 Patrick K. Quoika1,2*
Patrick K. Quoika1,2* Anna S. Kamenik2,3
Anna S. Kamenik2,3 Monica L. Fernández-Quintero2
Monica L. Fernández-Quintero2 Martin Zacharias1
Martin Zacharias1 Klaus R. Liedl2
Klaus R. Liedl2- 1Center for Protein Assemblies (CPA), Physics Department, Chair of Theoretical Biophysics, Technical University of Munich, Munich, Germany
- 2Institute for General, Inorganic and Theoretical Chemistry, Center for Molecular Biosciences Innsbruck (CMBI), University of Innsbruck, Innsbruck, Austria
- 3Laboratory of Physical Chemistry, ETH Zurich, Zurich, Switzerland
Poly (N-isopropylacrylamide) (PNIPAM) is a famous representative of thermosensitive polymers. Thermosensitive polymers undergo a phase transition with lower critical solution temperature. Commonly, their phase behavior is linked to a conformational collapse above a certain temperature. This thermosensitive conformational transition is called Coil-Globule transition. In contrast, most other polymers usually show inverse temperature behavior, i.e., an upper critical solution temperature, corresponding to a Globule-Coil transition. Besides their numerous possible applications, thermosensitive polymers are of interest for fundamental research, because of similarities to macromolecular conformational transitions, e.g., protein folding. The counter-intuitive behavior of thermosensitive polymers is commonly associated with solvation effects. Thus, an accurate description of the solvent is crucial for the investigation of thermosensitive polymers in molecular simulations. Here, we investigate the influence of the in silico water model on the thermosensitive Coil-Globule transition in molecular dynamics simulations. To this end, we performed extensive atomistic simulations of the syndiotactic PNIPAM 20-mer at multiple temperatures with eight different water models–four of which are 3-point water models (TIP3P-type) and four are 4-point water models (TIP4P-type). We found that the thermosensitive Coil-Globule transition is strongly influenced by the water model in the simulations. Depending on the water model, the conformational ensemble of the polymer is shifted significantly, which leads to dramatically different results: The estimated transition temperature may span between 255 and 350 K. Consequently, depending on the description of the solvent, the physicochemical and mechanical properties of these polymers, e.g., the polymer-solvent affinity and persistence length, vary. These divergent results originate from the strength of interactions between polymer and solvent, but also on the bulk state of the solvent. Both these quantities vary between water models. We found that the Lennard-Jones interaction parameter ϵ of the water model correlates with the transition temperature of the polymer. Indeed, the quadrupole moment of the water model shows an even higher correlation with this quantity. Our results suggest a connection between the phase diagram of the solvent and the thermosensitive transition of the polymer.
1 Introduction
Thermosensitive polymers are defined by their atypical phase behavior. In contrast to most other polymers, they show a phase transition with lower critical solution temperature (LCST)–i.e., the solvent quality is bad above, and good below the transition temperature (Taylor and Cerankowski, 1975; Aseyev et al., 2011). Their fascinating phase behavior leads to numerous biomedical and non-biomedical applications of these polymers, such as gel actuators, drug delivery systems, or tissue engineering, amongst others (Hoffman, 1995; Pal et al., 2009). The phase behavior of the (possibly) most-famous representative of thermosensitive polymers, poly (N-isopropylacrylamide) (PNIPAM), has been discovered in the late 1960s (Heskins and Guillet, 1968). Yet, the molecular origin of its LCST behavior is still under debate. Commonly, it is associated with a conformational change of the polymer chains, i.e., the Coil-Globule transition. Accordingly, the thermosensitive Coil-Globule transition describes a conformational collapse of thermosensitive polymer chains above the LCST (Wu and Wang, 1998). We describe the Coil-Globule transition in more detail, including visual examples of both conformational states, in the supplementary information, section A and L. The Coil-Globule transition is not only interesting by itself, but–due to similarities to protein folding (hydrophobic collapse) (Dill, 1985; Inoue et al., 2019)–also facilitates an understanding of other macromolecular systems. Changes in the solvent conditions, such as salt, cosolute and cosolvent concentrations, strongly alter the thermosensitive behavior (Schild et al., 1991; Kunugi et al., 2002). Thus, it is widely accepted that thermosensitivity is linked to solvation effects (Hummer, 2007). Indeed, the connection between thermosensitivity and solvation is already implicitly contained the Flory-Huggins theory (Flory, 1953). In earlier publications, we have come to the conclusion that polymer-solvent interactions are of crucial importance for the energetic balance of the thermosensitive Coil-Globule transition (Quoika et al., 2020; Quoika et al., 2021), which is in agreement with the conclusions of other authors (Hummer, 2007; Bischofberger et al., 2015).
Molecular dynamics (MD) simulations enable the investigation of temperature-dependent conformational changes with atomic resolution (Deshmukh et al., 2013). However, it is challenging to investigate the thermosensitive Coil-Globule transition in conventional MD simulations, because of the slow timescales of the process (Boţan et al., 2016; Kang et al., 2016; García and Hasse, 2019; Quoika et al., 2021). Furthermore, simulations over a wide-range of temperatures are necessary, which leads to a multiplication of the computational effort–especially at low temperatures, where the dynamics are generally slow. While the thermosensitive Coil-Globule transitions has been investigated by means of MD simulations before, little effort has been put into the systematic analysis of the impact of the description of the explicit solvent in these simulations. Given the importance of the solvent for the process, clearly, the water model in molecular simulations may have an impact on the process. Recently, Tavagnacco et al. (2022) have shown the general impact of different water models in molecular simulations of the system. However, they only investigated two different water models and focused on the pressure dependence of the Coil-Globule transition. Here, we present a complementary study on the impact of different water models on the thermosensitive character of PNIPAM in molecular simulations.
In this study, we systematically investigate the influence of the water model on the thermosensitive Coil-Globule transition in MD simulations. To this end, we performed extensive simulations of the PNIPAM 20-mer in eight established water models at multiple temperatures. Half of these are 3-point water models and the other half are 4-point water models–namely OPC3, SPC, SPCE, TIP3P; and OPC, TIP4P-2005, TIP4P-EW, TIP4P-ICE, respectively (references are given in the methods section). To be able to isolate the impact of the solvent description, we used the same force field parameters of the polymer in all simulations (OPLS2005 force field). A similar approach was successfully followed by Hess and van der Vegt (2006) to analyze the interactions between amino acid analogues and water. Accordingly, we elucidate the effect of different water models on the physicochemical properties of thermosensitive polymers and the Coil-Globule transition and identify systematic differences. Finally, we provide an explanation of water model-dependent deviations in the simulations of thermosensitive polymers. Apart from the here-shown results, we also provide an extensive supplementary information (SI) with detailed discussion of the influence of the water model on the thermodynamics of the Coil-Globule transition.
2 Computational methods
2.1 Model system poly(N-isopropylacrylamide)
We used poly(N-isopropylacrylamide) (PNIPAM), a well-known representative of the class of thermosensitive polymers, as model system. This polymer has been discussed and implemented for numerous applications (Lanzalaco and Armelin, 2017; Bilardo et al., 2022). Besides, it has been the object of research in numerous studies–both experimental (Rizzi et al., 2021; Vdovchenko et al., 2021; Liu et al., 2022) and computational (Pang et al., 2010; Bruce et al., 2019; García and Hasse, 2019). We chose to simulate a 20-mer of PNIPAM, since it is long enough to show a Coil-Globule transition, while not being unnecessarily long. This pragmatic choice allowed for a better coverage of the conformational space than with longer polymer chains (Quoika et al., 2021).
2.2 Simulation setup
We performed extensive unbiased simulations of the polymer in pure water. Thus, we used a common and conventional setup for the simulations of thermosensitive polymers. We prepared extended conformations of syndiotactic 20-mers of PNIPAM, with the Maestro software package (Schrödinger LLC, 2019). Furthermore, we solvated these structures in cubic boxes of a side length of 7 nm in various water models (see below). This choice of box size was based on an estimation of the maximum possible polymer end-to-end distance. We explain this calculation in more detail in the SI. Furthermore, we show the distribution of end-to-end distances of the polymer in our simulations there. After a minimization and equilibration at the respective temperatures, we performed extensive independent simulations in the isothermal-isobaric ensemble. The length of these production runs was chosen depending on the water model (see below). All simulations have been carried out with the GROMACS MD-Simulation software package (Abraham et al., 2015). Independent of the water model we used the atomistic OPLS2005 force field for the polymer (Jorgensen et al., 1996; Kaminski et al., 2001). This force field is commonly used and has been established for simulations of PNIPAM (Walter et al., 2010; Podewitz et al., 2019; Quoika et al., 2021). All additional technical information is given in the SI, section B.
