 Hongyu Wang
Hongyu Wang Xiaoya Chang
Xiaoya Chang Dongping Chen
Dongping Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mech. Eng. , 27 August 2021
Sec. Engine and Automotive Engineering
Volume 7 - 2021 | https://doi.org/10.3389/fmech.2021.738354
This article is part of the Research Topic Particulate Matter Emissions from Conventional and Reformulated Fuel Combustion: Advances in Experiments and Simulations View all 9 articles
The surface reactivity of carbonaceous nanoparticles is revealed from the barrier height and reaction enthalpy of hydrogen abstraction reaction by H radicals computed at the M06-2X/6–311g(d,p)//B3LYP/6-311G(d,p) level of theory. Small polycyclic aromatic hydrocarbon (PAH) clusters are selected as the model system of carbonaceous nanoparticles. The PAHs considered are naphthalene, pyrene, coronene, ovalene and circumcoronene. Cluster sizes range from dimer to tetramer with a parallel or crossed configuration. All results show similar values as that of monomers, but naphthalene dimers with a crossed configuration yield a lower barrier height and reaction enthalpy by ∼2 kcal/mol. A minor size dependence is noticed in the series of naphthalene clusters where a larger cluster exhibits a smaller barrier height. Larger homogeneous PAH clusters in a size range of 1.1–1.9 nm are later generated to mimic nascent soot surface. It is found that the barrier height decreases with the increase in particle size, and the averaged values are ∼2 kcal/mol lower than that of monomers. More importantly, a larger particle shows a wider spread in barrier heights, and low barrier heights are seen in the surface shallow regions (e.g., surface pockets). The lowest barrier height of ∼8.5 kcal/mol is observed at a C-H site locating in a surface pocket. A set of model systems are built to reveal the underlying mechanism of reduction in barrier height. It is shown that the reduction is caused by local interactions between the neighboring atoms and the local curvature. Further analysis on the average localized ionization potential shows that larger particles have higher reactivity, further supporting our findings from the barrier height of hydrogen abstraction reactions. Therefore, it is concluded that the surface reactivity depends on the particle size and the most reactive sites always locate at the surface pockets.
Surface reaction is a key process in the mass growth of soot formation (Frenklach and Wang, 1991; Frenklach, 1996; Appel et al., 2000; Wang et al., 2015). For instance, the predicted particle size distributions in a stretch-stabilized ethylene flames are ultra-sensitive to the parameters in surface reactions (Camacho et al., 2017). Besides mass growth, surface reactions also account for soot oxidation (Camacho et al., 2015), which relates to the mitigation of soot emission in practical combustors. The oxidative reactivity of soot particles formed from conventional and reformulated fuels has complex impacts on the fuel consumption, engine performance, lubricant oil degradation in the road vehicles (Sharma et al., 2016). Recently, Lapuerta and the coworkers (Lapuerta et al., 2020) reviewed the soot reactivity on diesel filter regeneration, suggesting that surface properties, i.e., surface area, solid-gas reactions and soot porosity, determine the regeneration process.
In soot models, surface reactions were usually treated as the analogy of gas-phase reactions, i.e., a reaction of polycyclic aromatic hydrocarbon (PAH) and radicals. However, not all soot surface can react with radicals at flame conditions, a steric parameter α was therefore proposed by Frenklach and Wang (1991) to account for the probability of successful collisions on the reactive sites. In the following works, parameter α was refitted against a wide range of experimental results, such as soot volume fraction (Frenklach and Wang, 1991; Dworkin et al., 2011; Khosousi and Dworkin, 2015), but none of the works agree with each other. Recently, we extracted the parameter α from microscopic representations of nascent soot using molecular dynamics method (Chen and Luo, 2020), and our work demonstrated that large uncertainties exist in the parameters of surface reactions, such as the steric parameter α and the active number density of active sites χi. The impact of the uncertainties on reaction rates are equivalent to the reduction of the energy barrier of surface reactions by 7.2 kcal/mol at 2000 K. It is worth noting that the barrier heights in surface reactions is never directly estimated and it is always assumed to be the same as the analogous gas-phase reaction in the last three decades.
The key pathway of mass growth in surface reaction is governed by the well-known HACA mechanism (Appel et al., 2000), where the Arrhenius parameters of hydrogen abstraction and C2H2 addition reactions are directly taken from the gas-phase reaction. The barrier heights (ΔE0) and reaction enthalpies (ΔH0) of hydrogen abstraction from planar PAHs with H radicals (Hou and You, 2017) are well benchmarked using the CCSD(T)/CBS level of theory. In particular, the ΔE0 and ΔH0 for benzene is 15.6 and 7.9 kcal/mol, respectively. Also, hydrogen abstraction reactions on aromatics exhibit a minor site dependence (Liu P. et al., 2019). In the formulation of ABF model (Appel et al., 2000), the reactive C-H site on particle surface locates at the aromatic bay. From density functional theory (DFT) calculations, C-H bonds locating at the aromatic bay exhibit a repulsion from the neighboring atoms, and the bond dissociation energy is typically reduced by 1–3 kcal/mol (Cioslowski et al., 1996). Therefore, Appel et al. (2000) reduced the activation energy of H abstraction reaction on particle surface by 3 kcal/mol to account for the above effect.
The potential energy surface (PES) of soot particle significantly differs from PAH molecules, which should impact the surface reactivity. The soot TEM images (Wan et al., 2018; Pfau et al., 2020) showed that the surface is made of various defects. In the catalysis community, it is well known that the crystal phase impacts the catalytic activity. Liu et al. (2017) reviewed the effects of crystal phase in Fischer-Tropsch catalysts, for example, cobalt, ruthenium, iron, iron carbides and nickel nanoparticles; the catalytic capacities show a clear dependence on the crystal phase. Similarly, in the low-temperature CO oxidation (Li et al., 2014), the catalytic activities of anatase is much higher than that of rutile in the tested conditions. Also, surface defects (e.g., kinks and steps) on the catalyst surface could provide active sites for surface reactions to enhance reactivity and even selectivity (Chen et al., 2019; Kopač et al., 2019). Although soot is a type of amorphous material and does not show a well-defined crystal structure, the knowledge derived from catalysis community makes us to question whether complex surface (local textural arrangements) of nascent soot impacts the reaction kinetics. This issue would justify the reduction in the surface reactions in the ABF model. Besides, surface reactions for mass growth always occur on the boundary of particles (Grančič et al., 2016) due to the geometric effect. By contrast, soot particles might undergo internal oxidation reactions under oxygen-rich environments (Sediako et al., 2017; Macián et al., 2019), and the complex potential energy surface inside a particle must impact the energy barrier to an extent. Nevertheless, the energy barrier of surface reaction should be lower than that occurring in the gas-phase reaction due to interactions from the neighboring components.
