 Celso R. C. Rêgo1*
Celso R. C. Rêgo1* Jörg Schaarschmidt1
Jörg Schaarschmidt1 Tobias Schlöder1
Tobias Schlöder1 Montserrat Penaloza-Amion1
Montserrat Penaloza-Amion1 Saientan Bag1
Saientan Bag1 Tobias Neumann2Timo Strunk2
Tobias Neumann2Timo Strunk2 Wolfgang Wenzel1
Wolfgang Wenzel1- 1Karlsruhe Institute of Technology (KIT), Institute of Nanotechnology, Eggenstein-Leopoldshafen, Germany
- 2Nanomatch GmbH, Karlsruhe, Germany
Establishing a fundamental understanding of the nature of materials via computational simulation approaches requires knowledge from different areas, including physics, materials science, chemistry, mechanical engineering, mathematics, and computer science. Accurate modeling of the characteristics of a particular system usually involves multiple scales and therefore requires the combination of methods from various fields into custom-tailored simulation workflows. The typical approach to developing patch-work solutions on a case-to-case basis requires extensive expertise in scripting, command-line execution, and knowledge of all methods and tools involved for data preparation, data transfer between modules, module execution, and analysis. Therefore multiscale simulations involving state-of-the-art methods suffer from limited scalability, reproducibility, and flexibility. In this work, we present the workflow framework SimStack that enables rapid prototyping of simulation workflows involving modules from various sources. In this platform, multiscale- and multimodule workflows for execution on remote computational resources are crafted via drag and drop, minimizing the required expertise and effort for workflow setup. By hiding the complexity of high-performance computations on remote resources and maximizing reproducibility, SimStack enables users from academia and industry to combine cutting-edge models into custom-tailored, scalable simulation solutions.
1 Introduction
In the Industry 4.0 context (Lasi et al., 2014), digital twins are an essential tool for companies based who’s R&D is based on scientific innovation (Posada et al., 2018). The digitalization of a system or a process provides vital information about the real-world scenario in real-time. This enables the efficient identification of bottlenecks, thereby speeding up the product development cycle (Zhu and Geng, 2013; Müller and Däschle, 2018) and, in consequence, saving R&D costs and shortening time-to-market (Mathew et al., 2017). In terms of the physical-chemical processes, the development of digital twins is gaining mainstream attention in the scientific community, especially in materials design (Wu et al., 2020; Ngandjong et al., 2021). In this field considerable efforts are made to build digital twins in order to screen and discover new functional materials e.g., for solar cells (Kim et al., 2021; Octavio de Araujo et al., 2021), batteries (Ponce et al., 2017; Bölle et al., 2019), thermoelectricity (Madsen, 2006; Yao et al., 2021), and catalysis (Mamun et al., 2019; Mamun et al., 2020). A prerequisite for building a useful digitaltwin is the availability of predictive simulation protocols. During the designing process of digital twins, a high level of complexity induces a significant interdisciplinary challenge, especially when the material characteristics need to be described by different scales of materials behavior, demanding, in many cases, multiscale methods. Schaarschmidt et al. (2021) show that workflow frameworks can address those challenges in computational materials design.
In addition to technical challenges, computational modeling of a complex physico-chemical process requires knowledge of different methods. These include, amongst others, density functional theory (DFT), molecular dynamics (MD), kinetic Monte-Carlo (KMC), and or finite element methods (FEM). Different techniques are employed depending on the studied phenomena and the scale at which the system is represented. However, in each method, models typically have many parameters and often require a meticulous manual setup to generate meaningful results. Many applications requiring multiscale or high-throughput calculations for a given system also include executing a large number of simulations. Handling these complex computational protocols with script-based approaches is challenging, especially in simulations where the numerical errors embedded in the codes need to be carefully controlled. Usually, multiple steps are required to handle errors, yet, these are often poorly documented and not standardized, making it challenging to keep track of, even for experienced computational experts, thereby limiting the reuse of these computational protocols even within the same group. Therefore, scientific workflows have been proposed to address these shortcomings and inefficiencies by providing automation, complexity reduction, high-performance computing (HPC) readiness, data reusability, data provenance, and reliability and resilience of formalized workflows. Workflows can describe a complex simulation protocol while only exposing a predefined set of relevant computational parameters to the end-user. Therefore, the general aim of workflow frameworks is to allow the end-users to focus on the science instead of spending time setting up and monitoring individual calculations. Several such frameworks have been proposed to leverage the scientific workflow benefits in the last decade. These include free and commercial solutions such as Fireworks (Jain et al., 2015), AiiDA (Pizzi et al., 2016; Huber et al., 2020; Uhrin et al., 2021), KNIME (Berthold et al., 2008), Pipeline Pilot (Warr, 2012), MyQueue (Hjorth Larsen et al., 2017; Mortensen et al., 2020), Pyiron (Janssen et al., 2019), or AFLOW (Curtarolo et al., 2012), and to name a few.
Next to reducing complexity, another major benefit of workflow frameworks is the improved reproducibility of formalized workflows. Reproducibility is a huge challenge for the scientific community: In 2016, researchers from fields such as biology, medicine, physics, chemistry, and engineering largely failed to reproduce their previously published experiments (Baker, 2016). The transition from theory + experiment to the theory + experiment + computer simulation paradigm (Rodrigues et al., 2021) imposes increasing challenges on the experimental and computational research. The advent of computer simulation in the theoretical sciences introduced further challenges regarding the reproducibility of scientific studies that are not present in purely analytical methods (Rodrigues et al., 2021). In a computational simulation study, five groups were asked to perform the same simulation tasks using eight codes with the same force fields. The initial results were highly inconsistent between the groups and simulation codes. Only after some iterations the outcomes started to become consistent (Schappals et al., 2017). This simple experiment shows that incorrect usage is, in most cases, the source of errors in simulations (Wong-ekkabut and Karttunen, 2016; DeFever et al., 2021). Thus, describing the full simulation in a formalized workflow ensures correct usage and consistency among identical and similar simulations.
As one approach to overcome the issue of reproducibility and leverage the advantages of reusability, transferability, and flexibility concepts, we discuss the SimStack workflow framework here. SimStack enables the rapid prototyping of complex simulation workflows with computational modules from various sources. The transfer of re-usable workflows between groups and researchers allow scientists to perform particular predefined simulations with the same quality as the computational expert who conceived and implemented it.
In this work, we present four workflow applications where SimStack has been employed. These workflows combine typical state-of-the-art methods of materials design to solve and deal with real problems and issues representative of those commonly encountered by researchers in the simulation field using a comprehensive range of methods. The SimStack concepts and their usage, features, and applicability to various fields are illustrated by the selected examples covering Umbrella Sampling, Exciton Dynamics, Dihedral Scan, and Emission spectra of organic molecules. The documentation of those workflows shows additional details on applying the SimStack framework features.
2 The Workflow Framework SimStack
The main goal of all major workflow frameworks is to capture the elements of a complex protocol and automate its execution. Depending on the implementation and target user group, expert knowledge is often required for using the framework, setting it up on local or remote compute resources, or incorporating new simulation methods. In many cases, easy-to-use frameworks are often limited in their flexibility and, therefore, hard to extend to the needs of a specific problem, while flexible frameworks are hard-to-use for inexperienced users. Here we introduce the SimStack framework https://simstack.de/, which addresses the issue by providing an easy-to-use flexible drag and drop graphical user interface (GUI), which is automatically generated for a given set of exposed parameters from a simple file in Extensible Markup Language (XML) format. The usage of the XML description of the user input coupled with a simple templating language enables computational experts and non-experts to provide a GUI for a particular application in a matter of minutes. SimStack connects to remote high-performance computing (HPC) resources and automates data transfer and execution of the entire workflow within the HPC environment. Thus, it facilitates the efficient implementation, adoption, and execution of complex and extensive simulation workflows and enables fast uptake of modeling techniques for advanced materials by researchers in academia and industry. SimStack is developed in a joint project by Nanomatch GmbH and the Karlsruhe Institute of Technology (KIT).
