 Line Aas Mortensen
Line Aas Mortensen Claus Bistrup
Claus Bistrup Helle Charlotte Thiesson
Helle Charlotte Thiesson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 24 November 2017
Sec. Nephrology
Volume 4 - 2017 | https://doi.org/10.3389/fmed.2017.00210
This article is part of the Research Topic Nephrotoxicity of Immune Checkpoint Inhibitors View all 6 articles
Calcineurin inhibitors have markedly reduced acute rejection rates in renal transplantation, thus significantly improved short-term outcome. The beneficial effects are, however, tampered by acute and chronic nephrotoxicity leading to interstitial fibrosis and tubular atrophy, which impairs long-term allograft survival. The mineralocorticoid hormone aldosterone induces fibrosis in numerous organs, including the kidney. Evidence from animal models suggests a beneficial effect of aldosterone antagonism in reducing calcineurin inhibitor-induced nephrotoxicity. This review summarizes current evidence of mineralocorticoid receptor antagonism in animal models of calcineurin inhibitor-induced nephrotoxicity and the results from studies of mineralocorticoid antagonism in renal transplant patients.
The introduction of calcineurin inhibitors (CNIs) in renal transplantation has markedly reduced the occurrence of acute rejection and graft failure (1, 2). The immunosuppressive effect of CNI is, however, counterbalanced by adverse long term effects on kidney function leading to histological changes (i.e., arteriolar hyalinosis, striped interstitial fibrosis, tubular atrophy, and glomerulosclerosis) referred to as interstitial fibrosis and tubular atrophy (IF/TA) (3).
The mineralocorticoid hormone aldosterone stimulates fibrogenesis in multiple organs. Evidence from animal and human studies indicates that antagonizing aldosterone signaling via the mineralocorticoid receptor (MR) by MR antagonists reduces CNI nephrotoxicity.
This review summarizes current evidence from animal studies and outlines the potential benefits of MR antagonism in kidney transplant patients.
Although structurally different, both cyclosporine (CsA) and tacrolimus exert their effect by inhibiting the activity of calcineurin, a calcium- and calmodulin-dependent phosphatase involved in the activation of T-lymphocytes. Complexes of CsA/cyclophylin or tacrolimus/FKBP12 bind competitively to calcineurin, thereby preventing the dephosphorylation and subsequent activation of nuclear factor of activated T-cells (NFAT). Activated NFAT promotes transcription of interleukin-2, which is pivotal for the activation of T-lymphocytes (4).
Nephrotoxicity has long been recognized as an adverse effect of CNI leading to chronic allograft failure and ultimately increased morbidity and mortality, mainly due to cardiovascular disease (5). Acute CNI nephrotoxicity is induced by vasoconstriction due to an imbalance between vasodilating and vasoconstricting factors and is reversible, whereas chronic CNI nephrotoxicity is considered to be irreversible. The proposed pathways of CNI nephrotoxicity are summarized in Figure 1. For an extensive review of CNI induced nephrotoxicity, see Ref. (3).
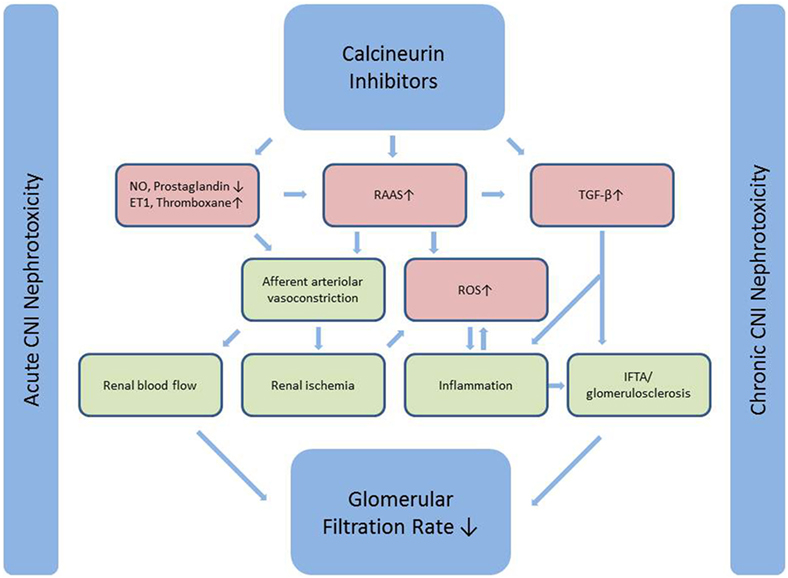
Figure 1. Calcineurin inhibitors induce afferent arteriolar vasoconstriction through an effect on both mediators of endothelial dysfunction and a direct stimulatory effect on the RAAS-system. Vasoconstriction leads to reduced renal blood flow (acute CNI nephrotoxicity) and renal ischemia, which ultimately leads to inflammation and fibrosis (chronic CNI nephrotoxicity). The latter is further induced by a direct stimulatory effect on the major pro-fibrotic cytokine TGF-β. Simplified from Naesens et al. (3). CNI, calcineurin inhibitor; NO, nitric oxide; ET1, endothelin 1; RAAS, renin-angiotensin-aldosterone system; TGF-β, transforming growth factor β; ROS, reactive oxygen species; IF/TA, interstitial fibrosis and tubular atrophy.
