 Sarah Svenningsen1
Sarah Svenningsen1 Parameswaran Nair1,2*
Parameswaran Nair1,2*
- 1Department of Medicine, McMaster University, Hamilton, ON, Canada
- 2St Joseph’s Healthcare, Hamilton, ON, Canada
Guidelines for the management of severe asthma do not emphasize the measurement of the inflammatory component of airway disease to indicate appropriate treatments or to monitor response to treatment. Inflammation is a central component of asthma and contributes to symptoms, physiological, and structural abnormalities. It can be assessed by a number of endotyping strategies based on “omics” technology such as proteomics, transcriptomics, and metabolomics. It can also be assessed using simple cellular responses by quantitative cytometry in sputum. Bronchitis may be eosinophilic, neutrophilic, mixed-granulocytic, or paucigranulocytic (eosinophils and neutrophils not elevated). Eosinophilic bronchitis is usually a Type 2 (T2)-driven process and therefore a sputum eosinophilia of greater than 3% usually indicates a response to treatment with corticosteroids or novel therapies directed against T2 cytokines such as IL-4, IL-5, and IL-13. Neutrophilic bronchitis represents a non-T2-driven disease, which is generally a predictor of response to antibiotics and may be a predictor to therapies targeted at pathways that lead to neutrophil recruitment such as TNF, IL-1, IL-6, IL-8, IL-23, and IL-17. Paucigranulocytic disease may not warrant anti-inflammatory therapy. These patients, whose symptoms may be driven largely by airway hyper-responsiveness may benefit from smooth muscle-directed therapies such as bronchial thermoplasty or mast-cell directed therapies. This review will briefly summarize the current knowledge regarding “omics-based signatures” and cellular endotyping of severe asthma and give an overview of segmentation of asthma therapeutics guided by the endotype.
Introduction
The definition of asthma has not changed in over 50 years (1). The variability in airflow that characterizes the disease may be driven by airway hyper-responsiveness or by airway inflammation that is one of the determinants of airway hyper-responsiveness (2). Despite this heterogeneity, guideline-based therapy with inhaled beta agonists and corticosteroids do not consider measurements of these individual components in routine clinical care. While the majority of asthmatics are responsive to guideline-based therapy and have reduced symptoms, improved quality-of-life, increased lung function as well as decreased exacerbation frequency (3), in approximately 5–10% of asthmatics, anticipated outcomes are not achieved (4). These patients with severe disease are responsible for the majority of indirect and direct asthma-related costs and economic burden. The advent of biologic therapies calls for a new paradigm of personalized medicine based on inflammatory endotype to better-inform who is most likely to benefit from specific targeted therapies.
Traditional asthma phenotyping (description of observable characteristics that do not provide an insight into the underlying pathobiology) classifies patients according to clinical characteristics such as exacerbating factors (allergens, exercise, and infections), age of onset, concomitant comorbidities (sinusitis and obesity), and response to treatment. More recently, unbiased clustering algorithms that have the capacity to incorporate a range of clinical variables (e.g., FEV1, BMI, ACQ, atopic status, and blood eosinophils) have been applied to large patient datasets to objectively identify clinical phenotypes of asthma. Such datasets include the Severe Asthma Research Program (SARP) (5), Airways Disease Endotyping for Personalized Therapeutics (ADEPT) (6), and Unbiased Biomarkers for the Prediction of Respiratory Disease Outcomes (U-BIOPRED) (6, 7) cohorts, which have been partitioned into up-to five clinical phenotypes. However, composites of observable characteristics do not provide us insight into mechanisms of persistent symptoms, physiological abnormalities, or inflammation, and therefore are of limited value to choose the appropriate biological agent. As opposed to phenotypes, characterizing severe asthmatics according to their endotype, a term applied to a “subtype of a condition that is defined by a distinct functional or pathophysiological mechanism,” may be more useful to directing therapy (8). This notion is strongly supported by the efficacy of biologic therapies that target-specific inflammatory mediators (e.g., IL-5) in well-defined patients characterized based on inflammatory biomarkers (9, 10). Currently, however, asthma management guidelines fail to adequately emphasize the measurement of the inflammatory component of airway disease (bronchitis).
Inflammatory endotype characterization should be considered a central component of the workup and management of severe asthma. Widespread acceptance of this notion, however, has been slow, perhaps because there is no consensus as to how to best identify asthma endotypes and what therapy to use for a given endotype. While novel omics-based signatures of severe asthma have emerged, they have not been evaluated clinically. We suggest that asthma endotype characterization can be reliably done on the nature of underlying airway inflammation assessed by sputum cytometry (1). This review aimed to summarize the current knowledge regarding cellular endotyping and novel “omics-based signatures” of severe asthma and give an overview of segmentation of asthma therapeutics guided by the inflammatory endotype. Molecular pathways and mechanisms associated with asthma endotypes were recently reviewed and therefore have not been discussed in detail here.
Inflammatory Endotypes
Wenzel et al. (11) defined two distinct inflammatory endotypes of severe, corticosteroid-dependent asthma based on the presence or absence of eosinophils in endobronchial biopsy and lavage. Since this landmark study, T-helper type 2 (Th2)-high and Th2-low have remained the most well recognized and described endotypes of severe asthma. The Th2-high endotype is characterized by the presence of eosinophilic airway inflammation, whereas the Th2-low endotype is usually characterized by neutrophilic or paucigranulocytic airway inflammation. While several non-invasive biomarkers exist for the detection of the Th2-high endotype [blood eosinophils, serum IgE, serum periostin, and exhaled nitric oxide (eNO)], sputum cytometry is currently the most clinically validated quantitative and responsive method to assess airway inflammation. Perceived difficulties in implementing this approach in routine clinical practice have limited its widespread use. In fact, as described by Lim and Nair (12), many easily accessible biomarkers with demonstrated clinical utility remain confined to the research arena or are only exploited in a small number of specialized academic centers. This may be due to the lack of measurement standardization, validated diagnostic or predictive thresholds, and evidence-based reference guidelines to inform how biomarkers should be used and interpreted in clinical practice. Cost and infrastructure constraints also limit generalizability of validated biomarkers. To overcome these issues, readily available or point-of-care diagnostic methods are welcomed. Quantitative cytometry of induced or spontaneous sputum is currently the most sensitive and specific non-invasive method to directly characterize the type and severity of airway inflammation in asthma (13). Importantly, standardized methodology for sputum induction, processing, and quantification (14, 15) is safe (16) and well tolerated by the majority of patients (17–19). The nature of bronchitis assessed by sputum cytometry can be stratified into four groups based on the percentage of sputum granulocytes: (1) eosinophilic, (2) neutrophilic, (3) mixed-granulocytic (eosinophils and neutrophils elevated), and (4) paucigranulocytic (eosinophils nor neutrophils elevated) (17, 20). In non-smoking healthy adults, Belda et al. established the 90th percentile for sputum eosinophil and neutrophil counts, reporting 1.1 and 64.4%, respectively, with a total cell count of 9.7 million cells/g (21). However, standardized stratification cutoffs have not been established in asthma and as a result have varied between studies. Proposed thresholds for sputum eosinophilia have ranged between >1.1 and 4% of the total cell count (17, 20, 22) with studies strongly suggesting that a cutoff of >3% is clinically relevant and can be used to guide treatment and reduce exacerbations (23). To indicate neutrophilia, thresholds >61% of the total cell count have been proposed (20). Overlooked by many centers, it is important to acknowledge that the presence of eosinophil free granules also indicates uncontrolled eosinophilic bronchitis (24). The prevalence of these groups have been reported in stable, severe, and exacerbated disease with the proportion of eosinophilic asthma ranging from 35 to 50% (17, 20). The groups differ with respect to their clinical characteristics and response to therapy. Patients with mixed-granulocytic asthma have more severe airflow obstruction, a higher frequency of exacerbations and daily wheeze, and increased health care utilization than patients with either eosinophilic or neutrophilic bronchitis alone (25). It is also important to note that asthmatics who exhibit concordant blood and sputum eosinophilia experience more airway symptoms than those with isolated blood or sputum eosinophilia alone (19).