Depending on the type of water model we simulated at a different range of temperatures. For 3-point water models we simulated between 220 and 340 K, whereas for 4-point water models, we simulated between 270 and 380 K. We made this decision based on, firstly, the transition temperature in the respective water model, and secondly, based on the phase diagram of the respective water model. The transition temperature turned out to be systematically lower in 3-point then in 4-point water models (see Results and Discussion). Since the transition temperature on 3-point water models was indeed mostly below the melting temperature of 4-point water models, we could not perform simulations in the same range of temperatures for 3- and 4-point waters models. Furthermore, due to the different temperature range of 3- and 4-point water models, we adapted different simulation lengths. As convergence generally takes longer at low temperatures, we simulated 12 μs in all simulations with 3-point water models and 8 μs in all simulations with 4-point water models.
2.3 Water models
We performed simulations of PNIPAM in eight different rigid water models. We selected four 3-point and four 4-point water models. Namely, we chose OPC3 (Izadi and Onufriev, 2016), SPC(Berendsen et al., 1981), SPCE (Berendsen et al., 1987) and TIP3P (Jorgensen, 1981) on the one hand; and OPC (Izadi et al., 2014), TIP4P-2005 (Abascal and Vega, 2005), TIP4P-EW (Horn et al., 2004) and TIP4P-ICE (Abascal et al., 2005) on the other hand. All these models have been verified to be useful computational models for water in macromolecular simulations. Investigating an even larger number of water models was simply infeasible in terms of computational cost.
In earlier studies, we found that the solvent is strongly bound to the polymer (Quoika et al., 2020). Furthermore, we reasoned that the solvent is thereby immobilized at the surface of the polymer. From that we concluded that the strongly bound water molecules in the solvation shell of the polymer lead to an entropic penalty. The absolute value of this penalty depends on the solvation shell volume, which is different for coil and globule conformations. Generally, it would be best to directly calculate the entropy of solvation of the two states and evaluate the difference. However, this is generally challenging to compute, especially for very flexible molecules or diverse molecular ensembles. We come back to this topic in the results and discussion section. It has been shown by Dzugutov (1996) that the bulk entropy in liquids can be related to the diffusion coefficient. This author showed a universal relationship between the diffusion coefficient and the two-particle entropy of monoatomic liquids. Thus, we assume that the bulk entropy of water in molecular simulations is similarly related to the translational diffusion coefficient, which has also been investigated by Saha and Mukherjee (2017). Hence, we calculated the translational diffusion coefficient of water as a measure of the bulk entropy of the solvent at different temperatures in different water models. We calculate the 3-dimensional diffusion coefficient with the Einstein relation:
where ⟨r(t)2⟩ is the mean squared displacement of water molecules in the simulation. We calculated the diffusion coefficient from separate simulations of pure water, with equivalent simulation setup as the simulations with polymer.
In the analysis of our results, we evaluated the correlation of the estimated Coil-Globule transition temperatures (see below) with various molecular descriptors of the water models. In the course, we also investigated the quadrupole moment as such descriptor. Generally, the quadrupole moment is a tensor. However, the quadrupole moment of water may be approximated by a single number, QT (Rick, 2004; Abascal and Vega, 2007; Niu et al., 2011; Stone, 2013; Izadi et al., 2014), which is sometimes referred to as the tetrahedral quadrupole moment (Carnie and Patey, 1982). In this common approximation, the linear component (along the dipole vector) of the traceless quadrupole tensor is neglected, as it is much smaller. More details may be found in the SI, section C, where we also provide a table with QT of the here-used water models.
2.4 Assignment of conformational states
To determine the equilibrium of the conformational transition, we have to assign arbitrary polymer conformations to either of the two conformational states, i.e., coil and globule. These states are defined by their radius of gyration, which is unambiguous with respect to the conformation. We show examples for coil and globule conformations in the SI, section A. To assign arbitrary conformations to one of the two states, we followed the same approach as in previous studies (Quoika et al., 2021). Accordingly, we classified polymer conformations by means of a two-state Markov model of the conformational transition of the polymer. These models were built, based on the radius of gyration and solvent accessible surface area (Abbott et al., 2015), as described in Podewitz et al. (2019); Quoika et al. (2020), Quoika et al. (2021).
2.5 Assessment of the Coil-Globule transition temperature
We wanted to compare the thermosensitive character of PNIPAM in different water models. Therefore, we evaluated the temperature-dependence of the Coil-Globule transition. To this end, we determined the Coil-Globule transition temperature, T*, in the simulations with water models. Thus, we estimated the equilibrium constant of the Coil-Globule transition at different temperatures and fitted the temperature-dependence according to the van’t Hoff equation (van’t Hoff, 1884), as in (Quoika et al., 2021). We provide an extensive explanation of this fitting procedure in the SI, section D. Hence, we ascribed every polymer conformation to either of the conformational states, i.e., coil or globule (see above). Subsequently, we calculated the equilibrium constant between these two states as
The equilibrium constant is related to the free energy of the process, by
We quantified the uncertainty of the estimated T* by trajectory splitting and leave-one-out cross-validation. The results of this leave-one-out cross-validation are shown in SI, section D.
2.6 Quantification of polymer-solvent interactions
We expected the polymer-solvent interactions to be of particular importance for the energetic balance of the Coil-Globule transition. Thus, we quantified these interactions for every water model. Hence, we calculated the mean potential energy between polymer and water in both conformational states (coil and globule) and computed the difference, as ΔHps(T). To this end, we followed the same approach as in previous publications (Quoika et al., 2020): To obtain this potential energy, we calculated the non-bonded interactions between polymer and solvent for all frames in our simulation. Furthermore, we separated the trajectory into coil and globule state (see above). Thus, we obtained a distribution of interaction energies for the two states. Further, we subtracted the mean interaction energies to obtain the difference between coil and globule states with respect to the polymer-solvent interactions. Analogously, we calculated the polymer-polymer and solvent-solvent interaction energies, ΔHpp and ΔHss. A similar approach has also been followed by Dalgicdir et al. (2017). Generally, this analysis corresponds to the thermodynamics at infinite dilution (see above).
Consistent with earlier studies we found that ΔHps only has a negligible dependence on temperature (within the investigated range of temperatures) as long as both states are reasonably well sampled at the respective temperature (Quoika et al., 2020). Thus, we determined the average interaction potential between water and polymer, ΔHps, as the median over all temperatures.
2.7 Polymer-solvent affinity–potential of mean force
We compared the polymer-solvent affinity in different water models. To this end, we calculated the potential of mean force between polymer and water as Boltzmann inversion of the radial distribution function (Chandler, 1987):
We analyzed the radial distribution function between all heavy atoms of the polymer and oxygen atoms of water. The resulting potential of mean force, therefore, originates from interactions between water and various polymer atom types, e.g., amide oxygen and nitrogen. The complete set of heavy-atoms of the polymer also includes the carbon atoms of the polymer. Since the carbon atoms are less polar than oxygen and nitrogen of the polymer, the interactions with water are much weaker.
Generally, the potential of mean force between the heavy atoms of the polymer and the solvent oxygen describes the solvent affinity of the surface of the polymer. Thus, it is related to the free energy of solvation. However, as we took a complex conformational ensemble of the polymer into account, the volume of the solvation shell varies in our simulation. Thus, it is challenging to estimate absolute values for solvation free energies from this quantity.
To enable a comparison between simulations, we integrated the potential of mean force up to a distance of 4.25 Å, which is the second local maximum in the radial distribution function of this set of atoms. Below, we refer to this integral of the potential of mean force as solvent affinity of the polymer surface, ω. Hence, we compared ω(T) in different water models. This value corresponds to the effective first solvation shell of the polymer at different temperatures. For clarity, we show this radial distribution function for an exemplary simulation in the SI, section F, specifically Supplementary Figures S13, S14. There, we also provide a more detailed explanation of this quantity.
2.8 Persistence length of the polymer
To compare the influence of the water model in the simulation on the mechanical properties of the polymer, we calculated the persistence length of the polymer. To this end, we followed the same approach as in earlier studies (Quoika et al., 2020). Accordingly, we calculated the persistence length from the mean bending angle of the backbone of the polymer. This calculation is explained in more detail in the SI, section E. As a result, we achieve the persistence length of the polymer at different temperatures with different water models. We compared these results to comprehend the temperature-dependence of the bending flexibility of the polymer with different water models.
