intricately linked, and oxidative injury and cellular stress drive apoptotic and senescence pathways that may contribute, at least in part, to both aging and to lung injury and fibrosis. Additional studies in IPF lung-derived cells or tissues are needed to establish the role of mtDNA damage in IPF.
Owing to the evolutionary origin of mitochondria as bacterial endosymbionts, mtDNA contains CpG-rich sequences, which have the potential to act as damage-associated molecular patterns and activate innate immune system mechanisms when they escape the mitochondrial milieu. This pathway may contribute to a pro-fibrotic microenvironment (49). Recently, it has been shown that IPF fibroblasts undergo glycolytic reprogramming associated with release of mtDNA into the cellular interstitium and ultimately into circulation. Furthermore, increased plasma levels of mtDNA were associated with an increase in all-cause mortality suggesting potential use of circulating mtDNA as a biomarker for progression or severity of IPF (50).
Sirtuins are Key Mediators of Mitochondrial Health Implicated in IPF
Deficiency of the mitochondrial NAD-dependent deacetylase, SIRT3, has been shown to confer susceptibility to epithelial cell apoptosis and fibrosis in both bleomycin and asbestos models of lung injury (48). SIRT3 is an active enzyme in ROS detoxification as well as mtDNA protection and repair pathways, and its dependence on NAD+ as a cofactor directly links SIRT3’s function to cellular bioenergetics and TCA cycle activity (51). In the mitochondrion, SIRT3 is implicated in regulation of two redox mechanisms. SIRT3 deacetylates and activates manganese superoxide dismutase (MnSOD) and isocitrate dehydrogenase 2 (IDH2) (52, 53). Low levels of SIRT3 result in increased acetylation and decreased MnSOD activity and an accumulation of mtROS and mtDNA oxidative damage (53). The matrix protein IDH2 is a key provider of the reducing agent, NADPH, necessary for proper functioning of protective antioxidant pathways (52). The mtDNA repair function of OGG1 is also regulated by SIRT3. In the absence of SIRT3, OGG1 is increasingly acetylated and deactivated resulting in unchecked mtDNA damage and lung epithelial cell apoptosis (44, 48).
SIRT3 knockout mice have also been shown to develop increased expression of TGF-β and fibrosis in multiple organs as they age. This has been shown to occur through increased acetylation and deactivation of glycogen synthase kinase-β and stabilization of profibrotic transcription factors, such as smad3 and β-catenin, independent of SIRT3’s effect on MnSOD activity (54). Multiple SIRT3-dependent pathways play important roles in preventing mitochondrial damage and vulnerability to fibrosis.
Another sirtuin, SIRT1, has been established to play a role in preventing both aging-related functional decline and age-related diseases such as IPF (55). SIRT1 is a nuclear NAD-dependent deacetylase, but one of its key functions is in modulating mitochondrial biogenesis and function through nuclear-to-mitochondrial signaling pathways (56). These pathways have demonstrated how nuclear DNA damage and telomere shortening can drive mitochondrial dysfunction. Telomerase reverse transcriptase deficiency and shortened telomeres are strongly linked with the development of pulmonary fibrosis, and this chromosomal shortening activates the DNA damage sensor and checkpoint inhibitor p53. Activated p53 represses expression of a key mediator of mitochondrial biogenesis, PPARγ coactivator-1α (PGC-1α) resulting in inefficient OXPHOS and increased ROS production (57). SIRT1 deacetylates and inactivates p53 restoring normal mitochondrial function (9, 56). The nuclear DNA damage sensor, poly[ADP-ribose] polymerase 1 (PARP-1), consumes NAD+ when activated and decreases activity of SIRT1 and PGC-1α. SIRT1 and PGC-1α have also been shown to translocate to the mitochondria and may assist in stabilization of mtDNA (16). Fasting and caloric restriction, long of interest in increasing longevity, and reducing functional decline with aging, increase SIRT1 protein levels and activity (51). SIRT1 translation is inhibited by the microRNA miR-34a, which is highly expressed in models of lung injury. SIRT1 deficiency results in increased acetylation of p53, which acts to induce expression of miR-34a, creating a self-reinforcing cycle and predisposing to development of fibrosis (58). In a bleomycin model of lung injury, upregulation of SIRT1 attenuated fibroblast activation and the development of fibrosis (59). Sirtuins clearly have complex functions in modulating mitochondrial biogenesis, function, ROS production, and mtDNA protection, and this area of investigation has potential to shed new light on the pathogenesis of IPF and other diseases of aging.