The development of soot molecular representations started from the homogeneous clusters of planar PAHs up to ∼8 nm in diameter (Chen et al., 2014; Chen et al., 2015b). Bowal et al. (2019a) extended the previous attempt and successfully created heterogenous clusters of planar PAHs, for example, a cluster with 50 circumcoronene and 50 coronene. Later, clusters with curved PAHs, i.e., corannulene, were generated from molecular dynamic simulations (Bowal et al., 2019b). Recently, heterogeneous clusters with cross-linked PAHs (Pascazio et al., 2019) were available using reactive molecular dynamic simulations. The applications of these model systems of soot can be seen in the solid-liquid morphology transition (Chen et al., 2014; Chen et al., 2015b), surface reactivity (Chen and Luo, 2020) and also optical properties (Liu C. et al., 2019; Chen and Wang, 2019b). In this work, we explore the energy barriers of hydrogen abstraction reactions from a set of model systems of soot by H radicals. The clusters with planar PAHs are selected as the model system to mimic the complex soot surface. The DFT method is used to compute the energy barriers of small PAH clusters with five PAHs, i.e., naphthalene, pyrene, coronene, ovalene and circumcoronene. A few PAH clusters with up to 15 monomers are generated to represent nascent soot, and their energy barriers for hydrogen abstraction reactions are estimated using a frozen algorithm. The size dependence of energy barriers is then discussed, and the location with reduced barrier heights are further examined in detail. Also, a set of model systems are built to reveal the physics behind the reductions in barrier heights. Last but not the least, the analysis of average local ionization potential is carried out to support our findings.
The dimer, trimer and tetramer of naphthalene and pyrene together with the dimers of coronene are generated from geometry optimizations at the B3LYP/6-311G(d,p) level of theory with an empirical dispersion correction (Grimme et al., 2010). The dimers of ovalene and circumcoronene are optimized at the B3LYP/6-31G(d) level of theory to reduce the computational cost. All optimized geometries are confirmed by analyzing vibrational frequencies, and the ZPE vibrational energy is scaled by 0.977 (Andersson and Uvdal, 2005). The optimized configurations of naphthalene, pyrene and coronene clusters from dimer to tetramer are shown in Figure 1, and configurations of other PAH clusters are included in the supplemental material (Supplementary Figure S1). The layer separation lies in the range of 3.3 Å and 3.5 Å depending on the size of monomers.
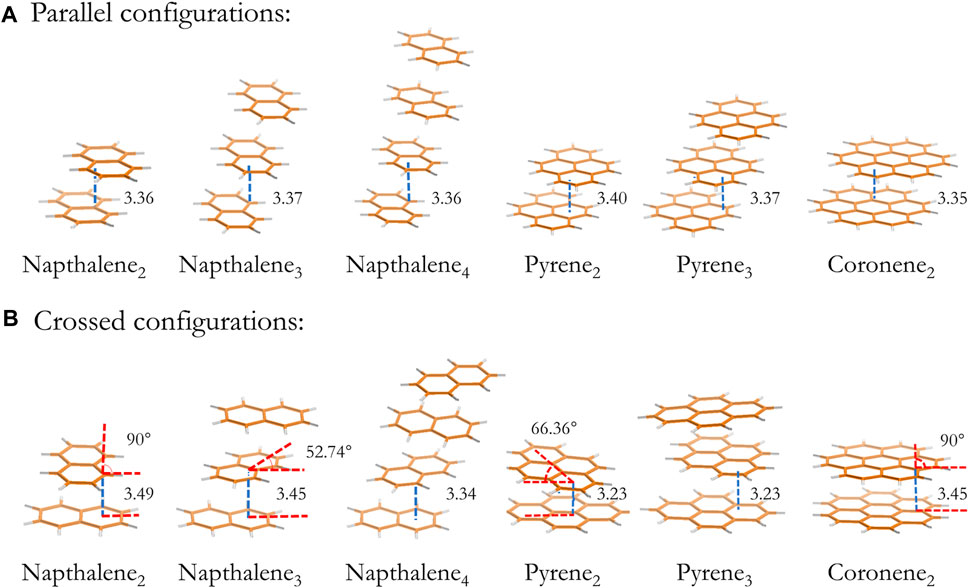
FIGURE 1. Optimized configurations of naphthalene clusters. The size of cluster from left to right is dimer to tetramer. The upper and lower panel presents a local minimum with a parallel (A) and a crossed (B) configuration, respectively. The layer separation and the rotation angle between planes are indicated on the figure.
Considering the multiple sites of surface reactions, an in-house python code is applied to generate initial configurations for both transition state and product optimization at each potential site for hydrogen abstraction reactions automatically. In particular, surface atoms are identified using the solvent-excluded surface with a probe size as 2.0 Å (Chen et al., 2015a; Chen and Luo, 2020). The barrier heights and reaction enthalpies (0 K) for hydrogen abstraction reactions are derived from single-point energy calculations at the M06-2X/6-311G(d,p) level of theory with an empirical dispersion correction (Grimme et al., 2010). The accuracy of our method was evaluated in a previous work (Hou and You, 2017), and the uncertainties compared to the CCSD(T)/CBS level of theory were less than 1 kcal/mol for the barrier heights and reaction enthalpies (0 K) of H abstraction reaction on the benzene and naphthalene. Gaussian 09 program is used to carry out the DFT calculations (Frisch et al., 2009).
The multi-configurational character of PAH clusters is investigated by considering T1 diagnostics at the CCSD(T)/cc-pVDZ level of theory. In particular, a naphthalene trimer with a parallel configuration is selected to react with H radical. All of the T1 diagnostics for the reactant, the product and the transition state are less than 0.02, which supports our application of the single-reference method for single-point energy calculations (Yu and Truhlar, 2014).
Besides the optimized structures, we generate six larger PAH clusters to mimic the complex surface of carbonaceous nanoparticles, including naphthalene10, naphthalene15, pyrene5, pyrene10, coroene5, and coronene10 clusters. The corresponding particle diameter is 1.47, 1.71, 1.31, 1.69, 1.46, and 1.87 nm, respectively. These clusters are equilibrated using MD simulations above the melting points of each configuration (Chen et al., 2014; Chen et al., 2015b), and the selected temperatures lie in the range of 500–1000 K. The potential functions for C-C, C-H and H-H interactions are described by the isoPAHAP potential (Totton et al., 2012). Note the configurations taken from MD trajectories correspond to a transient structure rather than a locally preferred structure. The morphology of these clusters (Figure 2) is consistent with the liquid-like structures in previous works (Chen et al., 2014; Chen et al., 2015b). An additional configuration (e.g., naphthalene15,2) is also included, and its results agree with those of naphthalene15 (see Supplementary Figure S2).