Attempts to prevent or reduce CNI nephrotoxicity in humans have focused on angiotensin antagonism or vasodilating agents. Although central in the hypothesized mechanism of CNI nephrotoxicity, studies targeting the effects of angiotensin II have not yielded the expected results on long-term allograft survival. One randomized clinical trial (RCT), although designed to evaluate the effect of angiotensin converting enzyme inhibitor (ACEI) ramipril on cardiovascular outcomes in renal transplant patients, did not show any difference in long-term renal function when compared to placebo (6). Similarly, the angiotensin receptor blocker (ARB) losartan did not have an effect on the composite endpoint of interstitial volume expansion and end-stage renal disease in 153 renal transplant patients after 5 years (7). Early studies indicated a beneficial effect of calcium channel antagonists in both short- (8) and long-term renal allograft function (9, 10); however, results have been somewhat conflicting [summarized in Ref. (3)] and have not translated into clinical practice. Whether the beneficial effect of calcium antagonists on renal function is mainly due to pre-renal factors or due to reduced renal fibrosis remains to be investigated. Studies of nitric oxide (NO) donors or vasodilatory prostanoids in humans and animal studies of anti-transforming growth factor β (TGF-β), antioxidants, statins, and magnesium have not shown a beneficial effect on kidney function (3). An alternative way to reduce CNI nephrotoxicity is via CNI minimization or complete CNI withdrawal; however, the majority of attempts have resulted in higher acute rejection rates (11). Of interest are the belatacept-protocols, showing superior graft function with belatacept for 7–10 years when compared with CsA despite higher rates of early acute rejection in the belatacept groups (12, 13). Adverse event rates were similar (12). The use of belatacept as an alternative to CNI in solid organ transplantation has been summarized in a recent review (14).
The relative contribution of CNI nephrotoxicity to late allograft failure has been the object of debate in recent years (15). Early reports indicated a prevalence of chronic CNI nephrotoxicity of almost 100% in renal allograft biopsies after 10 years (16), which was supported by the finding of IF/TA in a large proportion of kidney biopsies from non-renal transplant patients (17). Since then, standard therapy has changed from high dose CsA toward lower-dose tacrolimus (18). Induction therapy in combination with mycophenolate has made CNI minimization possible. A recent study by Nankivell et al. compared sequential kidney graft biopsies from the CsA era (1988–1998) with the tacrolimus era (1999–2012). These showed a lower prevalence of chronic histological lesions in the tacrolimus group, indicating lower nephrotoxicity of current protocols. However, both cellular and humoral acute rejection rates were significantly lower in the tacrolimus era (19) where mycophenolate had also replaced azathioprine. Hence, the superior results of the tacrolimus era might be more complex than merely CNI minimization and reflect both immunological and non-immunological advances.
Aldosterone regulates sodium and water balance via the MR in the kidney but is also involved in deleterious processes leading to fibrosis including vasoconstriction, inflammation, and oxidative stress (Figure 2). Upon aldosterone binding to the MR, the receptor translocates to the nucleus where it regulates gene transcription (20). To date, two steroidal MR antagonists have been approved for clinical use, namely spironolactone and eplerenone. Recommended dosages for both drugs are 25–50 mg daily in heart failure and up to 400 mg daily for spironolactone in hyperaldosteronism and liver cirrhosis with ascites. Spironolactone competitively binds to the MR with a high affinity, but also exerts antiandrogenic side effects through binding to the progesterone and androgen receptors (21). Eplerenone is more selective to the MR with fewer antiandrogenic side effects, but simultaneously has a lower affinity for the MR, hence is less potent (22). Much effort has gone into developing newer compounds with high affinity and high selectivity. The non-steroidal MR antagonist finerenone is currently undergoing phase III clinical trials in heart failure and diabetic kidney disease. Further, several other MR antagonists are in the pipeline (23).
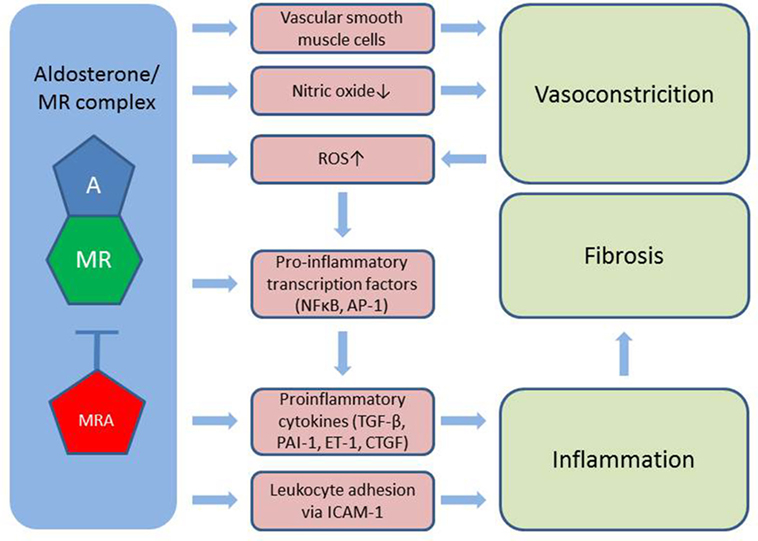
Figure 2. Aldosterone induces vasoconstriction via MR in vascular smooth muscle cells and through reduced bioavailability of nitric oxide. Also, aldosterone stimulates the formation of ROS further worsened by vasoconstriction. Activation of pro-inflammatory transcription factors as well as the direct stimulation of cytokines and leukocyte adhesion to the vessel wall leads to inflammation, which contributes to tissue fibrosis. A, aldosterone; MR, mineralocorticoid receptor; MRA, MR antagonist; ROS, reactive oxygen species; TGF-β, transforming growth factor β; PAI-1, plasminogen activator inhibitor 1; ET-1, endothelin 1; CTFG, colony transforming growth factor; ICAM-1, intercellular adhesion molecule 1; NFκβ, nuclear factor κβ; AP-1, activator protein 1.