As mentioned above, there is no consensus on the specific definition of inflammatory cellular endotypes. Based on our clinical experience of over 20 years in over 5,000 patients, a patient can be determined to have eosinophilic asthma if there is evidence of elevated sputum (>3% with or without degranulation) and/or blood eosinophils (≥400 cells/μL) on at least two occasions, and if treatment strategies aimed at suppressing eosinophils are effective in controlling symptoms and exacerbations (26). Likewise, a patient can be determined to have neutrophilic asthma if there is evidence of elevated neutrophils (≥64%) but not eosinophils (<3%) with an increased total cell count (≥9.7 million cells/g) on at least two occasions and if treatment strategies aimed at suppressing eosinophils are not effective in controlling symptoms and exacerbations. Patients can exhibit features of both the eosinophilic and neutrophilic endotype. A patient can be determined to have mixed-granulocytic asthma if there is evidence of both neutrophils and eosinophils, independently or concurrently, on at least two occasions. Finally, a patient can be determined to have paucigranulocytic asthma if there is no evidence of elevated eosinophils (<3%) or neutrophils (<64%) and if treatment strategies aimed at suppressing eosinophils and neutrophils are not effective in controlling symptoms and exacerbations. The workup and therapy regimen for these inflammatory endotypes have been described at the end of this review.
Novel Omics Endotyping Strategies
The maturation of omics-based technologies has facilitated the investigation of transcriptomics (27–33), proteomics (25, 34), and metabolomics (35, 36) to better understand the molecular characteristics of asthma, which have all been recently reviewed. Large multicenter initiatives are now ongoing, including the U-BIOPRED project (37), which aim to identify distinct severe asthma endotypes by integrating inflammatory biomarkers derived from “omics” and clinical data. Thus far, omics measurements have been utilized to (1) identify asthma endotypes (38), (2) identify genes related to inflammatory characteristics (39), and (3) to describe the molecular characteristics of clinical asthma phenotypes (6). Studies are required to investigate the clinical benefit of these more sophisticated and computationally intensive endotyping strategies, both to initiate the appropriate treatment and to longitudinally monitor responses to various anti-inflammatory (including biologics) therapies in asthma.
Transcriptomics
When compared with non-asthmatics, whole-genome expression in peripheral blood of severe asthmatics is different such that a severe asthma disease signature comprised of nearly 1,700 genes was identified by Bigler et al. (38). Within severe asthma, distinct gene signatures associated with eosinophilia, mast cells, and group 3 innate lymphoid cells have been identified in patients with adult-onset asthma when compared with patients with childhood-onset asthma (32). Beyond asthma versus non-asthma stratification, numerous studies (summarized in Table 1) have aimed to define transcriptomic endotypes of asthma by analyzing differential gene expression in bronchial epithelium (27) and induced sputum samples (28, 30, 31, 33). Woodruff and colleagues were the first to identify two evenly sized “Th2-high” and “Th2-low” subgroups of mild, steroid naive asthma based on unsupervised hierarchical clustering of the expression levels of three Th2 cytokine induced genes (POSTN, CLCA1, and SERPINB2) in bronchial epithelial brushings. These subgroups were different with respect to their expression levels of IL-5 and IL-13 within the airway, airway hyper-responsiveness, IgE, blood and airway eosinophilia, and reticular basement membrane thickness. Not surprisingly, Th2-high gene expression was predictive of corticosteroid responsiveness as clinically relevant improvements in FEV1 following 8 weeks of fluticasone use were only observed in the Th2-high group (27). Wilson et al. (39) identified seven genes (COX-2, ADAM-7, SLCO1A2, TMEFE2, and TRPM-1, and two unnamed genes) in bronchial brushing samples with expression levels that were moderately correlated with submucosal eosinophils, suggesting that they may also predict corticosteroid responsiveness. Given the limited clinical applicability of invasive bronchoscopic samples, Woodruff and colleagues went on to evaluate the variable expression of 14 genes relevant to Th2 inflammation in induced sputum samples (30). Generating a quantitative composite metric of IL-4, IL-5, and IL-13 gene expression, termed the “Th2 gene mean,” the study population was again dichotomized into Th2-high (70%) and Th2-low (30%) subgroups (30). When compared with the Th2-low cluster, the Th2-high cluster had higher eNO levels as well as sputum and blood eosinophil counts (30). It is notable that sputum (AUC = 0.89) and peripheral blood (AUC = 0.89) eosinophil counts alone, but not eNO (AUC = 0.76), performed well as biomarkers of Th2-high asthma as assessed by the sputum cell “Th2 gene mean” (30). Acknowledging the limitations of analyzing pre-selected genes, Baines et al. (28) subjected whole-genome gene expression profiles from induced sputum of adults with stable asthma to unsupervised hierarchical cluster analysis. Three distinct transcriptional asthma phenotypes (TAPs) were identified that had similarities to previously defined sputum inflammatory phenotypes of eosinophilic (TAP1), neutrophilic (TAP2), and paucigranulocytic (TAP3) asthma (28). In fact, 92% of the differentially expressed genes between the TAPs overlapped when the population was grouped according to sputum quantitative cell count (eosinophilic, neutrophilic, and paucigranulocytic). The same investigators subsequently identified a sputum gene expression signature comprised of six genes (CLC, CPA3, DNASE1L3, IL1B, ALPL, and CXCR2) that was able to discriminate asthmatics according to their inflammatory endotype and predict ICS response (29). Of most interest, the six-gene expression signature outperformed the ability of sputum eosinophil count to predict corticosteroid response (FEV1 responder vs. non-responder; AUC = 91.5) (29). Somewhat contradictory to the findings of Baines et al., when unbiased hierarchical clustering was performed on 508 genes that were differentially expressed between severe asthmatics with and without eosinophilic airway inflammation confirmed by sputum cytometry, one Th2-high and two non-Th2 transcriptome-associated clusters (TACs) were defined (33). When examining the distribution of the three TACs according to sputum inflammatory endotype, two TACs were associated with eosinophilic (TAC1 or TAC3) and neutrophilic (TAC2 or TAC3) inflammation, suggesting that two distinct transcriptional signatures are associated with both sputum eosinophilia and neutrophilia (33). Similarly, Yan et al. (31) did not detect significant between cluster differences in sputum cell differentials suggesting an imperfect association with Th2 pathophysiology. Taken together, conflicting evidence surrounds the association between transcriptomic endotypes and sputum quantitative cell count.