Metabolic Reprogramming in IPF
Fibrotic lung tissue in IPF has been shown to have increased metabolic activity as demonstrated by increased FDG uptake on PET scan, and increased PET activity correlates with disease progression and mortality (60). Cell-type specific metabolic changes occur in the IPF lung (Figure 2). Type II alveolar epithelial cells have downregulation of genes involved in lipid synthesis and metabolism identified through single cell RNA sequencing (61). Fibroblasts have been shown to undergo a metabolic shift away from the highly efficient method of ATP production, OXPHOS, to the less efficient method of glycolysis despite adequate oxygen to continue OxPhos (62–67). Additionally, alveolar macrophages in fibrotic lung tissue increase fatty acid oxidation in response to the shift to glycolysis (66). A similar metabolic shift was initially observed in cancer cells and was eponymously dubbed the Warburg effect after its discoverer (68). The Warburg-like metabolic reprogramming observed in IPF cells results in increased glucose uptake and an accumulation of TCA cycle metabolites and byproducts that act as signaling mechanisms. Kottmann and colleagues demonstrated that glycolytic flux increases lactate production and lowers the local tissue pH resulting in increased activation of TGF-β, stabilization of the transcription factor HIF-1α, and increased transcription of lactate dehydrogenase 5 (LDH5), which synergizes with TGF-β to induce differentiation of fibroblasts to myofibroblasts (62). LDH5 produces lactate, which can be exported by monocarboxylate transporter (MCT)-4 and may be taken up by adjacent cells expressing MCT-1. Shuttled lactate can be oxidized to pyruvate providing additional energy through the TCA cycle and driving OXPHOS. This phenomenon is termed the reverse Warburg effect and promotes fibroblast proliferation and increased levels of mtROS.
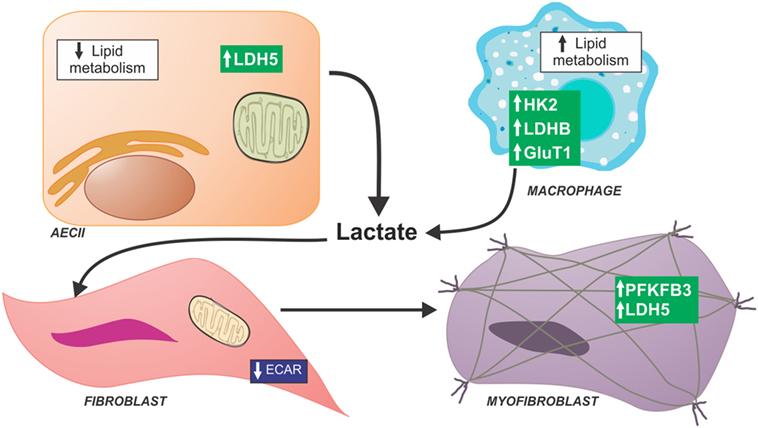
Figure 2. Schematic of metabolic dysfunction in lung fibrosis. Idiopathic pulmonary fibrosis (IPF) lung have a generalized decrease in late stages of glycolysis compared with normal lungs; however, increased levels of lactate were found in IPF lungs suggesting that all products of glycolysis are shuttled toward lactate production. Several studies have been focused in metabolic alterations of specific cell compartments in the IPF lung. IPF lung fibroblasts show decreased glycolytic function determined by extracellular acid production. IPF myofibroblasts have been reported to have higher expression of glycolytic enzymes and lactate content. Single cell RNA sequence data in alveolar epithelial cells show low expression of enzymes necessary for lipid metabolism. By contrast, alveolar macrophages isolated from bleomycin-treated mice show glycolytic reprogramming and increase fatty acid oxidation. Abbreviations: AECII, type II alveolar epithelial cell; GluT1, glucose transporter 1; HK2, hexokinase 2; LDH5, lactate dehydrogenase 5; LDHB, lactate dehydrogenase B; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3.