Mineralocorticoid receptors are expressed in endothelial- and vascular smooth muscle cells, cardiomyocytes, mesangial cells, and podocytes in the kidney, adipocytes and a number of circulating cells, e.g., monocytes, macrophages, and dendritic cells (24). Aldosterone induces vasoconstriction, particularly under conditions of endothelial damage, likely through a direct action on vascular smooth muscle cells (25), and further reduces bioavailability of the potent vasodilator NO through MR-dependent mechanisms (26). This imbalance between vasoconstriction and vasodilation is commonly referred to as endothelial dysfunction and is associated with increased cardiovascular morbidity and mortality (25). MR antagonism has shown promising results in large scale RCTs in congestive heart failure (27, 28) reducing both morbidity and all-cause mortality. In addition to endothelial dysfunction, aldosterone stimulates the generation of reactive oxygen species by increasing the activity of nicotinamide adenine dinucleotide phosphate oxidase and by reducing the expression of glucose-6-phosphate dehydrogenase. This leads to the formation of superoxide and hydrogen peroxide, which in turn activate pro-inflammatory transcription factors nuclear factor kappa B (Nfκβ) and activator protein 1. Also, the aldosterone/MR complex directly stimulates pro-inflammatory growth factors and cytokines [e.g., TGF-β, plasminogen activator inhibitor 1 (PAI-1), endothelin 1, and colony transforming growth factor (CTGF)]. MR located in endothelial cells stimulates expression of the intercellular adhesion molecule 1, which facilitates the adhesion of leukocytes to the endothelium (24). The inflammatory environment hereby created plays a major part in initiating fibrogenesis (29).
Several clinical studies have evaluated the use of MR antagonists in chronic kidney disease. Results have been summarized in two systematic reviews including 27 and 19 studies, respectively, comprising 33 different trials. Most trials investigated the addition of MR antagonists to ACEI or ARB. Currie et al. reported a reduction of protein excretion of 38.7 ± 21.5% in any measure of protein/albumin excretion including all 19 trials associated with MR antagonism, which could be explained by a reduction of blood pressure (BP) (30). Bolignano et al. reported significantly reduced protein excretion rates for both spironolactone and eplerenone groups in addition to ACEI/ARB when compared to placebo (31). None of the included studies were powered to evaluate the effect on long term kidney function.
Cyclosporine A directly stimulates renin synthesis in the juxtaglomerular apparatus in rats and humans and further increases plasma renin activity (PRA) in rats, but this finding has not been reproduced in humans (32). On the contrary, early reports indicated that CsA reduced PRA and did not alter aldosterone levels in both healthy controls (33) and renal transplant patients (34). This difference between rats and humans could reflect physiological differences between species, but could also be related to differences in dosage and duration of treatment (32). A recent RCT measured PRA and plasma aldosterone at baseline and annually for 5 years in kidney transplant patients randomized to losartan or placebo. The majority of PRA and plasma aldosterone measurements in both groups were within normal range. Interestingly, higher aldosterone levels were associated with inferior graft function (35). It has been hypothesized that intrarenal RAAS contributes to CNI nephrotoxicity (32). Eplerenone reduces fibrosis and renal mRNA expression of collagen, TGF-β, and PAI-1 in adrenalectomized rats, indicating an influence of locally produced aldosterone (36). The importance of intrarenal activation of RAAS needs further investigation. As outlined in Figures 1 and 2, many deleterious processes related to aldosterone are represented in the chain of events leading to acute and chronic CNI nephrotoxicity explaining the rationale to investigate the effect of MR antagonism in CNI nephrotoxicity.
Acute CNI nephrotoxicity is characterized by vasoconstriction of the afferent arteriole leading to a decreased renal blood flow (RBF) and subsequently reduced glomerular filtration. Several groups have demonstrated an effect of MR antagonism preventing the reduction of RBF following CsA (Table 1). Perez-Rojas et al. demonstrated a reduction of RBF in CsA-treated rats. This effect was abolished by spironolactone, which also increased glomerular filtration rates (GFR) (37). The beneficial effect of spironolactone on kidney function in acute CNI nephrotoxicity was already highlighted in the 1980s (38–40). Similar results have been achieved using eplerenone (41). Recent evidence points toward MR in vascular smooth muscle cells to play a role in the vasoconstriction associated with acute CNI nephrotoxicity (42). Regarding structural changes related to acute CNI nephrotoxicity, Nielsen et al. demonstrated a reduction of tubular hyaline vacuolization and arteriolar depositions by eplerenone (41). Also, McAuley et al. found that spironolactone significantly decreased urinary tubular damage marker N-acetyl-beta-d-glucosaminidase (38).
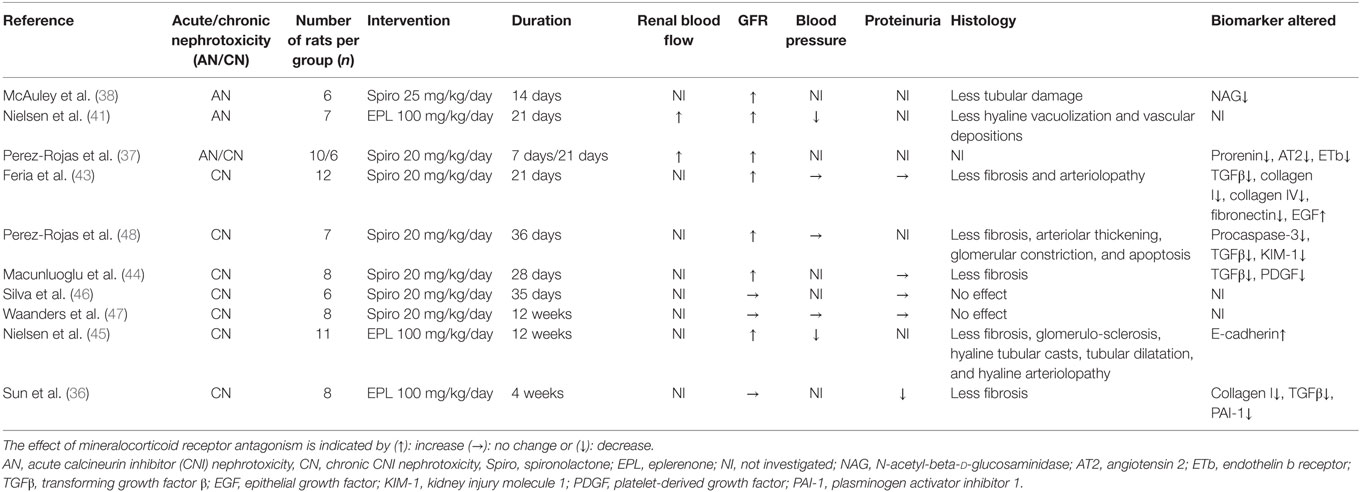
Table 1. Included animal studies.