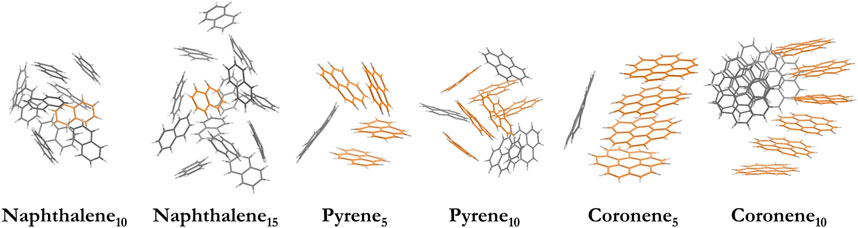
FIGURE 2. Model systems of nascent soot, including naphthalene10, naphthalene15, pyrene5, pyrene10, coroene5, and coronene10 clusters. The orange stack highlights the maximum stacks in the configurations.
The saddle point for hydrogen abstraction reactions on the models of nascent soot (Figure 2) is investigated by considering only the H radical and the substituted hydrogen atom during the optimization process, while other atoms are frozen to reduce the computational costs by at least one order of magnitude. The computational error of the current frozen scheme is examined using a naphthalene tetramer, a pyrene trimer and a coronene dimer with a parallel configuration (Table 1). The full optimized transition state calculations at the M06-2X/6-311G(d,p) level of theory are treated as the benchmark to evaluate the uncertainty in our frozen algorithm. The selected frozen scheme overestimates the barrier height by ∼2.50 kcal/mol on average, which is mainly due to the zero-point energy (ZPE) correction (1.75 kcal/mol). Although the errors in the frozen scheme can be reduced to the level of ZPE corrections by applying a distance threshold of 3 Å to include more atoms in the optimization, it is still not recommended because the searching algorithm of transition states would pull back the neighbouring hydrogen atoms to the equilibrated position of C-H bonds and significantly reduce the overall barrier heights. Note that the clusters in this work are snapshots sampled at high temperatures, and the neighbouring hydrogen atoms in C-H bonds might sit far away from the equilibrated position. It is evident that the frozen scheme with 3 Å as the critical criteria yields unphysical barrier heights as 3.5 kcal/mol for the napthalene15 cluster (Supplementary Figure S3).

TABLE 1. The errors in energy barriers of transition states using different frozen schemes and the respective ZPE corrections.a
We report DFT calculations of barrier heights and reaction enthalpies (0 K) for hydrogen abstraction reactions at all potential sites of small naphthalene, pyrene, coronene, ovalene and circumcoronene clusters as shown in Figure 3. The results show that the calculated ΔE0 are very close to that of monomers (e.g., 16.5 kcal/mol of benzene), and the dimer data falls in the similar range as a previous work (Hou and You, 2017). A naphthalene dimer with a crossed configuration yields a smaller value as 14.6 kcal/mol, while the ΔE0 of its counterpart with a parallel configuration is 15.9 kcal/mol. By contrast, the crossed configuration of coronene dimers have a higher ΔE0 than the parallel configuration. Other cases show no differences considering both isomers. The obvious difference between reaction barriers of crossed and parallel naphthalene dimers can be attributed the significant geometry change in the transition state (see the configurations in Figure 3). For a naphthalene dimer with a crossed configuration, it rotates along the central plane by ∼30° to access the transition state. However, no such rotation is seen in the trimer and tetramer. One interesting trend should be emphasized here; ΔE0 is weakly dependent on the particle size. As the size grows from dimer to tetramer of naphthalene, a reduced ΔE0 is observed, and pyrene cases follow this trend. Although the reduction is minor (∼1 kcal/mol), this raises a question whether this dependence can be extrapolated to larger sizes.
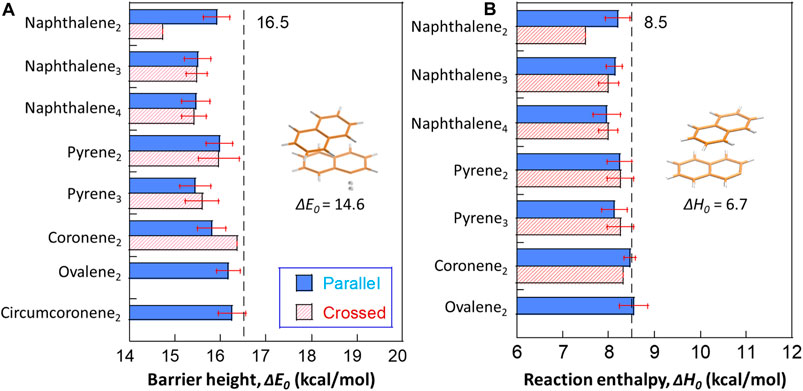
FIGURE 3. The barrier heights (A) and reaction energies (B) for hydrogen abstraction from small PAH clusters by H radicals. The transition state of a naphthalene dimer with the lowest barrier height among all optimized configurations is highlighted in Figure 3A. The structure showing the least reaction enthalpy for H abstraction reaction is included in Figure 3B. The dashed lines refer to the average barrier heights and reaction energies of optimized monomers at the M06-2X/6-311G(d,p) level of theory as 16.5 and 8.5 kcal/mol, respectively. The red error bars refer to one standard deviation from the results on different sites within the particular case.
For reaction enthalpies (0 K), most of the computed values are close to the values of monomers (e.g., 8.5 kcal/mol). Again, naphthalene dimers with a crossed configuration exhibit the lowest reaction enthalpy as 6.7 kcal/mol. It was found in previous works (Cioslowski et al., 1996; Hou and You, 2017) that the hydrogens locating at the bay site exhibit repulsive forces weakening the C-H bonds by 1–3 kcal/mol. However, the reduction in naphthalene dimers with a crossed configuration does not share the same physical reason. In our case, the layer separation is 3.49 Å (Figure 1), falling in the weak attraction region, and thus the neighboring hydrogens do not experience repulsions. Instead, an obvious intermolecular deformation is seen in the product of abstraction reaction, and this preferred complex yields a reduced energy resulting the reduction in the overall ΔH0. Also, no size dependence is observed in reaction enthalpies.