Primary myofibroblasts derived from IPF lungs and lung fibroblasts treated with TGF-β demonstrate increased lactate contents. Additionally, IPF myofibroblasts showed increase in expression of glycolytic enzymes including 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKB3), an enzyme that increases levels of fructose-2,6-bisphosphate and drives glycolysis (63). Glycolytic reprogramming in fibroblasts is, at least partially, driven by TGF-β. Treating normal fibroblasts with TGF-β has been shown to increase transcription of phosphofructokinase (PFK) and hexokinase 2, two key rate-limiting glycolytic enzymes (63). Increased lactate production may be due to shunting of glycolytic products toward anaerobic metabolism due to concurrent mitochondrial dysfunction.
By contrast, a recent study comparing IPF lung tissue to normal human lung using a combined metabolomic and microarray analysis of key metabolic enzymes showed a decrease in the late-stage glycolytic products, fructose 1,6-bisphosphate and phosphoenolpyruvate, and a decrease in PFK and PFKFB3, suggesting an overall reduction in glycolysis in IPF whole lung tissue (69). These findings are in concordance with recent observations in fibroblasts derived from IPF lungs. IPF fibroblasts trended toward having a lower rate of glycolysis, as measured by extracellular acidification rate, and had lesser glycolytic flux in response to TGF-β stimulation compared to normal lung fibroblasts (43). The latter finding may represent a diminished capacity to respond to TGF-β after chronic exposure to this profibrotic cytokine in vivo.
Despite these differences, it is known that TGF-β can mediate metabolic reprogramming. TGF-β induces expression of the facultative glucose transporter, glucose transporter 1 (GLUT1), via canonical Smad and PDGFR signaling resulting in increased cellular glucose uptake. Inhibiting GLUT1 upregulation is protective against bleomycin-induced fibrosis (64), and GLUT1 activation of AMPK is integral to fibroblast activation (67). Since metabolic reprogramming appears to be a critical step in promoting fibroblast-to-myofibroblast differentiation, preventing this shift is an intriguing target in attenuating IPF. However, further studies are needed to confirm the metabolic changes in different cell compartments in the IPF lung and determine how these alterations might modify disease progression.
Mitochondrial Dysfunction and Senescence
Inducing senescence and apoptosis in fibroblasts are important mechanisms in the wound healing response after injury (70); on the contrary, the aberrant deposition of extracellular matrix seen in IPF is associated with an accumulation of apoptosis-resistant senescent cells (39, 43, 71, 72). Mitochondrial dysfunction has been shown to induce cellular senescence, a process termed mitochondrial dysfunction-associated senescence. In addition, mitophagy defects seem to contribute to a cellular senescence phenotype, which we have observed in the setting of mitochondrial dysfunction induced by PINK1 deficiency in vivo (28). Dysfunctional mitochondria are less efficient at oxidizing NADH to NAD+ resulting in a reduced NAD+/NADH ratio, activation of AMPK and p53 resulting in a senescent phenotype (73). Senescent IPF lung epithelial cells, fibroblasts, and myofibroblasts secrete a cell type-specific profile of pro-fibrotic and pro-inflammatory cytokines known as the senescence-associated secretory phenotype (SASP) (72, 74). Senescent fibroblasts exhibit an increased level of the ROS-generating enzyme Nox4 and low levels of the key mediator of cellular antioxidant response pathways, NFE2-related factor 2, and this redox imbalance plays a role in maintaining the senescent phenotype. Treatment with a Nox inhibitor restores the redox balance and results in a decrease in senescent cells and a reduction in fibrosis (18). In vitro, mouse model, and ex vivo studies in human IPF lung cells have all shown improvement in fibrosis with treatment with a combination of antioxidants (quercetin) and senolytic agents (dasatinib) (72, 74). This approach shows promise as a therapeutic option.
