 Swati Mohan
Swati Mohan Yuanbing Mao
Yuanbing Mao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mater. , 22 September 2020
Sec. Energy Materials
Volume 7 - 2020 | https://doi.org/10.3389/fmats.2020.00259
This article is part of the Research Topic Carbon- and Inorganic-based Nanostructures for Energy Applications View all 10 articles
Perovskite oxides are an important and effective class of mixed oxides which play a significant role in the fields of energy storage and conversion systems. Here we present a series of cobaltite perovskite LaCoO3 particles which have been doped with 0, 5, 10, 20, and 30% of Sr2+ and have been synthesized by a combined sol–gel and molten-salt synthesis procedure, which provides a regular morphology of the particles. These Sr2+-doped LaCoO3 particles have been characterized by powder X-ray diffraction, Raman spectroscopy, infrared spectroscopy, X-ray photoelectron spectroscopy, and scanning electron microscopy. Moreover, these Sr2+ doped LaCoO3 particles have been demonstrated as efficient catalysts for oxygen evolution reaction (OER) based on the measured specific capacitance, total charge, most accessible charge, electrochemically active surface area, and roughness factor using rotating disk and rotating ring-disk electrode techniques. The 30% Sr2+-doped LaCoO3 sample shows enhanced electrocatalytic OER activity in 0.5 M H2SO4 media compared to the LaCoO3 samples doped with 0, 5, 10, and 20% Sr2+. Among all five LaCoO3 samples, the doped LaCoO3 samples demonstrate better OER activity than the undoped sample.
Growing environmental and geopolitical problems have been arising from the shortage of fossil fuel, promoting efforts to develop cheap, ample, and eco-friendly materials for advanced energy conversion and storage systems. In water electrolysis, the overpotential requirement is a serious issue at which oxygen evolution reaction (OER) occurs (Huynh and Meyer, 2007; Yeo and Bell, 2011). Therefore, it is a significant challenge to design active electrocatalysts for water electrolysis to meet the demands of the sustainable energy-powered economy (Loi and Hummelen, 2013). On the other hand, the global use of noble metal oxides like ruthenium and iridium oxides, especially in the field of catalysis, requires us to minimize the use of these noble metal oxides and to introduce new types of materials, e.g., perovskite oxides. Perovskites are fascinating alternatives to noble metals because of their catalytic activity and cost-effectiveness (Gasteiger et al., 2005; Jörissen, 2006; Neburchilov et al., 2010).
Perovskite oxides have the general formula of ABO3, where A is lanthanide, alkaline-earth, or alkaline cations, and B is transition metal cations such as Fe, Mn, Co, Cr, or Ti. Perovskite oxides can be customized with chemical and physical properties due to the exceptional tunability of their structure and composition. They are a type of mixed oxide and are a class of materials that show a remarkable electronic structure, redox behavior, ionic and electronic activity, and thermal stability (Pena and Fierro, 2001; Schaak and Mallouk, 2002; Royer et al., 2014; Zhu et al., 2014). Perovskites with multiple A or B cations of difference sizes and valences have distortion in their crystal structure. Doping of A or B cations of perovskite with elements of Ce, Ba, or Sr creates oxygen vacancies and increases oxygen mobility (Nitadori and Misono, 1985; Nitadori et al., 1986; de la Cruz et al., 2001; Mefford et al., 2014). In perovskites, the cations present on A and B sites play a crucial role in altering its electronic structure and tuning catalytic properties (JO’M et al., 1983; Bockris and Otagawa, 1984; Vojvodic and Nørskov, 2011; Mueller et al., 2015).