Several aspects of chronic CNI nephrotoxicity in rat models have been investigated. Most studies initiate MR antagonists in parallel with CsA treatment, thus investigating if chronic kidney damage can be prevented (36, 37, 43–47). One study, however, investigated whether MR antagonism was beneficial after development of IF/TA. This showed improvement of GFR, fibrosis, and apoptosis, while all parameters further deteriorated in the non-MR antagonist group (=CsA-group) (48).
Similar to acute CNI nephrotoxicity, CsA has been shown to reduce RBF in rat models of chronic CNI nephrotoxicity, as indirectly indicated by constriction of glomeruli. This constriction was partly reverted by spironolactone (48). The molecular background for the vasoconstriction was investigated in sodium-depleted rats (37). They found an upregulation of prorenin by CsA, reverted by spironolactone. This study further demonstrated that the addition of spironolactone resulted in downregulation of the renal expression of endothelin A receptors and upregulation of endothelin B receptors, favoring vasodilation. Spironolactone had no influence on endothelin expression, adenosine pathways, and cyclooxygenase 2 mRNA levels (37). Taken together, this indicates that the beneficial effect of MR antagonism on renal vasoconstriction in chronic CNI nephrotoxicity involves both RAAS and endothelin pathways.
The majority of studies on chronic CNI nephrotoxicity in rats demonstrate a beneficial effect of MR antagonism on GFR (37, 43–45, 48). This relates to reduced renal vasoconstriction, but a likely contribution is the reduction of structural changes. One study failed to reach significance evaluating GFR based on s-creatinine levels after 5 weeks of spironolactone (46). In this study, blood concentrations of CsA were substantially lower than in similar studies and all kidneys were morphologically normal.
To our knowledge, only one study has investigated the effect of MR antagonsim in a model of renal transplantation in rats (47). In this study, 16 rats had kidney grafts from the same strain (Dark agouti—isografted) and 14 rats had kidney grafts from a different strain (Dark Agouti to Wistar Furth—allografted). All animals received CsA and were randomized to spironolactone 20 mg/kg/day or vehicle. After 12 weeks, there was no difference in GFR between vehicle and spironolactone groups in both iso- and allografted animals as evaluated by creatinine clearance. The allograft model was developed to mimic the histologic changes seen in chronic rejection in humans (49), thereby reflecting both immunologic and non-immunologic allograft damage. The observation that there was no fibrosis in kidneys from isografted animals despite CsA-treatment and extensive fibrosis in allografted animals highlights an immunological fibrogenesis rather than induced by CsA.
In contrast to CNI treatment in humans, rat models of CNI nephrotoxicity are not generally associated with hypertension. Accordingly, the majority of studies in chronic CNI nephrotoxicity did not find any significant effect of neither CNI treatment nor MR antagonist on BP by invasive (48) or non-invasive measurements (43, 47). One group, however, did find a significant increase in both daytime and nighttime BP in the CsA group compared to controls evaluated by intraarterial measurements in conscious animals. Eplerenone reduced BP during the resting period (45).
Only one of the reported studies of chronic CNI nephrotoxicity showed reduced proteinuria during MR antagonism (36). One study even reported a tendency for a lower grade of proteinuria in the CsA group compared to vehicle and spironolactone groups (43). This observation could be partly explained by the simultaneous reduction of GFR but could also reflect differences between rats and humans.
Feria et al. found less tubulointerstitial fibrosis in the spironolactone group (43). This was later confirmed by Macunluoglu et al. with all samples in the CsA-group displaying moderate to severe interstitial fibrosis (>25% affected area) compared to only one-fourth of samples in the spironolactone-group (44). Nielsen et al. showed increased volume fraction of renal interstitium in the CsA-group compared to controls, which was reduced in the eplerenone-group (45). Perez-Rojas et al. investigated the effect of MR antagonism initiated after established chronic CNI nephrotoxicity. After 18 days of CsA treatment, kidney sections from six rats displayed 20.2% tubulointerstitial fibrosis. Interestingly, spironolactone, when initiated at day 18, seemed to slow the progression of fibrosis at day 36 compared to CsA alone (48). Feria et al. and Nielsen et al. further demonstrated less arteriolopathy in groups treated with MR antagonists compared with CsA (43, 45).
In addition to histological changes in chronic CNI nephrotoxicity, focus has been on identifying markers of fibrosis. The molecular regulation of the fibrotic processes is an intricate cascade of cytokines and growth factors leading to increased transcription of constituents of fibrosis, i.e., collagen I, III, and fibronectin. CsA increases several fibrogenic markers in models of chronic CNI nephrotoxicity. One of the major pro-fibrotic cytokines is TGF-β acting via (1) the canonical pathway to activate transcription factors Smad2 and Smad3, which, in complex with Smad4, act to induce the transcription of pro-fibrotic molecules or (2) the non-canonical, Smad-independent pathways activating mitogen-activated protein kinases, c-Jun terminal kinase, p38, and extracellular-signal regulated kinase to increase the transcription of other pro-fibrotic cytokines as well as collagens and fibronectin (50, 51). Several studies have demonstrated increased renal mRNA levels of TGF-β in response to CsA, an increase that was attenuated by MR antagonsim (36, 43, 48). Macunluoglu et al. demonstrated a similar effect of MR antagonists by immunostaining of renal tissue (44).
