 Noémie Simon
Noémie Simon Alexandre Hertig
Alexandre Hertig
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 05 August 2015
Sec. Pathology
Volume 2 - 2015 | https://doi.org/10.3389/fmed.2015.00052
This article is part of the Research Topic Organ Fibrosis: Triggers, Pathways and Cellular Plasticity View all 7 articles
Renal proximal tubular cells are the most energy-demanding cells in the body. The ATP that they use is mostly produced in their mitochondrial and peroxisomal compartments, by the oxidation of fatty acids. When those cells are placed under a biological stress, such as a transient hypoxia, fatty acid oxidation (FAO) is shut down for a period of time that outlasts injury, and carbohydrate oxidation does not take over. Facing those metabolic constraints, surviving tubular epithelial cells exhibit a phenotypic switch that includes cytoskeletal rearrangement and production of extracellular matrix proteins, most probably contributing to acute kidney injury-induced renal fibrogenesis, thence to the development of chronic kidney disease. Here, we review experimental evidence that dysregulation of FAO profoundly affects the fate of tubular epithelial cells, by promoting epithelial-to-mesenchymal transition, inflammation, and eventually interstitial fibrosis. Restoring physiological production of energy is undoubtedly a possible therapeutic approach to unlock the mesenchymal reprograming of tubular epithelial cells in the kidney. In this respect, the benefit of the use of fibrates is uncertain, but new drugs that could specifically target this metabolic pathway, and, hopefully, attenuate renal fibrosis merit future research.
Renal fibrosis is the final common pathway to all chronic kidney diseases (CKD), suggesting that a biological switch is activated in any context of injury, suppressing noble renal functions long-term and reactivating embryonic-like mesenchymal ones (1). Such a switch would obviously be a major source of new therapeutics.
Although the consequence of renal fibrosis is the loss of epithelial functions, it is defined morphologically in organs by an expansion of the connective tissue (swelling of interstitial myofibroblasts and excess of deposition of extracellular matrix). At the tubular cell scale, it consists of an atrophic type lesion with a thickening of basement membranes and a flattened epithelium, suggesting dedifferentiation. However, evidence has accumulated to show that proximal tubular epithelial cells (PTC) are not uninvolved in the process of interstitial fibrogenesis: placed under biological stress, they can undergo phenotypic changes, acquire active mesenchymal functions, and even proliferate (2), and hence, contributing to the synthesis of extracellular matrix proteins and perpetuating fibrosis (3, 4). As epithelial cells have two equilibrium states (epithelial during homeostasis, mesenchymal in pathological situations), some authors often refer to this bi-stability as epithelial-to-mesenchymal transition (EMT) (5), analogous with the profound phenotypic changes observed in primary epiblasts during embryogenesis (or at the edge of a carcinoma). However, the term is confusing, since in embryos and tumors, EMT is essential to disperse cells. Whether EMT becomes so intense in adult fibrotic kidneys that epithelial cells leave their tubular structure and provide de novo myofibroblasts has been vigorously debated (6, 7). Over the last decade, experimental research has shown that pericytes, not epithelial cells, are considered the most significant source of such de novo myofibroblasts (8), and the contribution of EMT in organ fibrogenesis is seen as local rather than diffuse, i.e., relevant within tubular structures (9). However, the process in humans is unknown, and EMT-like biological changes might significantly contribute, even at the local level.
Hypoxia is one of the many biological stresses that may tip the balance toward a mesenchymal program. Segment 3 renal PTC (the pars recta, located at the corticomedullary junction) in particular are very hypoxia sensitive. This is because of a combination of the locally lowest oxygen pressure in this anatomic segment on the one hand (10, 11), with a high-energy consumption linked to highly specialized ATP-consuming transporters on the other (12, 13). Like most of the highly metabolic cells, the preferred energy fuel is the one with the highest ATP production: fatty acid oxidation (FAO) (14). The aim of this review is to provide an insight into how down-regulation of FAO observed during acute kidney injury (AKI) – not only during ischemic AKI but also in other experimental conditions – precedes EMT and constitutes a major alteration of cell metabolism, which drives the mesenchymal transition. Drugs promoting or restoring FAO are therefore promising.
Recently, the concept of AKI has been redefined. This term now encompasses a range of renal impairment, from even small changes in function (serum creatinine or urinary output) to the necessity for renal replacement therapy. AKI can be secondary to a large spectrum of causes. Histological features of AKI often include lesions of acute tubular necrosis (ATN), a renal lesion that was long thought to be fully reversible. However, this paradigm shifted some years ago toward the notion that the repair of ATN could be “maladaptive” and initiate fibrogenesis at a molecular level, even when morphology had (at least transiently) returned to normal (15). Similarly, increasing evidence in kidney transplantation suggests that ischemic episodes are connected to transplant fibrosis (16). Mechanisms at stake in the interconnection between AKI and CKD are an intense area of research. At present, three major abnormalities have been found to be associated with the fibrotic outcome of a transient AKI: (a) the epigenetic silencing of RASAL1, a proliferation inhibitor, in myofibroblasts; (b) the cell cycle arrest in G2/M in tubular epithelial cells (the G2/M phase is where the epithelial cell function is closer to a mesenchymal one); and (c) down-regulation of FAO in tubular epithelial cells (14, 17, 18). These mechanisms are not exclusive of each other.
A fatty acid (FA) is a carboxylic acid with a long aliphatic (as opposed to aromatic) tail. It can be produced by FA synthesis or by hydrolysis of triglycerides or phospholipids; conversely, a triglyceride is a storage (triester) form of FA. Triglyceride overload leads to lipid droplet formation. Historical studies by Weidemann and Krebs were the first to report that in dog kidneys these droplets can be used in case of scarcity. Thus, FAO may help PTC to adapt to energy demand (19).