Due to their slow reaction kinetics, both OER and oxygen reduction reaction (ORR) need electrocatalysts. Pt and Pt-alloys are the most essential catalysts for ORR while the Ir and Ru-based materials are good OER catalysts (Gupta et al., 2009; Slanac et al., 2012; Cui et al., 2013; Mefford et al., 2016). Many research studies based on perovskites for OER and ORR have been focused on structural and electronic properties of the surface or bulk (Otagawa and Bockris, 1983; Bockris and Otagawa, 1984; Matsumoto and Sato, 1986). Over the past 40 years, La0.8Sr0.2CoO3 has been studied to give different mechanistic theories as an active catalyst for ORR (Meadowcroft, 1970). For example, Hong et al. (2015) summarized the OER activity by showing correlations between the surface and bulk properties of metal oxides and their electrocatalytic activities. This study showed that the substitution of La2+ by Sr2+ to maintain the perovskite structure following the effects of vacancy defects, covalency, and oxygen exchange on the electrode surface during the electrocatalysis of OER. LSCO (LaSrCoO3) electrocatalysis in alkaline media showed good structural stability, electrolytic corrosion resistance, and high activities for ORR and OER (Jasem and Tseung, 1979; Gorlin and Jaramillo, 2010; McCrory et al., 2013).
There are several procedures to synthesize Sr2+-doped LaCoO3, such as solid-state synthesis (Li et al., 2002), sol–gel (Patel and Patel, 2012), chemical vapor deposition (CVD; Armelao et al., 2005), spray-freeze drying (Lee et al., 2006), aqueous gel-casting technique (Cheng et al., 2008), thermal decompositions (Kaituo et al., 2014), precipitation (Singh and Rakesh, 2009), combustion (Luo and Liu, 2007), low pressure plasma synthesis (Rousseau et al., 2007), etc. In terms of morphology control of products, many of these methods are not sufficient, including the solid-state route and sol–gel method. Molten-salt synthesis method is a simple, reliable, eco-friendly, and cost-effective method when compared to the methods mentioned above. To obtain undoped and Sr2+-doped LaCoO3 particles with a controlled and regular morphology, we introduced a new combination of sol-gel and molten-salt synthesis procedure in this work. Furthermore, we studied their catalytic applications of OER.
Specifically, we successfully synthesized uniform LaCoO3 particles with 0, 5, 10, 20, and 30% Sr2+ nominal doping levels using our facile and reliable method. We characterized their composition, morphology, and structure by powder X-ray diffraction (XRD), Raman spectroscopy, infrared spectroscopy, X-ray photoelectron spectroscopy, X-ray energy dispersive spectroscopy, and scanning electron microscopy. More importantly, their electrocatalytic performance for OER in acidic 0.5 M H2SO4 media was investigated systematically using rotating disk and rotating ring-disk electrode techniques in terms of specific capacitance, total charge, most accessible charge, electrochemically active surface area, and roughness factor. The 30% Sr2+-doped LaCoO3 sample showed the highest electrocatalytic OER activity compared to the LaCoO3 samples doped with 0, 5, 10, and 20% Sr2+. Among all five LaCoO3 samples, the doped LaCoO3 samples demonstrate a better OER activity than the undoped sample. Therefore, this study proves that the combined sol–gel and molten-salt synthesis method is a novel and desirable method to prepare LaCoO3 particles with a uniform morphology and enhanced OER activity in acidic media.
The Sr2+-doped LaCoO3 samples were prepared as follows using the combined sol–gel and molten-salt method synthesis process. Specifically, in a representative synthesis, a stoichiometric amount of La(NO3)3⋅6H2O, Sr(NO3)3 and Co(NO3)2⋅6H2O on a 1 mM scale were added into 10 ml distilled water. After stirring for 2 h at room temperature, polyvinyl alcohol (PVA) aqueous solution (20 wt%) was added. After further stirring, the obtained sol was dried in an oven at 80°C for 2 h, followed by 120°C on a hot plate overnight. During the molten-salt synthesis step, 0.23 g dried gel of each of the five samples was first mixed with 60 mM of NaNO3 + KNO3 mixture (1:1) and then ground together in a mortar and pestle for 20 min (Zuniga et al., 2018). The resulting mixture was transferred into a crucible and kept at 700°C for 6 h with a ramp-up rate of 15° min–1 and cooling-down rate of 10°C min–1. In the final step, the annealed products were washed with deionized water several times and dried in an oven at 60°C overnight. The prepared LaCoO3 samples doped with 0, 5, 10, 20, and 30% Sr2+ were denoted as LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30%, respectively.