Future Directions: Targeting Mitochondria as an Antifibrotic Therapy
Research has extensively shown that both aging and IPF are associated with alterations in cellular bioenergetics and mitochondrial homeostasis. Many of these mitochondrial pathways, especially those involved in maintenance of redox balance, mtDNA protection, and reversal of senescence, have shown promise as therapeutic targets in attenuating or ameliorating fibrosis in vitro and in animal models of pulmonary fibrosis. These early investigations of the role of altered bioenergetics in the development of fibrotic lung disease pave the way for translational research to implicate these pathways in human IPF. Other pathways, specifically those involved in maintaining normal mitophagy and turnover of dysfunctional mitochondria and mitochondrial-targeted antioxidants, present potential avenues for altering the natural history of pulmonary fibrosis but require further investigation to identify a point of intervention. Few potential interventions appear ready for human trials to restore mitochondrial biogenesis and mitophagy in alveolar epithelial cells and use of combination senolytic and antioxidant agents to remove senescent cells from injured lung and prevent propagation of SASP signaling (75). These approaches benefit from a history of therapeutic use and safety in treatment of human disease.
There is considerably more to learn about the role of mitochondrial dysfunction in the development and maintenance of the pro-fibrotic conditions that drive IPF, regarding the interplay between mitochondrial and nuclear signaling in overall cellular dysfunction, telomere shortening, and mtDNA damage, and the varying effects of mitochondrial dysfunction in the different cell types (epithelial cells, macrophages, fibroblasts, and myofibroblasts) involved in the development of IPF. Additionally, cellular perturbations seen with normal aging such as decreased macroautophagy and cellular proteostasis have the potential to incite mitochondrial dysfunction and changes in cellular bioenergetics. Development of animal models capable of isolating defects in mitochondrial homeostasis to a particular cell type may be revelatory of the role each cell type plays and shed light on the optimal targets for intervention. Particular attention to the effects of mitochondrial dysfunction on telomere length in the resident progenitor cells of the lungs responsible for regenerating the alveolar epithelium may present novel approaches for preventing stem cell exhaustion in IPF.
While many of these pathways have shown potential as therapeutic targets in isolation, an integrated approach to the bioenergetic and homeostatic changes that are seen with aging and IPF, such as telomere shortening, reduced mitochondrial biogenesis, increased ROS production, and increased nuclear and mtDNA damage, is essential to determining the pathways and therapeutic targets that are most likely to affect the natural history of IPF.
Author Contributions
DZ wrote the manuscript along with MB. The manuscript was conceptualized, supervised and modified by ALM and MR. All authors offered intellectual contribution and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
ALM is funded by NIH R01 HL131789A; the Aging Institute, University of Pittsburgh; the Institute for Transfusion Medicine; and the Hemophilia Center for Western Pennsylvania. MR is funded by NIH R01 HL123766-01A1.
References
1. Khan SS, Singer BD, Vaughan DE. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell (2017) 16(4):624–33. doi:10.1111/acel.12601
2. Pardo A, Selman M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann Am Thorac Soc (2016) 13(Suppl_5):S417–21. doi:10.1513/AnnalsATS.201605-341AW
3. Mora AL, Bueno M, Rojas M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin Invest (2017) 127(2):405–14. doi:10.1172/JCI87440
4. Okamura DM, Pennathur S. The balance of powers: redox regulation of fibrogenic pathways in kidney injury. Redox Biol (2015) 6:495–504. doi:10.1016/j.redox.2015.09.039