Fatty acid oxidation occurs in the mitochondrial and peroxisomal compartments. FAs must first be supplied, either by extracellular uptake through the FA transport protein CD36 (20), or by in situ cytosolic synthesis, or by the deacylation of cellular phospholipids under the action of phospholipase A2 (PLA2). Second, FAs has to be transported from the cytosol to the respective organelles in order to be oxidized and provide the cell with ATP (Figure 1). The outer membranes of the mitochondrion and the peroxisome are not permeable to long-chain FA, so FAs need to use a specific transporter called the carnitine shuttle. For this to occur, they need to be “activated” by coenzyme A in the cytosol, under the action of an acyl-CoA synthetase, which is located on the outer membrane of the organelle. The resulting long-chain acyl-CoA products will then interact with a carnitine molecule to regenerate coenzyme A and produce a long-chain acyl carnitine (LCAC), to which the outer membrane is readily permeable. This step also requires the rate-limiting enzyme of the carnitine shuttle, the carnitine palmitoyl-transferase 1 (CPT-1), similarly located on the outer membrane. LCAC is eventually able to cross the inner (impermeable) membrane thanks to the carnitine-acyl-carnitine translocase. The carnitine palmitoyl-transferase 2 then ensures a reverse reaction regenerating the carnitine molecule using coenzyme A, resulting again in an acyl-CoA product, which will undergo β-oxidation in the peroxisome and the mitochondrion. The system is complex but still economic in that the carnitine molecule will be transported back to the cytoplasm by the same shuttle. Oxidation (the loss of an electron) then occurs because electron carriers flavine adenine dinucleotide (FAD) and nicotine adenosine dinucleotide (NAD) will accept an electron from acyl-CoA, and hence, be reduced to FADH and NADH, respectively. Since these reactions occur close to the inner membrane, where the electron transfer chain is located, FAD and NAD are instantly regenerated. The term β-oxidation refers to the position of the carbon group being oxidized. The energy yield of FA β-oxidation is very high, with an average of 106 ATP equivalents per FA, as opposed to 36 during the oxidation of carbohydrates.
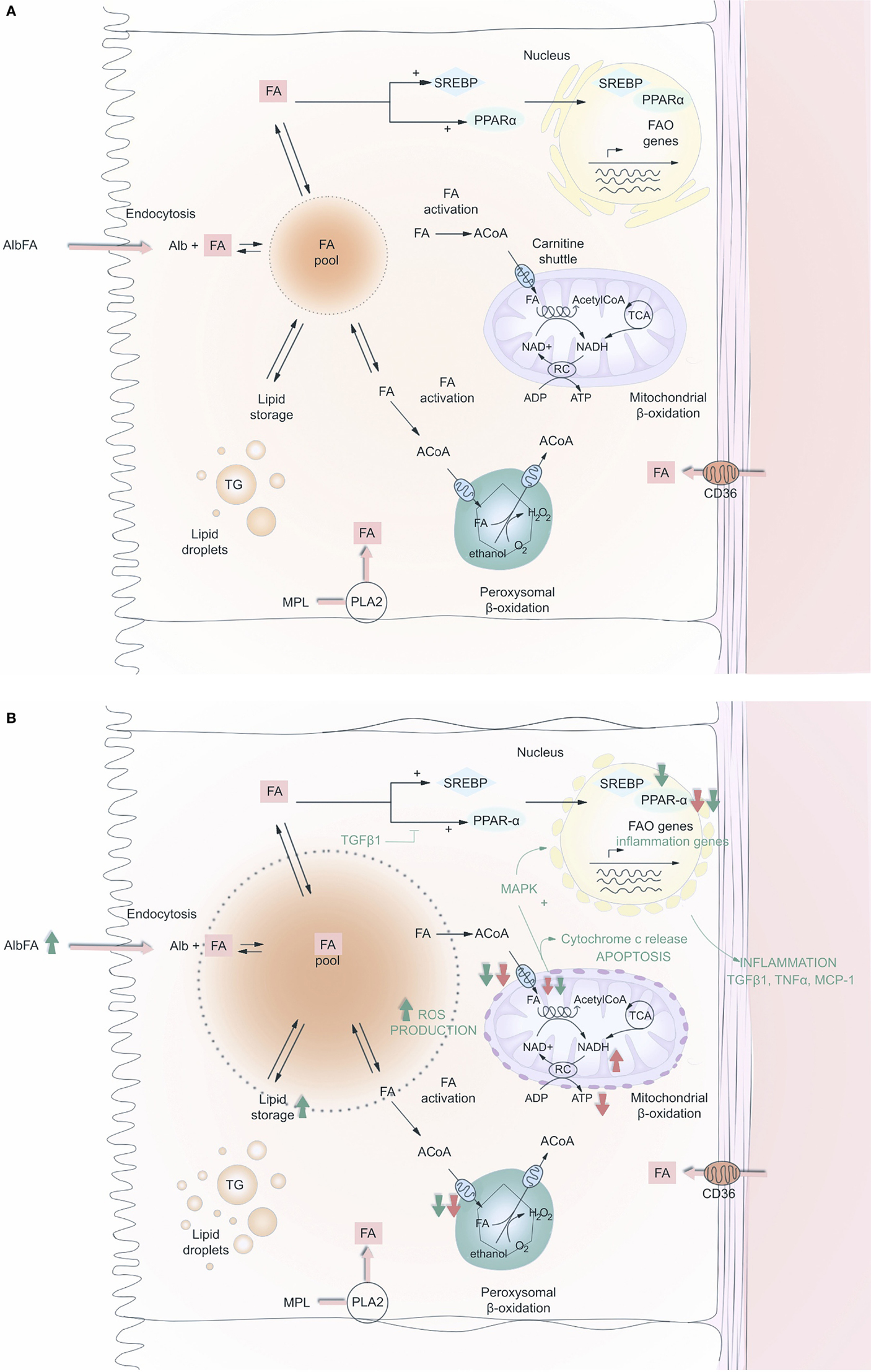
Figure 1. Fatty acid (FA) metabolism in renal PTC before (A) and after (B) acute kidney injury. FA may enter the cell either at the apical or at the baso-lateral side, free or albumin bound. They may also be produced after hydrolysis of membrane phospholipids, by phospholipase A2. Intracellular FA is then routed to anabolic or catabolic pathways; FA is stored in the global triglyceride pool or oxidized in mitochondria or peroxisome to produce ATP. The carnitine shuttle gives access to the matrix of these two organelles. FAO enzymes are positively retro-controlled by FA accumulation at the transcriptional level by the activation of SREBP1c and PPAR-α. On (B), red and green arrows indicate what is being down-regulated (down arrows) or up-regulated (up arrows) during AKI and fibrosis, respectively. Abbreviations: FA, fatty acid; CoA, CoenzymeA; ACoA, acyl-CoenzymeA; Alb, albumin; SREBP, sterol regulatory element-binding protein-1c; PPAR-α, peroxisome proliferator activated receptor-alpha; MAPK, mitogen-activated protein kinase; FAO, fatty acid oxidation; MPL, membrane phospholipid; ROS, reactive oxygen species; NAD, nicotine adenosine dinucleotide; RC, respiratory chain; PLA2, phospholipase A2; TG, triglyceride; TGF-β1, transforming growth factor β1; ADP, adenosine diphosphate; ATP, adenosine triphosphate; TNF α, tumor necrosis factor α; MCP-1, monocyte chemo-attractant protein-1.