3 Results and discussion
3.1 Transition temperature in different water models
We show the estimated Coil-Globule transition temperatures, T*, in different water models in Figure 1. While the thermosensitivity is generally reproduced in all of the here-used water models, we found that T* varies significantly. Thus, the apparent thermosensitive character of the polymer strongly depends on the water model in the simulations, which agrees with other studies (Tavagnacco et al., 2022). We find that T* is generally higher for 4-point water models than for 3-point water models. Furthermore, we notice a particularly high uncertainty for the estimation of T* in TIP3P. Unfortunately, T* is particularly low in this water model. It is challenging to achieve highly accurate results at such low temperatures, because of the increasingly slow transition time scales. Generally, we invested significantly more simulation time than most other studies with atomic resolution and our results are consistent with prior publications (Kang et al., 2016; Adroher-Benítez et al., 2017; Quoika et al., 2020; Quoika et al., 2021).
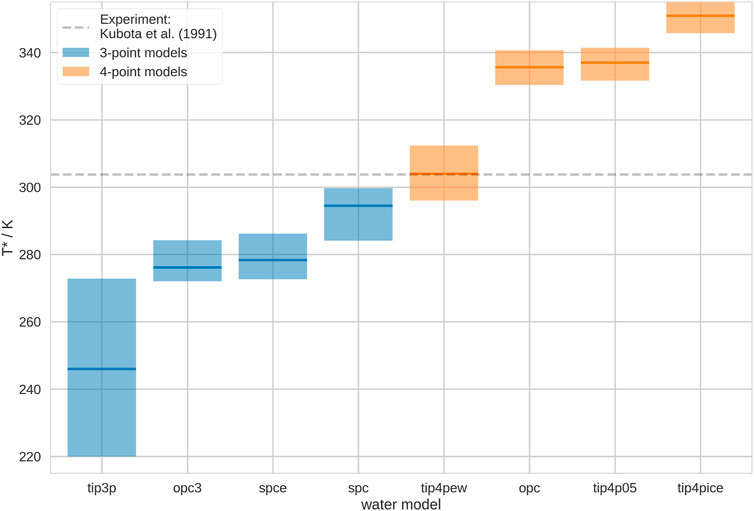
FIGURE 1. Coil-Globule transition temperature, T*, in different water models. We show the mean transition temperature with a min./max. uncertainty estimate from leave-one-out cross-validation. The water models are ordered according to mean T*. 3-point water models are shown in blue, 4-point water models are shown in orange. These results correspond to infinite dilution. As grey dashed line, we show the experimental transition temperature T* = 303.74 ± 0.05 K, as reported by Kubota et al. (1990).
Our results suggest that the water model may shift the temperature-dependent conformational ensemble of the polymer. Such behavior has only rarely been discussed in the literature (Rukmani et al., 2019). Clearly, such water model-dependent shift in the ensemble does not need to occur for all macromolecules in molecular simulations. However, similar effects may occur for processes that are particularly sensitive to hydration effects, alike the (thermosensitive) Coil-Globule transition. For example, Anandakrishnan et al. (2019) found that protein folding processes are sensitive to the water model in molecular simulations, and also other authors came to a similar conclusion (Best and Mittal, 2010). Furthermore, the behavior of intrinsically disordered proteins (IDPs) has been shown to be significantly influenced by the water model in molecular simulations (Henriques et al., 2015; Piana et al., 2015; Shabane et al., 2019). Moreover, Calvelo et al. (2021) describe an effect of water models on transmembrane cyclic peptide nanotubes.
Comparing our results with literature values, we notice that Tavagnacco et al. (2018) also performed simulations of the Coil-Globule transition of PNIPAM in TIP4P-ICE. However, they observed a significantly different transition temperature. They report a value of T* = 303 K, thus, almost 50 K lower than our result. However, there are a few decisive differences between their simulations and ours. Firstly, they used an atactic polymer chain in their simulation. It has been shown that the tacticity may have a significant effect on the thermosensitive character of polymers Ito and Ishizone (2006); Chiessi and Paradossi (2016). Secondly, they used a modified version of the OPLS2005 force field, as implemented by Siu et al. (2012), which may shift the transition temperature. Lastly, they simulated at a different degree of polymerization (DP), i.e., they simulated a 30-mer instead of a 20-mer. It has been shown in experiments that there is a dependence of the thermosensitive character on the DP. Shan et al. (2009) found that higher DP leads to lower transition temperatures, which would be consistent with the comparison of our results with Tavagnacco et al. (2018).
We compare our results with the measurements of Kubota et al. (1990), who performed light scattering experiments of PNIPAM. They report T* = 30.59°C ± 0.05 °C. Based on our results, in combination with the OPLS2005 force field, TIP4P-EW seems to reproduce the experimental behavior best. However, this does not allow general statements of the quality of this water model for the reproduction of thermosensitive conformational transitions. The here-observed results originate from the interplay of polymer force field and water model. As soon as another polymer force field is combined with TIP4P-EW, T* may shift (Walter et al., 2010; Kamath et al., 2013). We picked OPLS 2005, as it is a general purpose force field, which has been established for this system. However, there is evidence that these polymer force field parameters may need further optimization (Dalgicdir and van der Vegt, 2019) (further discussion below). This does not change the conclusions of our study, as we wanted to make a point about the generally significant impact of the choice of water model on such processes as the thermosensitive Coil-Globule transition.
Apart from the water model, the polymer force field parameters may also have a critical impact on T* in molecular simulations. For example, Dalgicdir and van der Vegt (2019) showed that a change in the partial charges of the polymer leads to a significant shift of T* of the PNIPAM 40-mer in SPCE. More specifically, they found that scaling the partial charges of the OPLS force field by a factor of 1.31 leads to such shift. Unfortunately, they do not report a precise value for the shift of T* due to this change of the charges, but it should be significantly more than 20 K (Without this modification they did not find a thermosensitive transition at all, within their investigated range of temperatures.) Their scaling approach was inspired by studies on the driving force of the pressure-induced aggregration of PNIPAM (Mochizuki et al., 2016a) and of the liquid–liquid phase separation of N-isopropylpropionamide, (Mochizuki et al., 2016b). We want to stress that these authors scaled all partial charges of the polymer, since their goal was to investigate the effect of the polar interactions on the aggregation behavior of these polymers. We would argue that the partial charges on the amide group are most important for the Coil-Globule transition of PNIPAM, because of the hydrogen bond interaction sites. Therefore, the partial charges of these atoms, namely amide-nitrogen and amide-oxygen, are presumably most critical. In our framework of explanation, a scaling of the partial charges of the polymer would lead to a change in two components of the enthalpy, namely ΔHpp and ΔHps. (Further discussion of these components of the transition enthalpy is given below.) Accordingly, such scaling would lead to stronger polymer-polymer interactions, which drive the transition. However, at the same time, it would lead to stronger polymer-solvent interactions, which prohibit the transitions. Which of these effects would be stronger is challenging to predict. Our assumption would be that an increase in partial charges leads to higher T*, since ΔHps is generally larger in magnitude. This assumption is in-line with the results of Dalgicdir and van der Vegt (2019). Up to now, we did not investigate the effect of the polymer force field on T*. Unfortunately, a systematic investigation of the force field parameters requires a lot of computational effort, since many long simulations are necessary (depending on the desired temperature-resolution). Furthermore, to make general statements, multiple parameter sets would need to be tested.
3.2 Persistence length
We found that the persistence length, P, and thus, the mechanical properties of the polymer, depend on the water model as well. We show P at different temperatures for different water models in Figure 2. Here, we show the fitting lines of P in dependence of temperature. The raw data and the underlying linear fits are shown in Supplementary Figure S12. For all water models, P decreases, as temperature increases. Accordingly, the flexibility of the polymer is generally higher at high temperatures, which agrees with the expectation (Bresler and Frenkel, 1943; Geggier et al., 2011). Furthermore, we notice that P is systematically higher in 4-point than in 3-point water models.