Six larger PAH clusters with sizes from 1.3 to 1.9 nm in diameter are further selected to mimic the surface of nascent soot (Figure 2). The reported energy barriers are deducted by 2.5 kcal/mol as a correction taken from the frozen algorithm. The average ΔE0 of all six cases fall in the range of 14.4–15.1 kcal/mol, and again, it is evident that the ΔE0 of the abstraction reaction shows a minor size dependence. A larger particle has a wider spread in the ΔE0. For example, the sigma value of the NAP15 cluster is roughly doubled compared to the smaller counterpart. The magnitude of spread correlates with surface complexity, and a larger surface usually exhibits more textual arrangements. It is found that only ∼10% of all calculated cases have a ΔE0 higher than 16.5 kcal/mol, suggesting that the surface reactivity of soot always higher than that of monomers. Also highlighted in Figure 4B is the spatial distribution of ΔE0 on each cluster. We classify the data into two cases for discussion, e.g., ΔE0>13.5 kcal/mol and ΔE0 < 13.5 kcal/mol. It is shown that most of the edge hydrogens on stacks fall in the former cases, and their reactivity is close to the monomer. At some surface shallow regions (e.g., pockets (Chen et al., 2015a)), the ΔE0 is reduced by at least 3 kcal/mol, which cannot be explained by the molecular repulsion in monomers (Cioslowski et al., 1996; Hou and You, 2017). In the original ABF model (Appel et al., 2000), a reduction of 3 kcal/mol is applied to account for the effect of molecular repulsion. Our results show that the ΔE0 of abstraction reactions at the surface pockets would require further reduction. For example, the lowest ΔE0 seen in all cases is ∼9.0 kcal/mol (about the half of the barrier seen in monomers). Together with the weak dependence on the particle size, we would expect a lower ΔE0 if extrapolating the particle size to larger values. However, the selected clusters in this work are smaller than 2 nm in diameter. It is still questionable whether the weak dependence of surface reactivity on the particle size can be extracted to larger soot particles. This size dependence of surface reactivity still requires future investigations.
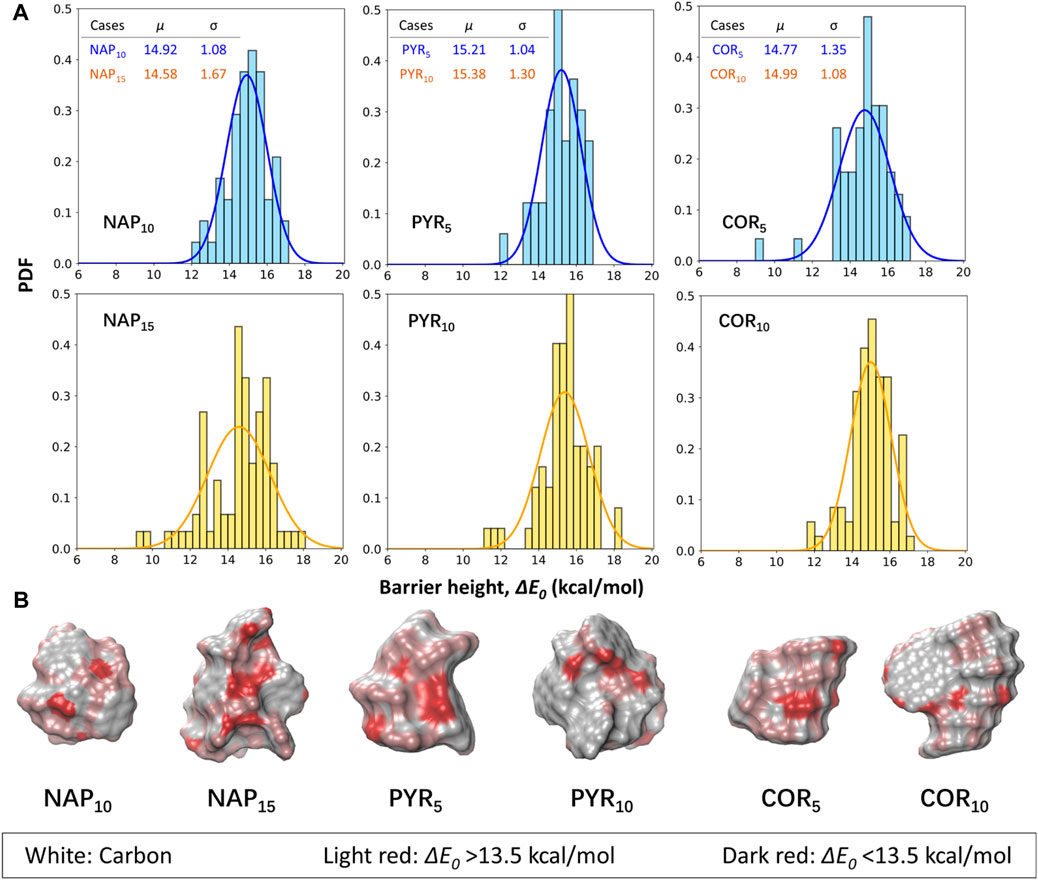
FIGURE 4. (A) The barrier heights for hydrogen abstractions from selected naphthalene (NAP), pyrene (PYR) and coronene (COR) clusters by H radicals. The blue bars represent the results of the smaller clusters, while the larger ones are illustrated by yellow bars. A correction of 2.5 kcal/mol is included to account for the errors in the frozen algorithm. μ and σ represent the mean and standard deviation of the fitted gaussian distributions. (B) The spatial distributions of barrier heights on each surface hydrogen atoms. The white, light red and dark red refer to surface carbons, surface hydrogens with ΔE0 > 13.5 kcal/mol, and surface hydrogens with ΔE0 < 13.5 kcal/mol, respectively.
Although a Gaussian distribution is fitted in Figure 4, we do not argue that the underlying distribution must be Gaussian-like. Instead, obvious failure can be noticed from both the cases of pyrene and coronene clusters. The fitted distributions here serve as the guidance to quantitively compare the different cases. Figure 5 includes all data from the six simplified soot models. The reported barrier height is 14.96 ± 1.32 kcal/mol. In all data, the number of cases with a barrier height lower than 13.5 kcal/mol is 12.15%. The detailed distributions of ΔE0 for each case are included in Supplementary Figure S4. Most of the cases are well covered by the treatment in original ABF model (Appel et al., 2000), however, the importance of these low-value barrier height remains uncertain for future reference.
