 Liangshuang Fan
Liangshuang Fan Hengyu Yang2,3
Hengyu Yang2,3 Guofeng Xie
Guofeng Xie- 1School of Physics and Optoelectronics, Xiangtan University, Xiangtan, China
- 2School of Materials Science and Engineering, Hunan University of Science and Technology, Xiangtan, China
- 3Hunan Provincial Key Laboratory of Advanced Materials for New Energy Storage and Conversion, Xiangtan, China
Recently, monolayer of triphosphides (e.g., InP3, SnP3, and GaP3) attracts much attention due to their good thermoelectric performance. Herein, we predict a novel triphosphide monolayer AsP3 and comprehensively investigate its thermoelectric properties by combining first-principles calculations and semiclassical Boltzmann transport theory. The results show that AsP3 monolayer has an ultralow thermal conductivity of 0.36 and 0.55 Wm K−1 at room temperature along the armchair and zigzag direction. Surprisingly, its maximum Seebeck coefficient in the p-type doping reaches 2,860 µVK−1. Because of the ultralow thermal conductivity and ultrahigh Seebeck coefficient, the thermoelectric performance of AsP3 monolayer is excellent, and the maximum ZT of p-type can reach 3.36 at 500 K along the armchair direction, which is much higher than that of corresponding bulk AsP3 at the same temperature. Our work indicates that the AsP3 monolayer is the promising candidate in TE applications and will also stimulate experimental scientists’ interest in the preparation, characterization, and thermoelectric performance tuning.
Introduction
Thermoelectric (TE) materials are regarded as potential candidates to alleviate the energy and environment crisis by recycling waste heat (Elsheikh et al., 2014; Champier, 2017; Gao et al., 2016). However, TE technology is currently facing two crucial issues: high production cost and low conversion efficiency (El Chaar et al., 2011). For the conversion efficiency of a TE material, it is characterized by the dimensionless figure of merit ZT = S2σT/κ (Ding et al., 2016; Gandi and Schwingenschlögl, 2014), where S is the Seebeck coefficient, σ is the electronic conductivity, T is the absolute temperature, and κ is the thermal conductivity, respectively. Over the past few decades, great progress has been achieved in enhancing the ZT values through certain strategies (Ouyang et al., 2019; Gayner and Kar, 2016), such as nanostructuring, quantum confinement, and the “phonon-glass electron-crystal” (Zhang et al., 2020; Hicks and Dresselhaus, 1993a; Hicks and Dresselhaus, 1993b). Therefore, searching “electronic crystal-phonon glass” thermoelectric materials has great significance (Jiang et al., 2019). Due to the development of nanotechnology, new thermoelectric materials are constantly being discovered in recent years. Phosphorene sheds light on two-dimensional (2D) layered materials in TE applications (Fei et al., 2014). However, the chemical degradation upon exposure to ambient conditions seriously limits its practical application (Wood et al., 2014). Scientists make great effort to find novel materials with puckered layered structure similar to phosphorene, which can not only retain the excellent electronic properties of phosphorene, but also overcome the chemical instability. Very recently, 2D triphosphides which include a combination of phosphorus and selective elements from Group III, IV, and V attract much attention because of their outstanding thermoelectric performance. For example, Ouyang et al. (2018) predicted the ZT value up to 2.06 along the armchair direction of InP3 monolayer at room temperature. Zhu et al. (2019) indicated that the ZT value of monolayer SnP3 was 3.46 along the armchair direction at 500 K. Single-layer SbP3 (Sun et al., 2020) and GeP3 (Jing et al., 2017) are also predicted as good TE materials.
In this work, utilizing first-principles calculations and the Boltzmann transport equation, we explore stability, phonon/electron, and TE properties of the AsP3 monolayer. Our calculated results show that the AsP3 monolayer exhibits ultrahigh Seebeck coefficient and ultralow thermal conductivity, and the maximum ZT of p-type AsP3 monolayer can reach 3.36 at 500 K along the armchair direction. This work reveals that AsP3 is a promising candidate for TE applications.
Methods
The TE properties of single-layer AsP3 are calculated using the first-principles and density functional theory (DFT) as performed in the Vienna ab initio simulation package (VASP) (Blöchl, 1994). We set up a vacuum layer with a thickness of 20 Å along the z-direction and use the vdW-DF2 method to correct interaction force between the layers (Klimeš et al., 2009). The generalized gradient approximation (GGA) and the Perdew−Burke−Ernzerhof (PBE) are adopted for processing the exchange-correlation functional (Perdew et al., 1996; Kresse and Furthmüller, 1996). A plane wave basis with a cutoff energy of 500 eV and a 6 × 6 × 1-k-mesh are used for the structural relaxation of the Brillouin zone (BZ). The convergence criterion for complete relaxation of all geometric structures is that the total energy change is <10−7 eVÅ−1 and the residual force is <0.01 eVÅ−1.