Several studies have reported on a triglyceride overload following endotoxic, toxic, and ischemic kidney injury (21, 22). FA accumulation is observed after ischemic AKI (23), most probably because of a lack of oxidative substrates to accomplish FAO (24). In the mitochondrion, the rate-controlling step of the FAO is the oxidative reaction catalyzed by 3-hydroxyacyl-CoA dehydrogenase. As explained above, and in Figure 1A, this enzyme requires NAD as oxidant under its oxidized form (NAD+), which is regenerated by the respiratory chain located in the inner mitochondrial membrane in normoxic conditions, as oxygen acts as the terminal electron acceptor. Likewise, acyl-CoA oxidase, the key enzyme for β-oxidation in the peroxisome, uses oxygen as a substrate to perform FAO. Therefore, hypoxia dramatically decreases the NAD+/NADH ratio. Short of fundamental substrates for the critical steps of FAO, FAO also dramatically decreases. Reperfusion should theoretically lead to the restoration of a functional FAO in the aftermath of an acute cellular stress. However, Gulati et al. demonstrated that in the peroxisome compartment, FAO enzymes were not only inhibited during the ischemic phase but also during reperfusion, because of a proteolytic degradation process, particularly affecting the acyl-CoA oxidase (25). In mitochondria, ischemia/reperfusion injury (IRI) also leads to a decline in the activity of CPT-1, the rate-limiting enzyme of the carnitine shuttle; therefore, cutting off FA supplies in the mitochondrial matrix (26). Feldkamp et al. also observed that PTC isolated from rabbit kidneys exhibit a free FA accumulation during hypoxia and reperfusion, and an ATP decrease during hypoxia that outlasts re-oxygenation (27). Although it is reasonable to think that in the absence of its usual fuel (FA), PTC would turn to glucose oxidation to produce ATP even at a lower rate, and resume even minimal epithelial functions, the sustained alteration of FAO is not followed by such a take over in the context of AKI, either in vitro or in vivo (14). With prolonged ATP shortage, proliferation and mesenchymal reprograming of epithelial cells could thus be an energy efficient route to survival. Resetting the tools for ATP production, and in particular for FAO, is thus a promising approach. Of note, whether this metabolic pathway is generally necessary for pericytes, or more particularly enables them to maintain a stable phenotype instead of becoming fibroblasts, is not known.
Peroxisome proliferator activated receptor-alpha (PPAR-α) is a transcription factor predominantly expressed in metabolically very active tissues, such as renal PTC, and has been shown to control FAO. In homeostasis, endogenous levels of FA act directly on PPAR-α as natural activators of this ligand-activated receptor superfamily member. PPAR-α increases the transcription of genes encoding FAO enzymes, and also acts upstream by stimulating cellular FA uptake through the modulation of the FA translocase CD36 (28). Conversely, during AKI, PPAR-α mRNA, and its DNA binding activity were found to decrease, as was the availability of its tissue specific co-activator PPAR-γ co-activator-1a (PPARGC1A) (29–31). Kang et al. have reported that transforming growth factor β1 (TGF-β1), a major player in kidney fibrosis, and a master inducer of EMT, can inhibit PPAR-α and PPARGC1A, key transcription factors of FAO genes. It logically results in a down-regulation of CPT-1 and triglyceride overload. How TGF-β1 suppresses PPAR-α, and PPARGC1A seems to be epigenetically regulated. MicroRNA-21 (miR-21), a downstream target of Smad3 (32), is able to silence PPAR-α (33). Strikingly, anti-miR-21 failed to suppress renal fibrosis in PPAR-α−/− mice, incidentally underlining the major role of PPAR-α/FAO in the process of renal fibrogenesis. In addition, chromatin immunoprecipitation (CHiP) studies revealed that Smad3 can bind to an intronic area of the PPARGC1A promoter, at a position where the DNA is enriched in the histone mark H3K4me1 (read: mono-methylation of lysine 4 in histone 3, a mark usually associated with activation of transcription). By overlapping with this region, Smad3 could thus impede epigenetic activation of PPARGC1A (14) (Figure 2).