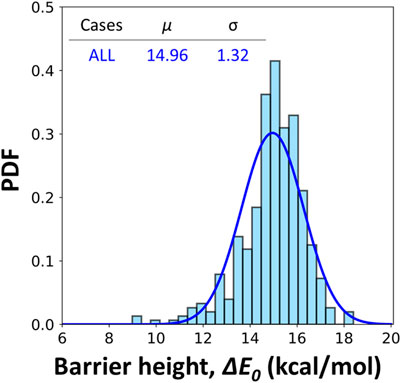
FIGURE 5. Fitted gaussian distribution of barrier heights for hydrogen abstractions by H radicals including NAP10, NAP15, PYR5, PYR10, COR5 and COR10 clusters. A correction of 2.5 kcal/mol is included to account for the errors in the frozen algorithm. μ and σ represents the mean and standard deviation of the fitted distributions.
We then take a step further to investigate the reduction in ΔE0. Model systems of COR2NAP1 clusters are generated with three different arrangements (Figure 6): 1) “perfect parallel”, 2) a stack of three PAHs with the angle between the upper coronene and naphthalene as 15° (termed as the “outward”), 3) a stack of three PAHs with the angle between the upper coronene and naphthalene as 15° (termed as the “inward”). In all cases, the layer separation between planes is kept as 3.3 Å and the lower coronene is the mirror image of the upper one. The frozen algorithm with a distance threshold of 3 Å is again used to calculate the barrier heights of COR2NAP1 clusters. It is found that the reduction of barrier height depends on the relative distance between the naphthalene and the other coronene molecules. A reduction in the barrier height is observed when moving naphthalene inward in all three cases. The local curvature is also an important factor; the “inward” cases yield a reduction of ∼4 kcal/mol, while the maximum reduction in the “outward” is ∼0.5 kcal/mol. In the “inward” cases, the distance between atoms in coronene and the abstracted site could be as low as 2.6 Å, which is much smaller than that of other cases. At such conditions, the abstracted site exhibits a strong repulsive force from neighboring species, and this corresponds to the bay site (Cioslowski et al., 1996; Hou and You, 2017) causing a reduction in barrier height. However, both “perfect parallel” and “outward” cases lie in the attraction regions, and the repulsion mechanism cannot explain the moderate reductions in barrier height. This phenomenon may relate to the delocalization of pi electrons which weakens the C-H bonds or the formation of weak hydrogen bonds (<4 kcal/mol). Although this set of model systems does not yield a large reduction as ∼8 kcal/mol, it reveals the fundamental reason in the reductions of barrier height. Considering larger particles with more complex surface, we expect to see a wide range of possible configurations, which might be the analogies of the simplified cases.
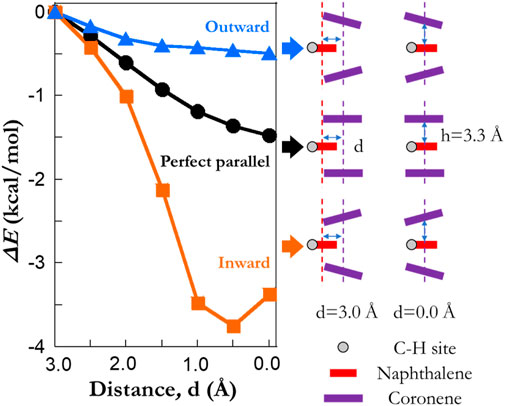
FIGURE 6. The impact of local textural arrangements on the barrier heights of hydrogen abstraction reactions by a H radical. A model system with two coronene molecules and one naphthalene is considered. The blue, black and orange lines refer to the results of a “outward”, a “perfect parallel” and an “inward” configuration, respectively. The rotation angle for both “outward” and “inward” cases is 15 degree. “C-H site” highlights the abstracted hydrogen atoms from model systems. Note that the results are extracted using the frozen scheme.
Average local ionization energy (ALIE) (Sjoberg et al., 1990) describes the site reactivity of electrophilic or radical attack on aromatic species. Here, we include the ALIE analysis to examine the surface reactivity of our model systems. The ALIE is computed as
where
The location of the minimum ALIE is first discussed as it always refers to the most reactive sites. Figure 7 shows the spatial distribution of ALIE values within NAP and PYR clusters. The ALIE values are projected onto the iso-surface of electron density as 0.001 Å−3. For the NAP monomer, the minimum ALIE locates on the top and bottom of the pi-plane, and the alpha site exhibits a slight higher reactivity, which is consistent with a previous work (Hou and You, 2017). Further increasing the PAH size to a tetramer, we see a minor decrease in the minimum ALIE. Also, it is noted that the parallel and crossed isomers share almost the same minimum ALIE, and the difference is within 0.03 eV. The minimum ALIE of NAP10 and NAP15 clusters is 7.40 and 7.24 eV, respectively. According to a recent analysis of various aromatic species using ALIE (Martin et al., 2019), the most reactive sites in both NAP10 and NAP15 clusters shares similar chemical reactivity as localized π-radicals, which can cause potential crosslinking reactions. For PYR clusters, the changes in the minimum ALIE follow the same trend, and the value is reduced from 7.8 to 7.4 eV considering a PYR10 cluster. In both cases, the minimum ALIE always locates at the surface pockets highlighting the enhanced reactivity within pockets. This is consistent with the indications from ΔE0 (Figure 6). Furthermore, it is interesting to note that the minimum ALIE of particles have a size dependence and a larger particle shows a lower minimum value. Therefore, it is evident that the surface reactivity of nascent soot is proportional to the particle size.
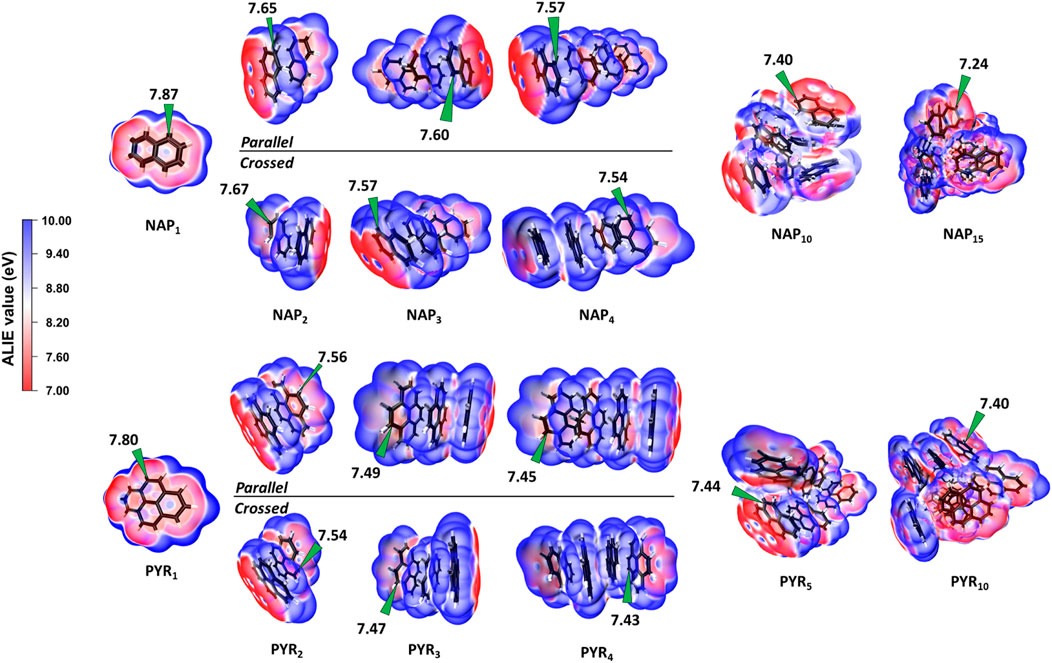
FIGURE 7. Local ionization energy of NAP and PYR clusters from monomer up to 15 components calculated at the B97D/6-311G(d,p) level of theory. The minimums of ALIE values are highlighted on the plot. The both parallel and crossed configurations are considered for dimers, trimers and tetramers.