2.1 SimStack Concept
As shown in Figure 1, the SimStack workflow framework is based on a lightweight client-server concept. The client provides a GUI for the end-user to construct, modify, and configure the workflows, submit the workflow to the server component on remote HPC resources, monitor submitted workflows, and browse and retrieve the generated data. Each workflow comprises various building blocks with predefined control elements for a given computational task. The tasks represent discrete steps in the execution of the workflow and are called Workflow Active Nodes (WaNos) within SimStack. The core component of a WaNo is an XML file describing the expected input, configurable parameters, the output generated by the WaNo, and the code to be executed. By drag and drop, the end-user can quickly create a new workflow from the available building blocks or adapt existing workflows to generate a custom-tailored solution for a scientific problem. In order to incorporate the user input, SimStack employs the templating engine Jinja (https://jinja.palletsprojects.com). With this templating approach, specific parameters can be exposed easily via the GUI and included as command line parameters or into script and input file templates, turning a static script into a user-configurable building block with a graphical interface within minutes. This concept enables the simple incorporation of any arbitrary software or script routinely used on HPC resources. Multiple compute backends can be configured within the client. Upon submission of a workflow, the client transfers the data via an SSH connection to the SimStack server on the connected remote machine. The SimStack server subsequently processes the workflow and coordinates the submission of the individual tasks via the local scheduling software and data transfer between the workflow elements. In order to provide broad compatibility with HPC backends, the SimStack server can communicate with all major schedulers and runs as a process of the individual user. Consequently, SimStack can be deployed and used without administrative access to the compute resource.
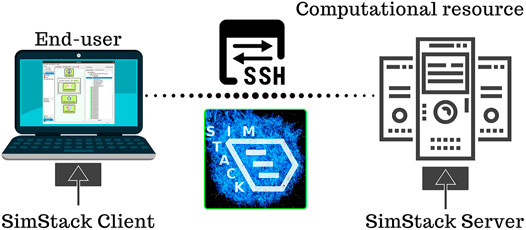
FIGURE 1. The SimStack workflow framework is based on a client-server concept connected via the secure shell (SSH) protocol. The end-user designs and sets up the workflow within the SimStack client on a local machine. The workflow and all required input are transferred to the HPC resource via SSH upon submission. The SimStack server process on the HPC resource subsequently generates a single job for each step of the workflow and manages the execution of these jobs through the local scheduling software.
SimStack aims to be as simple as possible. Every workflow on the HPC resource is processed within a dedicated directory labeled with a submission timestamp and the workflow name. Every WaNo inside this workflow is referenced with a unique path in this directory and is also referenced by a unique ID (UID). The generated data remains at the HPC resource. From within the client, the user can browse this data on the remote resource within a hierarchical structure, view images, text files, and download specific files to his local machine if needed. Besides this, each WaNo can include an automated report in HyperText Markup Language (HTML), providing a concise summary of each workflow step.
2.2 SimStack Documentation
In order to guide users, documentation is made available, continuously updated, and extended at https://simstack.readthedocs.io. The documentation includes instructions on Client installation and configuration, a tutorial exploring the main SimStack features and functionalities like branching workflows and parallelizing high-throughput tasks. The developer section guides how software can be integrated into SimStack via WaNos to build custom workflows or make its own developments available to the community as a SimStack component. It furthermore provides a reference guide for the WaNo XML syntax and available tags. Beyond the documentation, the user can also find an exemplary WaNo available at https://github.com/KIT-Workflows/Test-WaNo. Figure 2 illustrates how the XML tags of this WaNo are translated into input fields of the GUI of SimStack.
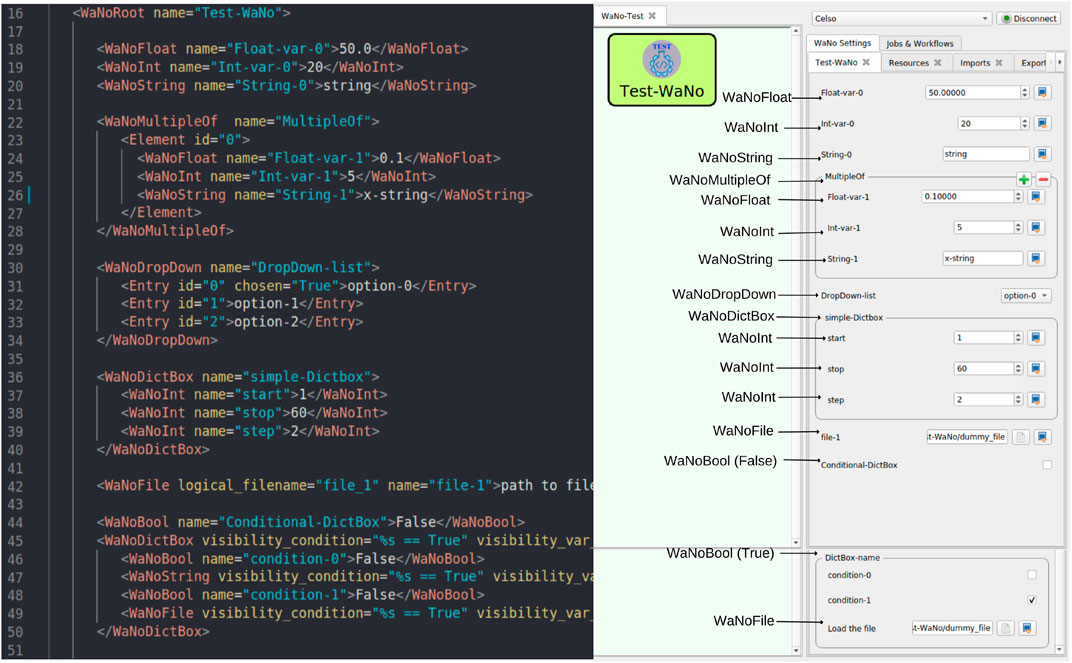
FIGURE 2. This WaNo example shows the XML file and its correspondent GUI. The right side displays the XML file with the tags available within the SimStack workflow framework. On the left side, the arrows associate the tags used to generate the field and variable types of the GUI. The visibility of the second WaNoDictBox in the XML is coupled to the Boolean variable Conditional-DictBox. In this example, the executable is the python script test-script.py.
Workflows designed and pre-configured by experts can be shared with non-expert users, enabling those to conduct high-level simulations with the same quality as expert users. Additionally, all data generated can be made discoverable and accessible either in public or private repositories meeting the FAIR principles (Wilkinson et al., 2016). Finally, all the WaNos and workflows built in the SimStack framework can be extensible, locally tested, shared between researchers, and made transparent regarding their dependencies (Thompson et al., 2020). These features minimize the barrier to transferring scientific state-of-the-art modeling approaches from experts (e.g., academic researchers) to non-experts (e.g., industrial users), thereby boosting the uptake of virtual design approaches.
3 Workflows
This section illustrates the application of the SimStack framework with different workflows implemented within SimStack. Four different exemplary workflows were selected to demonstrate the broad applicability of Simstack and its main features and concepts. The Umbrella Sampling workflow computes the binding free energy of the adsorption of a molecule on surfaces by chaining structure builders, MD code, umbrella sampling, and weighted histogram analysis methods. In the Exciton Dynamics workflow, we present a multiscale simulation approach combining DFT, forcefield-based molecular modeling, and KMC approaches to generate a digital twin of OLED devices. In this workflow, we translate molecular properties to the device scale to determine their impact on the efficiency and lifetime of OLED devices. The Dihedral Scan workflow calculates the dihedral energy potential obtained from MC and DFT calculations, which can be used to parametrize forcefields for MC and MD simulations. The Emission spectra of organic molecules workflow computes fluorescent, phosphorescent, and Thermally Activated Delayed Fluorescence (TADF) molecules to determine their emission wavelength by combing DFT and TDDFT methods.
3.1 Umbrella Sampling
Knowledge about the binding free energy of molecules to different surfaces is of enormous importance in a great variety of applications from natural and engineering sciences (Wagner et al., 2021; Rauwolf et al., 2021; Bag et al., 2021). Umbrella Sampling (US) simulation (Wagner et al., 2021; Rauwolf et al., 2021; Kästner, 2011; Bag et al., 2020; Suyetin et al., 2022) is one of the widely used methods for this purpose. However, performing a US simulation to evaluate the binding free energy (to a given surface) of an arbitrary small molecule requires a complicated simulation routine as depicted in Figure 3A. Starting with a molecular model, one needs to first generate the forcefield parameters for the molecule. The molecular model has to be combined with the predefined surface model thereafter and the combined system has to be solvated and charge neutralized. After equilibrating this system, one has to make many copies of the system for different distances (reaction coordinate) of the molecule from the surface. Each individual system will then be subjected to an equilibration and a production run and the histograms of the reaction coordinates will be collected. In the end, all these histograms have to be analyzed using the Weighted Histogram Analysis Method (WHAM) (Kumar et al., 1992) to get the binding free energy. Therefore, we designed a workflow using the SimStack framework features, to implement the complicated US simulation routine for the calculation of binding free energy of arbitrary small molecules to predefined (silica/graphene) surfaces. The structure of the SimStack Workflow is illustrated in Figure 3B.