The Boltzmann transport theory and rigid band approximation are used to evaluate the electronic transport properties, which are carried out in the BoltzTrap software with a dense k-mesh 40 × 40 × 1 (Madsen and Singh, 2006). This method has achieved good agreement between theoretical calculations and experiments for many TE materials (Bilc et al., 2006; Xu et al., 2010; Parker and Singh, 2010). For the phonon transport properties, it can be obtained by solving the Boltzmann transport equation in the ShengBTE packages with 50 × 50 × 1 k-points (Li et al., 2014). The phonon dispersions and the harmonic second-order interaction force constants (2nd IFCs) are calculated by using VASP and PHONOPY software (Togo and Tanaka, 2015), in which the 3 × 3 × 1 k-mesh within 4 × 4 × 1 supercell is adopted. The third-order IFCs (3rd IFCs) is obtained by using the 3 × 3 × 1 supercell with the finite-difference method (Li et al., 2012), which considers the interaction including the seventh nearest neighbor atoms.
Results and Discussion
Atomic and Electronic Structures
Based on calculations of the first-principles, the AsP3 monolayer exhibits a hexagonal honeycomb configuration with space group
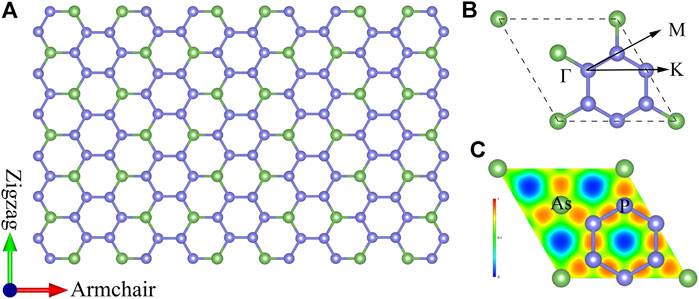
FIGURE 1. (A) Top view of atomic structure, (B) corresponding first Brillouin zone path, and (C) electronic local function for monolayer AsP3.
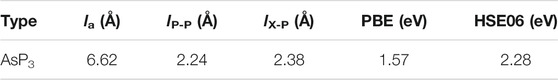
TABLE 1. Calculated lattice constants (la), bond lengths of P–P (lP–P) and X–P (lX–P), and bandgap based on PBE and HSE06.
We study the electrical band structures as well as the corresponding density of states (DOS) of AsP3 monolayer, as presented in Figure 2A. We find that the AsP3 monolayer is an indirect bandgap semiconductor, its valence band maximum (VBM) is locating at the Γ point, whereas the conduction band minimum (CBM) is located between the Γ and M points. The accurate bandgap of 2.28 eV is obtained by using the Herd–Scuseria–Emzerhof hybrid functional (HSE06) (Heyd et al., 2003). In the vicinity of Fermi level, the two highest valence bands (VBs) exhibit overlap at the Γ point, which benefits their electrical transport characteristics (Ding et al., 2018). The DOS further demonstrates that the edges of the VB and CB are mainly coming from the P atoms, which can also be observed in Figure 2B, where the partial charge density provides that the charge is mainly concentrated near P atoms. Meantime, a peak shape of DOS is acquired in monolayer AsP3, which is regarded as the electronic transmission characteristic of high TE performance (Peng et al., 2014).
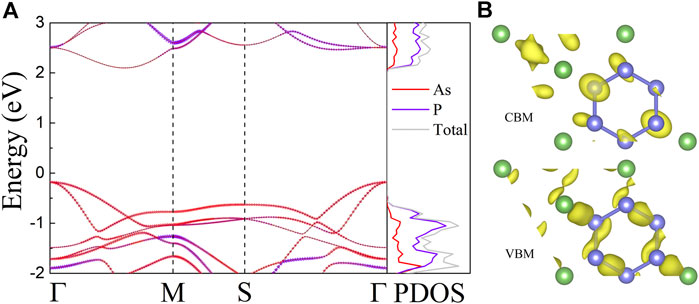
FIGURE 2. (A) Calculated orbital-resolved band structures and corresponding DOS of AsP3 monolayer. The contribution from As (red) and p (purple) atoms weight the colors of band structures. (B) The partial charge densities of DOS.