The samples were characterized by powder XRD on a Rigaku-MiniflexTM II X-ray diffractometer with Cu Kα1 radiation (λ = 0.15406 nm). The XRD data were collected using a scanning mode in the 2θ range from 20° to 80° with a scanning step size of 0.04° and a scanning rate of 4.0° min–1. Raman scattering data was recorded by employing the back-scattering geometries on a Bruker SENTERRA RAMAN microscope with an objective of 20× of an optical microscope. The excitation line (785 nm) of an Ar+ laser beam was focused to a spot size of 5 μm with a laser power of 25 mW. The spectral resolution range used was 3–5 cm–1 with an integration time of 100 s. Infrared spectra were recorded on a Thermal Nicolet Nexus 470 spectrometer. The morphology of the LSCO samples was observed by a field emission scanning electron microscope (SEM, Carl Zeiss Sigma VP FESEM) equipped with a field emission gun operated at 5 kV.
Three-electrode electrochemical cell configuration was used (Mohan and Mao, 2018; Mao et al., 2019). The working electrode was the catalyst-coated glassy carbon electrode with an area of 0.5024 cm2; a platinum wire was used as the counter electrode and the Ag/AgCl electrode as the reference electrode. To prepare the working electrode, we first prepared the catalyst ink by dispersing 1 mg of the LSCO catalyst powder as active material in 362 μL water and 57 μL Nafion solution by sonication for 20 min. In the next step, we took 42 μL of the prepared catalyst ink, cast it onto the surface of the glassy carbon electrode which corresponds to 0.38 mg/cm2, and dried it in a vacuum oven overnight. The electrochemical measurements, including cyclic voltammogram (CV) and linear sweep voltammogram (LSV), were performed in 0.5 N H2SO4(aq) electrolyte which was purged with nitrogen gas for 10 min prior to the electrochemical measurements to remove the dissolved air from the electrolyte toward the OER using an Autolab potentiostat/galvanostat (PGSTAT302) with Nova 10.11 software. Current density was evaluated as a function of applied voltage from the range of 0.4–1 V versus the Ag/AgCl at scan rates of 5, 10, 20, 50, 70, and 100 mV/s for CV characterization. Linear sweep voltammogram was done at a scan rate of 5 mV/s for all five catalysts in terms of OER.
Figure 1A shows the XRD patterns of the synthesized samples of LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30%. The observed reflections could be perfectly indexed with the peaks of (0 1 2), (1 1 0), (1 0 4), (2 0 2), (0 0 6) (0 2 4), (1 2 2), and (2 1 4) on the basis of tetragonal crystal structure (JCPDS file #251060). The XRD of all five catalysts were found to be crystalline in nature. In addition to XRD data, evidence regarding the chemical composition of the synthesized products was obtained from IR spectra (Figure 1B). The observed FTIR absorption band at ∼603 cm–1 could be assigned to the vibration of the Co–O bond in an octahedral coordination while the band observed at ∼551 cm–1 for La(Sr)–O bond stretching. Furthermore, the vibration of the La–O bond in a dodecahedral coordination was confirmed by the presence of a strong peak at 425 cm–1 (Khalil, 2003; Mahmood et al., 2013; Agilandeswari and Ruban Kumar, 2014).