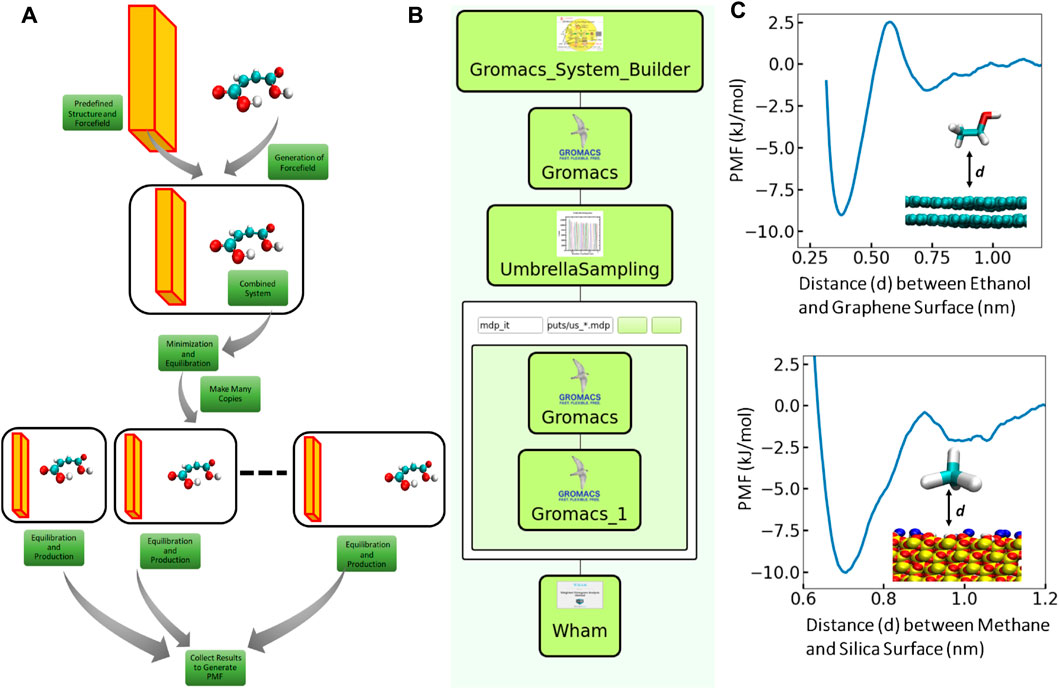
FIGURE 3. (A) Schematic diagram illustrating potential of mean force (PMF) calculation of an arbitrary small molecule to a given surface using US. (B) Structure of the SimStack workflow designed to accomplish the complex PMF calculation described in Figure 3A. We combined four different WaNo to build the complete workflow. (C) PMF profiles calculated using the workflow. PMF profile of ethanol adsorbtion on Graphene are shown in the top panel and the PMF profile between methane and Silica are shown in the bottom panel. Here we have used two layers of Graphene to model the Graphene surface. The atomistic models of the small molecules (ethanol and methane) and the surfaces are shown in the inset.
Here, we combine four different WaNos: 1) GromacsSystemBuilder, 2) Umbrella Sampling 3) Gromacs and 4) Wham. The features and function of the different WaNos in this workflow are described as follows:
1) GromacsSystemBuilder: The WaNo prepares the necessary input files for a Gromacs run (Van Der Spoel et al., 2005). It takes the “pdb” file of the small molecule as input and combines it with the predefined graphene/Silica surface. To generate the forcefield (FF) parameters for the small molecule, the WaNo uses the AmberTools software package (Case et al., 2016). The FF parameters for the surface are also preloaded along with their structure. The combined system is further solvated in water and charge neutralised using standard Gromacs commands (Van Der Spoel et al., 2005). In the end all necessary input files for the Gromacs run are generated. Input: pdb (*pdb) of the small molecule. Output: Gromacs input files (*gro, *top, and *ndx).
2) Umbrella Sampling: The WaNo generates the snippet of the specific gromacs run parameter file for all the US windows. This snippet can be read by the Gromacs WaNo and run the US. The users are supposed to provide the description of the reaction coordinates as input and the WaNo creates all the Windows for the US run. Input: Description of reaction coordinates and umbrella specification. Output: All Umbrella sampling windows (with all the Gromacs input files) and their specific MD run parameter (*mdp) file.
3) Gromacs: This is simply a WaNo to run Gromacs (Van Der Spoel et al., 2005). Input: i) *gro file, ii) *top file, iii) *ndx file, iv) The Gromacs MD run parameters, v) If the gromacs run is an umbrella sampling run then the custom umbrella sampling inputs, vi) Custom forcefield files called in the *top file. Output: i) The binary run parameter file for gromacs (*tpr), ii) The equilibrated system Geometry (*gro). iii) In case of US run, the additional files for the histogram (pullf/pullx files).
4) Wham: This WaNo collects the output from the US run and generates the potential of mean force (PMF). Input: files for Histogram (pullf/pullx files) generated after US. Output: Free energy Curve.
We further use this developed workflow to calculate the free energy of binding of various small molecules to the surfaces (Graphene and Silica). In Figure 3C we show the PMF profile from two such calculations: ethanol binding on Graphene and methane binding on Silica. The free energy of binding of ethanol to the Graphene is ∼ 8 kJ/mol while the corresponding free energy between methane and Silica is ∼ 10 kJ/mol. Although the free energy of binding is very similar for both the systems, the PMF profile for methane (to the Silica) is wider around minima which indicates strong binding affinity of methane in comparison to ethanol. It is evident from 3 (c) that ethanol can come much closer to the Graphene than methane can come to the Silica. Both of the surfaces show essentially no interaction when the molecules are more than 1 nm away from the surface.
3.2 Exciton Dynamics
Modern organic light emitting diodes (OLED) consist of multiple layers of small organic molecules (Li et al., 2017; Wong and Zysman-Colman, 2017; Lee et al., 2019; Zou et al., 2020). To achieve optimal performance and long lifetime of these devices, molecular properties of materials used in a single OLED need to be carefully aligned. While the vast chemical spaces opens the prospect of employing “perfect” material combinations in an OLED, the identification of suitable material pairings via experimental trial and error is time consuming and costly, and especially in the area of blue pixels, OLEDs have to date not been able to exploit their full potential (Scholz et al., 2015; Song and Lee, 2017). One fundamental reason for this shortcoming is the complexity connected to tuning charge carrier and exciton dynamics in OLEDs, which in turn determine efficiency and lifetime: The full system dynamics is a complex consequence of a multitude of microscopic processes (charge hops between molecules, formation of excitons, and excitonic loss processes, etc.) that are determined by microscopic molecular properties (Friederich et al., 2016; Friederich et al., 2017). Further, these properties change when molecules are embedded in thin films, depending on their exact environment, and are therefore hardly accessible experimentally (Bag et al., 2019; Li et al., 2019). To support experimental R&D by deriving fundamental understanding of how microscopic properties determine device performance by triggering and balancing a zoo of microscopic processes, we developed a multiscale simulation approach translating molecular properties to the device scale. This workflow consists of four basic steps, illustrated in Figure 4A. In the first step, customized force-fields are derived for all molecules involved. Subsequently, we run a simulation protocol mimicking physical vapor deposition to generate digital twins of thin films with atomistic resolution. In a third step we perform a full quantum chemical electronic structure analysis of molecules in the thin film morphology to compute molecular properties required for the simulation of charge carrier and exciton dynamics, taking into account environmental effects. Ultimately, we conduct KMC simulations in LightForge, resulting in all-particle trajectories for further analysis of the system dynamics.