Electrical Transport Properties
Based on the deformation potential (DP) theory (Bardeen and Shockley, 1950), the carrier mobility of AsP3 monolayer can be calculated by the formula:
where m*, κB, El, and C2D are the effective mass, the Boltzmann constant, the DP constant, and the 2D elastic constants, respectively. These parameters and the relaxation time
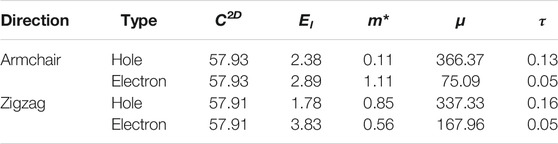
TABLE 2. Calculated elastic modulus C2D (J m−2), DP constant El (eV), effective mass m* (me), carrier mobility μ (cm−2 V−1 s−1), and relaxation time τ (ps) for electron and hole in AsP3 monolayer at 300 K.
We obtain the electrical transport properties of AsP3 monolayer via calculating the semiclassical Boltzmann transport equation (May et al., 2009), involving Seebeck coefficient S, electrical conductivity σ, electrical thermal conductivity κe, and power factor (PF), which are important parameters for evaluating the TE properties. The rigid band approximation theory is adopted to simulate the influence of doping on electron transport. It is assumed that the band structure of the n-type and p-type systems does not change in shape under light doping, whereas the Fermi level moves up and down (Singh, 2007; Parker and Singh, 2011). Positive and negative μ correspond to the n-type and p-type doping. Here, the S, σ, and PF concerning chemical potential μ can be computed by employing the Fermi–Dirac distribution function fμ (Parker and Singh, 2011) as:
where V is the volume of the primitive cell, and the transport distribution function
where N0 is the number of q points sampling, τ is the electronic relaxation time, and ν is electronic velocity.
Figures 3A,B are the Seebeck coefficient S of AsP3 monolayer as a function of µ. The downward trend of the S with the temperature gradually slows down as the temperature increases. Surprisingly, the AsP3 monolayer shows ultrahigh S compared to other triphosphates with high TE performance (InP3 (Ouyang et al., 2018), SnP3 (Zhu et al., 2019), and CaP3 (Zhu et al., 2020a)). At room temperature, the S of AsP3 monolayer reaches 2,860 µVK−1 along the zigzag direction and 2,800 µVK−1 along the armchair direction. The double degeneracy of the valence band at the Г point and the steep electronic density of states near the Fermi level effectively increase the S.
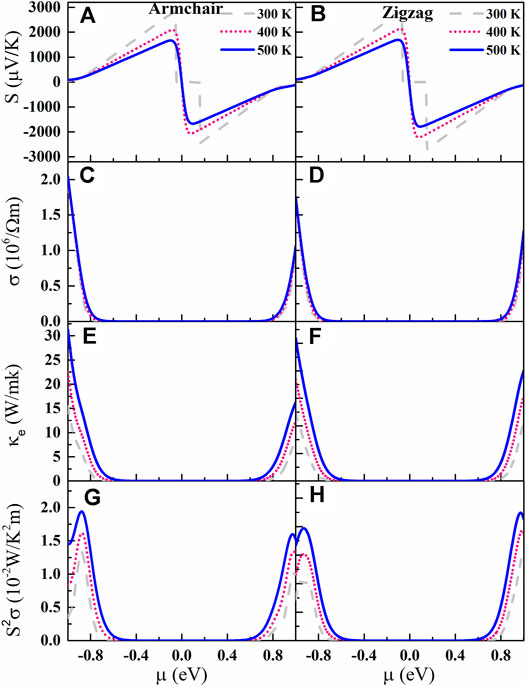
FIGURE 3. (A)–(H) Seebeck coefficients, electrical conductivity, electrical thermal conductivity, and power factor vs. chemical potential µ at different temperatures (300, 400, and 500 K).
The electrical conductivity σ vs. μ is shown in Figures 3C,D. It is found that the p-type σ is visibly larger than that of the n-type for AsP3 monolayer, due to the higher mobility and relaxation time of hole. Since electrons submit the Fermi–Dirac distribution, the gradient of σ around VBM and CBM tends to be flat with increasing temperature. According to the Wiedemann–Franz law, the κe is proportional to the σ: κe= LσT, where L is the Lorenz number
Based on the obtained S and σ, we calculate the PF as shown in Figures 3G,H. Similar to σ, the PF of p-type AsP3 monolayer is significantly higher than that of n-type, which means that it is a potential p-type TE material.
Phonon Transport Properties
The phonon spectrum and projected density of state (PhDOS) of the AsP3 monolayer are illustrated in Figure 4A. No virtual phonon modes are observed in phonon dispersion, which indicates that the AsP3 monolayer is dynamically stable. The phonon spectrum of AsP3 contains three phonon branches (ZA, TA, and LA) and twenty-one optical branches. The low-frequency optical branch and the acoustic branch of the AsP3 monolayer are softened and overlapped, leading to the strong acoustic–optic interaction (Gao et al., 2018). Besides, the flat-frequency band over the entire frequency range can lead to smaller phonon group velocity (Ma et al., 2016; Ma et al., 2018). It can be seen from the corresponding PhDOS that the phonon mode mainly contains As atoms in the low-frequency region (below 3 THz), while the high-frequency region (over 12 THz) is contributed by the P atom.