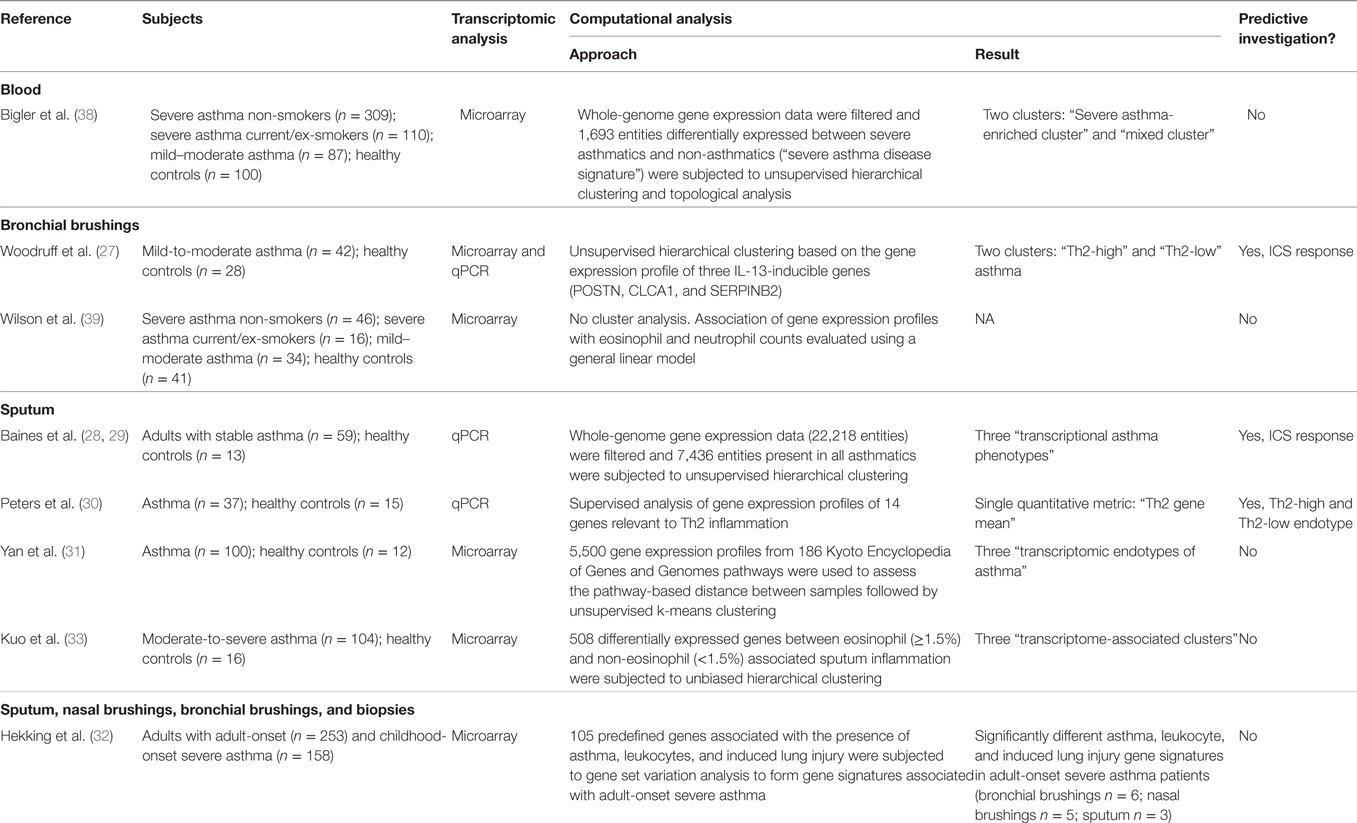
Table 1. Summary of transcriptomics studies.
Proteomics
While gene expression studies have dominated the omics landscape, it is their translated products, the proteins, which mediate airway inflammation in asthma by regulating cell recruitment. Numerous studies, limited by sample size, have investigated the “proteomic profile” of asthma in bronchoalveolar lavage fluid (BALF) (40, 41), bronchial biopsy (42), and sputum supernatants (43, 44). One relatively large study by SARP investigators (34) focused on 18 cytokines detectable in BALF and discriminated mild-to-moderate and severe asthmatics into four groups based on cytokine expression. The groups were independent of corticosteroid use and phenotypically distinct with respect to BALF cellularity and lung function. In another study, Hastie et al. (25) investigated the hypothesis that sputum inflammatory granulocytes define phenotypic subgroups of asthma with different patterns of inflammatory proteins. Protein microarray data of induced sputum were stratified by sputum cell differential (eosinophilic: ≥2% eos and <40% neuts; neutrophilic: <2% eos and ≥40% neuts; mixed-granulocytic: ≥2% eos and ≥40% neuts; paucigranulocytic: <2% eos and <40% neuts) and revealed differentially increased inflammatory proteins between the groups (25). Of note, 19 inflammatory mediators were significantly elevated in those asthmatics with neutrophilic bronchitis, a subset of which were correlated with neutrophil counts.
Metabolomics
Metabolomics, the exploration of biochemical molecules derived from metabolic processes, was recently reviewed for its applications in asthma (36). Studies to date strongly suggest that metabolic profiles measured in exhaled breath, urine, plasma, and serum may be applied as a point-of-care tool to discriminate asthma endotypes (36). Of most interest, “breathomics” (45) profiles volatile organic compounds in exhaled breath using an electronic nose and has demonstrated ability to discriminate asthmatics from healthy controls (46–48). Electronic nose “breathprints” are correlated with the percentage of sputum eosinophils (48, 49). One highly relevant study concluded that exhaled breath analysis outperformed the ability of both eNO and the percentage of sputum eosinophils to predict corticosteroid response in asthmatics from whom corticosteroids had been withdrawn (48).