5. Bueno M, Lai Y-C, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest (2015) 125(2):521–38. doi:10.1172/JCI74942
6. Schirone L, Forte M, Palmerio S, Yee D, Nocella C, Angelini F, et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid Med Cell Longev (2017) 2017:3920195. doi:10.1155/2017/3920195
7. Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest (2000) 106(7):847–56. doi:10.1172/JCI10268
8. Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab (2007) 5(2):151–6. doi:10.1016/j.cmet.2007.01.008
9. Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature (2011) 470(7334):359–65. doi:10.1038/nature09787
10. López-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Exp Gerontol (2008) 43(9):813–9. doi:10.1016/j.exger.2008.06.014
11. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell (2006) 127(6):1109–22. doi:10.1016/j.cell.2006.11.013
12. Short KR, Vittone JL, Bigelow ML, Proctor DN, Rizza RA, Coenen-Schimke JM, et al. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes (2003) 52(8):1888–96. doi:10.2337/diabetes.52.8.1888
13. Sinha RA, Singh BK, Zhou J, Wu Y, Farah BL, Ohba K, et al. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy (2015) 11(8):1341–57. doi:10.1080/15548627.2015.1061849
14. Lesmana R, Sinha RA, Singh BK, Zhou J, Ohba K, Wu Y, et al. Thyroid hormone stimulation of autophagy is essential for mitochondrial biogenesis and activity in skeletal muscle. Endocrinology (2016) 157(1):23–38. doi:10.1210/en.2015-1632
15. Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med (2017) 24(1):39–49. doi:10.1038/nm.4447
16. Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell (2014) 157(4):882–96. doi:10.1016/j.cell.2014.03.026
17. Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax (2010) 65(8):733–8. doi:10.1136/thx.2009.113456
18. Bernard K, Logsdon NJ, Miguel V, Benavides GA, Zhang J, Carter AB, et al. NADPH oxidase 4 (Nox4) suppresses mitochondrial biogenesis and bioenergetics in lung fibroblasts via a nuclear factor erythroid-derived 2-like 2 (Nrf2)-dependent pathway. J Biol Chem (2017) 292(7):3029–38. doi:10.1074/jbc.M116.752261
19. Ding W-X, Yin X-M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem (2012) 393(7):547–64. doi:10.1515/hsz-2012-0119
20. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ (2013) 20(1):31–42. doi:10.1038/cdd.2012.81
21. Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys (2007) 462(2):245–53. doi:10.1016/j.abb.2007.03.034
22. Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the Yin and Yang of cell death control. Circ Res (2012) 111(9):1208–21. doi:10.1161/CIRCRESAHA.112.265819
23. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol (2011) 12(1):9–14. doi:10.1038/nrm3028
24. Kobayashi K, Araya J, Minagawa S, Hara H, Saito N, Kadota T, et al. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J Immunol (2016) 197(2):504–16. doi:10.4049/jimmunol.1600265
25. Sosulski ML, Gongora R, Danchuk S, Dong C, Luo F, Sanchez CG. Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFβ1. Aging Cell (2015) 14(5):774–83. doi:10.1111/acel.12357
26. Kim S-J, Cheresh P, Jablonski RP, Williams DB, Kamp DW. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int J Mol Sci (2015) 16(9):21486–519. doi:10.3390/ijms160921486
27. Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity (2016) 44(3):582–96. doi:10.1016/j.immuni.2016.01.001
28. Bueno M, Brands J, Voltz L, Fiedler K, Mays B, St Croix C, et al. ATF3 represses PINK1 gene transcription in lung epithelia to control mitochondrial homeostasis. Aging Cell (2018). doi:10.1111/acel.12720
29. Hawkins A, Guttentag SH, Deterding R, Funkhouser WK, Goralski JL, Chatterjee S, et al. A non-BRICHOS SFTPC mutant (SP-CI73T) linked to interstitial lung disease promotes a late block in macroautophagy disrupting cellular proteostasis and mitophagy. Am J Physiol Lung Cell Mol Physiol (2015) 308(1):L33–47. doi:10.1152/ajplung.00217.2014
30. Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS One (2015) 10(3):e0121246. doi:10.1371/journal.pone.0121246
31. Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct (2012) 2012:646354. doi:10.1155/2012/646354
32. Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A (1998) 95(20):11715–20. doi:10.1073/pnas.95.20.11715
33. Chandel NS, Vander Heiden MG, Thompson CB, Schumacker PT. Redox regulation of p53 during hypoxia. Oncogene (2000) 19(34):3840–8. doi:10.1038/sj.onc.1203727
34. Chandel NS, Trzyna WC, McClintock DS, Schumacker PT. Role of oxidants in NF-B activation and TNF-gene transcription induced by hypoxia and endotoxin. J Immunol (2000) 165(2):1013–21. doi:10.4049/jimmunol.165.2.1013
35. Nemoto S, Takeda K, Yu ZX, Ferrans VJ, Finkel T. Role for mitochondrial oxidants as regulators of cellular metabolism. Mol Cell Biol (2000) 20(19):7311–8. doi:10.1128/MCB.20.19.7311-7318.2000
36. Rhee SG, Bae YS, Lee SR, Kwon J. Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE (2000) 2000(53):pe1. doi:10.1126/stke.2000.53.pe1
38. Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang J-Y, et al. SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3β. Mol Cell Biol (2015) 36(5):678–92. doi:10.1128/MCB.00586-15
39. Thannickal VJ. Mechanistic links between aging and lung fibrosis. Biogerontology (2013) 14(6):609–15. doi:10.1007/s10522-013-9451-6
40. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi:10.1016/j.cell.2013.05.039
41. Friguet B. Oxidized protein degradation and repair in ageing and oxidative stress. FEBS Lett (2006) 580(12):2910–6. doi:10.1016/j.febslet.2006.03.028
42. Yoon Y-S, Lee J-H, Hwang S-C, Choi KS, Yoon G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene (2005) 24(11):1895–903. doi:10.1038/sj.onc.1208262
43. Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, et al. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol (2017) 313(6):L1164–73. doi:10.1152/ajplung.00220.2017
44. Kim S-J, Cheresh P, Jablonski RP, Morales-Nebreda L, Cheng Y, Hogan E, et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic Biol Med (2016) 101:482–90. doi:10.1016/j.freeradbiomed.2016.11.007
45. Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A (1997) 94(2):514–9. doi:10.1073/pnas.94.2.514
46. Fujita M, Mizuta Y, Ikegame S, Ouchi H, Ye Q, Harada E, et al. Biphasic effects of free radical scavengers against bleomycin-induced pulmonary fibrosis. Pulm Pharmacol Ther (2008) 21(5):805–11. doi:10.1016/j.pupt.2008.06.006
47. Hallberg BM, Larsson N-G. TFAM forces mtDNA to make a U-turn. Nat Struct Mol Biol (2011) 18(11):1179–81. doi:10.1038/nsmb.2167
48. Jablonski RP, Kim S-J, Cheresh P, Williams DB, Morales-Nebreda L, Cheng Y, et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J (2017) 31(6):2520–32. doi:10.1096/fj.201601077R
49. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
50. Ryu C, Sun H, Gulati M, Herazo-Maya JD, Chen Y, Osafo-Addo A, et al. Extracellular mitochondrial DNA is generated by fibroblasts and predicts death in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2017) 196(12):1571–81. doi:10.1164/rccm.201612-2480OC
51. Kincaid B, Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci (2013) 5:48. doi:10.3389/fnagi.2013.00048
52. Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem (2012) 287(17):14078–86. doi:10.1074/jbc.M112.355206
53. Chen Y, Zhang J, Lin Y, Lei Q, Guan K-L, Zhao S, et al. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep (2011) 12(6):534–41. doi:10.1038/embor.2011.65
54. Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal (2014) 20(10):1646–54. doi:10.1089/ars.2013.5482
55. Tang BL. Sirt1 and the mitochondria. Mol Cells (2016) 39(2):87–95. doi:10.14348/molcells.2016.2318
56. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature (2005) 434(7029):113–8. doi:10.1038/nature03354
57. Sui B, Hu C, Jin Y. Mitochondrial metabolic failure in telomere attrition-provoked aging of bone marrow mesenchymal stem cells. Biogerontology (2016) 17(2):267–79. doi:10.1007/s10522-015-9609-5