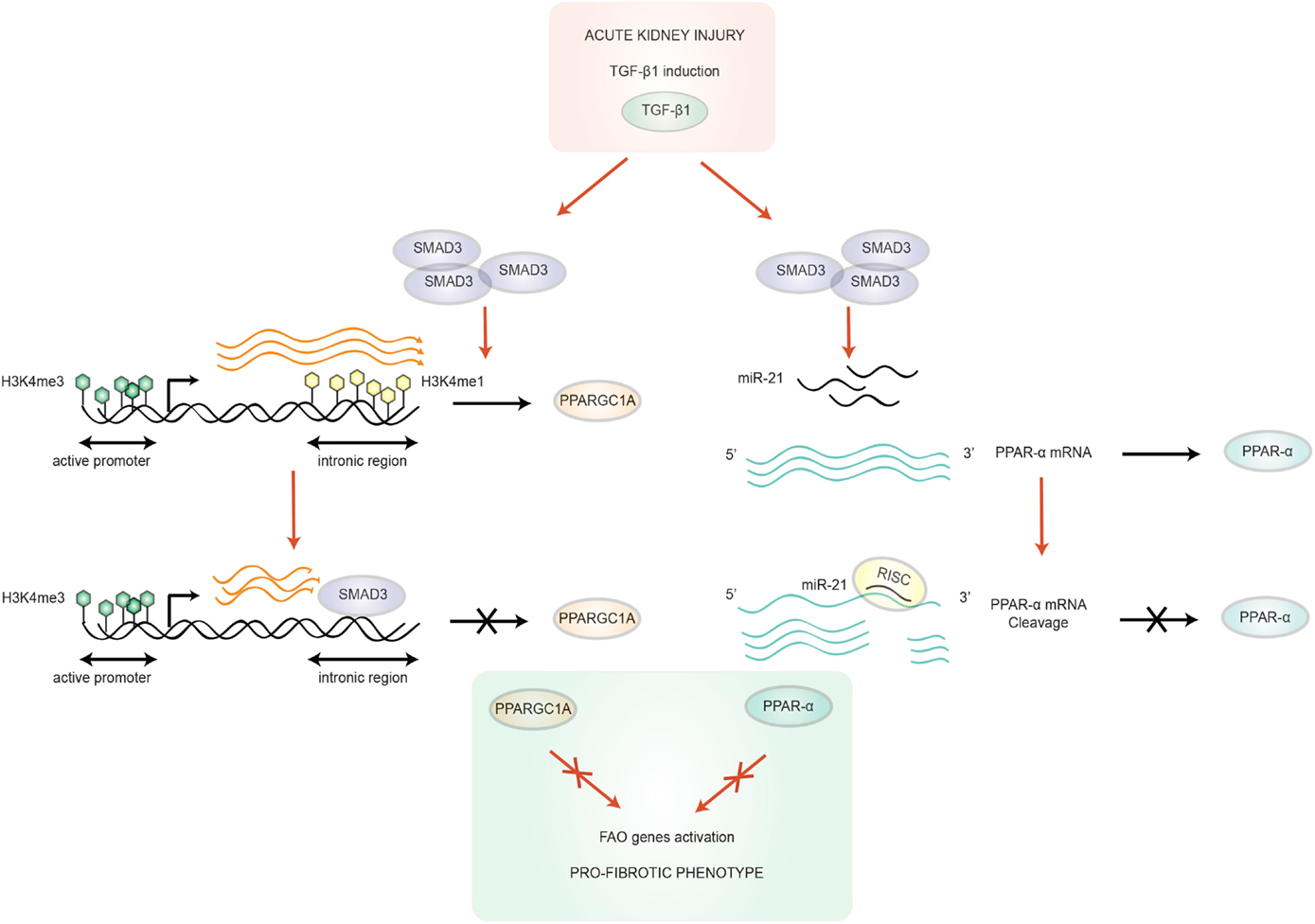
Figure 2. Transforming growth factor-β1/SMAD3 mediated-regulation of FAO. TGF-β1 is up-regulated after acute kidney injury, which activates SMAD3, which in turn can bind to an intronic region of the PPARGC1A gene. SMAD3 binding overlaps with the active enhancer histone tail modification H3K4me1 of this sequence, resulting in the blocking of the progression transcription machinery. In addition, this region is also annotated as an active enhancer in human kidney PTC (14). SMAD3 can also target PPAR-α, the other key regulator gene of FAO, through microRNA (miR-21) overexpression. miR-21 silences PPAR-α by recognition of an octamer sequence complementary to miR-21 seed region in the 3′UTR of PPAR-α mRNA (32). These two mechanisms cooperate in the acquisition of a pro-fibrotic phenotype. Abbreviations: PPAR-α, peroxisome proliferator activated receptor-alpha; PPARGC1A, PPAR-γ co-activator-1a; RISC, RNA-induced silencing complex; TGF-β1, transforming growth factor β1.