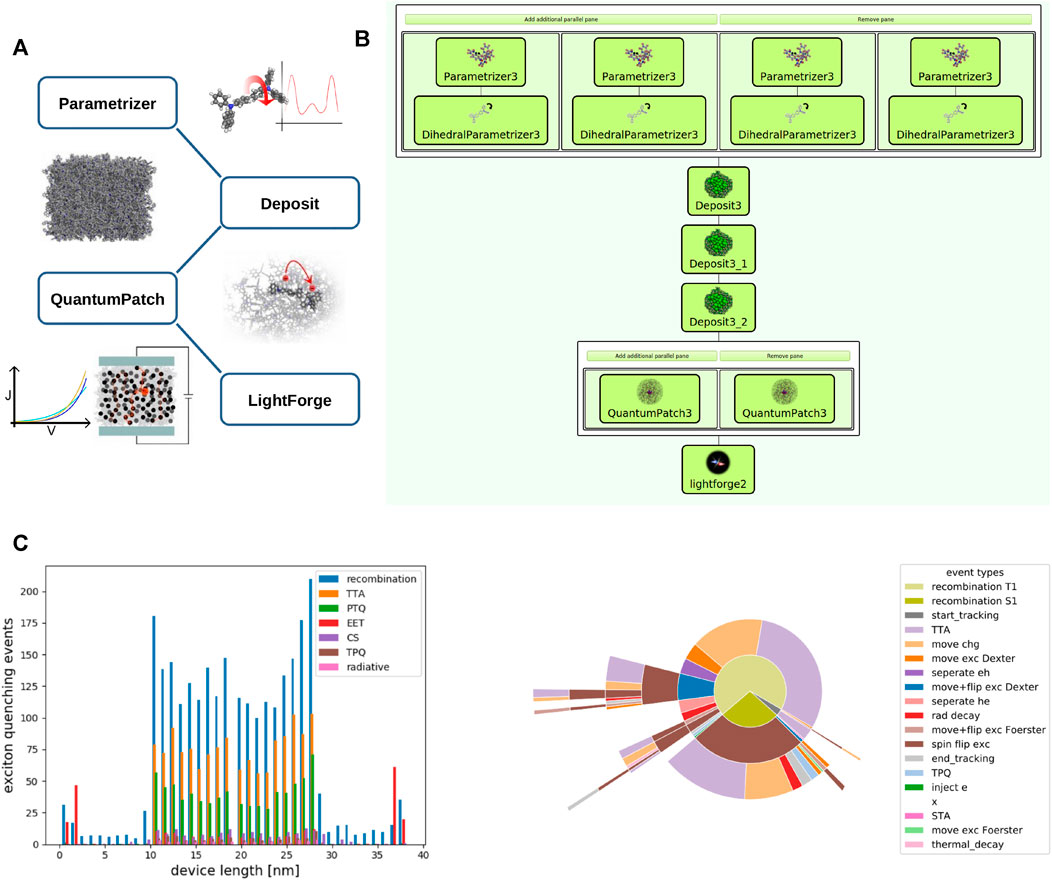
FIGURE 4. (A) Illustration of the multiscale-OLED workflow: Parametrizer: computation of customized molecule specific force-fields; Deposit: simulation of physical vapor deposition to generate digital twins of thin films with atomistic resolution. QuantumPatch: full quantum chemical electronic structure analysis of molecules in the thin film morphology; LightForge: KMC simulations of the system dynamics. (B) The simulation workflow for a three-layer OLED consisting of HTL, EML (mixed guest-host system) and ETL. (C) Exemplified output of LightForge. Left panel: spatial distribution of major excitonic events in the three-layer OLED indicating triplet-triplet annihilation (TTA) as major loss channel. Right panel: Exciton lifecycles in this OLED.
To enable the efficient analysis of a variety of OLEDs with different layer setups and materials we integrated all simulation modules in the workflow platform SimStack. The full workflow for a specific OLED is constructed via drag and drop and may be saved for later re-use. Figure 4B depicts the workflow exemplified for a three-layer OLED, comprising a hole-transport layer (HTL), an emission layer consisting of a host material and an emitter, and an electron-transport layer (ETL): In the first layer we compute customized forcefields for all four materials using “parallel” panels. In addition to the Parametrizer module, we use the DihedralParametrizer module to account for flexibility of molecules. The outputs of each parallel panel (i.e., the forcefield files of a single material) are then passed to the respective deposit modules, where we first deposit the HTL (Deposit3), followed by the deposition of host and emitter of the EML (Deposit3_1) and the deposition of the ETL (Deposit3_2). In each deposition step we define the molecular input from the respective DihedralParametrizer module(s), the size of the simulation box, number of molecules to be deposited and, in the case of the EML, concentrations of the molecular mix, along with certain simulation parameters. Note that each deposited morphology is passed to the next deposition step as a substrate so that the output of the last deposition is a three-layer morphology. Using this three-layer morphology as input, we conduct two independent (and therefore parallel) QuantumPatch computations: We compute electronic couplings in the left panel and energy level distributions in the right panel. Both are required by LightForge to compute rate distributions for microscopic processes. For simplicity, other key quantities such as transition dipoles and further input for quenching rates are set manually in LightForge.
An output of a corresponding parametric simulation using a phosphorescent emitter is exemplified in Figure 4C. The left panel depicts the spatial distribution of major excitonic events, i. e. the count of exciton formation and quenching events over the device cross section. Here we see that most excitons are, as expected, generated in the EML (“recombination”). Further, we find that the major loss channel in the EML is triplet-triplet annihilation (TTA). As this process occurs at high exciton densities, we can derive from this simulation that a reduction of emitter concentration in the EML may increase efficiency. The left panel of Figure 4C depicts the averaged exciton life-cycle for this system. Read from the inside out, we find that almost all singlets (generated by “recombination S1”) undergo a triplet conversion (“spin flip exc”) before they are quenched by triplet- or polaron-quenching (“TTA” and “move chg” respectively).
In this study we implemented a multiscale workflow to simulate charge-carrier and exciton dynamics in multilayer OLEDs in the workflow platform SimStack. This workflow consists of 14 simulation modules with models for different time and length scales. A corresponding manual execution of this workflow via manual file transfer and submission of each individual module would eliminate the advantage that computer simulations pose in OLED design, as it would be time consuming and prone to errors. Instead, the implementation via SimStack provides a re-usable solution that can be adapted within minutes to various OLED setups (different number of layers, layer thicknesses, materials and material combinations, etc.) to maximize the impact of virtual design in OLED development. The exemplified output of this workflows demonstrates how this type of simulation can aid experimental R&D by deriving design rules, in this case reducing emitter concentration.
3.3 Dihedral Scan
It is imperative to perform preliminary optimization steps to generate reliable atomic models and then calculate Physico-chemical properties by applying Molecular Dynamics or Monte Carlo simulations. While it is frequent to use Quantum calculations such as DFT to obtain molecular conformations with high accuracy, depending on the molecule complexity, this approach could lead to local energy conformations. In many molecules, such as conjugated compounds, the most critical term that governs their energetic profiles are their dihedral movements, which configurations could influence their optical absorption and emission properties and their performance during MD simulations (Wildman et al., 2016). Studying different torsions for a given molecule is sensible before performing any parametrization. Dihedral scans using low-level theory calculations can determine global and local energy configurations before applying a final higher-level refinement calculation and reducing the computational cost in search of desired structures. In our recent paper (Penaloza-Amion et al., 2022) we report how the study of dihedrals using DFT scan calculations on a dimer of poly cis-transoid (4-carboxyphenyl) acetylene gave structural insights regarding the clockwise and counterclockwise helical screw-sense.
Following our previous approach, we created the Dihedral-Scan workflow (Montserrat Penaloza-Amion, 2022) (https://github.com/KITWorkflows/Dihedral-Scan) to support the study of torsions for all-atom molecule models as a preliminary step for further studies such as MD or MC simulations. Our workflow consists of the following WaNos: 1) SIMONA-DHscan (Penaloza-Amion, 2022), 2) Range-It, 3) For each loop, 4)UnpackMol, 5) DFT-Turbomole and 6) Table-Generator. As shown in Figure 5A, the first step is to perform a dihedral screening with SIMONA-DHscan. SMILE code or structure coordinates in PDB format are allowed. Using SIMONA (Strunk et al., 2012; Penaloza-Amion et al., 2021) all possible torsions are identified, and dihedral scans on all dihedrals are performed individually. Each scan consists of the arbitrary rotation of the torsion selected and optimizing adjacent torsions using the metropolis MC algorithm. The calculation of the total energy of each configuration generated is based on the Coulomb and Lenard-Jones terms from the General Amber forcefield (GAFF) (Ozpinar et al., 2010). Parameters such as molecule net charge and rotation steps are provided manually. Finally, each scan calculation is scored by the energy difference of the maximum and minimum energies to reveal which torsion has the most significant energy influence in the tested molecule. The outputs provided are 1) scoring list and plots of all torsion profiles calculated, 2) compressed file with all the configurations for the best-scored torsion, and 3) input list with atoms Ids for best dihedral scored for further DFT calculations. The next step is to perform a high-level calculation with DFT-Turbomole 5) on all the structures provided by SIMONA-DHscan. Steps (2), (3), and 4) are needed to support the workflow in extracting the structures inputs and performing parallel calculations of each point for the dihedral profile. Finally, output data can be collected with Table-Generator WaNo to generate an out file containing the data needed to plot the final energy profile, as can be seen in Figure 5C.