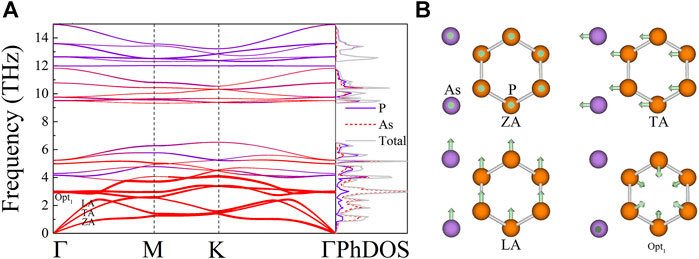
FIGURE 4. (A) Phonon spectra and phonon density of states (PhDOS) for AsP3, (B) the corresponding vibrational modes of the acoustic phonon branches (ZA, TA, and LA), and the lowest optical branch (Opt1) near and at the Γ point.
Figure 4B depicts the corresponding vibration modes near the Γ point of the acoustic branch (ZA, TA, and LA) and the lowest optical branch (Opt1) to display the characteristics of the phonon mode more intuitively. It can be seen that the vibration of the three-phonon branch occurs along the in-plane (TA and LA) or outside the plane (ZA), which is a property of natural vibration. Interestingly, it is observed that As atoms of the Opt1 branch have in-plane opposite motions relative to their adjacent As atoms, while P atoms have in-plane antiphase motions. This phenomenon can significantly block heat transport, thereby reducing the thermal conductivity of the lattice (Ding et al., 2015).
Based on the 2nd and 3rd IFCs, the κl is calculated by the self-consistent iterative method. The calculation formula of κl is as follows:
where CV, ν, and τqλ are phonon model specific heat, group velocity, and relaxation time, respectively. As shown in Figure 5A, we plot the κl of AsP3 monolayer vs. temperature. The inherent κl is obviously proportional to 1/T, which is attributed to the inherent enhancement of phonon–phonon scattering (Liu et al., 2018). Excitingly, the AsP3 monolayer shows ultralow thermal conductivity. At room temperature, κl along the armchair direction (0.36 Wm−1 K−1) is lower than that along the zigzag direction (0.55 Wm−1 K−1). We explore the relationship between κl and frequency to describe the contribution of different phonons. Figure 5B shows that acoustic branches (below 2 THz) along armchair (zigzag) direction contribute about 75% (86%) to κl.
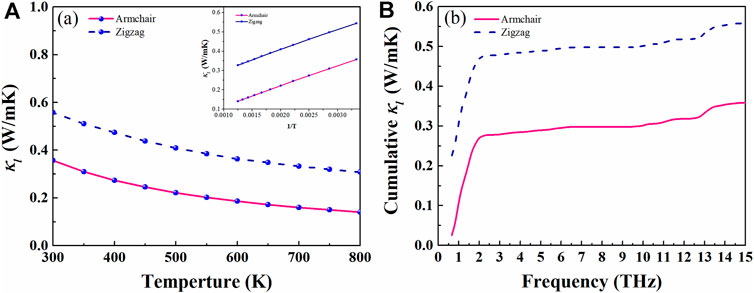
FIGURE 5. Lattice thermal conductivity of AsP3 as a function of temperature (A) and frequency (B). The insets of (A) is lattice thermal conductivity as a function of T−1.
The phonon group velocity is an important coefficient to evaluate the heat transferability, which can be obtained by the following equation:
The corresponding group velocity of AsP3 monolayer is plotted in Figure 6A. We can see that the group velocity in the low-frequency area is distinctly larger than that of the high-frequency area. It exhibits a smaller average group velocity about 1.91 km s−1, which is related to the flat phonon dispersions and low cutoff frequency.