Important Considerations and Future Studies
There are numerous limitations to clustering techniques applied in the studies discussed above; therefore, these results should be interpreted with caution. One such concern is cluster stability. Specifically, clusters identified at a specific point in time may not be reproducible at subsequent time points. Longitudinal stability assessment of the clusters that have been described is required to understand how they behave over time and in response to treatment and/or exacerbations. Similarly, different clusters may be identified across different asthma populations and therefore validation of cluster classification in independent cohorts of asthmatic patients is necessary to understand generalizability. These important considerations were evaluated for the first time by Loza et al. (6) who defined four severe asthma phenotypes in two independent severe asthma cohorts (ADEPT and U-BIOPRED), performing both external validation and longitudinal stability assessments. Cluster number and composition may also be influenced by the clustering methodology applied. Finally, it is also important to consider statistical robustness as the majority of studies to date define clusters comprised of a small number of asthma patients and hence have limited statistical power.
None of the omics-signatures discussed above have been translated to clinical practice as prognostic or predictive tests. In fact, the clinical utility of these signatures is currently unknown as the majority of studies to date have been observational and hypothesis generating. The ultimate potential of a biomarker-based clinical test is dependent on its analytical and clinical validity in addition to its clinical utility. A 30-point checklist of criteria, developed by McShane et al., should be considered to gauge the potential clinical utility of the omics-based signatures discussed in this review (50). Important considerations include those related to the specimen and assays used, and the appropriateness of the statistical methods used to develop and validate the signature (50). Studies are now required to address the paucity of evidence concerning (1) the longitudinal variability of omics data and endotypes and (2) the responsiveness of omics to therapies. Randomized clinical trials (RCTs) will be necessary to definitively confirm the clinical utility of novel omics-based signatures and design consideration for biomarker RCTs have been proposed (51). Specifically, intervention studies will be necessary to shed light on the capacity of these signatures to direct personalized therapy and to adjust doses of drugs during exacerbations in severe asthma. The qualitative nature of omics is an obvious limitation, therefore the development and application of qualitative metrics are certainly desirable. Similar to that described by Hinks et al. (52), multi-dimensional clinicopathobiologic clustering should also be considered to maximally leverage the measurements available to tertiary care clinicians. It is clear however that omics-based endotypes have similar molecular, physiological, and clinical characteristics to the inflammatory endotypes of eosinophilic, neutrophilic, mixed-granulocytic, and paucigranulocytic asthma.
Inflammatory Endotype-Guided Therapeutic Strategy
In this section, we focus our discussion on the segmentation of therapy in severe asthma [as defined by the European Respiratory Society/American Thoracic Society (4)] guided by inflammatory endotype. We support the notion that the specific nature of bronchitis reveals the underlying mechanism driving the bronchitis and therefore predicts therapy response. As summarized in Figure 1, our therapeutic strategy is dependent on the type of bronchitis (assessed using quantitative cytometry in induced sputum) (53) and severity of airway hyper-responsiveness (assessed using methacholine inhalation challenge). We emphasize the importance of identifying the particular component of airways disease that drives symptoms in each patient prior to therapy selection. Furthermore, the components of airways disease should be assessed and subsequently reassessed to optimize therapy such that symptom and inflammation control is achieved. Careful endotyping is probably not necessary to manage most patients with asthma. This strategy has not been shown to reduce exacerbations in patients with mild asthma (reference our own Jayaram et al., study). It is currently recommended only for patients with more severe asthma.
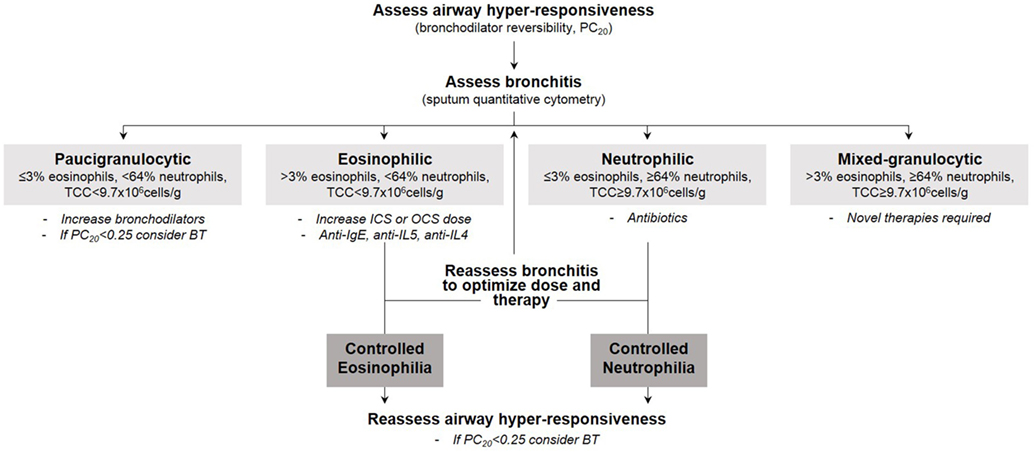
Figure 1. Our therapeutic strategy in severe asthma guided by inflammatory endotype and severity of airway hyper-responsiveness.
Management of the T2-High Endotype
Although most commonly referred to as Th2-high, type 2 (T2)-high has emerged as a more appropriate and inclusive term given the involvement of numerous cell types (including type 2 innate lymphoid cells and natural killer cells) that are outside the originally described Th2 cell population (54). Eosinophilic bronchitis indicates a T2-driven mechanism and is usually steroid responsive. When bronchitis is eosinophilic in nature with a differential cell count of more than 3% (or eosinophil-free granules are observed), inhaled corticosteroids should be initiated and the dose titrated to keep the sputum eosinophil count below 3%. In situations where high-dose inhaled corticosteroids do not control sputum eosinophilia, oral corticosteroids are required. In corticosteroid-treated patients, absent eosinophils may suggest that the current steroid dose is unwarranted and therefore should be reduced to avoid over-treatment. In RCTs in adults, moderate-to-severe asthmatics managed by normalizing induced sputum eosinophils had significantly reduced exacerbations and hospital admissions when compared with those managed by national asthma guidelines (23, 55). When this strategy was applied clinically in 52 OCS-dependent asthmatics, maintained symptom control, reduced exacerbations, and preserved lung function was observed over 5 years (56). Novel biologic therapies should be considered for their steroid sparing effect, also for the minority of patients who are corticosteroid-insensitive (unresolved sputum eosinophilia despite high doses of oral corticosteroids) (57).