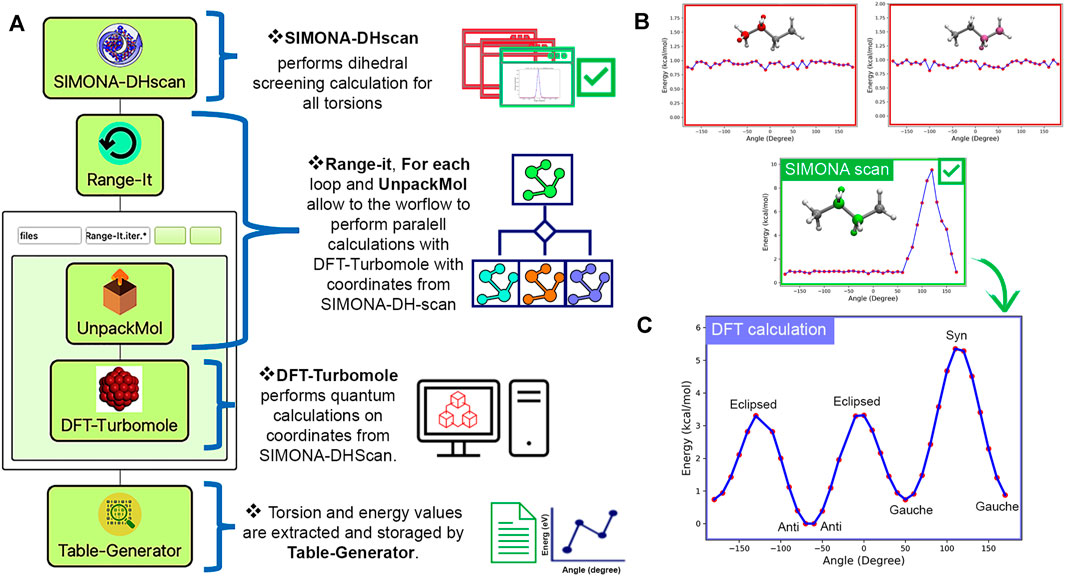
FIGURE 5. (A) Scheme representing the dihedral scan workflow using SIMONA-DHscan, Range-It, ForEach loop, UnpackMol, DFT-Turbomole, and Table-Generator WaNos. (B) Dihedral profiles of n-butane calculated with SIMONA-DHscan. The best profile is highlighted in green, and the negative scores are red. (C) Dihedral profile output obtained after using Dihedral-Scan workflow for n-butane showing syn, eclipsed, gauche, and anti configurations.
To illustrate the Dihedral-Scan workflow, we calculated the dihedral energy profile for n-butane (Figure 5B,C). The n-butane structure is generated by providing a SMILE code in SIMONA-DHscan WaNo. SIMONA identified three dihedrals, providing their respective dihedral profiles (5 (b)). Each SIMONA simulation is performed using the dihedral scan protocol explained before. After the identification of the best-scored torsion (5 (b, green)), the coordinates used to generate the SIMONA dihedral profile are used to feed the quantum calculation using Range-It, UnpackMol, ForEach loop, and DFT-Turbomole WaNos. Each configuration was optimized using the hybrid B3LYP functional (Becke, 1993a; Becke, 1993b) and def2-SV(P) basis set (Zheng et al., 2011). The data obtained after using Table-Generator to extract the angle and total energy values indicate that our workflow can identify the torsion that has the biggest influence on the configuration of n-butane. Additionally, after the refinement calculations using DFT, the energy profile of n-butane reveals the syn, eclipsed, gauche, and anti configurations (5 (c)). Our results showed that Dihedral-Scan could identify torsions, score them, and perform quantum calculations that support future MD or MC simulations.
3.4 Emission Spectra of Organic Molecules
Luminescent molecules have found widespread applications as emitter molecules in OLED devices in which the recombination of electrons an holes leads to the formation of exciton which can—after migration to an emitter molecule—relax to the ground state by emitting a photon. Several types of emitter molecules exist in so far three generation of OLEDs based on fluorescent, phosphorescent and TADF molecule with their respective advantages and drawbacks. When designing new emitter molecules, one important factor (next to other equally important ones as for example the accessibility and stability) is the emission wavelength which corresponds to the color of the molecules.
The computational procedure to determine the emission wavelength of a molecule consists of several DFT and TDDFT calculation steps including structure optimizations of the ground and first excited state as well as the calculation of electronic excitations for both optimized structures. While for most molecules, this task is a routine one for an expert on the underlying DFT code, this is in general not the case for the average user. Furthermore, the repetition of this task for a large set of molecules is time-consuming and prone to errors when done manually even by an experienced user.
We therefore developed a workflow for the execution of this procedure which requires nothing more than the structure of the molecules as an input while other parameters of the (TD) DFT calculation such as e.g., the functional, the basis set or the type of excited states can be easily adjusted if necessary. The workflow is able to loop over a large number of molecules for screening purposes, and also gives the additional option of calculating the ground state IR spectrum of each molecule.
The workflow is structured as follows: The first WaNo (“Prepare-Screening”) creates input files in Gaussian style from a given list of SMILES codes or an archive file containing several structure files. These input files have the advantage over a simple xyz format of containing the desired charge and multiplicity of the molecule and therefore allow to easily calculate spectra for ions as well. After this preparatory WaNo which furthermore gives the options of choosing the type of calculated spectrum, a sequence of DFT calculations is performed using the “DFT-Turbomole” WaNo for each structure file. The first two steps consist of a preoptimization of the structures BP86/def-SV(P) level followed by an optimization at the B3LYP-D3/def2-TZVP level of theory which will be used throughout all the following calculations of the workflow. Depending on the choices made in the initial WaNo, the workflows continues with a DFT frequency analysis, the calculation of the electronic excitation spectrum, and finally an optimization of the first (nth) excited state followed by an electronic excitation spectrum for the structure of the excited state. The final WaNo in the workflow (“Plot-Spectra”) reads in the results from the previous Turbomole calculations which are saved in yml format and plots the Spectra. Figure 6 shows the structure of the workflow (a) as well as an example input file containing the SMILES codes for the three geminal diones Benzil, Biacetyl and 1,2-Cyclohexanedione (b) which was used to generate the UV/Vis-spectrum plots (c).