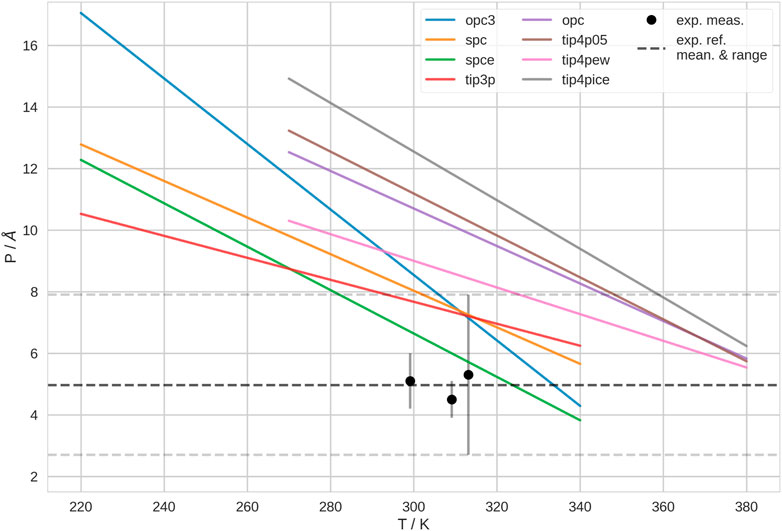
FIGURE 2. Persistence length, P, of PNIPAM at different temperatures, T, in different water models. 3-point water models are listed in the left column of the legend; 4-point water model are listed in the middle column of the legend. Here, we show the fitting lines, of linear fits of P in dependence on T. The raw data with the underlying fits is shown in Supplementary Figure S12. We show the experimentally determined persistence lengths of PNIPAM as black points (Kutnyanszky et al., 2012). As dashed lines, we show average and range of the experimental measurements as guide to the eye.
Generally, the experimental determination of P is not trivial. Indeed, the literature data for P of PNIPAM is inconsistent and controversial (Zhu and Chen, 2019). The results depend on many factors, such as experimental technique, but also solvent conditions and especially polymer preparation. Sometimes, authors compare P of PNIPAM in hydrogels with P of single polymer chains. We believe that this is not a fair comparison. Thus, we decided to compare with Kutnyanszky et al. (2012), as they measure single molecules of PNIPAM in pure water. They performed single molecule force spectroscopy experiments on PNIPAM and determined P = 5 ± 2.6 Å. Furthermore, they found that P does not change significantly between 26°C–40°C (≃ 299–313 K) or at various solvent conditions. Thus, their reported average value is derived from measurements in various solvents. In Figure 2, we show their measurements in pure water for reference. Accordingly, Kutnyanszky et al. (2012) found that the persistence length is not significantly different below and above the transition temperature. Unfortunately, these authors did not measure outside of this temperature range. Also, we did not find any other experimental study where P of PNIPAM was determined over a wider range of temperatures. Thus, it is not clear if the temperature-dependence of P is stronger in our simulations than in experiment, eventually. According to this comparison, the polymer is stiffer in our simulations than in experiments, i.e., the polymer shows significantly higher P, over a wide temperature range. In 3-point water models, P is only in the experimental range above 300 K. Whereas, in 4-point water models, this only happens above 330 K and higher. Hence, with respect to P, the 3-point water models generally yield better agreement with the experimentally determined property of the polymer.
3.3 Polymer-solvent interactions
In Figure 3, we show the polymer-solvent affinity, ω, as quantified by the potential of mean force in the first effective solvation shell. We find that ω is generally low for 4-point water models, which translates to high affinity (lower potential of mean force). Thus, the polymer has, e.g., a particularly high affinity to TIP4P-ICE, while it has a particularly low affinity to TIP3P, which is consistent with the results of Tavagnacco et al. (2022). Of course, the polymer-solvent affinity also depends on the polymer force field (see above). Furthermore, we realize that the ranking of ω follows almost the same order as T* in Figure 1.
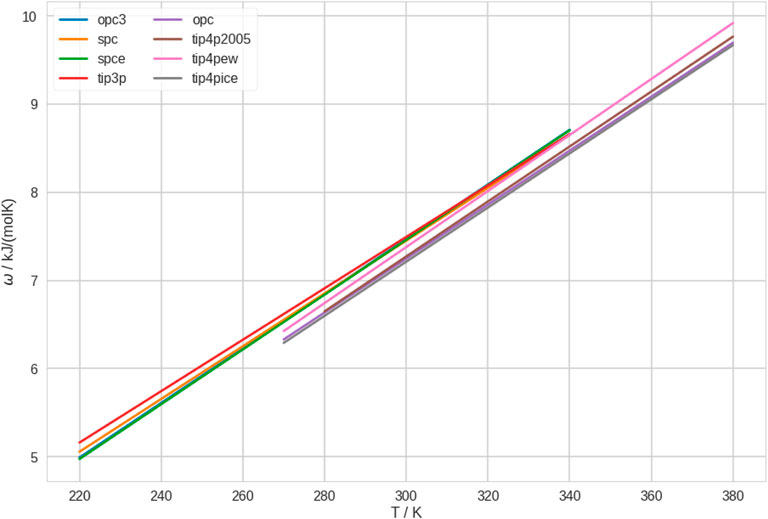
FIGURE 3. Polymer-solvent affinity, ω, in different water models. We quantified this property by the potential of mean force between polymer and solvent. For clarity, we only show the fitting lines here. The independent linear fits with the raw data can be found in Supplementary Figure S15.
In addition, we investigated ω of the polymer in different conformational states. To this end, we calculated the radial distribution function between polymer and solvent separately for the conformational ensembles of coil and globule at all temperatures for all water models. Analogously, we calculated the potential of mean force and integrated the first effective solvation shell. These results are shown in Supplementary Figures S16, S17. We found that ω is generally lower in coil conformations than in globule conformations, for all water models at all temperatures. While this does not yield a discrimination between water models, it confirms our conclusions from earlier studies that coil conformations are stabilized by strong interactions with the solvent. The preferential interactions between water and coil conformations leads to the thermosensitive character of the polymer.
Furthermore, we show the average ΔHps in Figure 4. We notice that ΔHps generally disfavors the Coil-Globule transition. This agrees with our expectation, since the favorable polymer-solvent interactions decrease as the solvation shell volume shrinks. Furthermore, we calculated equivalent quantities for the polymer-polymer and solvent-solvent interactions, ΔHpp and ΔHss. Generally, we expect the water model to have an influence on ΔHps and ΔHss, whereas we expect ΔHpp to be approximately equal in all water models. We found that ΔHpp and ΔHss favor the Coil-Globule transition, which agrees with our expectation. Furthermore, we found that in rough approximation, ΔHpp ≃ΔHss. Accordingly, the polymer-polymer and the solvent-solvent interactions are similar in magnitude, i.e., polymer-polymer contacts are approx. equally strong than solvent-solvent contacts. This relation is really only approximately true. (More detailed explanation in Quoika et al. (2020) and SI, section L). As a trend, we found that polymer-polymer interactions are actually slightly more favorable. Depending on the water model, this effect is stronger or less strong. Again, this is consistent with our findings in previous studies and agrees with our expectations (Quoika et al., 2020). In contrast, the polymer-solvent interactions are higher in magnitude, in the order of, -ΔHps ≈ ΔHpp+ΔHss. A more detailed discussion can be found in the SI, section L, and in Quoika et al. (2020). Hence, in sum ΔH is small, but not zero. It is known from experiments that Coil-Globule transition of PNIPAM is an endothermic process, ΔH > 0 (Tiktopulo et al., 1994). Consistently, we found that ΔH > 0 in all here-used water models.
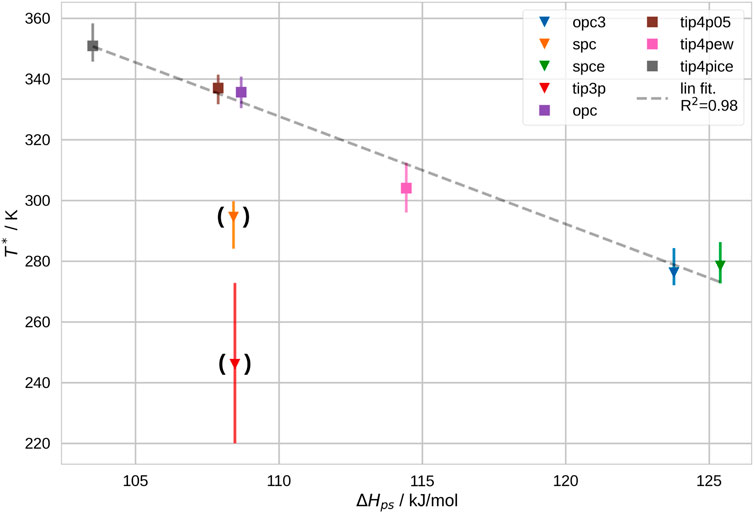
FIGURE 4. Difference between coil and globule states in interaction potential between polymer and solvent, ΔHps, vs. T*. We show a dashed trend line for this correlation in grey. Two points–TIP3P and SPC–are not taken into account for this trend. These results correspond to infinite dilution. The here-shown values correspond to the mean change in potential energy between polymer and solvent, during the Coil-Globule transition of PNIPAM 20-mers in infinitely dilute solution.