In accordance with findings regarding TGF-β, CsA increased renal mRNA levels of both collagen I (36, 43) and fibronectin (43). This increase was mitigated by MR antagonism. CsA also increased α-smooth muscle actin—a marker of activated fibroblasts, although these levels were independent of MR antagonism (45), and the renal mRNA levels of several other profibrotic cytokines, i.e., platelet-derived growth factor (PDGF) (44), CTGF, and PAI-1 (36) all of which participate in the profibrotic cascade involving TGF-β. Concomitant MR antagonism significantly reduced levels of PDGF (44), and PAI-1, but not CTGF (36).
Tubular atrophy associated with renal fibrosis is partly due to apoptosis of tubular epithelial cells. Perez-Rojas et al. found a significant reduction of apoptotic nuclei in renal cortex by addition of spironolactone to CsA. This was paralleled by a reduction of apoptosis-marker procaspase 3 in the spironolactone group. Also, they demonstrated an increase in both cortical and urinary mRNA levels of kidney injury molecule 1 (KIM-1) in response to CsA and subsequent reduction by spironolactone (48).
Epithelial growth factor (EGF) stimulates regeneration of the tubular epithelium after ischemic damage and Feria et al. found a significant reduction of cortical EGF caused by CsA. Spironolactone diminished this reduction when added to CsA, but interestingly when administered alone also significantly reduced cortical EGF levels compared with vehicle (43). Nielsen et al. demonstrated a reduction of the epithelial marker E-Cadherin in response to CsA, which was attenuated by eplerenone (45). Taken together, this indicates a protective effect of MR antagonists on tubular epithelial cells. Whether this is mainly due to prevention of TGF-β-induced apoptosis and the reduction of ischemic damage to the kidney as a result of higher RBF remains to be established. None of the identified animal studies have addressed the potential effect of MR antagonism in reducing inflammation and endothelial dysfunction. These pathways may contribute to the renoprotection conferred by MR antagonists. For an overview of the included studies, see Table 1.
Despite evidence from animal studies, very few studies have investigated the effect of MR antagonists preventing CNI nephrotoxicity in humans. This is partly due to concerns regarding hyperkalemia related to MR antagonism in patients with impaired renal function and concomitant CNI treatment. This issue was addressed in a recent publication testing the safety of eplerenone 25 mg/day for 8 weeks in a group of 31 kidney transplant patients treated with CsA. Median eGFR was 41 mL/min. A slight increase in serum-potassium was detected already after 2 days of treatment. Nine patients had at least one episode of mild hyperkalemia (>5.0 mmol/L) but only one patient experienced moderate hyperkalemia (>5.5 mmol/L). No patients were withdrawn from the study due to hyperkalemia. There was no change in systolic BP, body weight, or s-bicarbonate after 8 weeks of treatment (52).
One single-blind RCT has investigated the effect of MR antagonism in a pediatric kidney transplant population. Twenty-four children with chronic allograft nephropathy were randomized to eplerenone or placebo for 24 months. Although underpowered to provide definite evidence, there was a tendency toward lower levels of tubulointerstitial fibrosis in the eplerenone group after 24 months (p = 0.06), but no significant changes in albuminuria, BP and eGFR. No difference was detected in urinary KIM-1, heat-shock protein 72 (HSP-72), 8-hydroxy-2-deoxyguanosine (marker of oxidative stress), or serum TGF-β (53).
The effect of MR antagonism on proteinuria has been addressed in an open label study of 11 proteinuric kidney transplant patients, who already received both ACEI and ARB. This study found a significant reduction in protein excretion from 4.4 to 2.7 g/day after the first month of treatment with spironolactone 25 mg/day, an effect that was sustained for the full 6 months of intervention. There was no difference in BP or GFR during this period (54).
The proposed effect of MR antagonists to reduce renal ischemia and thereby oxidative stress was investigated in a double-blind RCT including 20 kidney transplant recipients randomized to spironolactone 25 mg or placebo on the day before transplantation and 3 days after. The aim was to investigate the effect of MR antagonism on oxidative stress relating to ischemia/reperfusion injury. Urinary hydrogen peroxide (H2O2) levels were significantly lower in the spironolactone group on the fifth postoperative day. There was no difference in tubular damage markers KIM-1, interleukin 18, or HSP-72. However, three patients in the spironolactone group displayed delayed graft function and subsequent biopsies revealed acute rejection in two of these. In the placebo group, one patient had rejection. It is worth noticing that B-Tac trough levels were possibly lower in the spironolactone group (3.65 vs. 6.05 ng/mL), although this was not significant. A difference in B-tacrolimus concentration could account for the greater rejection rate in the spironolactone group, but could also contribute to lower H2O2 levels, since tacrolimus reduces RBF, thereby inducing oxidative stress (55). The included human studies are summarized in Table 2.
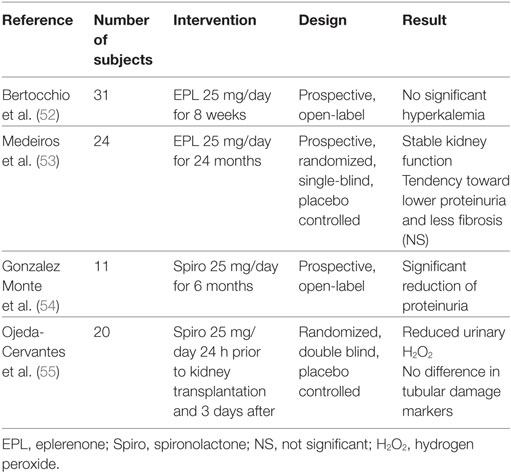
Table 2. Included studies in renal transplant patients.