Figure 8 presents the correlation between ALIE values and surface area of each atom including both hydrogen and carbon atoms. The surface area is calculated from the iso-surface of electron density as 0.001 Å−3 (Figure 7) (Bader et al., 1987). The ALIE values cluster into two groups in general (Figure 8); one group locates in the range of 0–5 Å2, while the other group clusters around 15 Å2. From the magnitude of surface area, we can classify the former group as the atoms in the surface pocket resulting the limited exposure. By contrast, the latter one refers to a group of bulk atoms, e.g., non-edge carbon in a PAH molecule. The atoms in the surface pocket show reduced ALIE values, indicating that the reactivity is higher than the bulk atoms. This finding supports our statement made from the analysis of reaction kinetics (Figure 6).
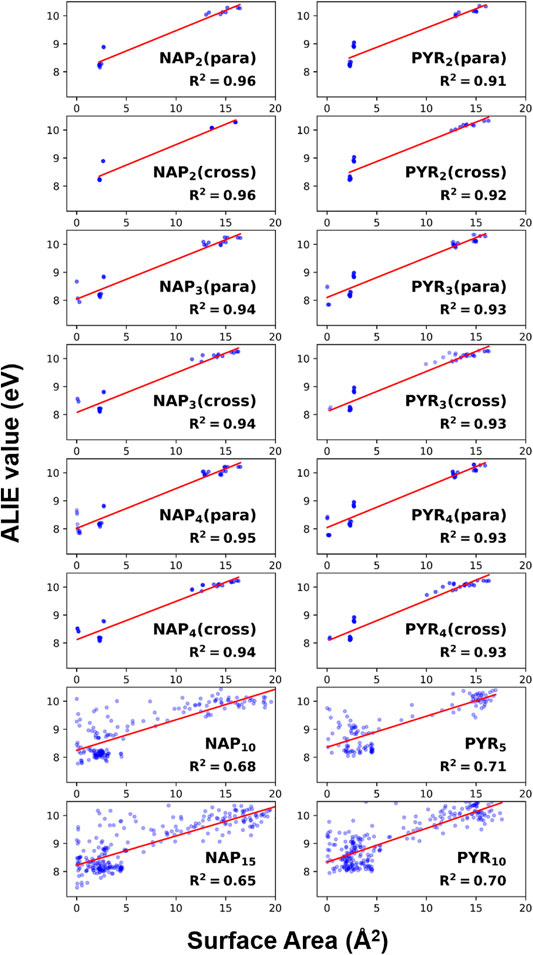
FIGURE 8. The correlation of ALIE values with surface area of each atom. The red lines represent linear fittings, and the fitting quality is illustrated by R2 shown in the plots.
Besides, the reactivity of aromatics can be interpreted using HOMO–LUMO gap (Randić, 2003; Chen and Wang, 2019a). A lower HOMO–LUMO gap implies a higher reactivity. As we have known that HOMO-LUMO gaps of homogeneous clusters decrease with the growth in the particle size, which can be well captured by the quantum confinement effect (Liu C. et al., 2019; Chen and Wang, 2019b). As a result, the indications from HOMO-LUMO gaps further support our finding in both ΔE0 and ALIE that larger nascent soot particles have a higher surface reactivity.
Homogeneous clusters are used here as an example to mimic the nascent soot surface. It is well known that nascent soot has a complex molecular composition. For example, recent works reveal that PAH species in nascent soot carry functional groups including aliphatic and oxygenated groups using high resolution AFM (Commodo et al., 2019; Schulz et al., 2019). We expect that functional groups should have a minor impact on the surface reactivity owing to their weak interactions with radicals. Our finding here can be extended to explain the reactivity change in soot aging (Kholghy et al., 2016), where nascent soot has more irregular disordered surface pockets and thus its reactivity prevails. It is revealed that the most reactive sites locates in surface pockets, but the dynamics nature of pockets (Chen et al., 2015a) would impact reaction coordinates for surface reactions. However, this effect requires further investigations to establish a dynamic picture of surface reactions.
We compute the barrier height and reaction enthalpy of hydrogen abstraction reaction on small naphthalene, pyrene, coronene, ovalene and circumcoronene clusters by H radicals. Both clusters with a parallel and crossed configuration shows similar results as that of monomers, except naphthalene dimers with a crossed configuration having a reduced barrier height and reaction enthalpy. Moreover, it is shown that the barrier height is weakly proportional to the particle. Further calculations on larger PAH clusters (e.g., 1.1–1.9 nm) providing extra evidence to support the size dependence. In addition, surface sallow regions exhibit the lowest barrier height of hydrogen abstraction reaction, and the lowest value is ∼8.5 kcal/mol, which is about the half of PAH monomers.
A set of model system is then generated to investigate the fundamental reason behind the reduction in barrier height for larger particles. It is found that the reduction in barrier height is attributed to the intermolecular interactions between neighboring atoms and the local curvature. Further ALIE analysis on NAP10 and NAP15 clusters supports the observations of size dependence in surface reactivity, where larger particles show a lower minimum ALIE value, indicating higher reactivity. Besides, it is shown that the minimum ALIE always locates in the surface pockets, in consistent with the results from barrier height. Therefore, it is concluded that the surface reactivity exhibits a size dependence, which is not previously noticed.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
HW prepared the data in the manuscript and performed the detailed analysis, CY prepared several sets of data. DC wrote the manuscript and designed the project.