A genomic study of hepatocellular carcinomas showed that down-regulation of critical genes involved in FAO (hydroxyl-acyl-CoA dehydrogenase and acyl-coA oxidase) correlates to a “dedifferentiation” state of tumoral tissue, which also involves up-regulation of SNAIL, a major regulator of EMT (34). More recently, a study revealed in a human renal cell line (HK2) that lipid accumulation and FAO decrease precede glucose-induced morphological changes by 48 h, in addition to the cytoskeletal switch typical of EMT [loss of E-cadherin and acquisition of alpha-smooth muscle actin (α-SMA)]. Although this does not prove a causal relationship, it is interesting to note that the silencing of acetyl-coA carboxylase 2 (ACC2), the enzyme catalyzing the carboxylation of acetyl-coA into malonyl-coA, a potent inhibitor of CPT-1, abrogated glucose-induced morphological changes, cytoskeletal switch, and increased the rate of FA (35). Kang et al. showed that a global down-regulation of genes involved in FA metabolism was also found ex vivo in human fibrotic kidneys compared to controls. They also reported that PTC treated by the CPT-1 inhibitor Etomoxir undergo morphologic and genomic changes, with the expression of more mesenchymal genes, such as ACTA2 (encoding α-SMA), VIM encoding vimentin, an intermediate filament, and COL1A1 and COL3A1 encoding fibrillary collagens (14). Thus, accumulation of lipids and/or decreased FAO could participate in the mesenchymal reprograming of epithelial cells in the kidney, a non-lipogenic tissue. We will now briefly discuss why the former, taken alone, is not considered a sufficient trigger of EMT.
“Lipotoxicity” is defined by FA accumulation on non-adipose tissues. Lipid accumulation reflects an imbalance between FA utilization and FA supplies, as in the case of FAO inhibition, and because of triglyceride buffer FA excess, overload is often visible as lipid droplets. In human diseases as well as in animal models, lipid deposition has repeatedly been observed in PTC (36) (in glomerulonephritis models, for example, glomerular injury allows PTC to reabsorb albumin/FA complexes, eventually resulting in cell FA accumulation).
Fatty acid accumulation could play a role in the systemic manifestations of “metabolic” pathologies, such as diabetes mellitus and obesity, where free or albumin-bound FA blood levels are increased (37), CD36 is overexpressed due to glucose exposure, and FA synthesis also increases [in OVE26 and Akita type 1 diabetic mice, animals exhibit an increase in sterol regulatory element-binding protein-1c (SREBP-1c) mRNA, a protein known to up-regulate FA synthase and acetyl-coA carboxylase]. This does not necessarily increase FAO; however, for example, acetyl-coA carboxylase inhibits FAO, and malonyl-coA inhibits CPT-1, so overall excess in FA might eventually inhibit FAO (38, 39). This hypothesis is also supported by protection of SREBP1c KO from high-fat diet-associated tubulo-interstitial injuries (38).
In addition, excess of FA may impact epithelial cells independently from the FAO pathway. Albumin-bound FAs have been reported to activate PPAR-δ, dose-dependently, and alter mitochondrial function, leading to cytochrome c release and caspase 3 activation (40, 41), and in vivo, to enhance tubular inflammation via a pro-inflammatory metabolite (42). Soumura et al. also showed that palmitic acid treatment induces an up-regulation of the expression of monocyte chemo-attractant protein-1 (MCP-1) and leads to activation of two pro-inflammatory pathways thanks to phosphorylation of MAPKs (ERK, p38, and JNK) and IκB, thus promoting the nuclear translocation of NF-κB (43). Katsoulieris et al. not only corroborated these results but also demonstrated that palmitic acid overload in a renal PTC model induces endoplasmic reticulum (ER) stress (44). ER stress, activated by unfolded or misfolded proteins or protein trafficking, is a well-known phenomenon in the development and progression of kidney disease. In this case, palmitic acid can cause ER stress by H2O2 production and C/EBP homologous protein (CHOP) expression (44). Oxidative and ER stress, and apoptosis unite to create a pro-inflammatory state in the vicinity of renal PTC (45). Last, knocking-out the scavenger receptor CD36 results in a reduction of activated NF-κB and oxidative stress level (46) in mice subjected to unilateral ureteral obstruction and a high-fat diet. Overall, lipotoxicity probably exists and contributes to epithelial injury, either directly through the activation of an apoptotic signal or indirectly by promoting the influx of inflammatory cells, a major factor in fibrosis.
Nevertheless, there is some doubt regarding the importance of lipid overload per se. Although cell-specific overexpression of CD36 in tubular epithelial cells from mice leads to lipid accumulation by the age of 8 weeks, it is not sufficient to drive spontaneous renal fibrogenesis, and more importantly, it does not enhance the susceptibility to renal fibrosis in two different animal models (diabetic nephropathy and folic acid nephropathy) (14). Within epithelial cells, it is thus thought that peroxisomal/mitochondrial defects in energy production are more detrimental than the lipid accumulation in the cytoplasm.