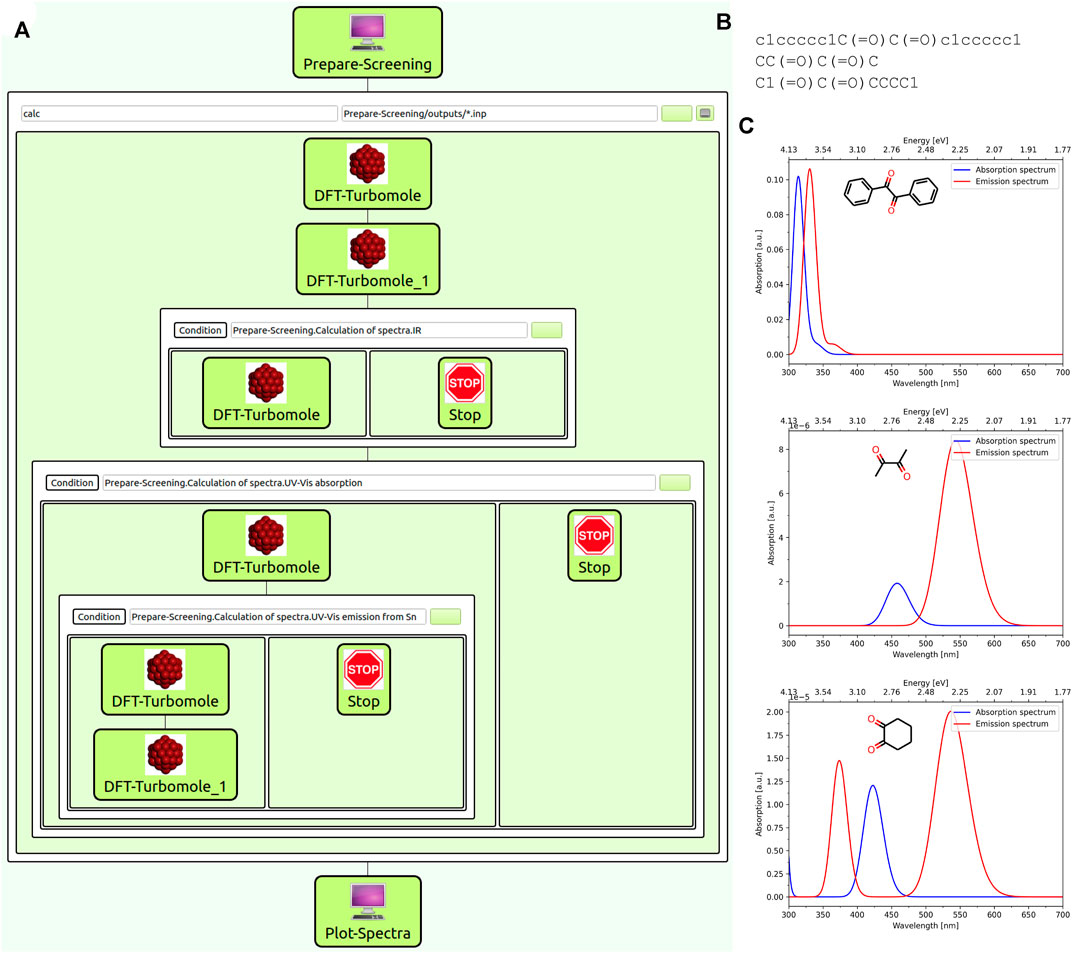
FIGURE 6. (A) Structure of the SimStack workflow for the calculation of UV/Vis emission spectra: The “Prepare-Screening” WaNo allows to choose the types of calculated spectra and creates input files for each molecule. The Following “DFT-Turbomole” WaNos perform the actual DFT and TDDFT calculation after which all information is gathered by the final “Plot-Spectra” WaNo which creates png files for each calculated IR or UV/Vis spectrum. (B) SMILES input file. (C) Calculated UV/Vis spectra at B3LYP/def2-TZVP level.
4 Conclusion and Perspectives
The presented workflow framework SimStack enables rapid prototyping of multi-module simulation workflows to design, implement, and test simulation protocols for various applications. The workflow design steps are carried out interactively via an easy-to-use flexible GUI. Simulation modules from any source are incorporated into SimStack as a simple file XML format, exposing a limited set of application-specific parameters to the end-user. This format enables computational experts and non-experts to provide a GUI for a particular application in a matter of minutes. SimStack connects to remote HPC resources and automates data transfer and execution of the simulation to and from the HPC environment. Pre-defined workflows can be saved for later re-use and transferred among users, enabling a high level of reproducibility and transferability of simulation protocols. This enables the transfer of state-of-the-art scientific simulation approaches from experts to non-experts, boosting the uptake of multiscale modeling approaches.
To show the usage of Simstack features, we presented four workflows tackling different scientific problems. These are real problems frequently found by researchers when designing simulation protocols. Those workflows make heavy use of the ForEach, AdvancedFor, If, and Parallel controls, which are responsible for branching the workflow and parallelize the simulation in the HPC resources. Currently, several WaNos and workflows using standard codes and libraries are supported in SimStack. Such as; SIMONA https://github.com/KIT-Workflows/Dihedral-Scan, VASP (Kresse and Joubert, 1999) https://github.com/KIT-Workflows/DFT-VASP, Quantum Espresso (Giannozzi et al., 2017) https://github.com/KIT-Workflows/DFT-QE, Turbomole (Ahlrichs et al., 1989) https://github.com/KIT-Workflows/DFT-Turbomole, ORCA (Neese, 2011), DFTB + (Hourahine et al., 2020) https://github.com/KIT-Workflows/DFTB-Neural-Net, Fenics (Alnaes et al., 2015) https://github.com/KIT-Workflows/Fenics-mesh, ASE (Hjorth Larsen et al., 2017), PymatGen (Ong et al., 2013), https://github.com/KIT-Workflows/Format-Converter and many in-house solutions https://github.com/KIT-Workflows/Table-Generator. All the WaNos where the same methods are embedded are code interoperable, e.g., workflow running periodic DFT simulations can run with VASP or Quantum Espresso.
Next to the available features and capabilities of SimStack, the software is continuously updated and extended. One of the main upcoming features is the capability to fully or partially restart a workflow.
DATA AVAILABILITY STATEMENT
Additional information on the workflows are available: \textbf(Umbrella Sampling) \url(https://github.com/KIT-Workflows/Umbrella-Sampling), \textbf(Exciton Dynamics) \url(https://github.com/KIT-Workflows/Exciton-Dynamics), \textbf(Dihedral Scan) \url(https://github.com/KIT-Workflows/Dihedral-Scan), and \textbf(Emission spectra of organic molecules) \url(https://github.com/KIT-Workflows/Spectrum-Screening). Documentation related to installing and tutorials of SimStack client are available on \url(https://www.simstack.de/) and \url(https://simstack.readthedocs.io/). SimStack is available at \url(https://www.simstack.de/) in the downloads section.
AUTHOR CONTRIBUTIONS
TS, TN, JS, WW, and CR: SimStack development team; TN and TS: WaNo development (Parametrizer3, DihedralParametrizer3, Deposit3, QuatumPatch3, and lightforge2), researching and writing of the paper; SB and JS: WaNo development (Gromacs_System_Builder, Gromacs, UmbrellaSampling, and Wham), researching and writing of the paper. TS WaNo and Workflow development (Prepare-Screening, DFT-Turbomole, Plot-Spectra, and Spectrum-Screening), writing of the paper; MP-A and CR: WaNo development (SIMONA-DHscan, Range-It, and UnpackMol), researching and writing. CR, JS, and WW: concepts organization, researching, funding acquisition, paper writing, and review.
FUNDING
JS, WW, and CR thank the German Federal Ministry of Education and Research (BMBF) for financial support of the project Innovation-Platform MaterialDigital (www.materialdigital.de) through project funding FKZ no: 13XP5094A. MP-A acknowledge the financial support by the Deutscher Akademischer Austauschdienst (DAAD) within the scholarship for Doctoral Research (91683960). TS further acknowledges funding by the “Virtual materials design” (VirtMat) initiative at KIT within the Joint Lab VMD funded by the Helmholtz society.
Conflict of Interest
TN, TS, and WW are shareholders of Nanomatch GmbH, which intends to commercialize SimStack for commercial usage.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahlrichs, R., Bär, M., Häser, M., Horn, H., and Kölmel, C. (1989). Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 162, 165–169. doi:10.1016/0009-2614(89)85118-8
Alnaes, M. S., Blechta, J., and Wells, G. N. (2015). The FEniCS Project Version 1.5. Archive Numer. Softw. 3, 20553. doi:10.11588/ans.2015.100.20553
Bag, S., Friederich, P., Kondov, I., and Wenzel, W. (2019). Concentration Dependent Energy Levels Shifts in Donor-Acceptor Mixtures Due to Intermolecular Electrostatic Interaction. Sci. Rep. 9. doi:10.1038/s41598-019-48877-9
Bag, S., Rauwolf, S., Schwaminger, S. P., Wenzel, W., and Berensmeier, S. (2021). Dna Binding to the Silica: Cooperative Adsorption in Action. Langmuir 37, 5902–5908. doi:10.1021/acs.langmuir.1c00381
Bag, S., Rauwolf, S., Suyetin, M., Schwaminger, S. P., Wenzel, W., and Berensmeier, S. (2020). Buffer Influence on the Amino Acid Silica Interaction. ChemPhysChem 21, 2347–2356. doi:10.1002/cphc.202000572
Baker, M. (2016). 1,500 Scientists Lift the Lid on Reproducibility. Nature 533, 452–454. doi:10.1038/533452a
Becke, A. D. (1993). A New Mixing of Hartree-Fock and Local Density‐functional Theories. J. Chem. Phys. 98, 1372–1377. doi:10.1063/1.464304
Becke, A. D. (1993). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Berthold, M. R., Cebron, N., Dill, F., Gabriel, T. R., Kötter, T., Meinl, T., et al. (2008). “KNIME: The Konstanz Information Miner,” in Data Analysis, Machine Learning and Applications (Berlin: Springer Berlin Heidelberg), 319–326. doi:10.1007/978-3-540-78246-9_38
Bölle, F. T., Mathiesen, N. R., Nielsen, A. J., Vegge, T., Lastra, J. M. G., and Castelli, I. E. (2019). Autonomous Discovery of Materials for Intercalation Electrodes. Batteries & Supercaps 3. doi:10.26434/chemrxiv.9971054.v1
Case, D., Betz, R., Botello-Smith, W., Cerutti, D., Cheatham, T., Darden, T., et al. (2016). Ambertools 16. San Francisco: University of California.