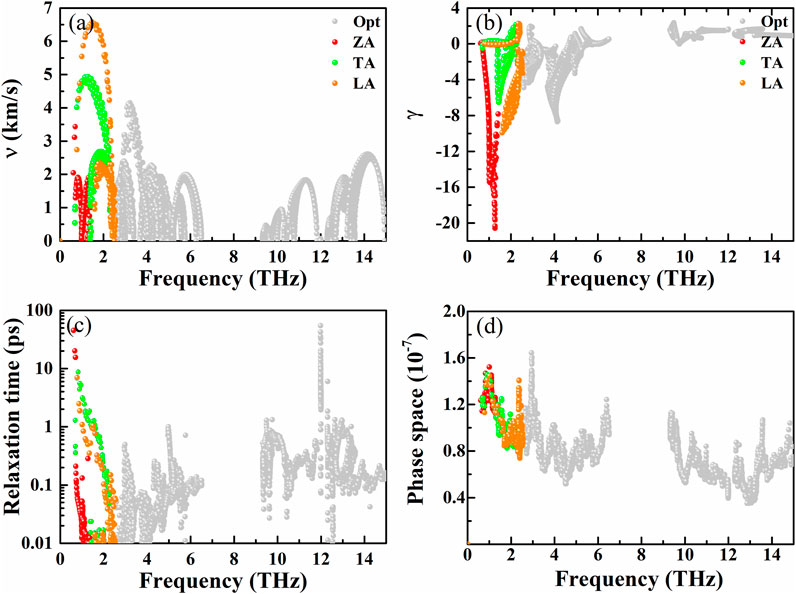
FIGURE 6. (A) Phonon group velocity, (B) Grüneisen parameters, (C) phonon relaxation time, and (D) three-phonon scattering phase space as a function of frequency for AsP3, respectively.
To determine the other origin of ultralow κl of AsP3 monolayer, we calculate the Grüneisen parameter γ, which can be used to describe the anharmonic interaction of crystals. It can be described by the following equation:
Generally, the larger |γ| represents the stronger anharmonicity and the lower κl. As presented in Figure 6B, one can see a larger |γ| in the low-frequency region, and the average of |γ| is about 16.17, indicating the existence of strong phonon–phonon scattering.
Figure 6C is the phonon relaxation time vs. frequency of AP3 monolayer. At 300 K, our calculated phonon relaxation time of AsP3 monolayer (4.3 ps) can be compared with the single-layer KAgS (4.05 ps) (Zhu et al., 2020b), which is an important factor for the low κl of AsP3 monolayer. The three-phonon scattering phase space (P3) is the available space for the three-phonon scattering process (Li and Mingo, 2015), which can further analyze the source of lower κl. As shown in Figure 6D, the AsP3 monolayer possesses a larger P3 over the entire frequency region, which allows stronger phonon–phonon scattering and weaker heat transport.
Thermoelectric Figure of Merit
Combining the electrical transport coefficients and thermal conductivity, we can estimate the ZT values of AsP3, as displayed in Figure 7. The p-type doped AsP3 monolayer exhibits a higher ZT than the n-type one, which is caused by larger PF of p-type. The maximum ZT values of p-type doped AsP3 monolayer at 500 K can reach 3.36 and 2.92 along the armchair and zigzag direction, while corresponding ZT values at 300 K is 2.22 and 1.84, much higher than that of bulk AsP3 (∼0.5 at 300 K) (Duan et al., 2019), which is mainly caused by the larger S and lower κl of AsP3 single-layered structure by dimensionality reduction. The maximum ZT value of AsP3 monolayer at 500 K is comparable with other 2D triphosphides, such as n-type doped InP3 (ZT = 3.23) (Ouyang et al., 2018), p-type doped SnP3 (ZT = 3.46) (Zhu et al., 2019), and p-type doped SbP3 (ZT = 3.5) (Sun et al., 2020). Besides, the maximum ZT values can be achieved when the carrier concentration is between 1 × 1010 and 1011 cm−2. Generally speaking, if the ZT value is close to 3, it is deemed a good TE material (Harman et al., 2002), therefore AsP3 monolayer can be a potential candidate for TE applications.
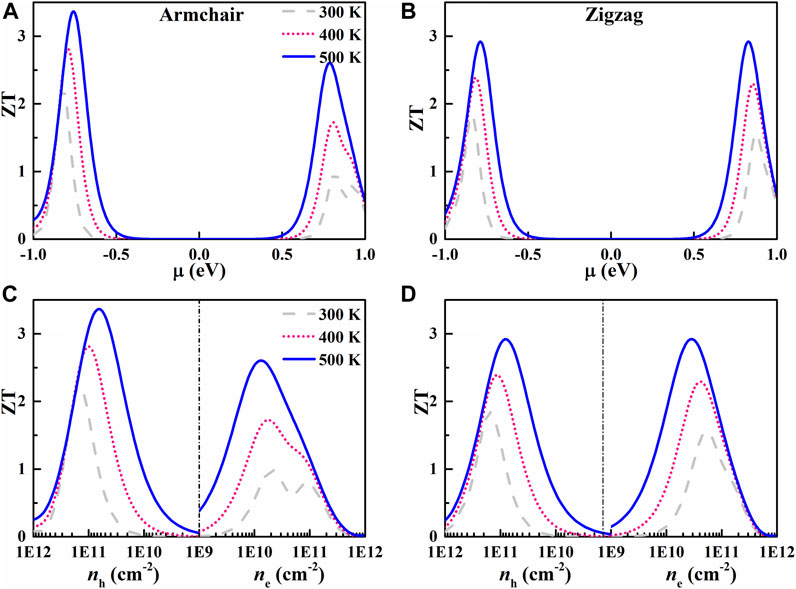
FIGURE 7. ZT as a function of the chemical potential (A) and carrier concentration (B) of the AsP3 monolayer.