To date, all biologic therapies that have been approved and the majority of those in development aim to target T2 inflammation [recently reviewed (58)] and are therefore directed toward the T2-high eosinophilic asthma endotype. Detailed illustrations of asthma pathobiology and the mechanism of action of targeted therapies are provided in recent reviews (59, 60). Anti-IgE [omalizumab (61)] therapy was the first approved monoclonal antibody and is effective in patients with allergic asthma, confirmed by a positive skin prick test and serum IgE levels ≥30 IU/mL. Two anti-IL-5 therapies are approved [mepolizumab (62) and reslizumab (63)] and one is in phase 3 development [benralizumab (64, 65)] for use in severe eosinophilic asthma. With the potential to block both IL-4 and IL-13, one anti-IL-4 receptor alpha therapy is currently in phase 3 development (dupilumab) following successful phase 2b trials (66, 67). The efficacy of strategies targeting IL-13 alone [lebrikizumab (68) and tralokinumab (69)] is inconclusive as only modest clinical benefit has been shown. Drugs that target both IL-4 and IL-13 signaling (e.g., dupilumab) have reported more clinically relevant effects in phase 2 clinical programs. The reason for this difference is not immediately obvious. Perhaps one of the reasons may be related to the lack of accurate endotyping to identify patients in whom IL-13-mediated biology was not the dominant pathobiology of asthma. Selecting patients with significant airway hyper-responsiveness and mucus secretion may have demonstrated greater clinical effect. Other novel therapies that are currently being investigated include anti-thymic stromal lymphopoietin (70), IL-33 blocking agents (71), and prostaglandin antagonists (CRTH2) (72). A general scheme to choose the appropriate monoclonal antibody based on simple clinical features (e.g., clinical history of allergy, severity of asthma based on the dose of corticosteroids, and readily available biomarkers such as blood and sputum eosinophils and fraction of eNO) is shown in Figure 2. This is based on our clinical experience, the lack of evidence supporting omalizumab in prednisone-dependent asthmatics (73), the lack of efficacy of mepolizumab 100 mg subcutaneous in patients with persistent sputum eosinophilia despite high-dose inhaled and oral corticosteroid use (74), and the efficacy of benralizumab (75) and reslizumab (76) in severe prednisone-dependent patients. However, this approach needs to be prospectively validated in clinical trials.

Figure 2. General scheme to choose the appropriate monoclonal antibody based on simple clinical features.
Management of the T2-Low Endotype
Neutrophilic bronchitis with a raised total cell count is suggestive of a non-T2-driven disease and is not usually steroid-responsive, but instead a predictor of response to antibiotics. Macrolides, including clarithromycin (77) and azithromycin (78), have demonstrated effectiveness in non-eosinophilic asthmatics. To identify the causative pathogen that may help to direct antibiotic therapy, molecular microbiology, and extended cultures, including 16 s deep sequencing, should be considered in those patients with recurrent infective exacerbations. Logically, neutrophilic bronchitis may be a predictor of therapies targeted at pathways that lead to neutrophil recruitment such as TNF, IL-1, IL-6, IL-8, IL-23, and IL-17. Several small molecule anti-neutrophilic biologics have been developed, although there are currently no active phase 3 trials. Such molecules include CXCR2 antagonists (79, 80), 5-lipoxygenase-activating protein inhibitors (81), anti-IL-17 (82), and anti-TNFα (83). It is evident that few therapeutic options exist for these patients; therefore, intense study of the underlying mechanisms contributing to the T2-low endotype is urgent. Similarly, despite the severity of their disease, currently there are no treatment options for patients with mixed-granulocytic asthma. In fact, no interventions have been evaluated for this specific inflammatory endotype although preliminary evidence suggests therapies targeting the IL-6 pathway may be beneficial (84).
Asthmatics with paucigranulocytic bronchitis may not warrant anti-inflammatory therapy as symptoms in these patients may be driven solely by smooth muscle dysfunction (airway hyper-responsiveness). Therefore, these patients may benefit from smooth muscle-directed therapies such as additional bronchodilators and long-acting anti-muscarinic antagonists, mast-cell directed therapies, or in the most severe cases, bronchial thermoplasty. Our clinical experience suggests that bronchial thermoplasty is indicated when severe airway hyper-responsiveness (PC20 < 0.25) and frequent exacerbations persist despite absent or controlled airway inflammation (85); however, clinical trials are required to confirm this hypothesis. Bronchial thermoplasty after therapy has been optimized to control eosinophilic and/or neutrophilic inflammation has been previously described by Cox et al. (85) for those patients with persistent symptoms. Although the mechanism of action is uncertain, bronchial thermoplasty aims to attenuate airway smooth muscle mass through the delivery of localized thermal energy (86).
Concluding Remarks
One of the major challenges of respiratory medicine is the management of patients with severe asthma. Identifying a specific endotype may have profound implications on advanced targeted therapy selection and intern clinical outcomes. It should be acknowledged that the mere presence of a particular gene, protein or a cell do not necessarily make them a therapeutic target or disease biomarker. Teasing out “association” from “causality” is of paramount importance. Koch’s postulates, described in 1890, remain a useful benchmark to establish whether there is causal relationship between a particular molecular or cellular observation and a disease presentation. Persistence of a particular cell type or cytokine and their temporal association with exacerbation and resolution provide strong evidence to support a causative role. A consensus as to how to best identify asthma endotypes and what therapy to use for a given endotype is now required. In the meantime, quantitative cell counts in sputum provide the most reliable method to identify T2 (most eosinophilic) and non-T2 (most neutrophilic) inflammatory processes.
Author Contributions
PN conceived the idea. SS prepared the first draft. PN and SS edited and reviewed the manuscript and have approved the final version of the manuscript.
Conflict of Interest Statement
PN has received consultancy fees from AstraZeneca, Boehringer Ingelheim, Sanofi, Teva, Knopp, Theravance, and Roche; research support from GlaxoSmithKline, AstraZeneca, Sanofi, Boehringer Ingelheim, Roche, and Novartis; and lecture fees from Roche, AstraZeneca, and Novartis.
Funding
PN is supported by the Frederick E. Hargreave Teva Innovation Chair in Airway Diseases. SS is supported by CIHR and CRRN postdoctoral fellowship awards.