Evidence underlines the need for sufficiently powered RCTs to evaluate the effect of MR antagonism in the kidney transplant population. Three ongoing trials were identified (https://ClinicalTrials.gov): (I) RCT (NCT02490904) investigating the effect of eplerenone for four days in relation to kidney transplantation in 132 patients on short- and long-term kidney function; (II) open label study (NCT01510795) evaluating the chronic BANFF-scores in 40 patients after 6 months of spironolactone as compared with a retrospective control group, and (III) RCT investigating the effect of spironolactone for 3 years in 170 patients with regards to preserving kidney function and reducing renal fibrosis (NCT01602861).
Calcineurin inhibitor causes renal fibrosis likely due to altered hemodynamics and a direct stimulatory effect on fibrogenic factors. Rat models of acute and chronic CNI nephrotoxicity have shown beneficial effects of MR antagonism in preserving GFR and reducing fibrosis. This is likely due to an increased RBF attenuating ischemic damage to the kidneys, in combination with a direct inhibitory action on pro-fibrotic cytokines. Few human studies have addressed this hypothesis. Improving long-term allograft survival remains a key challenge in renal transplantation with the potential to improve quality of life, morbidity, and mortality of the affected individuals. Results of future and ongoing clinical trials may clarify if aldosterone antagonism can prevent or improve CNI nephrotoxicity in humans.
LM wrote the article, CB and HT reviewed and revised the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACEI, angiotensin converting enzyme inhibitors; ARB, angiotensin receptor blockers; BP, blood pressure; CNI, calcineurin inhibitor; CsA, cyclosporine A; CTGF, colony transforming growth factor; EGF, epithelial growth factor; GFR, glomerular filtration rate; H2O2, hydrogen peroxide; HSP-72, heat-shock protein 72; ICAM-1, intercellular adhesion molecule 1; IF/TA, interstitial fibrosis and tubular atrophy; KIM-1, kidney injury molecule 1; MR, mineralocorticoid receptor; NADP, nicotinamide adenine dinucleotide phosphate; NFAT, nuclear factor of activated T-cells; Nfκβ, nuclear factor kappa B; PAI-1, plasminogen activator inhibitor 1; PDGF, platelet-derived growth factor; PRA, plasma renin activity; RAAS, renin-angiotensin-aldosterone system; RBF, renal blood flow; RCT, randomized clinical trial; TGF-β, transforming growth factor β.
1. Calne RY, White DJ, Thiru S, Evans DB, McMaster P, Dunn DC, et al. Cyclosporin A in patients receiving renal allografts from cadaver donors. Lancet (1978) 2(8104–5):1323–7. doi:10.1016/S0140-6736(78)91970-0
2. Starzl TE, Todo S, Fung J, Demetris AJ, Venkataramman R, Jain A. FK 506 for liver, kidney, and pancreas transplantation. Lancet (1989) 2(8670):1000–4. doi:10.1016/S0140-6736(89)91014-3
3. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol (2009) 4(2):481–508. doi:10.2215/CJN.04800908
4. Kapturczak MH, Meier-Kriesche HU, Kaplan B. Pharmacology of calcineurin antagonists. Transplant Proc (2004) 36(2 Suppl):25S–32S. doi:10.1016/j.transproceed.2004.01.018
5. Stoumpos S, Jardine AG, Mark PB. Cardiovascular morbidity and mortality after kidney transplantation. Transpl Int (2015) 28(1):10–21. doi:10.1111/tri.12413
6. Paoletti E, Bellino D, Marsano L, Cassottana P, Rolla D, Ratto E. Effects of ACE inhibitors on long-term outcome of renal transplant recipients: a randomized controlled trial. Transplantation (2013) 95(6):889–95. doi:10.1097/TP.0b013e3182827a43
7. Ibrahim HN, Jackson S, Connaire J, Matas A, Ney A, Najafian B, et al. Angiotensin II blockade in kidney transplant recipients. J Am Soc Nephrol (2013) 24(2):320–7. doi:10.1681/ASN.2012080777
8. van Riemsdijk IC, Mulder PG, de Fijter JW, Bruijn JA, van Hooff JP, Hoitsma AJ, et al. Addition of isradipine (Lomir) results in a better renal function after kidney transplantation: a double-blind, randomized, placebo-controlled, multi-center study. Transplantation (2000) 70(1):122–6.