We found that ΔHps correlates well with T*, apart from two outliers–i.e., SPC and TIP3P. These are the two least modern models in our set. Our explanation is that the bulk solvent is described differently in these two models, which has an impact on the free energy of the Coil-Globule transition as outlined in the following. In earlier studies, we concluded that a strong immobilization of water molecules at the polymer surface leads to an entropic penalty (Quoika et al., 2020; Quoika et al., 2021). Consequently, the magnitude of this entropic penalty, depends on the immobilization with respect to bulk solvent. To comprehend and compare the magnitude of this entropic penalty, we quantified the lifetime of hydrogen bonds in pure solvent. To this end, we followed an approach described by van der Spoel et al. (2006), which is described in more detail in the SI, section G. Consistently, we found that the lifetime of hydrogen bonds in SPC and TIP3P is lower than in all other water models. These results are shown in Supplementary Figure S18. Another argument for our hypothesis is that the diffusion coefficients of SPC and TIP3P vary from the other water models, as it is known that entropy is linked to the diffusion coefficient (Dzugutov, 1996; Saha and Mukherjee, 2017). We show the diffusion coefficient of different water models at various temperatures in Supplementary Figure S19 and an Arrhenius plot of the diffusion coefficient in Supplementary Figure S20. Indeed, the lifetimes of hydrogen bonds in pure solvent and the diffusion coefficients agree very well. Furthermore, Pascal et al. (2012) also found that the standard molar entropy of SPC and TIP3P is particularly high. The deviation of bulk solvent entropy in these two water models leads to a shift in the energetic balance of the Coil-Globule transition: The magnitude of the change in solvation entropy associated with the Coil-Globule transition is shifted in comparison to the other water models. As a consequence, the balance between entropy and enthalpy is shifted, which leads to a shift in the transition temperature. The absolute shift is challenging to predict, since solvation entropy and enthalpic contributions change simultaneously. However, the shown deviations agree with our expectation: The particularly high bulk entropy in SPC and TIP3P lead to a higher entropic penalty for the stabilization of the coil. Accordingly, the transition temperature is lower in these two water models. We included a more detailed discussion of this effect in the SI, section L.
Unfortunately, it is challenging to quantitatively verify our hypothesis that the change in solvation entropy associated with the Coil-Globule transition varies between water models. To this end, we would need to calculate the difference in solvation entropy of both conformational states (coil and globule) at different temperatures in different water models. While there are multiple tools available to compute solvation thermodynamics, these usually require fixed solute conformations (Nguyen et al., 2012; Velez-Vega et al., 2015; Heinz and Grubmüller, 2021; Waibl et al., 2022). However, in earlier studies, we found that both conformational states are conformationally diverse (Quoika et al., 2021). Thus, we would need to perform a thorough thermodynamic analysis–including solvation thermodynamics–of many conformations of the polymer for both states to obtain reasonable ensemble averages. This procedure would need to be repeated at multiple temperatures with multiple water models. All in all, this would be a complex and costly analysis, which is, unfortunately, beyond scope of this article.
We want to emphasize, that ΔHps does not strictly translate into the general strength of the interactions between polymer and solvent (Hps). While the interactions between polymer and solvent, Hps, may be comparably strong for one particular water model, the mean difference in polymer-solvent interactions, ΔHps, between coil and globule ensemble may be less strong. Generally, we found that the interactions between polymer and 4-point water models are stronger than between polymer and 3-point water models. Still, we found that the ranking of Hps shows almost the same ranking as T*, similar as for ΔHps.
3.4 Correlation between the Coil-Globule transition temperature and force field parameters of the water model
To comprehend the decisive difference between water models for the thermosensitive Coil-Globule transition, we were looking for a descriptor that correlates well with T*. We already developed the understanding that the shift of T* originates from an interplay of polymer-solvent and solvent-solvent interactions. Since we did not change the force field parameters of the polymer, we investigated the molecular properties of the different water models to this end. In the following, we discuss the outcome of this investigation.
3.4.1 Charge and dipole moment
Since the dominant interactions between polymer and solvent, i.e., hydrogen bonds, are of electrostatic nature, we investigated the correlation between the partial charge on the hydrogen atoms of the water models and T*, Supplementary Figure S21. We found a noticeable, yet weak correlation. Accordingly, this quantity does not yield a straightforward description for the shift of T*, neither. Nevertheless, we believe that the partial charges of the water model are decisive for the Coil-Globule transition, however not only the values, but also the (relative) position of the charges. We give some additional discussion on the impact of the partial charges of the water models on the energetic balance of the Coil-Globule transition in the SI section L. Furthermore, we investigated the correlation of the dipole moment of the water models to T*, Supplementary Figure S22. Surprisingly, we did not find any substantial correlation.
3.4.2 Lennard-Jones interaction parameters
Further, we investigated the correlation between T* and the Lennard-Jones (LJ) parameters of the water models. We found that, while ϵ shows a noticable trend, σ does not show any significant correlation with T*. This is not surprising, since σ is very similar for all investigated water models. The correlation of σ and T* may be observed in SI Supplementary Figure S23. We show the correlation between ϵ and T* in Figure 5. We recognize that the majority of the water models scatter at low values for ϵ, between 630 and 680 J/mol and low T*, between 250 and 305 K. The biggest outlier in this group is TIP3P as it generally leads to the lowest T* by far. We believe that this deviation stems from the description of the bulk solvent with this water model (see above; Section 3.3). In contrast, OPC TIP4P-2005 and TIP4P-ICE, have significantly higher ϵ and also lead to significantly higher transition temperatures. In comparison, TIP4P-EW has a much lower ϵ than the other 4-point water models and thus, also produces a lower T*. Furthermore, we notice that TIP4P-2005 and OPC show a very similar transition temperature, despite having rather different ϵ. This deviation may possibly be explained by the difference in bulk entropy of these water models. It is challenging to verify this hypothesis, yet, our analysis of the lifetimes of hydrogen bonds and of the diffusion coefficient in bulk solvent hint in the same direction (SI sections G and H).
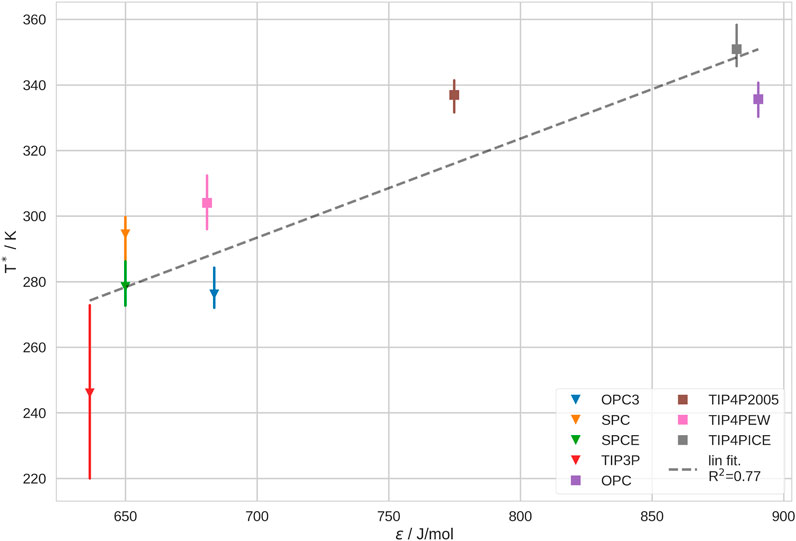
FIGURE 5. Coil-Globule transition temperature, T*, in different water models vs. the Lennard-Jones parameter, ϵ, of the respective water models. We show the estimated mean and min./max. of T* from leave-one-out cross-validation. 3-point water models are depicted as triangles, 4-point water models as squares.
In sum, our results indicate that the Lennard-Jones interaction parameter, ϵ, of the water model has a significant impact on the thermosensitive Coil-Globule transition. Yet, ϵ alone does not yield a reliable prediction of T*. Our observations are in good agreement with studies of conformational ensembles of intrinsically disordered proteins (IDPs). Shabane et al. (2019) found that water models with higher ϵ favor less compact conformations of IDPs. Also, Piana et al. (2015) came to a similar conclusion.