Proximal tubular epithelial cell-specific PPAR-α overexpression in mice was found to be sufficient to maintain FAO and conferred protection against IRI (47). Agonists of PPAR-α have been proposed for therapeutic use to prevent cisplatin-induced AKI, free FA accumulation, and ischemia–reperfusion injury (30, 48, 49). The first class tested was fibrates, with mixed results. Bezafibrate displays a protective effect against apoptosis in a cellular model and attenuates intracellular free FA accumulation (48). Takahashi et al. showed in a high-fat diet model that pre-treatment with clofibrate at a low dose (but not at a high dose) protects against free FA toxicity (30). PPAR-α agonists administered 5 days before injury in a rat model of renal IRI were found to regulate acyl-coA oxidase at the transcriptional and protein levels and to attenuate tubular necrosis. In a bilateral ischemia rat model, it has also been reported that PPAR-α agonists, clofibrate, fenofibrate, and WY14643, reduce renal dysfunction and inflammation related to IRI (49, 50). Of note, in the case of fenofibrate, protective effect was lost in PPAR-α−/− (49, 51). The effect of these reno-protective mechanisms was partially elucidated by Tanaka and co-workers, who demonstrated in a high-fat diet mice model that fenofibrate not only enhances lipolysis by overexpressing CPT-1, acyl-coA oxidase, and medium-chain acyl-CoA dehydrogenase but also inhibits the expression of the pro-fibrotic factors plasminogen activator inhibitor-1 (PAI-1) and MCP-1 (52). In a hypertension rat model fed with a high-fat diet, fenofibrate treatment induced PPAR-α expression and decreased apoptosis (53). Despite these promising data, the efficacy of fibrates in the clinic is so far limited to the reduction of albuminuria in type 2 diabetic patients. However, this data should be interpreted with caution, since the primary endpoint of the study was not fulfilled. No additional benefit was demonstrated with Gemfibrozil in two different large cohort studies (54, 55). Importantly, however, a recent meta-analysis concluded that fibrates played a preventive role in cardiovascular events in CKD patients, and confirmed albuminuria reduction (56). The effects of fibrates on the progression rate of CKD are still unknown since no study has yet been designed for this primary outcome (57).
Another approach to increase FAO would be to facilitate the transport of FA into the mitochondrial matrix. A treatment combining carnitine and 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR), which indirectly activates CPT-1 through the adenosine monophosphate-activated protein kinase (AMPK), was found to improve renal function after IRI, suggesting that FAO can be artificially stimulated (58). Propionyl l-carnitine also seems to be a promising candidate in IRI lesion prevention, through refeeding the carnitine shuttle and indirectly the Krebs cycle (59).
Fatty acid oxidation is unsurprisingly shut down during oxygen deprivation, a major cause of AKI. The observation that FAO does not properly resume after the injury suggests that tubular epithelial cells have to cope with a sustained lack of energy (Figure 1B). This could be one reason among others why cells switch from a highly demanding epithelial phenotype, to a more economical, mesenchymal one, and why even a transient AKI increases the risk of developing CKD. Lipid accumulation resulting from FAO stunning probably also contributes indirectly to enhanced fibrogenesis, by promoting inflammation. It is reasonable to assume that unlocking the state of metabolic sideration of PTC would permit the rescue of an epithelial phenotype, and reversion to pro-fibrotic mesenchymal functions. Playing a major role in the regulation of FAO, the PPAR-α/PPARGC1A ensemble is a reasonable therapeutic target for the future.
NS wrote the draft and designed the figures. AH suggested the plan of the review and finalized the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Ms. Pauline Prévoté for the design of the Figures. AH is the recipient of an Interface contract with the French National Institute for Health and Medical Research (INSERM).
1. Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol (2005) 17(1):17–25. doi: 10.1681/ASN.2005070757
2. Nadasdy T, Laszik Z, Blick KE, Johnson DL, Burst-Singer K, Nast C, et al. Human acute tubular necrosis: a lectin and immunohistochemical study. Hum Pathol (1995) 26(2):230–9. doi:10.1016/0046-8177(95)90042-X
3. Rastaldi MP, Ferrario F, Giardino L, Dell’Antonio G, Grillo C, Grillo P, et al. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int (2002) 62(1):137–46. doi:10.1046/j.1523-1755.2002.00430.x
4. Xu-Dubois Y-C, Baugey E, Peltier J, Colombat M, Ouali N, Jouanneau C, et al. Epithelial phenotypic changes are associated with a tubular active fibrogenic process in human renal grafts. Hum Pathol (2013) 44(7):1251–61. doi:10.1016/j.humpath.2012.10.010
5. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119(6):1420–8. doi:10.1172/JCI39104
6. Galichon P, Finianos S, Hertig A. EMT-MET in renal disease: should we curb our enthusiasm? Cancer Lett (2013) 341(1):24–9. doi:10.1016/j.canlet.2013.04.018
7. Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest (2014) 124(6):2299–306. doi:10.1172/JCI72267
8. Humphreys BD, Lin S-L, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol (2010) 176(1):85–97. doi:10.2353/ajpath.2010.090517
9. Galichon P, Hertig A. Epithelial to mesenchymal transition as a biomarker in renal fibrosis: are we ready for the bedside? Fibrogenesis Tissue Repair (2011) 4:11. doi:10.1186/1755-1536-4-11
10. Venkatachalam MA, Bernard DB, Donohoe JF, Levinsky NG. Ischemic damage and repair in the rat proximal tubule: differences among the S1, S2, and S3 segments. Kidney Int (1978) 14(1):31–49. doi:10.1038/ki.1978.87
11. Shanley PF, Brezis M, Spokes K, Silva P, Epstein FH, Rosen S. Hypoxic injury in the proximal tubule of the isolated perfused rat kidney. Kidney Int (1986) 29(5):1021–32. doi:10.1038/ki.1986.102
12. Sugden MC, Holness MJ, Donald E, Lall H. Substrate interactions in the short- and long-term regulation of renal glucose oxidation. Metabolism (1999) 48(6):707–15. doi:10.1016/S0026-0495(99)90169-5
13. Meyer C, Nadkarni V, Stumvoll M, Gerich J. Human kidney free fatty acid and glucose uptake: evidence for a renal glucose-fatty acid cycle. Am J Physiol (1997) 273(3):E650–4.