9. Kuypers DR, Neumayer HH, Fritsche L, Budde K, Rodicio JL, Vanrenterghem Y, et al. Calcium channel blockade and preservation of renal graft function in cyclosporine-treated recipients: a prospective randomized placebo-controlled 2-year study. Transplantation (2004) 78(8):1204–11. doi:10.1097/01.TP.0000137793.23371.42
10. Midtvedt K, Hartmann A, Foss A, Fauchald P, Nordal KP, Rootwelt K, et al. Sustained improvement of renal graft function for two years in hypertensive renal transplant recipients treated with nifedipine as compared to lisinopril. Transplantation (2001) 72(11):1787–92. doi:10.1097/00007890-200112150-00004
11. Camilleri B, Bridson JM, Halawa A. Calcineurin inhibitor-sparing strategies in renal transplantation: where are we? A comprehensive review of the current evidence. Exp Clin Transplant (2016) 14(5):471–83. doi:10.6002/ect.2015.0283
12. Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and long-term outcomes in kidney transplantation. N Engl J Med (2016) 374(4):333–43. doi:10.1056/NEJMoa1506027
13. Vincenti F, Blancho G, Durrbach A, Grannas G, Grinyo J, Meier-Kriesche HU, et al. Ten-year outcomes in a randomized phase II study of kidney transplant recipients administered belatacept 4-weekly or 8-weekly. Am J Transplant (2017):1–9. doi:10.1111/ajt.14452
14. Kumar D, LeCorchick S, Gupta G. Belatacept as an alternative to calcineurin inhibitors in patients with solid organ transplants. Front Med (2017) 4:60. doi:10.3389/fmed.2017.00060
15. Naesens M, Lerut E. Calcineurin inhibitor nephrotoxicity in the era of antibody-mediated rejection. Transplantation (2016) 100(8):1599–600. doi:10.1097/TP.0000000000001244
16. Nankivell BJ, Borrows RJ, Fung CL, O’Connell PJ, Chapman JR, Allen RD. Calcineurin inhibitor nephrotoxicity: longitudinal assessment by protocol histology. Transplantation (2004) 78(4):557–65. doi:10.1097/01.TP.0000128636.70499.6E
17. Schwarz A, Haller H, Schmitt R, Schiffer M, Koenecke C, Strassburg C, et al. Biopsy-diagnosed renal disease in patients after transplantation of other organs and tissues. Am J Transplant (2010) 10(9):2017–25. doi:10.1111/j.1600-6143.2010.03224.x
18. Ekberg H, Tedesco-Silva H, Demirbas A, Vitko S, Nashan B, Gurkan A, et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med (2007) 357(25):2562–75. doi:10.1056/NEJMoa067411
19. Nankivell BJ, P’Ng CH, O’Connell PJ, Chapman JR. Calcineurin inhibitor nephrotoxicity through the lens of longitudinal histology: comparison of cyclosporine and tacrolimus eras. Transplantation (2016) 100(8):1723–31. doi:10.1097/TP.0000000000001243
20. Fuller PJ, Young MJ. Mechanisms of mineralocorticoid action. Hypertension (2005) 46(6):1227–35. doi:10.1161/01.HYP.0000193502.77417.17
21. Ochs HR, Greenblatt DJ, Bodem G, Smith TW. Spironolactone. Am Heart J (1978) 96(3):389–400. doi:10.1016/0002-8703(78)90052-2
22. Stier CT Jr. Eplerenone: a selective aldosterone blocker. Cardiovasc Drug Rev (2003) 21(3):169–84. doi:10.1111/j.1527-3466.2003.tb00114.x
23. Yang J, Young MJ. Mineralocorticoid receptor antagonists-pharmacodynamics and pharmacokinetic differences. Curr Opin Pharmacol (2016) 27:78–85. doi:10.1016/j.coph.2016.02.005
24. Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol (2013) 9(8):459–69. doi:10.1038/nrneph.2013.110
25. Moss ME, Jaffe IZ. Mineralocorticoid receptors in the pathophysiology of vascular inflammation and atherosclerosis. Front Endocrinol (2015) 6:153. doi:10.3389/fendo.2015.00153
26. Fels J, Oberleithner H, Kusche-Vihrog K. Menage a trois: aldosterone, sodium and nitric oxide in vascular endothelium. Biochim Biophys Acta (2010) 1802(12):1193–202. doi:10.1016/j.bbadis.2010.03.006
27. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med (1999) 341(10):709–17. doi:10.1056/NEJM199909023411001
28. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med (2003) 348(14):1309–21. doi:10.1056/NEJMoa030207
29. Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol (2011) 7(12):684–96. doi:10.1038/nrneph.2011.149
30. Currie G, Taylor AH, Fujita T, Ohtsu H, Lindhardt M, Rossing P, et al. Effect of mineralocorticoid receptor antagonists on proteinuria and progression of chronic kidney disease: a systematic review and meta-analysis. BMC Nephrol (2016) 17(1):127. doi:10.1186/s12882-016-0337-0
31. Bolignano D, Palmer SC, Navaneethan SD, Strippoli GF. Aldosterone antagonists for preventing the progression of chronic kidney disease. Cochrane Database Syst Rev (2014) 4:CD007004. doi:10.1002/14651858.CD007004.pub3
32. Lee DB. Cyclosporine and the renin-angiotensin axis. Kidney Int (1997) 52(1):248–60. doi:10.1038/ki.1997.328
33. Weir MR, Klassen DK, Shen SY, Sullivan D, Buddemeyer EU, Handwerger BS. Acute effects of intravenous cyclosporine on blood pressure, renal hemodynamics, and urine prostaglandin production of healthy humans. Transplantation (1990) 49(1):41–7. doi:10.1097/00007890-199001000-00009
34. Bantle JP, Nath KA, Sutherland DE, Najarian JS, Ferris TF. Effects of cyclosporine on the renin-angiotensin-aldosterone system and potassium excretion in renal transplant recipients. Arch Intern Med (1985) 145(3):505–8. doi:10.1001/archinte.1985.00360030153026
35. Issa N, Ortiz F, Reule SA, Kukla A, Kasiske BL, Mauer M, et al. The renin-aldosterone axis in kidney transplant recipients and its association with allograft function and structure. Kidney Int (2014) 85(2):404–15. doi:10.1038/ki.2013.278
36. Sun QL, Li M, Rui HL, Chen YP. Inhibition of local aldosterone by eplerenone reduces renal structural damage in a novel model of chronic cyclosporine A nephrotoxicity. J Renin Angiotensin Aldosterone Syst (2015) 16(2):301–10. doi:10.1177/1470320314561248
37. Perez-Rojas JM, Derive S, Blanco JA, Cruz C, Martinez de la Maza L, Gamba G, et al. Renocortical mRNA expression of vasoactive factors during spironolactone protective effect in chronic cyclosporine nephrotoxicity. Am J Physiol Renal Physiol (2005) 289(5):F1020–30. doi:10.1152/ajprenal.00166.2005
38. McAuley FT, Whiting PH, Thomson AW, Simpson JG. The influence of enalapril or spironolactone on experimental cyclosporin nephrotoxicity. Biochem Pharmacol (1987) 36(5):699–703. doi:10.1016/0006-2952(87)90721-0
39. Thomson AW, McAuley FT, Whiting PH, Simpson JG. Angiotensin-converting enzyme inhibition or aldosterone antagonism reduces cyclosporine nephrotoxicity in the rat. Transplant Proc (1987) 19(1 Pt 2):1242–3.