Curtarolo, S., Setyawan, W., Hart, G. L. W., Jahnatek, M., Chepulskii, R. V., Taylor, R. H., et al. (2012). Aflow: An Automatic Framework for High-Throughput Materials Discovery. Comput. Mater. Sci. 58, 218–226. doi:10.1016/j.commatsci.2012.02.005
DeFever, R. S., Matsumoto, R. A., Dowling, A. W., Cummings, P. T., and Maginn, E. J. (2021). MoSDeF Cassandra: A Complete python Interface for the Cassandra Monte Carlo Software. J. Comput. Chem. 42, 1321–1331. doi:10.1002/jcc.26544
Friederich, P., Coehoorn, R., and Wenzel, W. (2017). Molecular Origin of the Anisotropic Dye Orientation in Emissive Layers of Organic Light Emitting Diodes. Chem. Mater. 29, 9528–9535. doi:10.1021/acs.chemmater.7b03742
Friederich, P., Meded, V., Poschlad, A., Neumann, T., Rodin, V., Stehr, V., et al. (2016). Molecular Origin of the Charge Carrier Mobility in Small Molecule Organic Semiconductors. Adv. Funct. Mater. 26, 5757–5763. doi:10.1002/adfm.201601807
Giannozzi, P., Andreussi, O., Brumme, T., Bunau, O., Buongiorno Nardelli, M., Calandra, M., et al. (2017). Advanced Capabilities for Materials Modelling with Quantum Espresso. J. Phys. Condens. Matter 29, 465901. doi:10.1088/1361-648x/aa8f79
Hjorth Larsen, A., Jørgen Mortensen, J., Blomqvist, J., Castelli, I. E., Christensen, R., Dułak, M., et al. (2017). The Atomic Simulation Environment-A Python Library for Working with Atoms. J. Phys. Condens. Matter 29, 273002. doi:10.1088/1361-648x/aa680e
Hourahine, B., Aradi, B., Blum, V., Bonafé, F., Buccheri, A., Camacho, C., et al. (2020). DFTB+, a Software Package for Efficient Approximate Density Functional Theory Based Atomistic Simulations. J. Chem. Phys. 152, 124101. doi:10.1063/1.5143190
Huber, S. P., Zoupanos, S., Uhrin, M., Talirz, L., Kahle, L., Häuselmann, R., et al. (2020). AiiDA 1.0, a Scalable Computational Infrastructure for Automated Reproducible Workflows and Data Provenance. Sci. Data 7, 300. doi:10.1038/s41597-020-00638-4
Jain, A., Ong, S. P., Chen, W., Medasani, B., Qu, X., Kocher, M., et al. (2015). FireWorks: a Dynamic Workflow System Designed for High‐throughput Applications. Concurrency Computat.: Pract. Exper. 27, 5037–5059. doi:10.1002/cpe.3505
Janssen, J., Surendralal, S., Lysogorskiy, Y., Todorova, M., Hickel, T., Drautz, R., et al. (2019). Pyiron: An Integrated Development Environment for Computational Materials Science. Comput. Mater. Sci. 163, 24–36. doi:10.1016/j.commatsci.2018.07.043
Kim, D., Muckley, E. S., Creange, N., Wan, T. H., Ann, M. H., Quattrocchi, E., et al. (2021). Exploring Transport Behavior in Hybrid Perovskites Solar Cells via Machine Learning Analysis of Environmental‐Dependent Impedance Spectroscopy. Adv. Sci. 8, 2002510. doi:10.1002/advs.202002510
Kresse, G., and Joubert, D. (1999). From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 59, 1758–1775. doi:10.1103/physrevb.59.1758
Kumar, S., Rosenberg, J. M., Bouzida, D., Swendsen, R. H., and Kollman, P. A. (1992). The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 13, 1011–1021. doi:10.1002/jcc.540130812
Lasi, H., Fettke, P., Kemper, H.-G., Feld, T., and Hoffmann, M. (2014). Industry 4.0. Bus Inf. Syst. Eng. 6, 239–242. doi:10.1007/s12599-014-0334-4
Lee, J.-H., Chen, C.-H., Lee, P.-H., Lin, H.-Y., Leung, M.-k., Chiu, T.-L., et al. (2019). Blue Organic Light-Emitting Diodes: Current Status, Challenges, and Future Outlook. J. Mater. Chem. C 7, 5874–5888. doi:10.1039/C9TC00204A
Li, J., Duchemin, I., Roscioni, O. M., Friederich, P., Anderson, M., Da Como, E., et al. (2019). Host Dependence of the Electron Affinity of Molecular Dopants. Mater. Horiz. 6, 107–114. doi:10.1039/C8MH00921J
Li, Y., Liu, J.-Y., Zhao, Y.-D., and Cao, Y.-C. (2017). Recent Advancements of High Efficient Donor-Acceptor Type Blue Small Molecule Applied for OLEDs. Mater. Today 20, 258–266. doi:10.1016/j.mattod.2016.12.003
Madsen, G. K. H. (2006). Automated Search for New Thermoelectric Materials: The Case of LiZnSb. J. Am. Chem. Soc. 128, 12140–12146. doi:10.1021/ja062526a
Mamun, O., Winther, K. T., Boes, J. R., and Bligaard, T. (2019). High-throughput Calculations of Catalytic Properties of Bimetallic alloy Surfaces. Sci. Data 6, 76. doi:10.1038/s41597-019-0080-z
Mamun, O., Winther, K. T., Boes, J. R., and Bligaard, T. (2020). A Bayesian Framework for Adsorption Energy Prediction on Bimetallic alloy Catalysts. Npj Comput. Mater. 6. doi:10.1038/s41524-020-00447-8
Mathew, K., Montoya, J. H., Faghaninia, A., Dwarakanath, S., Aykol, M., Tang, H., et al. (2017). Atomate: A High-Level Interface to Generate, Execute, and Analyze Computational Materials Science Workflows. Comput. Mater. Sci. 139, 140–152. doi:10.1016/j.commatsci.2017.07.030
[Dataset] Montserrat Penaloza-Amion, C. R. C. R. (2022). Kit-workflows/dihedral. Available at: https://github.com/kit-workflows/dihedral-scan.
Mortensen, J., Gjerding, M., and Thygesen, K. (2020). Myqueue: Task and Workflow Scheduling System. Joss 5, 1844. doi:10.21105/joss.01844
Müller, J. M., and Däschle, S. (2018). Business Model Innovation of Industry 4.0 Solution Providers towards Customer Process Innovation. Processes 6, 260. doi:10.3390/pr6120260
Ngandjong, A. C., Lombardo, T., Primo, E. N., Chouchane, M., Shodiev, A., Arcelus, O., et al. (2021). Investigating Electrode Calendering and its Impact on Electrochemical Performance by Means of a New Discrete Element Method Model: Towards a Digital Twin of Li-Ion Battery Manufacturing. J. Power Sourc. 485, 229320. doi:10.1016/j.jpowsour.2020.229320
Octavio de Araujo, L., Sabino, F. P., Rêgo, C. R. C., and Guedes-Sobrinho, D. (2021). Bulk Rashba Effect Splitting and Suppression in Polymorphs of Metal Iodine Perovskites. J. Phys. Chem. Lett. 12, 7245–7251. doi:10.1021/acs.jpclett.1c02048
Ong, S. P., Richards, W. D., Jain, A., Hautier, G., Kocher, M., Cholia, S., et al. (2013). Python Materials Genomics (Pymatgen): A Robust, Open-Source python Library for Materials Analysis. Comput. Mater. Sci. 68, 314–319. doi:10.1016/j.commatsci.2012.10.028
Ozpinar, G. A., Peukert, W., and Clark, T. (2010). An Improved Generalized AMBER Force Field (GAFF) for Urea. J. Mol. Model. 16, 1427–1440. doi:10.1007/s00894-010-0650-7
Penaloza-Amion, M., C Rego, C. R., and Wenzel, W. (2022). Local Electronic Charge Transfer in the Helical Induction of Cis-Transoid Poly (4-carboxyphenyl) Acetylene by Chiral Amines. J. Chem. Inf. Model. 62, 544. doi:10.1021/acs.jcim.1c01347
[Dataset] Penaloza-Amion, M. (2022). Montserratamion/SIMONA-DHscan. Available at: https://github.com/montserratamion/simona-dhscan.