3.5 Coil-Globule transition temperature correlates with quadrupole moment of water model
Finally, we found that the quadrupole moment of the water models, QT, shows a good correlation to T*. This is shown in Figure 6. We explain this result by the fact that QT captures the strength of interactions between polymer and solvent, but also in bulk solvent. It is known that QT is linked to the phase diagram of (in silico) water (Abascal and Vega, 2007). Hence, we hypothesize that the Coil-Globule transition thermodynamics may generally be linked to the phase diagram of the solvent (which may not only be pure water). Unfortunately, complete phase diagrams are not available for all investigated water models (by the time we write this manuscript). Generally, the accurate determination of the phase diagram of water is not trivial in molecular simulations (Sanz et al., 2004; Xiong et al., 2020). Thus, it is beyond scope of this study to obtain the phase diagrams of the newer water models (OPC and OPC3). We want to remark that, while most of the investigated water models reproduce the properties of water decently well at 300 K, their performance varies at substantially different temperatures. Clearly, the parametrization of a rigid water model that performs well over a wide range of temperatures is challenging, since water shows many remarkable properties (Gallo et al., 2016). Possibly, flexible and/or polarizable water models may perform better over wide temperature ranges (Lamoureux et al., 2003; Ren and Ponder, 2004). However, as such simulations are inherently much more costly, this was infeasible for our study, due to the necessity of extensive sampling.
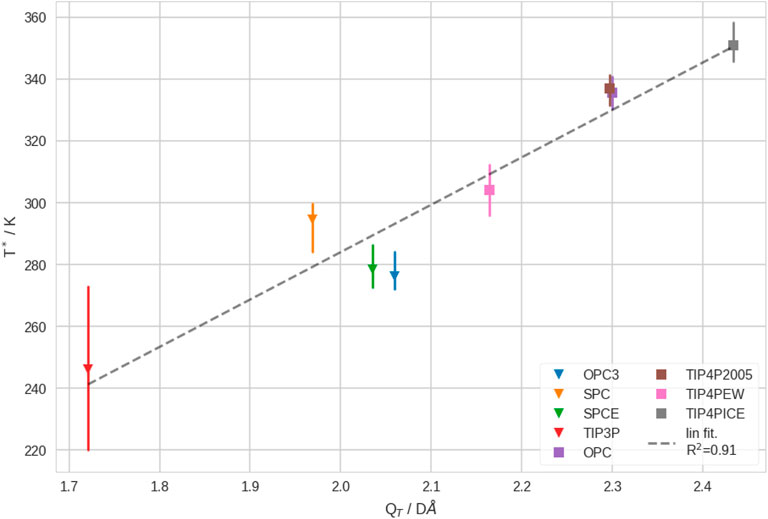
FIGURE 6. Transition temperature, T*, vs. the quadrupole moment, QT, of different water models. We show the estimated mean and min./max. of T*, from leave-one-out cross-validation. Furthermore, we show 3-point water model as triangles and 4-point water models as squares. These results correspond to infinite dilution.
4 Conclusion
While the thermosensitivity is generally preserved, the water model substantially changes the thermosensitive character of polymers in molecular simulations: Only due to the water model, the Coil-Globule transition temperature of PNIPAM may shift up to almost 100 K, i.e., between 255–350 K. Generally, we found that the polymer has higher solvent affinity in coil conformations than in globule conformations. This is true for all water models and this is also the reason for the consistent qualitative reproduction of the thermosensitivity. Thus, we reconfirmed the conclusions of our previous studies that the solvent stabilizes coil conformations at low temperatures. Furthermore, we found that, depending on the water model, the physicochemical properties of the polymer–such as, persistence length and polymer-solvent affinity–vary. Interestingly, we noticed systematic differences between 3-point and 4-point water models in that respect.
As a rule, the transition temperature correlates well with the strength of polymer-solvent interactions. There are two exceptions from this rule, namely SPC and TIP3P. These are the least modern water models in our set. We hypothesize that these two models describe the bulk solvent significantly different than the other water models in our set. Our study of the bulk diffusion and the lifetime of hydrogen bonds in bulk solvent support this hypothesis. This would lead to divergent solvation entropy, which is important for the energetic balance of the thermosensitive Coil-Globule transition. Furthermore, we found that water models with stronger Lennard-Jones interaction parameters favor extended conformations of the polymer, which is in agreement with studies on IDPs. Ultimately, we found that the quadrupole moment of the water model generally correlates well with the thermosensitive transition temperature. This leads to the assumption that the thermosensitivity of the polymer is linked to the phase diagram of the solvent.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
PQ performed simulations, analyses and writing. AK and MF-Q supported the analyses and writing process. KL and MZ provided infrastructure and partly supervised the project.
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No. 764958. Additionally, This work was supported by the Austrian Science Fund (FWF), project Nos. P34518 and P30737. Moreover, the authors gratefully acknowledge the scientific support and HPC resources provided by the Erlangen National High Performance Computing Center (NHR@FAU) of the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) under the NHR project b118bb. NHR funding is provided by federal and Bavarian state authorities. NHR@FAU hardware is partially funded by the German Research Foundation (DFG)—440719683.
Acknowledgments
We would like to thank Dr. Yin Wang for technical support throughout this project. Furthermore, we would like to thank both our reviewers for very constructive and helpful criticism. We believe that their comments have indeed helped a lot to improve this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmats.2023.1005781/full#supplementary-material
References
Abascal, J. L. F., Sanz, E., García Fernández, R., and Vega, C. (2005). A potential model for the study of ices and amorphous water: TIP4P/Ice. J. Chem. Phys. 122, 234511. doi:10.1063/1.1931662
Abascal, J. L. F., and Vega, C. (2005). A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 123, 234505. doi:10.1063/1.2121687
Abascal, J. L. F., and Vega, C. (2007). The water forcefield: Importance of dipolar and quadrupolar interactions. J. Phys. Chem. C 111, 15811–15822. doi:10.1021/jp074418w
Abbott, L. J., Tucker, A. K., and Stevens, M. J. (2015). Single chain structure of a poly(N -isopropylacrylamide) surfactant in water. J. Phys. Chem. B 119, 3837–3845. doi:10.1021/jp511398q
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25. doi:10.1016/j.softx.2015.06.001
Adroher-Benítez, I., Moncho-Jordá, A., and Odriozola, G. (2017). Conformation change of an isotactic poly (N -isopropylacrylamide) membrane: Molecular dynamics. J. Chem. Phys. 146, 194905. doi:10.1063/1.4983525
Anandakrishnan, R., Izadi, S., and Onufriev, A. V. (2019). Why computed protein folding landscapes are sensitive to the water model. J. Chem. Theory Comput. 15, 625–636. doi:10.1021/acs.jctc.8b00485
Aseyev, V., Tenhu, H., and Winnik, F. M. (2011). Non-ionic thermoresponsive polymers in water. Adv. Polym. Sci. 242, 29–89. doi:10.1007/12_2010_57
Berendsen, H. J. C., Grigera, J. R., and Straatsma, T. P. (1987). The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271. doi:10.1021/j100308a038
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., and Hermans, J. (1981). Interaction models for water in relation to protein hydration. Dordrecht, Netherlands: Springer, 331–342. doi:10.1007/978-94-015-7658-1_21
Best, R. B., and Mittal, J. (2010). Protein simulations with an optimized water model: Cooperative helix formation and temperature-induced unfolded state collapse. J. Phys. Chem. B 114, 14916–14923. doi:10.1021/jp108618d
Bilardo, R., Traldi, F., Vdovchenko, A., and Resmini, M. (2022). Influence of surface chemistry and morphology of nanoparticles on protein corona formation. WIREs Nanomedicine Nanobiotechnology 14, e1788. doi:10.1002/wnan.1788
Bischofberger, I., Calzolari, D. C. E., De Los Rios, P., Jelezarov, I., and Trappe, V. (2015). Hydrophobic hydration of poly-N-isopropyl acrylamide: A matter of the mean energetic state of water. Sci. Rep. 4, 4377. doi:10.1038/srep04377