This paper is supported by the project of State Key Laboratory of Explosion Science and Technology (Beijing Institute of Technology). The project number is ZDKT21-0. DC also acknowledges the support by the National Natural Science Foundation of China (No. 21961122007 and 51806016).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmech.2021.738354/full#supplementary-material
Andersson, M. P., and Uvdal, P. (2005). New Scale Factors for Harmonic Vibrational Frequencies Using the B3LYP Density Functional Method with the Triple-ζ Basis Set 6-311+G (D, P). The J. Phys. Chem. A 109 (12), 2937–2941. doi:10.1021/jp045733a
Appel, J., Bockhorn, H., and Frenklach, M. (2000). Kinetic Modeling of Soot Formation with Detailed Chemistry and Physics: Laminar Premixed Flames of C2 Hydrocarbons. Combustion and Flame 121 (1), 122–136. doi:10.1016/S0010-2180(99)00135-2
Bader, R. F. W., Carroll, M. T., Cheeseman, J. R., and Chang, C. (1987). Properties of Atoms in Molecules: Atomic Volumes. J. Am. Chem. Soc. 109 (26), 7968–7979. doi:10.1021/ja00260a006
Bowal, K., Martin, J. W., Misquitta, A. J., and Kraft, M. (2019b). Ion-Induced Soot Nucleation Using a New Potential for Curved Aromatics. Combustion Sci. Tech. 1, 1–19. doi:10.1080/00102202.2019.1565496
Bowal, K., Martin, J. W., and Kraft, M. (2019a). Partitioning of Polycyclic Aromatic Hydrocarbons in Heterogeneous Clusters. Carbon 143, 247–256. doi:10.1016/j.carbon.2018.11.004
Camacho, J., Singh, A. V., Wang, W., Shan, R., Yapp, E. K. Y., Chen, D., et al. (2017). Soot Particle Size Distributions in Premixed Stretch-Stabilized Flat Ethylene-Oxygen-Argon Flames. Proc. Combustion Inst. 36 (1), 1001–1009. doi:10.1016/j.proci.2016.06.170
Camacho, J., Tao, Y., and Wang, H. (2015). Kinetics of Nascent Soot Oxidation by Molecular Oxygen in a Flow Reactor. Proc. Combustion Inst. 35 (2), 1887–1894. doi:10.1016/j.proci.2014.05.095
Chen, D., Akroyd, J., Mosbach, S., and Kraft, M. (2015a). Surface Reactivity of Polycyclic Aromatic Hydrocarbon Clusters. Proc. Combustion Inst. 35 (2), 1811–1818. doi:10.1016/j.proci.2014.06.140
Chen, D., Akroyd, J., Mosbach, S., Opalka, D., and Kraft, M. (2015b). Solid-liquid Transitions in Homogenous Ovalene, Hexabenzocoronene and Circumcoronene Clusters: A Molecular Dynamics Study. Combustion and Flame 162 (2), 486–495. doi:10.1016/j.combustflame.2014.07.025
Chen, D., and Luo, K. H. (2020). Reactive Sites on the Surface of Polycyclic Aromatic Hydrocarbon Clusters: A Numerical Study. Combustion and Flame 211, 362–373. doi:10.1016/j.combustflame.2019.09.034
Chen, D., Totton, T. S., Akroyd, J. W. J., Mosbach, S., and Kraft, M. (2014). Size-dependent Melting of Polycyclic Aromatic Hydrocarbon Nano-Clusters: A Molecular Dynamics Study. Carbon 67, 79–91. doi:10.1016/j.carbon.2013.09.058
Chen, D., and Wang, H. (2019a). HOMO-LUMO Energy Splitting in Polycyclic Aromatic Hydrocarbons and Their Derivatives. Proc. Combustion Inst. 37 (1), 953–959. doi:10.1016/j.proci.2018.06.120
Chen, D., and Wang, H. (2019b). HOMO-LUMO Gaps of Homogeneous Polycyclic Aromatic Hydrocarbon Clusters. J. Phys. Chem. C 123 (45), 27785–27793. doi:10.1021/acs.jpcc.9b08300
Chen, S., Xiong, F., and Huang, W. (2019). Surface Chemistry and Catalysis of Oxide Model Catalysts from Single Crystals to Nanocrystals. Surf. Sci. Rep. 74 (4), 100471. doi:10.1016/j.surfrep.2019.100471
Cioslowski, J., Liu, G., Martinov, M., Piskorz, P., and Moncrieff, D. (1996). Energetics and Site Specificity of the Homolytic C−H Bond Cleavage in Benzenoid Hydrocarbons: An Ab Initio Electronic Structure Study. J. Am. Chem. Soc. 118 (22), 5261–5264. doi:10.1021/ja9600439
Commodo, M., Kaiser, K., De Falco, G., Minutolo, P., Schulz, F., D'Anna, A., et al. (2019). On the Early Stages of Soot Formation: Molecular Structure Elucidation by High-Resolution Atomic Force Microscopy. Combustion and Flame 205, 154–164. doi:10.1016/j.combustflame.2019.03.042
Dworkin, S. B., Zhang, Q., Thomson, M. J., Slavinskaya, N. A., and Riedel, U. (2011). Application of an Enhanced PAH Growth Model to Soot Formation in a Laminar Coflow Ethylene/air Diffusion Flame. Combustion and Flame 158 (9), 1682–1695. doi:10.1016/j.combustflame.2011.01.013
Frenklach, M. (1996). On Surface Growth Mechanism of Soot Particles. Symp. (International) Combustion 26 (2), 2285–2293. doi:10.1016/S0082-0784(96)80056-7
Frenklach, M., and Wang, H. (1991). Detailed Modeling of Soot Particle Nucleation and Growth. Symp. (International) Combustion 23 (1), 1559–1566. doi:10.1016/s0082-0784(06)80426-1
Frisch, M., Trucks, G., Schlegel, H., Scuseria, G., Robb, M., Cheeseman, J., et al. (2009). Gaussian 09, Revision D. 01. Wallingford CT: Gaussian, Inc.