References
1. Hargreave FE, Nair P. The definition and diagnosis of asthma. Clin Exp Allergy (2009) 39(11):1652–8. doi:10.1111/j.1365-2222.2009.03321.x
2. Hargreave FE, Parameswaran K. Asthma, COPD and bronchitis are just components of airway disease. Eur Respir J (2006) 28(2):264–7. doi:10.1183/09031936.06.00056106
3. Adams NP, Bestall JC, Jones P, Lasserson TJ, Griffiths B, Cates CJ. Fluticasone at different doses for chronic asthma in adults and children. Cochrane Database Syst Rev (2008) 4:CD003534. doi:10.1002/14651858.CD003534.pub3
4. Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J (2014) 43(2):343–73. doi:10.1183/09031936.00202013
5. Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med (2010) 181(4):315–23. doi:10.1164/rccm.200906-0896OC
6. Loza MJ, Djukanovic R, Chung KF, Horowitz D, Ma K, Branigan P, et al. Validated and longitudinally stable asthma phenotypes based on cluster analysis of the ADEPT study. Respir Res (2016) 17(1):165. doi:10.1186/s12931-016-0482-9
7. Lefaudeux D, De Meulder B, Loza MJ, Peffer N, Rowe A, Baribaud F, et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J Allergy Clin Immunol (2016) 139(6):1797–807. doi:10.1016/j.jaci.2016.08.048
8. Lotvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol (2011) 127(2):355–60. doi:10.1016/j.jaci.2010.11.037
9. Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med (2009) 360(10):973–84. doi:10.1056/NEJMoa0808991
10. Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med (2009) 360(10):985–93. doi:10.1056/NEJMoa0805435
11. Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med (1999) 160(3):1001–8. doi:10.1164/ajrccm.160.3.9812110
12. Lim HF, Nair P. Airway inflammation and inflammatory biomarkers. Semin Respir Crit Care Med (2017) 38. doi:10.1055/s-0037-1606217
13. Korevaar DA, Westerhof GA, Wang J, Cohen JF, Spijker R, Sterk PJ, et al. Diagnostic accuracy of minimally invasive markers for detection of airway eosinophilia in asthma: a systematic review and meta-analysis. Lancet Respir Med (2015) 3(4):290–300. doi:10.1016/S2213-2600(15)00050-8
14. Pizzichini E, Pizzichini MM, Efthimiadis A, Evans S, Morris MM, Squillace D, et al. Indices of airway inflammation in induced sputum: reproducibility and validity of cell and fluid-phase measurements. Am J Respir Crit Care Med (1996) 154(2 Pt 1):308–17. doi:10.1164/ajrccm.154.2.8756799
15. Djukanovic R, Sterk PJ, Fahy JV, Hargreave FE. Standardised methodology of sputum induction and processing. Eur Respir J Suppl (2002) 37:1s–2s. doi:10.1183/09031936.02.00000102
16. Vlachos-Mayer H, Leigh R, Sharon RF, Hussack P, Hargreave FE. Success and safety of sputum induction in the clinical setting. Eur Respir J (2000) 16(5):997–1000. doi:10.1183/09031936.00.16599700
17. D’Silva L, Hassan N, Wang HY, Kjarsgaard M, Efthimiadis A, Hargreave FE, et al. Heterogeneity of bronchitis in airway diseases in tertiary care clinical practice. Can Respir J (2011) 18(3):144–8. doi:10.1155/2011/430317
18. D’Silva L, Neighbour H, Gafni A, Radford K, Hargreave F, Nair P. Quantitative sputum cell counts to monitor bronchitis: a qualitative study of physician and patient perspectives. Can Respir J (2013) 20(1):47–51. doi:10.1155/2013/248167
19. Schleich FN, Chevremont A, Paulus V, Henket M, Manise M, Seidel L, et al. Importance of concomitant local and systemic eosinophilia in uncontrolled asthma. Eur Respir J (2014) 44(1):97–108. doi:10.1183/09031936.00201813
20. Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology (2006) 11(1):54–61. doi:10.1111/j.1440-1843.2006.00784.x
21. Belda J, Leigh R, Parameswaran K, O’Byrne PM, Sears MR, Hargreave FE. Induced sputum cell counts in healthy adults. Am J Respir Crit Care Med (2000) 161(2 Pt 1):475–8. doi:10.1164/ajrccm.161.2.9903097
22. Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, Pavord ID. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax (2002) 57(10):875–9. doi:10.1136/thorax.57.10.875
23. Jayaram L, Pizzichini MM, Cook RJ, Boulet LP, Lemiere C, Pizzichini E, et al. Determining asthma treatment by monitoring sputum cell counts: effect on exacerbations. Eur Respir J (2006) 27(3):483–94. doi:10.1183/09031936.06.00137704
24. Nair P, Hargreave FE. Measuring bronchitis in airway diseases: clinical implementation and application: airway hyperresponsiveness in asthma: its measurement and clinical significance. Chest (2010) 138(2 Suppl):38S–43S. doi:10.1378/chest.10-0094
25. Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, et al. Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol (2010) 125(5):1028–36.e13. doi:10.1016/j.jaci.2010.02.008
26. Aleman F, Lim HF, Nair P. Eosinophilic endotype of asthma. Immunol Allergy Clin North Am (2016) 36(3):559–68. doi:10.1016/j.iac.2016.03.006
27. Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med (2009) 180(5):388–95. doi:10.1164/rccm.200903-0392OC
28. Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol (2011) 127(1):153–60, 160.e1–9. doi:10.1016/j.jaci.2010.10.024
29. Baines KJ, Simpson JL, Wood LG, Scott RJ, Fibbens NL, Powell H, et al. Sputum gene expression signature of 6 biomarkers discriminates asthma inflammatory phenotypes. J Allergy Clin Immunol (2014) 133(4):997–1007. doi:10.1016/j.jaci.2013.12.1091
30. Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV. Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J Allergy Clin Immunol (2014) 133(2):388–94. doi:10.1016/j.jaci.2013.07.036
31. Yan X, Chu JH, Gomez J, Koenigs M, Holm C, He X, et al. Noninvasive analysis of the sputum transcriptome discriminates clinical phenotypes of asthma. Am J Respir Crit Care Med (2015) 191(10):1116–25. doi:10.1164/rccm.201408-1440OC
32. Hekking PP, Loza MJ, Pavlidis S, de Meulder B, Lefaudeux D, Baribaud F, et al. Pathway discovery using transcriptomic profiles in adult-onset severe asthma. J Allergy Clin Immunol (2017). doi:10.1016/j.jaci.2017.06.037
33. Kuo CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J (2017) 49(2). doi:10.1183/13993003.02135-2016