Boţan, V., Ustach, V., Faller, R., and Leonhard, K. (2016). Direct phase equilibrium simulations of NIPAM oligomers in water. J. Phys. Chem. B 120, 3434–3440. doi:10.1021/acs.jpcb.6b00228
Bresler, S. E., and Frenkel, J. I. (1943). Character of the thermal motion of long organic chains, with reference to the elastic properties of rubber. Rubber Chem. Technol. 16, 1–16. doi:10.5254/1.3540099
Bruce, E. E., Bui, P. T., Rogers, B. A., Cremer, P. S., and van der Vegt, N. F. A. (2019). Nonadditive ion effects drive both collapse and swelling of thermoresponsive polymers in water. J. Am. Chem. Soc. 141, 6609–6616. doi:10.1021/jacs.9b00295
Calvelo, M., Lynch, C. I., Granja, J. R., Sansom, M. S. P., and Garcia-Fandiño, R. (2021). Effect of water models on transmembrane self-assembled cyclic peptide nanotubes. ACS Nano 15, 7053–7064. doi:10.1021/acsnano.1c00155
Carnie, S., and Patey, G. (1982). Fluids of polarizable hard spheres with dipoles and tetrahedral quadrupoles integral equation results with application to liquid water. Mol. Phys. 47, 1129–1151. doi:10.1080/00268978200100822
Chiessi, E., and Paradossi, G. (2016). Influence of tacticity on hydrophobicity of poly(N -isopropylacrylamide): A single chain molecular dynamics simulation study. J. Phys. Chem. B 120, 3765–3776. doi:10.1021/acs.jpcb.6b01339
Dalgicdir, C., Rodríguez-Ropero, F., and van der Vegt, N. F. A. (2017). Computational calorimetry of PNIPAM cononsolvency in water/methanol mixtures. J. Phys. Chem. B 121, 7741–7748. doi:10.1021/acs.jpcb.7b05960
Dalgicdir, C., and van der Vegt, N. F. A. (2019). Improved temperature behavior of PNIPAM in water with a modified OPLS model. J. Phys. Chem. B 123, 3875–3883. doi:10.1021/acs.jpcb.9b01644
Deshmukh, S. A., Li, Z., Kamath, G., Suthar, K. J., Sankaranarayanan, S. K., and Mancini, D. C. (2013). Atomistic insights into solvation dynamics and conformational transformation in thermo-sensitive and non-thermo-sensitive oligomers. Polymer 54, 210–222. doi:10.1016/j.polymer.2012.11.009
Dill, K. A. (1985). Theory for the folding and stability of globular proteins. Biochemistry 24, 1501–1509. doi:10.1021/bi00327a032
Dzugutov, M. (1996). A universal scaling law for atomic diffusion in condensed matter. Nature 381, 137–139. doi:10.1038/381137a0
Gallo, P., Amann-Winkel, K., Angell, C. A., Anisimov, M. A., Caupin, F., Chakravarty, C., et al. (2016). Water: A tale of two liquids. Chem. Rev. 116, 7463–7500. doi:10.1021/acs.chemrev.5b00750
García, E. J., and Hasse, H. (2019). Studying equilibria of polymers in solution by direct molecular dynamics simulations: poly(N-isopropylacrylamide) in water as a test case. Eur. Phys. J. Special Top. 227, 1547–1558. doi:10.1140/epjst/e2018-800171-y
Geggier, S., Kotlyar, A., and Vologodskii, A. (2011). Temperature dependence of DNA persistence length. Nucleic Acids Res. 39, 1419–1426. doi:10.1093/nar/gkq932
Heinz, L. P., and Grubmüller, H. (2021). Per|Mut: Spatially resolved hydration entropies from atomistic simulations. J. Chem. Theory Comput. 17, 2090–2098. doi:10.1021/acs.jctc.0c00961
Henriques, J., Cragnell, C., and Skepö, M. (2015). Molecular dynamics simulations of intrinsically disordered proteins: Force field evaluation and comparison with experiment. J. Chem. Theory Comput. 11, 3420–3431. doi:10.1021/ct501178z
Heskins, M., and Guillet, J. E. (1968). Solution properties of poly(N-isopropylacrylamide). J. Macromol. Sci. Part A - Chem. 2, 1441–1455. doi:10.1080/10601326808051910
Hess, B., and van der Vegt, N. F. A. (2006). Hydration thermodynamic properties of amino acid analogues: A systematic comparison of biomolecular force fields and water models. J. Phys. Chem. B 110, 17616–17626. doi:10.1021/jp0641029
Hoffman, A. S. (1995). intelligent” polymers in medicine and biotechnology. Macromol. Symp. 98, 645–664. doi:10.1002/masy.19950980156
Horn, H. W., Swope, W. C., Pitera, J. W., Madura, J. D., Dick, T. J., Hura, G. L., et al. (2004). Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 120, 9665–9678. doi:10.1063/1.1683075
Hummer, G. (2007). Water pulls the strings in hydrophobic polymer collapse. Proc. Natl. Acad. Sci. 104, 14883–14884. doi:10.1073/pnas.0706633104
Inoue, M., Hayashi, T., Hikiri, S., Ikeguchi, M., and Kinoshita, M. (2019). Mechanism of globule-to-coil transition of poly(N-isopropylacrylamide) in water: Relevance to cold denaturation of a protein. J. Mol. Liq. 292, 111374. doi:10.1016/j.molliq.2019.111374
Ito, M., and Ishizone, T. (2006). Living anionic polymerization ofN-methoxymethyl-n-isopropylacrylamide: Synthesis of well-defined poly(n-isopropylacrylamide) having various stereoregularity. J. Polym. Sci. A Polym. Chem. 44, 4832–4845. doi:10.1002/pola.21583
Izadi, S., Anandakrishnan, R., and Onufriev, A. V. (2014). Building water models: A different approach. J. Phys. Chem. Lett. 5, 3863–3871. doi:10.1021/jz501780a
Izadi, S., and Onufriev, A. V. (2016). Accuracy limit of rigid 3-point water models. J. Chem. Phys. 145, 074501. doi:10.1063/1.4960175
Jorgensen, W. L., Maxwell, D. S., and Tirado-Rives, J. (1996). Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236. doi:10.1021/ja9621760
Jorgensen, W. L. (1981). Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 103, 335–340. doi:10.1021/ja00392a016
Kamath, G., Deshmukh, S. A., Baker, G. A., Mancini, D. C., and Sankaranarayanan, S. K. R. S. (2013). Thermodynamic considerations for solubility and conformational transitions of poly-N-isopropyl-acrylamide. Phys. Chem. Chem. Phys. 15, 12667. doi:10.1039/c3cp44076a
Kaminski, G. A., Friesner, R. A., Tirado-Rives, J., and Jorgensen, W. L. (2001). Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 105, 6474–6487. doi:10.1021/jp003919d
Kang, Y., Joo, H., and Kim, J. S. (2016). Collapse–swelling transitions of a thermoresponsive, single poly(N -isopropylacrylamide) chain in water. J. Phys. Chem. B 120, 13184–13192. doi:10.1021/acs.jpcb.6b09165
Kubota, K., Shouei, F., and Ando, I. (1990). Solution properties of poly(n-isopropylacrylamide) in water. Polym. J. 22, 15–20. doi:10.1295/polymj.22.15
Kunugi, S., Tada, T., Tanaka, N., Yamamoto, K., and Akashi, M. (2002). Microcalorimetric study of aqueous solution of a thermoresponsive polymer, poly(n-vinylisobutyramide) (PNVIBA). Polym. J. 34, 383–388. doi:10.1295/polymj.34.383
Kutnyanszky, E., Embrechts, A., Hempenius, M. A., and Vancso, G. J. (2012). Is there a molecular signature of the lcst of single pnipam chains as measured by afm force spectroscopy? Chem. Phys. Lett. 535, 126–130. doi:10.1016/j.cplett.2012.03.070
Lamoureux, G., MacKerell, A. D., and Roux, B. (2003). A simple polarizable model of water based on classical Drude oscillators. J. Chem. Phys. 119, 5185–5197. doi:10.1063/1.1598191
Lanzalaco, S., and Armelin, E. (2017). Poly(N-isopropylacrylamide) and copolymers: A review on recent progresses in biomedical applications. Gels 3, 36. doi:10.3390/gels3040036
Liu, P., Freeley, M., Zarbakhsh, A., and Resmini, M. (2022). Adsorption of soft NIPAM nanogels at hydrophobic and hydrophilic interfaces: Conformation of the interfacial layers determined by neutron reflectivity. J. Colloid Interface Sci. 623, 337–347. doi:10.1016/j.jcis.2022.05.010
Mochizuki, K., Sumi, T., and Koga, K. (2016a). Driving forces for the pressure-induced aggregation of poly(n-isopropylacrylamide) in water. Phys. Chem. Chem. Phys. 18, 4697–4703. doi:10.1039/c5cp07674a