Grančič, P., Martin, J. W., Chen, D., Mosbach, S., and Kraft, M. (2016). Can Nascent Soot Particles Burn from the inside? Carbon 109, 608–615. doi:10.1016/j.carbon.2016.08.025
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 132 (15), 154104. doi:10.1063/1.3382344
Hou, D., and You, X. (2017). Reaction Kinetics of Hydrogen Abstraction from Polycyclic Aromatic Hydrocarbons by H Atoms. Phys. Chem. Chem. Phys. 19 (45), 30772–30780. doi:10.1039/C7CP04964A
Kholghy, M. R., Veshkini, A., and Thomson, M. J. (2016). The Core-Shell Internal Nanostructure of Soot - A Criterion to Model Soot Maturity. Carbon 100, 508–536. doi:10.1016/j.carbon.2016.01.022
Khosousi, A., and Dworkin, S. B. (2015). Soot Surface Reactivity during Surface Growth and Oxidation in Laminar Diffusion Flames. Combustion and Flame 162 (12), 4523–4532. doi:10.1016/j.combustflame.2015.09.005
Kopač, D., Likozar, B., and Huš, M. (2019). Catalysis of Material Surface Defects: Multiscale Modeling of Methanol Synthesis by CO2 Reduction on Copper. Appl. Surf. Sci. 497, 143783. doi:10.1016/j.apsusc.2019.143783
Lapuerta, M., Rodríguez–Fernández, J., Sánchez-Valdepeñas, J., and Science, C. (2020). Soot Reactivity Analysis and Implications on Diesel Filter Regeneration. Prog. Energ. Combustion Sci. 78, 100833. doi:10.1016/j.pecs.2020.100833
Li, J., Lu, G., Wu, G., Mao, D., Guo, Y., Wang, Y., et al. (2014). Effect of TiO2 crystal Structure on the Catalytic Performance of Co3O4/TiO2 Catalyst for Low-Temperature CO Oxidation. Catal. Sci. Technol. 4 (5), 1268–1275. doi:10.1039/C3CY01004J
Liu, C., Singh, A. V., Saggese, C., Tang, Q., Chen, D., Wan, K., et al. (2019a). Flame-formed Carbon Nanoparticles Exhibit Quantum Dot Behaviors. Proc. Natl. Acad. Sci. USA 116 (26), 12692–12697. doi:10.1073/pnas.1900205116
Liu, J.-X., Wang, P., Xu, W., and Hensen, E. J. M. (2017). Particle Size and Crystal Phase Effects in Fischer-Tropsch Catalysts. Engineering 3 (4), 467–476. doi:10.1016/J.ENG.2017.04.012
Liu, P., Li, Z., Bennett, A., Lin, H., Sarathy, S. M., and Roberts, W. L. (2019b). The Site Effect on PAHs Formation in HACA-Based Mass Growth Process. Combustion and Flame 199, 54–68. doi:10.1016/j.combustflame.2018.10.010
Lu, T., and Chen, F. (2012). Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 33 (5), 580–592. doi:10.1002/jcc.22885
Macián, V., Serrano, J. R., Piqueras, P., and Sanchis, E. J. (2019). Internal Pore Diffusion and Adsorption Impact on the Soot Oxidation in wall-flow Particulate Filters. Energy 179, 407–421. doi:10.1016/j.energy.2019.04.200
Martin, J. W., Hou, D., Menon, A., Pascazio, L., Akroyd, J., You, X., et al. (2019). Reactivity of Polycyclic Aromatic Hydrocarbon Soot Precursors: Implications of Localized π-Radicals on Rim-Based Pentagonal Rings. J. Phys. Chem. C 123 (43), 26673–26682. doi:10.1021/acs.jpcc.9b07558
Pascazio, L., Martin, J. W., Botero, M. L., Sirignano, M., D’Anna, A., and Kraft, M. (2019). Mechanical Properties of Soot Particles: The Impact of Crosslinked Polycyclic Aromatic Hydrocarbons. Combustion Sci. Tech. 1, 1–21. doi:10.1080/00102202.2019.1668380
Pfau, S. A., La Rocca, A., Fay, M. W., and Flame, M. (2020). Quantifying Soot Nanostructures: Importance of Image Processing Parameters for Lattice Fringe Analysis. Combustion and Flame 211, 430–444. doi:10.1016/j.combustflame.2019.10.020
Randić, M. (2003). Aromaticity of Polycyclic Conjugated Hydrocarbons. Chem. Rev. 103 (9), 3449–3606. doi:10.1021/cr9903656
Schulz, F., Commodo, M., Kaiser, K., De Falco, G., Minutolo, P., Meyer, G., et al. (2019). Insights into Incipient Soot Formation by Atomic Force Microscopy. Proc. Combustion Inst. 37 (1), 885–892. doi:10.1016/j.proci.2018.06.100
Sediako, A. D., Soong, C., Howe, J. Y., Kholghy, M. R., and Thomson, M. J. (2017). Real-time Observation of Soot Aggregate Oxidation in an Environmental Transmission Electron Microscope. Proc. Combustion Inst. 36 (1), 841–851. doi:10.1016/j.proci.2016.07.048
Sharma, V., Uy, D., Gangopadhyay, A., O'Neill, A., Paxton, W. A., Sammut, A., et al. (2016). Structure and Chemistry of Crankcase and Exhaust Soot Extracted from Diesel Engines. Carbon 103, 327–338. doi:10.1016/j.carbon.2016.03.024
Sjoberg, P., Murray, J. S., Brinck, T., and Politzer, P. (1990). Average Local Ionization Energies on the Molecular Surfaces of Aromatic Systems as Guides to Chemical Reactivity. Can. J. Chem. 68 (8), 1440–1443. doi:10.1139/v90-220
Totton, T. S., Misquitta, A. J., and Kraft, M. (2012). A Quantitative Study of the Clustering of Polycyclic Aromatic Hydrocarbons at High Temperatures. Phys. Chem. Chem. Phys. 14 (12), 4081–4094. doi:10.1039/C2CP23008A
Wan, K., Chen, D., and Wang, H. (2018). On Imaging Nascent Soot by Transmission Electron Microscopy. Combustion and Flame 198, 260–266. doi:10.1016/j.combustflame.2018.09.021
Wang, Y., Raj, A., and Chung, S. H. (2015). Soot Modeling of Counterflow Diffusion Flames of Ethylene-Based Binary Mixture Fuels. Combustion and Flame 162 (3), 586–596. doi:10.1016/j.combustflame.2014.08.016
Keywords: carbonaceous nanoparticles, DFT, energy barriers, H abstraction, surface reactions
Citation: Wang H, Chang X and Chen D (2021) Surface Reactivity of Carbonaceous Nanoparticles: The Importance of Surface Pocket. Front. Mech. Eng 7:738354. doi: 10.3389/fmech.2021.738354
Received: 08 July 2021; Accepted: 05 August 2021;
Published: 27 August 2021.
Edited by:
Hongsheng Guo, National Research Council Canada (NRC-CNRC), CanadaReviewed by:
Jianfei Xi, Nanjing Normal University, ChinaCopyright © 2021 Wang, Chang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongping Chen, ZGM1MTZAYml0LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.