14. Kang HM, Ahn SH, Choi P, Ko Y-A, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med (2014) 21(1):37–46. doi:10.1038/nm.3762
15. Lameire NH, Bagga A, Cruz D, De Maeseneer J, Endre Z, Kellum JA, et al. Acute kidney injury: an increasing global concern. Lancet (2013) 382(9887):170–9. doi:10.1016/S0140-6736(13)60647-9
16. Colvin RB. Chronic allograft nephropathy. N Engl J Med (2003) 349(24):2288–90. doi:10.1056/NEJMp038178
17. Bechtel W, McGoohan S, Zeisberg EM, Müller GA, Kalbacher H, Salant DJ, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med (2010) 16(5):544–50. doi:10.1038/nm.2135
18. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med (2010) 16(5):535–43; 1p following 143. doi:10.1038/nm.2144
19. Weidemann MJ, Krebs HA. The fuel of respiration of rat kidney cortex. Biochem J (1969) 112(2):149–66.
20. Iwao Y, Nakajou K, Nagai R, Kitamura K, Anraku M, Maruyama T, et al. CD36 is one of important receptors promoting renal tubular injury by advanced oxidation protein products. Am J Physiol Renal Physiol (2008) 295(6):F1871–80. doi:10.1152/ajprenal.00013.2008
21. Johnson ACM, Stahl A, Zager RA. Triglyceride accumulation in injured renal tubular cells: alterations in both synthetic and catabolic pathways. Kidney Int (2005) 67(6):2196–209. doi:10.1111/j.1523-1755.2005.00325.x
22. Zager RA, Johnson ACM, Hanson SY. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int (2005) 67(1):111–21. doi:10.1111/j.1523-1755.2005.00061.x
23. Ruidera E, Irazu CE, Rajagopalan PR, Orak JK, Fitts CT, Singh I. Fatty acid metabolism in renal ischemia. Lipids (1988) 23(9):882–4. doi:10.1007/BF02536209
24. Portilla D. Role of fatty acid beta-oxidation and calcium-independent phospholipase A2 in ischemic acute renal failure. Curr Opin Nephrol Hypertens (1999) 8(4):473–7. doi:10.1097/00041552-199907000-00012
25. Gulati S, Ainol L, Orak J, Singh AK, Singh I. Alterations of peroxisomal function in ischemia-reperfusion injury of rat kidney. Biochim Biophys Acta (1993) 1182(3):291–8. doi:10.1016/0925-4439(93)90071-8
26. Idrovo J-P, Yang W-L, Nicastro J, Coppa GF, Wang P. Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J Surg Res (2012) 177(1):157–64. doi:10.1016/j.jss.2012.05.053
27. Feldkamp T, Kribben A, Roeser NF, Senter RA, Weinberg JM. Accumulation of nonesterified fatty acids causes the sustained energetic deficit in kidney proximal tubules after hypoxia-reoxygenation. Am J Physiol Renal Physiol (2006) 290(2):F465–77. doi:10.1152/ajprenal.00305.2005
28. Lefebvre P. Sorting out the roles of PPAR in energy metabolism and vascular homeostasis. J Clin Invest (2006) 116(3):571–80. doi:10.1172/JCI27989
29. Portilla D, Dai G, Mcclure T, Bates L, Kurten R, Megyesi J, et al. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int (2002) 62(4):1208–18. doi:10.1111/j.1523-1755.2002.kid553.x
30. Takahashi K, Kamijo Y, Hora K, Hashimoto K, Higuchi M, Nakajima T, et al. Pretreatment by low-dose fibrates protects against acute free fatty acid-induced renal tubule toxicity by counteracting PPARα deterioration. Toxicol Appl Pharmacol (2011) 252(3):237–49. doi:10.1016/j.taap.2011.02.012
31. Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest (2011) 121(10):4003–14. doi:10.1172/JCI58662
32. Zhong X, Chung ACK, Chen H-Y, Meng X-M, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol (2011) 22(9):1668–81. doi:10.1681/ASN.2010111168
33. Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med (2012) 4(121):ra18–121. doi:10.1126/scitranslmed.3003205
34. Kato M. Reduction of fatty acid oxidation and responses to hypoxia correlate with the progression of de-differentiation in HCC. Mol Med Rep (2012) 7:1–6. doi:10.3892/mmr.2012.1201
35. Xu Y, Huang J, Xin W, Chen L, Zhao X, Lv Z, et al. Lipid accumulation is ahead of epithelial-to-mesenchymal transition and therapeutic intervention by acetyl-CoA carboxylase 2 silence in diabetic nephropathy. Metabolism (2014) 63(5):716–26. doi:10.1016/j.metabol.2014.02.010
36. Bobulescu IA. Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens (2010) 19(4):393–402. doi:10.1097/MNH.0b013e32833aa4ac
37. Wahba IM, Mak RH. Obesity and obesity-initiated metabolic syndrome: mechanistic links to chronic kidney disease. Clin J Am Soc Nephrol (2007) 2(3):550–62. doi:10.2215/CJN.04071206
38. Jiang T, Wang Z, Proctor G, Moskowitz S, Liebman SE, Rogers T, et al. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J Biol Chem (2005) 280(37):32317–25. doi:10.1074/jbc.M500801200
39. Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes (2006) 55(9):2502–9. doi:10.2337/db05-0603
40. Arici M. Stimulation of proximal tubular cell apoptosis by albumin-bound fatty acids mediated by peroxisome proliferator activated receptor-gamma. J Am Soc Nephrol (2003) 14(1):17–27. doi:10.1097/01.ASN.0000042167.66685.EA