Conclusion
In conclusion, we investigate the TE performance of AsP3 monolayer by a highly precise first-principles method. The calculated results show that AsP3 monolayer is an indirect bandgap semiconductor with a bandgap of 2.28 eV. It also possesses excellent Seebeck coefficients and electrical conductivity. Due to its lower values of phonon velocities (1.91 km s−1), larger Grüneisen parameters |γ| (16.17), as well as shorter phonon relaxation time (4.30 ps), AsP3 monolayer exhibits ultralow κl 0.36 Wm−1 K−1 (0.55 Wm−1 K−1) at room temperature in the armchair (zigzag) direction. At 500 K, the maximum ZT of the AsP3 monolayer can reach up to 3.36 (armchair direction) and 3.08 (zigzag direction) by the optimal p-type doping. These results prove the huge advantages of AsP3 single-layer film in high-efficiency conversion of heat energy.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
GX established the theoretical models and supervised the project, and LF and HY performed the calculations and data analysis.
Funding
This work was financially supported by National Natural Science Foundation of China (NSFC) (Grant No. 11874145 and 11474243).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bardeen, J., and Shockley, W. (1950). Deformation Potentials and Mobilities in Non-polar Crystals. Phys. Rev. 80, 72–80. doi:10.1103/physrev.80.72
Bilc, D. I., Mahanti, S. D., and Kanatzidis, M. G. (2006). Electronic Transport Properties of PbTe and AgPbmSbTe2+m Systems. Phys. Rev. B 74, 125202. doi:10.1103/physrevb.74.125202
Blöchl, P. E. (1994). Projector Augmented-Wave Method. Phys. Rev. B 50, 17953–17979. doi:10.1103/physrevb.50.17953
Champier, D. (2017). Thermoelectric Generators: A Review of Applications. Energ. Convers. Manage. 140, 167–181. doi:10.1016/j.enconman.2017.02.070
Ding, G., Gao, G. Y., Huang, Z., Zhang, W., and Yao, K. (2016). Thermoelectric Properties of monolayerMSe2(M = Zr, Hf): Low Lattice thermal Conductivity and a Promising Figure of merit. Nanotechnology 27, 375703. doi:10.1088/0957-4484/27/37/375703
Ding, G., Wang, C., Gao, G., Yao, K., Dun, C., Feng, C., et al. (2018). Engineering of Charge Carriers via a Two-Dimensional Heterostructure to Enhance the Thermoelectric Figure of merit. Nanoscale 10, 7077–7084. doi:10.1039/c7nr09029c
Ding, J., Xu, B., Lin, Y., Nan, C., and Liu, W. (2015). Lattice Vibration Modes of the Layered Material BiCuSeO and First Principles Study of its Thermoelectric Properties. New J. Phys. 17, 083012. doi:10.1088/1367-2630/17/8/083012
Duan, S., Cui, Y., Chen, X., Yi, W., Liu, Y., and Liu, X. (2019). Ultrahigh Thermoelectric Performance Realized in Black Phosphorus System by Favorable Band Engineering through Group VA Doping. Adv. Funct. Mater. 29, 1904346. doi:10.1002/adfm.201904346
El Chaar, L., lamont, L. A., and El Zein, N. (2011). Review of Photovoltaic Technologies. Renew. Sustain. Energ. Rev. 15, 2165–2175. doi:10.1016/j.rser.2011.01.004
Elsheikh, M. H., Shnawah, D. A., Sabri, M. F. M., Said, S. B. M., Hassan, M. H., Ali Bashir, M. B., et al. (2014). A Review on Thermoelectric Renewable Energy: Principle Parameters that Affect Their Performance. Renew. Sust. Energ. Rev. 30, 337–355. doi:10.1016/j.rser.2013.10.027
Fei, R., Faghaninia, A., Soklaski, R., Yan, J.-A., Lo, C., and Yang, L. (2014). Enhanced Thermoelectric Efficiency via Orthogonal Electrical and thermal Conductances in Phosphorene. Nano Lett. 14, 6393–6399. doi:10.1021/nl502865s
Gandi, A. N., and Schwingenschlögl, U. (2014). WS2As an Excellent High-Temperature Thermoelectric Material. Chem. Mater. 26, 6628–6637. doi:10.1021/cm503487n