Mochizuki, K., Sumi, T., and Koga, K. (2016b). Liquid–liquid phase separation of n-isopropylpropionamide aqueous solutions above the lower critical solution temperature. Sci. Rep. 6, 24657. doi:10.1038/srep24657
Nguyen, C. N., Kurtzman Young, T., and Gilson, M. K. (2012). Grid inhomogeneous solvation theory: Hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril. J. Chem. Phys. 137, 044101. doi:10.1063/1.4733951
Niu, S., Tan, M.-L., and Ichiye, T. (2011). The large quadrupole of water molecules. J. Chem. Phys. 134, 134501. doi:10.1063/1.3569563
Pal, K., Banthia, A. K., and Majumdar, D. K. (2009). Polymeric hydrogels: Characterization and biomedical applications. Des. Monomers Polym. 12, 197–220. doi:10.1163/156855509X436030
Pang, J., Yang, H., Ma, J., and Cheng, R. (2010). Solvation behaviors of N -isopropylacrylamide in water/methanol mixtures revealed by molecular dynamics simulations. J. Phys. Chem. B 114, 8652–8658. doi:10.1021/jp100743k
Pascal, T. A., Schärf, D., Jung, Y., and Kühne, T. D. (2012). On the absolute thermodynamics of water from computer simulations: A comparison of first-principles molecular dynamics, reactive and empirical force fields. J. Chem. Phys. 137, 244507. doi:10.1063/1.4771974
Piana, S., Donchev, A. G., Robustelli, P., and Shaw, D. E. (2015). Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 119, 5113–5123. doi:10.1021/jp508971m
Podewitz, M., Wang, Y., Quoika, P. K., Loeffler, J. R., Schauperl, M., and Liedl, K. R. (2019). Coil–globule transition thermodynamics of poly(N -isopropylacrylamide). J. Phys. Chem. B 123, 8838–8847. doi:10.1021/acs.jpcb.9b06125
Quoika, P. K., Fernández-Quintero, M. L., Podewitz, M., Hofer, F., and Liedl, K. R. (2021). Implementation of the freely jointed chain model to assess kinetics and thermodynamics of thermosensitive coil–globule transition by Markov states. J. Phys. Chem. B 125, 4898–4909. doi:10.1021/acs.jpcb.1c01946
Quoika, P. K., Podewitz, M., Wang, Y., Kamenik, A. S., Loeffler, J. R., and Liedl, K. R. (2020). Thermosensitive hydration of four acrylamide-based polymers in coil and globule conformations. J. Phys. Chem. B 124, 9745–9756. doi:10.1021/acs.jpcb.0c07232
Ren, P., and Ponder, J. W. (2004). Temperature and pressure dependence of the AMOEBA water model. J. Phys. Chem. B 108, 13427–13437. doi:10.1021/jp0484332
Rick, S. W. (2004). A reoptimization of the five-site water potential (TIP5P) for use with Ewald sums. J. Chem. Phys. 120, 6085–6093. doi:10.1063/1.1652434
Rizzi, E., Deligne, C., Dehouck, L., Bilardo, R., Sano, Y., Shimizu, F., et al. (2021). A triple culture cell system modeling the human blood-brain barrier. J. Vis. Exp. (171), e63134. doi:10.3791/63134
Rukmani, S. J., Kupgan, G., Anstine, D. M., and Colina, C. M. (2019). A molecular dynamics study of water-soluble polymers: Analysis of force fields from atomistic simulations. Mol. Simul. 45, 310–321. doi:10.1080/08927022.2018.1531401
Saha, D., and Mukherjee, A. (2017). Connecting diffusion and entropy of bulk water at the single particle level. J. Chem. Sci. 129, 825–832. doi:10.1007/s12039-017-1317-z
Sanz, E., Vega, C., Abascal, J. L. F., and MacDowell, L. G. (2004). Phase diagram of water from computer simulation. Phys. Rev. Lett. 92, 255701. doi:10.1103/PhysRevLett.92.255701
Schild, H. G., Muthukumar, M., and Tirrell, D. A. (1991). Cononsolvency in mixed aqueous solutions of poly(N-isopropylacrylamide). Macromolecules 24, 948–952. doi:10.1021/ma00004a022
Shabane, P. S., Izadi, S., and Onufriev, A. V. (2019). General purpose water model can improve atomistic simulations of intrinsically disordered proteins. J. Chem. Theory Comput. 15, 2620–2634. doi:10.1021/acs.jctc.8b01123
Shan, J., Zhao, Y., Granqvist, N., and Tenhu, H. (2009). Thermoresponsive properties of N -isopropylacrylamide oligomer brushes grafted to gold nanoparticles: Effects of molar mass and gold core size. Macromolecules 42, 2696–2701. doi:10.1021/ma802482e
Siu, S. W. I., Pluhackova, K., and Böckmann, R. A. (2012). Optimization of the OPLS-AA force field for long hydrocarbons. J. Chem. Theory Comput. 8, 1459–1470. doi:10.1021/ct200908r
Tavagnacco, L., Zaccarelli, E., and Chiessi, E. (2022). Modeling solution behavior of poly(N -isopropylacrylamide): A comparison between water models. J. Phys. Chem. B 126, 3778–3788. doi:10.1021/acs.jpcb.2c00637
Tavagnacco, L., Zaccarelli, E., and Chiessi, E. (2018). On the molecular origin of the cooperative coil-to-globule transition of poly(N -isopropylacrylamide) in water. Phys. Chem. Chem. Phys. 20, 9997–10010. doi:10.1039/C8CP00537K
Taylor, L. D., and Cerankowski, L. D. (1975). Preparation of films exhibiting a balanced temperature dependence to permeation by aqueous solutions—A study of lower consolute behavior. J. Polym. Sci. Polym. Chem. Ed. 13, 2551–2570. doi:10.1002/pol.1975.170131113
Tiktopulo, E. I., Bychkova, V. E., Ricka, J., and Ptitsyn, O. B. (1994). Cooperativity of the coil-globule transition in a homopolymer: Microcalorimetric study of poly(n-isopropylacrylamide). Macromolecules 27, 2879–2882. doi:10.1021/ma00088a031
van der Spoel, D., van Maaren, P. J., Larsson, P., and Tîmneanu, N. (2006). Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J. Phys. Chem. B 110, 4393–4398. doi:10.1021/jp0572535
van’t Hoff, M. J. H. (1884). Etudes de dynamique chimique. Recl. Des. Trav. Chim. Des. Pays-Bas 3, 333–336. doi:10.1002/recl.18840031003
Vdovchenko, A., Pearce, A. K., Freeley, M., O’Reilly, R. K., and Resmini, M. (2021). Effect of heterogeneous and homogeneous polymerisation on the structure of pNIPAm nanogels. Polym. Chem. 12, 6854–6864. doi:10.1039/D1PY01333E
Velez-Vega, C., McKay, D. J. J., Kurtzman, T., Aravamuthan, V., Pearlstein, R. A., and Duca, J. S. (2015). Estimation of solvation entropy and enthalpy via analysis of water oxygen–hydrogen correlations. J. Chem. Theory Comput. 11, 5090–5102. doi:10.1021/acs.jctc.5b00439
Waibl, F., Kraml, J., Fernández-Quintero, M. L., Loeffler, J. R., and Liedl, K. R. (2022). Explicit solvation thermodynamics in ionic solution: Extending grid inhomogeneous solvation theory to solvation free energy of salt–water mixtures. J. Computer-Aided Mol. Des. 36, 101–116. doi:10.1007/s10822-021-00429-y
Walter, J., Ermatchkov, V., Vrabec, J., and Hasse, H. (2010). Molecular dynamics and experimental study of conformation change of poly(N-isopropylacrylamide) hydrogels in water. Fluid Phase Equilibria 296, 164–172. doi:10.1016/j.fluid.2010.03.025
Wu, C., and Wang, X. (1998). Globule-to-Coil transition of a single homopolymer chain in solution. Phys. Rev. Lett. 80, 4092–4094. doi:10.1103/PhysRevLett.80.4092
Xiong, Y., Shabane, P. S., and Onufriev, A. V. (2020). Melting points of OPC and OPC3 water models. ACS Omega 5, 25087–25094. doi:10.1021/acsomega.0c02638
Keywords: thermosensitive polymer, PNIPAM, coil-globule transition, molecular dynamics simulations, in silico water models, solvation thermodynamics
Citation: Quoika PK, Kamenik AS, Fernández-Quintero ML, Zacharias M and Liedl KR (2023) Water model determines thermosensitive and physicochemical properties of poly(N-isopropylacrylamide) in molecular simulations. Front. Mater. 10:1005781. doi: 10.3389/fmats.2023.1005781
Received: 28 July 2022; Accepted: 02 January 2023;
Published: 19 January 2023.
Edited by:
Tristan Giesa, Constructure GmbH, GermanyReviewed by:
Ester Chiessi, University of Rome Tor Vergata, ItalyAlexey V. Onufriev, Virginia Tech, United States
Copyright © 2023 Quoika, Kamenik, Fernández-Quintero, Zacharias and Liedl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick K. Quoika, cGF0cmljay5xdW9pa2FAdHVtLmRl