58. Shetty SK, Tiwari N, Marudamuthu AS, Puthusseri B, Bhandary YP, Fu J, et al. p53 and miR-34a feedback promotes lung epithelial injury and pulmonary fibrosis. Am J Pathol (2017) 187(5):1016–34. doi:10.1016/j.ajpath.2016.12.020
59. Zeng Z, Cheng S, Chen H, Li Q, Hu Y, Wang Q, et al. Activation and overexpression of Sirt1 attenuates lung fibrosis via P300. Biochem Biophys Res Commun (2017) 486(4):1021–6. doi:10.1016/j.bbrc.2017.03.155
60. Justet A, Laurent-Bellue A, Thabut G, Dieudonné A, Debray M-P, Borie R, et al. [18F]FDG PET/CT predicts progression-free survival in patients with idiopathic pulmonary fibrosis. Respir Res (2017) 18(1):74. doi:10.1186/s12931-017-0556-3
61. Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight (2016) 1(20):e90558. doi:10.1172/jci.insight.90558
62. Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med (2012) 186(8):740–51. doi:10.1164/rccm.201201-0084OC
63. Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu R-M, et al. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med (2015) 192(12):1462–74. doi:10.1164/rccm.201504-0780OC
64. Andrianifahanana M, Hernandez DM, Yin X, Kang J-H, Jung M-Y, Wang Y, et al. Profibrotic up-regulation of glucose transporter 1 by TGF-β involves activation of MEK and mammalian target of rapamycin complex 2 pathways. FASEB J (2016) 30(11):3733–44. doi:10.1096/fj.201600428R
65. Xu J, Li J, Yu Z, Rao H, Wang S, Lan H. HMGB1 promotes HLF-1 proliferation and ECM production through activating HIF1-α-regulated aerobic glycolysis. Pulm Pharmacol Ther (2017) 45:136–41. doi:10.1016/j.pupt.2017.05.015
66. Xie N, Cui H, Ge J, Banerjee S, Guo S, Dubey S, et al. Metabolic characterization and RNA profiling reveal glycolytic dependence of pro-fibrotic phenotype of alveolar macrophages in lung fibrosis. Am J Physiol Lung Cell Mol Physiol (2017) 313(5):L834–44. doi:10.1152/ajplung.00235.2017
67. Cho SJ, Moon J-S, Lee C-M, Choi AMK, Stout-Delgado HW. Glucose transporter 1-dependent glycolysis is increased during aging-related lung fibrosis, and phloretin inhibits lung fibrosis. Am J Respir Cell Mol Biol (2017) 56(4):521–31. doi:10.1165/rcmb.2016-0225OC
68. Maher TM. Aerobic glycolysis and the Warburg effect. An unexplored realm in the search for fibrosis therapies? Am J Respir Crit Care Med (2015) 192(12):1407–9. doi:10.1164/rccm.201508-1699ED
69. Zhao YD, Yin L, Archer S, Lu C, Zhao G, Yao Y, et al. Metabolic heterogeneity of idiopathic pulmonary fibrosis: a metabolomic study. BMJ Open Respir Res (2017) 4(1):e000183. doi:10.1136/bmjresp-2017-000183
70. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell (2014) 31(6):722–33. doi:10.1016/j.devcel.2014.11.012
71. Chilosi M, Poletti V, Rossi A. The pathogenesis of COPD and IPF: distinct horns of the same devil? Respir Res (2012) 13:3. doi:10.1186/1465-9921-13-3
72. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun (2017) 8:14532. doi:10.1038/ncomms14532
73. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab (2016) 23(2):303–14. doi:10.1016/j.cmet.2015.11.011
74. Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J (2017) 50(2):1602367. doi:10.1183/13993003.02367-2016
Keywords: aging, mitochondrial dysfunction, lung fibrosis, bioenergetics, mitophagy, senescence
Citation: Zank DC, Bueno M, Mora AL and Rojas M (2018) Idiopathic Pulmonary Fibrosis: Aging, Mitochondrial Dysfunction, and Cellular Bioenergetics. Front. Med. 5:10. doi: 10.3389/fmed.2018.00010
Received: 06 September 2017; Accepted: 15 January 2018;
Published: 05 February 2018
Edited by:
Argyrios Tzouvelekis, Alexander Fleming Biomedical Sciences Research Center, GreeceReviewed by:
Bruno Crestani, Paris Diderot University, FranceEleni Papakonstantinou, Aristotle University of Thessaloniki, Greece
Copyright: © 2018 Zank, Bueno, Mora and Rojas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana L. Mora, YW5hbW9yYUBwaXR0LmVkdQ==;
Mauricio Rojas, cm9qYXNtQHVwbWMuZWR1