34. Brasier AR, Victor S, Boetticher G, Ju H, Lee C, Bleecker ER, et al. Molecular phenotyping of severe asthma using pattern recognition of bronchoalveolar lavage-derived cytokines. J Allergy Clin Immunol (2008) 121(1):30–7.e6. doi:10.1016/j.jaci.2007.10.015
35. McGeachie MJ, Dahlin A, Qiu W, Croteau-Chonka DC, Savage J, Wu AC, et al. The metabolomics of asthma control: a promising link between genetics and disease. Immun Inflamm Dis (2015) 3(3):224–38. doi:10.1002/iid3.61
36. Kelly RS, Dahlin A, McGeachie MJ, Qiu W, Sordillo J, Wan ES, et al. Asthma metabolomics and the potential for integrative omics in research and the clinic. Chest (2017) 151(2):262–77. doi:10.1016/j.chest.2016.10.008
37. Shaw DE, Sousa AR, Fowler SJ, Fleming LJ, Roberts G, Corfield J, et al. Clinical and inflammatory characteristics of the European U-BIOPRED adult severe asthma cohort. Eur Respir J (2015) 46(5):1308–21. doi:10.1183/13993003.00779-2015
38. Bigler J, Boedigheimer M, Schofield JP, Skipp PJ, Corfield J, Rowe A, et al. A severe asthma disease signature from gene expression profiling of peripheral blood from U-BIOPRED cohorts. Am J Respir Crit Care Med (2016) 195:(10):1279–80. doi:10.1164/rccm.201604-0866OC
39. Wilson SJ, Ward JA, Sousa AR, Corfield J, Bansal AT, De Meulder B, et al. Severe asthma exists despite suppressed tissue inflammation: findings of the U-BIOPRED study. Eur Respir J (2016) 48(5):1307–19. doi:10.1183/13993003.01129-2016
40. Wu J, Kobayashi M, Sousa EA, Liu W, Cai J, Goldman SJ, et al. Differential proteomic analysis of bronchoalveolar lavage fluid in asthmatics following segmental antigen challenge. Mol Cell Proteomics (2005) 4(9):1251–64. doi:10.1074/mcp.M500041-MCP200
41. Cederfur C, Malmstrom J, Nihlberg K, Block M, Breimer ME, Bjermer L, et al. Glycoproteomic identification of galectin-3 and -8 ligands in bronchoalveolar lavage of mild asthmatics and healthy subjects. Biochim Biophys Acta (2012) 1820(9):1429–36. doi:10.1016/j.bbagen.2011.12.016
42. O’Neil SE, Sitkauskiene B, Babusyte A, Krisiukeniene A, Stravinskaite-Bieksiene K, Sakalauskas R, et al. Network analysis of quantitative proteomics on asthmatic bronchi: effects of inhaled glucocorticoid treatment. Respir Res (2011) 12:124. doi:10.1186/1465-9921-12-124
43. Gharib SA, Nguyen EV, Lai Y, Plampin JD, Goodlett DR, Hallstrand TS. Induced sputum proteome in healthy subjects and asthmatic patients. J Allergy Clin Immunol (2011) 128(6):1176–84.e6. doi:10.1016/j.jaci.2011.07.053
44. Lee TH, Jang AS, Park JS, Kim TH, Choi YS, Shin HR, et al. Elevation of S100 calcium binding protein A9 in sputum of neutrophilic inflammation in severe uncontrolled asthma. Ann Allergy Asthma Immunol (2013) 111(4):268–75.e1. doi:10.1016/j.anai.2013.06.028
45. van der Schee MP, Paff T, Brinkman P, van Aalderen WM, Haarman EG, Sterk PJ. Breathomics in lung disease. Chest (2015) 147(1):224–31. doi:10.1378/chest.14-0781
46. Dragonieri S, Schot R, Mertens BJ, Le Cessie S, Gauw SA, Spanevello A, et al. An electronic nose in the discrimination of patients with asthma and controls. J Allergy Clin Immunol (2007) 120(4):856–62. doi:10.1016/j.jaci.2007.05.043
47. Montuschi P, Santonico M, Mondino C, Pennazza G, Mantini G, Martinelli E, et al. Diagnostic performance of an electronic nose, fractional exhaled nitric oxide, and lung function testing in asthma. Chest (2010) 137(4):790–6. doi:10.1378/chest.09-1836
48. van der Schee MP, Palmay R, Cowan JO, Taylor DR. Predicting steroid responsiveness in patients with asthma using exhaled breath profiling. Clin Exp Allergy (2013) 43(11):1217–25. doi:10.1111/cea.12147
49. Ibrahim B, Basanta M, Cadden P, Singh D, Douce D, Woodcock A, et al. Non-invasive phenotyping using exhaled volatile organic compounds in asthma. Thorax (2011) 66(9):804–9. doi:10.1136/thx.2010.156695
50. McShane LM, Cavenagh MM, Lively TG, Eberhard DA, Bigbee WL, Williams PM, et al. Criteria for the use of omics-based predictors in clinical trials. Nature (2013) 502(7471):317–20. doi:10.1038/nature12564
51. Freidlin B, McShane LM, Korn EL. Randomized clinical trials with biomarkers: design issues. J Natl Cancer Inst (2010) 102(3):152–60. doi:10.1093/jnci/djp477
52. Hinks TS, Brown T, Lau LC, Rupani H, Barber C, Elliott S, et al. Multidimensional endotyping in patients with severe asthma reveals inflammatory heterogeneity in matrix metalloproteinases and chitinase 3-like protein 1. J Allergy Clin Immunol (2016) 138(1):61–75. doi:10.1016/j.jaci.2015.11.020
53. Dasgupta A, Neighbour H, Nair P. Targeted therapy of bronchitis in obstructive airway diseases. Pharmacol Ther (2013) 140(3):213–22. doi:10.1016/j.pharmthera.2013.07.001
54. Robinson D, Humbert M, Buhl R, Cruz AA, Inoue H, Korom S, et al. Revisiting Type 2-high and Type 2-low airway inflammation in asthma: current knowledge and therapeutic implications. Clin Exp Allergy (2017) 47(2):161–75. doi:10.1111/cea.12880
55. Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet (2002) 360(9347):1715–21. doi:10.1016/S0140-6736(02)11679-5
56. Aziz-Ur-Rehman A, Dasgupta A, Kjarsgaard M, Hargreave FE, Nair P. Sputum cell counts to manage prednisone-dependent asthma: effects on FEV1 and eosinophilic exacerbations. Allergy Asthma Clin Immunol (2017) 13:17. doi:10.1186/s13223-017-0190-0
57. Nair P. Anti-interleukin-5 monoclonal antibody to treat severe eosinophilic asthma. N Engl J Med (2014) 371(13):1249–51. doi:10.1056/NEJMe1408614
58. Katial RK, Bensch GW, Busse WW, Chipps BE, Denson JL, Gerber AN, et al. Changing paradigms in the treatment of severe asthma: the role of biologic therapies. J Allergy Clin Immunol Pract (2017) 5(2S):S1–14. doi:10.1016/j.jaip.2016.11.029
59. Mukherjee M, Sehmi R, Nair P. Anti-IL5 therapy for asthma and beyond. World Allergy Organ J (2014) 7(1):32. doi:10.1186/1939-4551-7-32
60. Darveaux J, Busse WW. Biologics in asthma—the next step toward personalized treatment. J Allergy Clin Immunol Pract (2015) 3(2):152–60; quiz 161. doi:10.1016/j.jaip.2014.09.014