41. Ruggiero C, Elks CM, Kruger C, Cleland E, Addison K, Noland RC, et al. Albumin-bound fatty acids but not albumin itself alter redox balance in tubular epithelial cells and induce a peroxide-mediated redox-sensitive apoptosis. Am J Physiol Renal Physiol (2014) 306(8):F896–906. doi:10.1152/ajprenal.00484.2013
42. Kees-Folts D, Sadow JL, Schreiner GF. Tubular catabolism of albumin is associated with the release of an inflammatory lipid. Kidney Int (1994) 45(6):1697–709. doi:10.1038/ki.1994.222
43. Soumura M, Kume S, Isshiki K, Takeda N, Araki S, Tanaka Y, et al. Oleate and eicosapentaenoic acid attenuate palmitate-induced inflammation and apoptosis in renal proximal tubular cell. Biochem Biophys Res Commun (2010) 402(2):265–71. doi:10.1016/j.bbrc.2010.10.012
44. Katsoulieris E, Mabley JG, Samai M, Sharpe MA, Green IC, Chatterjee PK. Lipotoxicity in renal proximal tubular cells: relationship between endoplasmic reticulum stress and oxidative stress pathways. Free Radic Biol Med (2010) 48(12):1654–62. doi:10.1016/j.freeradbiomed.2010.03.021
46. Okamura DM, Pennathur S, Pasichnyk K, Lopez-Guisa JM, Collins S, Febbraio M, et al. CD36 regulates oxidative stress and inflammation in hypercholesterolemic CKD. J Am Soc Nephrol (2009) 20(3):495–505. doi:10.1681/ASN.2008010009
47. Li S, Nagothu KK, Desai V, Lee T, Branham W, Moland C, et al. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-α in mice confers protection during acute kidney injury. Kidney Int (2009) 76(10):1049–62. doi:10.1038/ki.2009.330
48. Nagothu KK, Bhatt R, Kaushal GP, Portilla D. Fibrate prevents cisplatin-induced proximal tubule cell death. Kidney Int (2005) 68(6):2680–93. doi:10.1111/j.1523-1755.2005.00739.x
49. Sivarajah A, Chatterjee PK, Hattori Y, Brown PA, Stewart KN, Todorovic Z, et al. Agonists of peroxisome-proliferator activated receptor-alpha (clofibrate and WY14643) reduce renal ischemia/reperfusion injury in the rat. Med Sci Monit (2002) 8(12):BR532–9.
50. Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD. Etomoxir-induced PPARα-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol (2000) 278(4):F667–75.
51. Patel NSA, di Paola R, Mazzon E, Britti D, Thiemermann C, Cuzzocrea S. Peroxisome proliferator-activated receptor-alpha contributes to the resolution of inflammation after renal ischemia/reperfusion injury. J Pharmacol Exp Ther (2009) 328(2):635–43. doi:10.1124/jpet.108.146191
52. Tanaka Y, Kume S, Araki S, Isshiki K, Chin-Kanasaki M, Sakaguchi M, et al. Fenofibrate, a PPARα agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int (2011) 79(8):871–82. doi:10.1038/ki.2010.530
53. Chung HW, Lim JH, Kim MY, Shin SJ, Chung S, Choi BS, et al. High-fat diet-induced renal cell apoptosis and oxidative stress in spontaneously hypertensive rat are ameliorated by fenofibrate through the PPAR-FoxO3a-PGC-1 pathway. Nephrol Dial Transplant (2012) 27(6):2213–25. doi:10.1093/ndt/gfr613
54. Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet (2005) 366(9500):1849–61. doi:10.1016/S0140-6736(05)67667-2
55. Cases A, Coll E. Dyslipidemia and the progression of renal disease in chronic renal failure patients. Kidney Int (2005) 68(S99):S87–93. doi:10.1111/j.1523-1755.2005.09916.x
56. Hottelart C, el Esper N, Achard JM, Pruna A, Fournier A. [Fenofibrate increases blood creatinine, but does not change the glomerular filtration rate in patients with mild renal insufficiency]. Nephrologie (1999) 20(1):41–4.
57. Jun M, Zhu B, Tonelli M, Jardine MJ, Patel A, Neal B, et al. Effects of fibrates in kidney disease. J Am Coll Cardiol (2012) 60(20):2061–71. doi:10.1016/j.jacc.2012.07.049
58. Decleves A-E, Zolkipli Z, Satriano J, Wang L, Nakayama T, Rogac M, et al. Regulation of lipid accumulation by AMK-activated kinase in high fat diet-induced kidney injury. Kidney Int (2013) 85(3):611–23. doi:10.1038/ki.2013.462
Keywords: fatty acid oxidation, epithelium, fibroblasts, acute kidney injury, chronic kidney diseases, fibrosis
Citation: Simon N and Hertig A (2015) Alteration of fatty acid oxidation in tubular epithelial cells: from acute kidney injury to renal fibrogenesis. Front. Med. 2:52. doi: 10.3389/fmed.2015.00052
Received: 28 May 2015; Accepted: 17 July 2015;
Published: 05 August 2015
Edited by:
Rick Mutsaers, University of Groningen, NetherlandsReviewed by:
Francesco Trapasso, University Magna Græcia of Catanzaro, ItalyCopyright: © 2015 Simon and Hertig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre Hertig, Hôpital Tenon, Urgences Néphrologiques et Transplantation Rénale, Université Pierre et Marie Curie, 4 rue de la Chine, Paris 75020, France,YWxleGFuZHJlLmhlcnRpZ0B1cG1jLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.