40. Iacona A, Rossetti A, Filingeri V, Orsini D, Cesarini A, Cervelli V, et al. Reduced nephrotoxicity and hepatoxicity in cyclosporin A therapy by enalapril and spironolactone in rats. Drugs Exp Clin Res (1991) 17(10–11):501–6.
41. Nielsen FT, Jensen BL, Marcussen N, Skott O, Bie P. Inhibition of mineralocorticoid receptors with eplerenone alleviates short-term cyclosporin A nephrotoxicity in conscious rats. Nephrol Dial Transplant (2008) 23(9):2777–83. doi:10.1093/ndt/gfn204
42. Amador CA, Bertocchio JP, Andre-Gregoire G, Placier S, Duong Van Huyen JP, El Moghrabi S, et al. Deletion of mineralocorticoid receptors in smooth muscle cells blunts renal vascular resistance following acute cyclosporine administration. Kidney Int (2016) 89(2):354–62. doi:10.1038/ki.2015.312
43. Feria I, Pichardo I, Juarez P, Ramirez V, Gonzalez MA, Uribe N, et al. Therapeutic benefit of spironolactone in experimental chronic cyclosporine A nephrotoxicity. Kidney Int (2003) 63(1):43–52. doi:10.1046/j.1523-1755.2003.00707.x
44. Macunluoglu B, Arikan H, Atakan A, Tuglular S, Ulfer G, Cakalagaoglu F, et al. Effects of spironolactone in an experimental model of chronic cyclosporine nephrotoxicity. Transplant Proc (2008) 40(1):273–8. doi:10.1016/j.transproceed.2007.11.025
45. Nielsen FT, Jensen BL, Hansen PB, Marcussen N, Bie P. The mineralocorticoid receptor antagonist eplerenone reduces renal interstitial fibrosis after long-term cyclosporine treatment in rat: antagonizing cyclosporine nephrotoxicity. BMC Nephrol (2013) 14:42. doi:10.1186/1471-2369-14-42
46. Silva FM, Miorin LA, Sens YA. The effect of aldosterone antagonist in experimental nephropathy induced by cyclosporine A. Ren Fail (2008) 30(4):439–42. doi:10.1080/08860220801947397
47. Waanders F, Rienstra H, Boer MW, Zandvoort A, Rozing J, Navis G, et al. Spironolactone ameliorates transplant vasculopathy in renal chronic transplant dysfunction in rats. Am J Physiol Renal Physiol (2009) 296(5):F1072–9. doi:10.1152/ajprenal.90643.2008
48. Perez-Rojas J, Blanco JA, Cruz C, Trujillo J, Vaidya VS, Uribe N, et al. Mineralocorticoid receptor blockade confers renoprotection in preexisting chronic cyclosporine nephrotoxicity. Am J Physiol Renal Physiol (2007) 292(1):F131–9. doi:10.1152/ajprenal.00147.2006
49. Kunter U, Floege J, von Jurgensonn AS, Stojanovic T, Merkel S, Grone HJ, et al. Expression of A20 in the vessel wall of rat-kidney allografts correlates with protection from transplant arteriosclerosis. Transplantation (2003) 75(1):3–9. doi:10.1097/01.TP.0000043982.08656.81
50. Vega G, Alarcon S, San Martin R. The cellular and signalling alterations conducted by TGF-beta contributing to renal fibrosis. Cytokine (2016) 88:115–25. doi:10.1016/j.cyto.2016.08.019
51. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol (2016) 12(6):325–38. doi:10.1038/nrneph.2016.48
52. Bertocchio JP, Barbe C, Lavaud S, Toupance O, Nazeyrollas P, Jaisser F, et al. Safety of Eplerenone for kidney-transplant recipients with impaired renal function and receiving cyclosporine A. PLoS One (2016) 11(4):e0153635. doi:10.1371/journal.pone.0153635
53. Medeiros M, Velasquez-Jones L, Hernandez AM, Ramon-Garcia G, Valverde S, Fuentes Y, et al. Randomized controlled trial of mineralocorticoid receptor blockade in children with chronic kidney allograft nephropathy. Clin J Am Soc Nephrol (2017) 12(8):1291–300. doi:10.2215/CJN.05300516
54. Gonzalez Monte E, Andres A, Polanco N, Toribio MJ, Santana R, Gutierrez Martinez E, et al. Addition of spironolactone to dual blockade of renin angiotensin system dramatically reduces severe proteinuria in renal transplant patients: an uncontrolled pilot study at 6 months. Transplant Proc (2010) 42(8):2899–901. doi:10.1016/j.transproceed.2010.08.024
Keywords: aldosterone, mineralocorticoid, cyclosporine A, tacrolimus, IF/TA, fibrosis
Citation: Mortensen LA, Bistrup C and Thiesson HC (2017) Does Mineralocorticoid Receptor Antagonism Prevent Calcineurin Inhibitor-Induced Nephrotoxicity? Front. Med. 4:210. doi: 10.3389/fmed.2017.00210
Received: 07 September 2017; Accepted: 08 November 2017;
Published: 24 November 2017
Edited by:
Michael L. Moritz, Children’s Hospital of Pittsburgh, United StatesReviewed by:
David W. Gjertson, University of California, Los Angeles, United StatesCopyright: © 2017 Mortensen, Bistrup and Thiesson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Line Aas Mortensen, bGluZS5tb3J0ZW5zZW5AcnN5ZC5kaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.