Penaloza-Amion, M., Sedghamiz, E., Kozlowska, M., Degitz, C., Possel, C., and Wenzel, W. (2021). Monte-carlo Simulations of Soft Matter Using Simona: A Review of Recent Applications. Front. Phys. 9, 83. doi:10.3389/fphy.2021.635959
Pizzi, G., Cepellotti, A., Sabatini, R., Marzari, N., and Kozinsky, B. (2016). AiiDA: Automated Interactive Infrastructure and Database for Computational Science. Comput. Mater. Sci. 111, 218–230. doi:10.1016/j.commatsci.2015.09.013
Ponce, V., Galvez-Aranda, D. E., and Seminario, J. M. (2017). Analysis of a Li-Ion Nanobattery with Graphite Anode Using Molecular Dynamics Simulations. J. Phys. Chem. C 121, 12959–12971. doi:10.1021/acs.jpcc.7b04190
Posada, J., Zorrilla, M., Dominguez, A., Simoes, B., Eisert, P., Stricker, D., et al. (2018). Graphics and media Technologies for Operators in Industry 4.0. IEEE Comput. Grap. Appl. 38, 119–132. doi:10.1109/mcg.2018.053491736
Rauwolf, S., Bag, S., Rouqueiro, R., Schwaminger, S. P., Dias-Cabral, A. C., Berensmeier, S., et al. (2021). Insights on Alanine and Arginine Binding to Silica with Atomic Resolution. J. Phys. Chem. Lett. 12, 9384–9390. doi:10.1021/acs.jpclett.1c02398
Rodrigues, J. F., Florea, L., de Oliveira, M. C. F., Diamond, D., and Oliveira, O. N. (2021). Big Data and Machine Learning for Materials Science. Discov. Mater. 1, 12. doi:10.1007/s43939-021-00012-0
Schaarschmidt, J., Yuan, J., Strunk, T., Kondov, I., Huber, S. P., Pizzi, G., et al. (2021). Workflow Engineering in Materials Design within the BATTERY 2030 + Project. Adv. Energ. Mater. 2021, 2102638. doi:10.1002/aenm.202102638
Schappals, M., Mecklenfeld, A., Kröger, L., Botan, V., Köster, A., Stephan, S., et al. (2017). Round Robin Study: Molecular Simulation of Thermodynamic Properties from Models with Internal Degrees of freedom. J. Chem. Theor. Comput. 13, 4270–4280. doi:10.1021/acs.jctc.7b00489
Scholz, S., Kondakov, D., Lüssem, B., and Leo, K. (2015). Degradation Mechanisms and Reactions in Organic Light-Emitting Devices. Chem. Rev. 115, 8449–8503. doi:10.1021/cr400704v
Song, W., and Lee, J. Y. (2017). Degradation Mechanism and Lifetime Improvement Strategy for Blue Phosphorescent Organic Light-Emitting Diodes. Adv. Opt. Mater. 5, 1600901. doi:10.1002/adom.201600901
Strunk, T., Wolf, M., Brieg, M., Klenin, K., Biewer, A., Tristram, F., et al. (2012). Simona 1.0: An Efficient and Versatile Framework for Stochastic Simulations of Molecular and Nanoscale Systems. J. Comput. Chem. 33, 2602–2613. doi:10.1002/jcc.23089
Suyetin, M., Bag, S., Anand, P., Borkowska-Panek, M., Gußmann, F., Brieg, M., et al. (2022). Modelling Peptide Adsorption Energies on Gold Surfaces with an Effective Implicit Solvent and Surface Model. J. Colloid Interf. Sci. 605, 493–499. doi:10.1016/j.jcis.2021.07.090
Thompson, M. W., Gilmer, J. B., Matsumoto, R. A., Quach, C. D., Shamaprasad, P., Yang, A. H., et al. (2020). Towards Molecular Simulations that Are Transparent, Reproducible, Usable by Others, and Extensible (TRUE). Mol. Phys. 118, e1742938. doi:10.1080/00268976.2020.1742938
Uhrin, M., Huber, S. P., Yu, J., Marzari, N., and Pizzi, G. (2021). Workflows in AiiDA: Engineering a High-Throughput, Event-Based Engine for Robust and Modular Computational Workflows. Comput. Mater. Sci. 187, 110086. doi:10.1016/j.commatsci.2020.110086
Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., and Berendsen, H. J. C. (2005). Gromacs: Fast, Flexible, and Free. J. Comput. Chem. 26, 1701–1718. doi:10.1002/jcc.20291
Wagner, R., Bag, S., Trunzer, T., Fraga-García, P., Wenzel, W., Berensmeier, S., et al. (2021). Adsorption of Organic Molecules on Carbon Surfaces: Experimental Data and Molecular Dynamics Simulation Considering Multiple Protonation States. J. Colloid Interf. Sci. 589, 424–437. doi:10.1016/j.jcis.2020.12.107
Warr, W. A. (2012). Scientific Workflow Systems: Pipeline Pilot and KNIME. J. Comput. Aided Mol. Des. 26, 801–804. doi:10.1007/s10822-012-9577-7
Wildman, J., Repiščák, P., Paterson, M. J., and Galbraith, I. (2016). General Force-Field Parametrization Scheme for Molecular Dynamics Simulations of Conjugated Materials in Solution. J. Chem. Theor. Comput. 12, 3813–3824. doi:10.1021/acs.jctc.5b01195
Wilkinson, M. D., Dumontier, M., Aalbersberg, I. J., Appleton, G., Axton, M., Baak, A., et al. (2016). The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 3, 18. doi:10.1038/sdata.2016.18
Wong, M. Y., and Zysman-Colman, E. (2017). Purely Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Adv. Mater. 29, 1605444. doi:10.1002/adma.201605444
Wong-ekkabut, J., and Karttunen, M. (2016). The Good, the Bad and the User in Soft Matter Simulations. Biochim. Biophys. Acta (Bba) - Biomembranes 1858, 2529–2538. doi:10.1016/j.bbamem.2016.02.004
Wu, B., Widanage, W. D., Yang, S., and Liu, X. (2020). Battery Digital Twins: Perspectives on the Fusion of Models, Data and Artificial Intelligence for Smart Battery Management Systems. Energy and AI 1, 100016. doi:10.1016/j.egyai.2020.100016
Yao, M., Wang, Y., Li, X., Sheng, Y., Huo, H., Xi, L., et al. (2021). Materials Informatics Platform with Three Dimensional Structures, Workflow and Thermoelectric Applications. Sci. Data 8, 236. doi:10.1038/s41597-021-01022-6
Zheng, J., Xu, X., and Truhlar, D. G. (2011). Minimally Augmented Karlsruhe Basis Sets. Theor. Chem. Acc. 128, 295–305. doi:10.1007/s00214-010-0846-z
Zhu, Q., and Geng, Y. (2013). Drivers and Barriers of Extended Supply Chain Practices for Energy Saving and Emission Reduction Among Chinese Manufacturers. J. Clean. Prod. 40, 6–12. doi:10.1016/j.jclepro.2010.09.017
Keywords: SimStack, workflows, Materials Design, Multiscale modelling, WaNos
Citation: Rêgo CRC, Schaarschmidt J, Schlöder T, Penaloza-Amion M, Bag S, Neumann T, Strunk T and Wenzel W (2022) SimStack: An Intuitive Workflow Framework. Front. Mater. 9:877597. doi: 10.3389/fmats.2022.877597
Received: 16 February 2022; Accepted: 11 April 2022;
Published: 31 May 2022.
Edited by:
Zhenyu Li, University of Science and Technology of China, ChinaReviewed by:
Kesong Yang, University of California, San Diego, United StatesThomas Hammerschmidt, Ruhr University Bochum, Germany
Copyright © 2022 Rêgo, Schaarschmidt, Schlöder, Penaloza-Amion, Bag, Neumann, Strunk and Wenzel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Celso R. C. Rêgo, Y2Vsc28ucmVnb0BraXQuZWR1