Gao, H. B., Huang, G. H., Li, H. J., Qu, Z. G., and Zhang, Y. J. (2016). Development of Stove-Powered Thermoelectric Generators: A Review. Appl. Therm. Eng. 96, 297–310. doi:10.1016/j.applthermaleng.2015.11.032
Gao, Z., Liu, G., and Ren, J. (2018). High Thermoelectric Performance in Two-Dimensional Tellurium: An Ab Initio Study. ACS Appl. Mater. Inter. 10, 40702–40709. doi:10.1021/acsami.8b11836
Gao, Z., Tao, F., and Ren, J. (2018). Unusually Low thermal Conductivity of Atomically Thin 2D Tellurium. Nanoscale 10, 12997–13003. doi:10.1039/c8nr01649f
Gayner, C., and Kar, K. K. (2016). Recent Advances in Thermoelectric Materials. Prog. Mater. Sci. 83, 330–382. doi:10.1016/j.pmatsci.2016.07.002
Harman, T. C., Taylor, P. J., Walsh, M. P., and LaForge, B. E. (2002). Quantum Dot Superlattice Thermoelectric Materials and Devices. Science 297, 2229–2232. doi:10.1126/science.1072886
Heyd, J., Scuseria, G. E., and Ernzerhof, M. (2003). Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 118, 8207–8215. doi:10.1063/1.1564060
Hicks, L. D., and Dresselhaus, M. S. (1993a). Effect of Quantum-Well Structures on the Thermoelectric Figure of merit. Phys. Rev. B 47, 12727–12731. doi:10.1103/physrevb.47.12727
Hicks, L. D., and Dresselhaus, M. S. (1993b). Thermoelectric Figure of merit of a One-Dimensional Conductor. Phys. Rev. B 47, 16631–16634. doi:10.1103/physrevb.47.16631
Jiang, X., Zhu, L., Li, B., and Yao, K. (2019). Thermoelectric Properties of Monolayer α-Te: Low Lattice thermal Conductivity and Extremely High Dimensionless Figure of merit. Phys. Let. A. 384, 126222. doi:10.1016/j.physleta.2019.126222
Jing, Y., Ma, Y., Li, Y., and Heine, T. (2017). GeP3: A Small Indirect Band Gap 2D Crystal with High Carrier Mobility and Strong Interlayer Quantum Confinement. Nano Lett. 17, 1833–1838. doi:10.1021/acs.nanolett.6b05143
Jonson, M., and Mahan, G. D. (1980). Mott’s Formula for the Thermopower and the Wiedemann-Franz Law. Phys. Rev. B 21, 4223–4229. doi:10.1103/physrevb.21.4223
Klimeš, J., Bowler, D. R., and Michaelides, A. (2009). Chemical accuracy for the van der Waals density functional. J. Phys. Condens. Matter 22, 022201. doi:10.1088/0953-8984/22/2/022201
Kresse, G., and Furthmüller, J. (1996). Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54, 11169–11186. doi:10.1103/physrevb.54.11169
Li, W., Carrete, J., A. Katcho, N., and Mingo, N. (2014). ShengBTE: A Solver of the Boltzmann Transport Equation for Phonons. Comput. Phys. Commun. 185, 1747–1758. doi:10.1016/j.cpc.2014.02.015
Li, W., Lindsay, L., Broido, D. A., Stewart, D. A., and Mingo, N. (2012). Thermal Conductivity of Bulk and Nanowire Mg2SixSn1−x Alloys from First Principles. Phys. Rev. B 86, 174307. doi:10.1103/physrevb.86.174307
Li, W., and Mingo, N. (2015). Ultralow Lattice thermal Conductivity of the Fully Filled Skutterudite YbFe4Sb12 Due to the Flat Avoided-Crossing Filler Modes. Phys. Rev. B 91, 144304. doi:10.1103/physrevb.91.144304
Liu, P. F., Bo, T., Xu, J., Yin, W., Zhang, J., Wang, F., et al. (2018). First-principles Calculations of the Ultralow thermal Conductivity in Two-Dimensional Group-IV Selenides. Phys. Rev. B 98, 235426. doi:10.1103/physrevb.98.235426
Ma, D., Ding, H., Meng, H., Feng, L., Shiomi, J., and Yang, N. (2016). Nano-cross-junction Effect on Phonon Transport in Silicon Nanowire Cages. Phys. Rev. B 94, 165434. doi:10.1103/physrevb.94.165434
Ma, D., Wan, X., and Yang, N. (2018). Unexpected thermal Conductivity Enhancement in Pillared Graphene Nanoribbon with Isotopic Resonance. Phys. Rev. B 98, 245420. doi:10.1103/physrevb.98.245420