61. Busse W, Buhl R, Fernandez Vidaurre C, Blogg M, Zhu J, Eisner MD, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol (2012) 129(4):983–9.e6. doi:10.1016/j.jaci.2012.01.033
62. Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med (2014) 371(13):1198–207. doi:10.1056/NEJMoa1403290
63. Castro M, Zangrilli J, Wechsler ME, Bateman ED, Brusselle GG, Bardin P, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med (2015) 3(5):355–66. doi:10.1016/S2213-2600(15)00042-9
64. Bleecker ER, FitzGerald JM, Chanez P, Papi A, Weinstein SF, Barker P, et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting beta2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet (2016) 388(10056):2115–27. doi:10.1016/S0140-6736(16)31324-1
65. FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, et al. Benralizumab, an anti-interleukin-5 receptor alpha monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (2016) 388(10056):2128–41. doi:10.1016/S0140-6736(16)31322-8
66. Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med (2013) 368(26):2455–66. doi:10.1056/NEJMoa1304048
67. Wenzel S, Castro M, Corren J, Maspero J, Wang L, Zhang B, et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting beta2 agonist: a randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet (2016) 388(10039):31–44. doi:10.1016/S0140-6736(16)30307-5
68. Hanania NA, Korenblat P, Chapman KR, Bateman ED, Kopecky P, Paggiaro P, et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): replicate, phase 3, randomised, double-blind, placebo-controlled trials. Lancet Respir Med (2016) 4(10):781–96. doi:10.1016/S2213-2600(16)30265-X
69. Brightling CE, Chanez P, Leigh R, O’Byrne PM, Korn S, She D, et al. Efficacy and safety of tralokinumab in patients with severe uncontrolled asthma: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med (2015) 3(9):692–701. doi:10.1016/S2213-2600(15)00197-6
70. Gauvreau GM, O’Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med (2014) 370(22):2102–10. doi:10.1056/NEJMoa1402895
71. Nabe T. Interleukin (IL)-33: new therapeutic target for atopic diseases. J Pharmacol Sci (2014) 126(2):85–91. doi:10.1254/jphs.14R12CP
72. Hall IP, Fowler AV, Gupta A, Tetzlaff K, Nivens MC, Sarno M, et al. Efficacy of BI 671800, an oral CRTH2 antagonist, in poorly controlled asthma as sole controller and in the presence of inhaled corticosteroid treatment. Pulm Pharmacol Ther (2015) 32:37–44. doi:10.1016/j.pupt.2015.03.003
73. Normansell R, Walker S, Milan SJ, Walters EH, Nair P. Omalizumab for asthma in adults and children. Cochrane Database Syst Rev (2014) 1:CD003559. doi:10.1002/14651858.CD003559.pub4
74. Sehmi R, Smith SG, Kjarsgaard M, Radford K, Boulet LP, Lemiere C, et al. Role of local eosinophilopoietic processes in the development of airway eosinophilia in prednisone-dependent severe asthma. Clin Exp Allergy (2016) 46(6):793–802. doi:10.1111/cea.12695
75. Nair P, Wenzel S, Rabe KF, Bourdin A, Lugogo NL, Kuna P, et al. Oral glucocorticoid-sparing effect of benralizumab in severe asthma. N Engl J Med (2017) 376(25):2448–58. doi:10.1056/NEJMoa1703501
76. Mukherjee M, Aleman F, Kjarsgaard M, Salter B, Nair G, LaVigne N, et al. Weight-adjusted intravenous reslizumab in severe asthma with inadequate response to fixed-dose subcutaneous mepolizumab. Am J Respir Crit Care Med (2017).
77. Simpson JL, Powell H, Boyle MJ, Scott RJ, Gibson PG. Clarithromycin targets neutrophilic airway inflammation in refractory asthma. Am J Respir Crit Care Med (2008) 177(2):148–55. doi:10.1164/rccm.200707-1134OC
78. Brusselle GG, Vanderstichele C, Jordens P, Deman R, Slabbynck H, Ringoet V, et al. Azithromycin for prevention of exacerbations in severe asthma (AZISAST): a multicentre randomised double-blind placebo-controlled trial. Thorax (2013) 68(4):322–9. doi:10.1136/thoraxjnl-2012-202698
79. Nair P, Gaga M, Zervas E, Alagha K, Hargreave FE, O’Byrne PM, et al. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy (2012) 42(7):1097–103. doi:10.1111/j.1365-2222.2012.04014.x
80. O’Byrne PM, Metev H, Puu M, Richter K, Keen C, Uddin M, et al. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med (2016) 4(10):797–806. doi:10.1016/S2213-2600(16)30227-2
81. Chaudhuri R, Norris V, Kelly K, Zhu CQ, Ambery C, Lafferty J, et al. Effects of a FLAP inhibitor, GSK2190915, in asthmatics with high sputum neutrophils. Pulm Pharmacol Ther (2014) 27(1):62–9. doi:10.1016/j.pupt.2013.11.007
82. Busse WW, Holgate S, Kerwin E, Chon Y, Feng J, Lin J, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med (2013) 188(11):1294–302. doi:10.1164/rccm.201212-2318OC
83. Wenzel SE, Barnes PJ, Bleecker ER, Bousquet J, Busse W, Dahlen SE, et al. A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med (2009) 179(7):549–58. doi:10.1164/rccm.200809-1512OC
84. Chu DK, Al-Garawi A, Llop-Guevara A, Pillai RA, Radford K, Shen P, et al. Therapeutic potential of anti-IL-6 therapies for granulocytic airway inflammation in asthma. Allergy Asthma Clin Immunol (2015) 11(1):14. doi:10.1186/s13223-015-0081-1
85. Cox C, Kjarsgaard M, Surette MG, Cox PG, Nair P. A multidimensional approach to the management of severe asthma: inflammometry, molecular microbiology and bronchial thermoplasty. Can Respir J (2015) 22(4):221–4. doi:10.1155/2015/459187
Keywords: endotype, severe asthma, omics, sputum cytometry, type 2-low asthma, type 2-high asthma
Citation: Svenningsen S and Nair P (2017) Asthma Endotypes and an Overview of Targeted Therapy for Asthma. Front. Med. 4:158. doi: 10.3389/fmed.2017.00158
Received: 11 May 2017; Accepted: 12 September 2017;
Published: 26 September 2017
Edited by:
Florence Emmanuelle Roufosse, Free University of Brussels, BelgiumReviewed by:
Bertrand De Meulder, European Institute for Systems Biology and Medicine (EISBM), FranceVirginie Doyen, Centre Hospitalier Universitaire Brugmann, Belgium
Copyright: © 2017 Svenningsen and Nair. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Parameswaran Nair, cGFyYW1lc0BtY21hc3Rlci5jYQ==