Madsen, G. K. H., and Singh, D. J. (2006). BoltzTraP. A Code for Calculating Band-Structure Dependent Quantities. Comput. Phys. Commun. 175, 67–71. doi:10.1016/j.cpc.2006.03.007
May, A. F., Toberer, E. S., Saramat, A., and Snyder, G. J. (2009). Characterization and Analysis of Thermoelectric Transport in N-type Ba8Ga16-xGe30+x. Phys. Rev. B 80, 125205. doi:10.1103/physrevb.80.125205
Ouyang, T., Jiang, E., Tang, C., Li, J., He, C., and Zhong, J. (2018). Thermal and Thermoelectric Properties of Monolayer Indium Triphosphide (InP3): a First-Principles Study. J. Mater. Chem. A. 6, 21532–21541. doi:10.1039/c8ta07012a
Ouyang, Y., Zhang, Z., Li, D., Chen, J., and Zhang, G. (2019). Emerging Theory, Materials, and Screening Methods: New Opportunities for Promoting Thermoelectric Performance. Annalen Der Physik 531, 1800437. doi:10.1002/andp.201800437
Parker, D., and Singh, D. J. (2010). High-temperature Thermoelectric Performance of Heavily Doped PbSe. Phys. Rev. B 82, 035204. doi:10.1103/physrevb.82.035204
Parker, D., and Singh, D. J. (2011). Potential Thermoelectric Performance from Optimization of Hole-Doped Bi2Se3. Phys. Rev. X 1, 021005. doi:10.1103/physrevx.1.021005
Peng, H., Kioussis, N., and Snyder, G. J. (2014). Elemental Tellurium as a Chiral P-type Thermoelectric Material. Phys. Rev. B 89, 195206. doi:10.1103/physrevb.89.195206
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Singh, D. J. (2007). Electronic and Thermoelectric Properties of CuCoO2: Density Functional Calculations. Phys. Rev. B 76, 085110. doi:10.1103/physrevb.76.085110
Sun, Z., Yuan, K., Chang, Z., Bi, S., Zhang, X., and Tang, D. (2020). Ultra-low thermal Conductivity and High Thermoelectric Performance of Two-Dimensional Triphosphides (InP3, GaP3, SbP3 and SnP3): a Comprehensive First-Principles Study. Nanoscale 12, 3330–3342. doi:10.1039/c9nr08679j
Togo, A., and Tanaka, I. (2015). First Principles Phonon Calculations in Materials Science. Scripta Materialia 108, 1–5. doi:10.1016/j.scriptamat.2015.07.021
Wood, J. D., Wells, S. A., Jariwala, D., Chen, K.-S., Cho, E., Sangwan, V. K., et al. (2014). Effective Passivation of Exfoliated Black Phosphorus Transistors against Ambient Degradation. Nano Lett. 14, 6964–6970. doi:10.1021/nl5032293
Xu, L., Zheng, Y., and Zheng, J. C. (2010). Thermoelectric Transport Properties of PbTe under Pressure. Phys. Rev. B 82, 195102. doi:10.1103/physrevb.82.195102
Zhang, Z., Ouyang, Y., Cheng, Y., Chen, J., Li, N., and Zhang, G. (2020). Size-dependent Phononic thermal Transport in Low-Dimensional Nanomaterials. Phys. Rep. 860, 1–26. doi:10.1016/j.physrep.2020.03.001
Zhu, X.-L., Liu, P.-F., Wu, Y.-Y., Zhang, P., Xie, G., and Wang, B.-T. (2020a). Significant Enhancement of the Thermoelectric Properties of CaP3 through Reducing the Dimensionality. Mater. Adv. 1, 3322–3332. doi:10.1039/d0ma00603c
Zhu, X.-L., Liu, P.-F., Zhang, J., Zhang, P., Zhou, W.-X., Xie, G., et al. (2019). Monolayer SnP3: an Excellent P-type Thermoelectric Material. Nanoscale 11, 19923–19932. doi:10.1039/c9nr04726c
Keywords: Seebeck coefficient, thermal conductivity, thermoelectric, first-principle calculations, transport
Citation: Fan L, Yang H and Xie G (2021) Thermoelectric Properties of Arsenic Triphosphide (AsP3) Monolayer: A First-Principles Study. Front. Mech. Eng 7:702079. doi: 10.3389/fmech.2021.702079
Received: 29 April 2021; Accepted: 21 May 2021;
Published: 11 June 2021.
Edited by:
Shiqian Hu, Yunnan University, ChinaReviewed by:
Dengke Ma, Nanjing Normal University, ChinaZhibin Gao, Xi’an Jiaotong University, China
Jie Chen, Tongji University, China
Copyright © 2021 Fan, Yang and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guofeng Xie, Z2Z4aWVAeHR1LmVkdS5jbg==