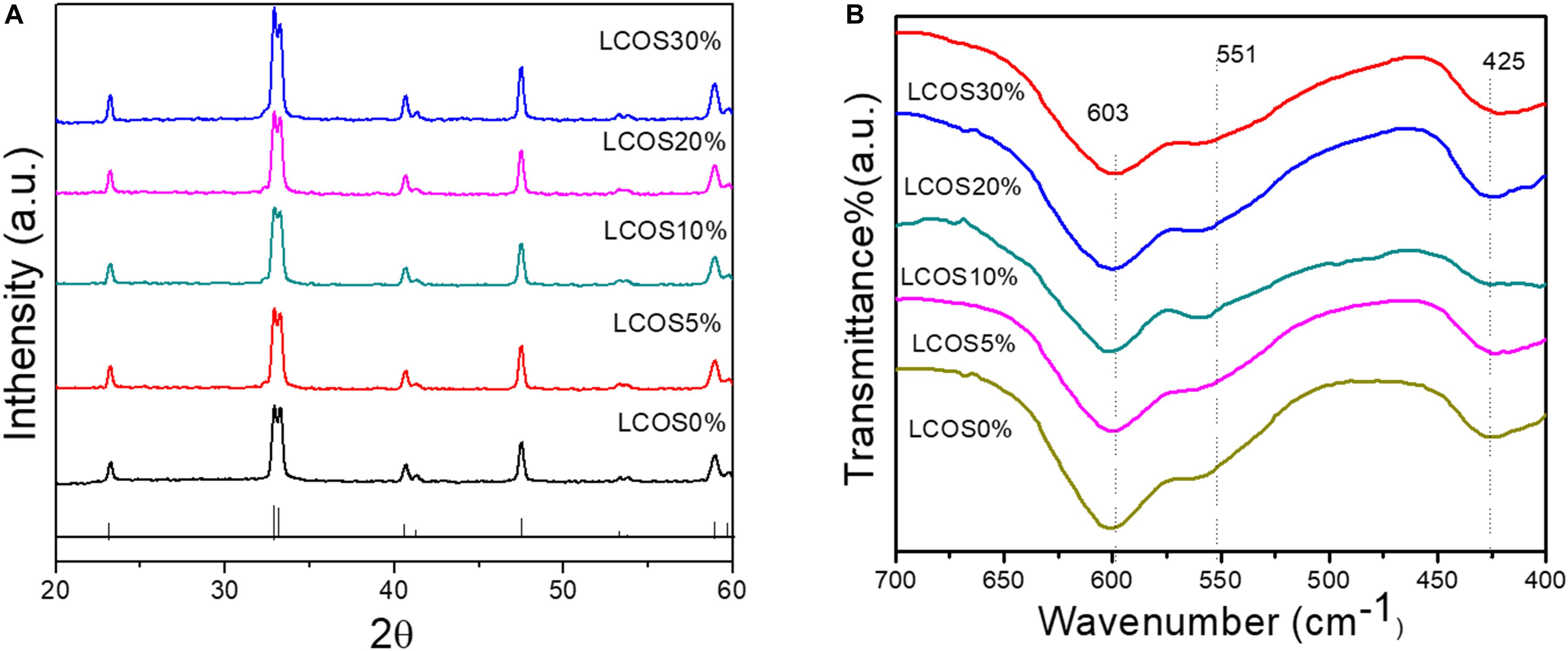
Figure 1. (A) Powder XRD patterns and (B) FTIR spectra of the LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% samples. The pattern shown at the bottom of (A) is the standard pattern (JCPDS # 251060) of LCO with a tetragonal crystal structure.
SEM images of the as-synthesized LSCO samples show the formation of submicron globules as first reported for LSCO (Figures 2A–E). We also prepared a LSCO0% sample by direct calcination of the sol-gel precursor (i.e., the dry gel was not treated with the molten-salt process) for comparison. As commonly observed from samples synthesized by the sol–gel process, the obtained LSCO0% product shows an irregular morphology and larger particles (Figure 2F). Therefore, in terms of product morphology, these SEM images confirm that our combined sol–gel and molten-salt synthesis process is better than the conventional sol–gel process.

Figure 2. SEM images of the LSCO samples: (A) LSCO0%, (B) LSCO5%, (C) LSCO10%, (D) LSCO20%, and (E) LSCO30% synthesized via the combined sol–gel and molten-salt procedure, and (F) LSCO0% via the conventional sol–gel process with direct calcination.
The CV curves of all LSCO catalysts modified as working electrodes (Figure 3A) exhibits a characteristic shape with the presence of the anodic and cathodic peak at a potential range of 0.4–1 V vs reversible hydrogen electrode (RHE), indicating a nonreversible reaction (Mohan and Mao, 2018; Mao et al., 2019). Also, the LSCO30% sample showed the highest current density and the LSCO0% showed the lowest current density among the five samples.
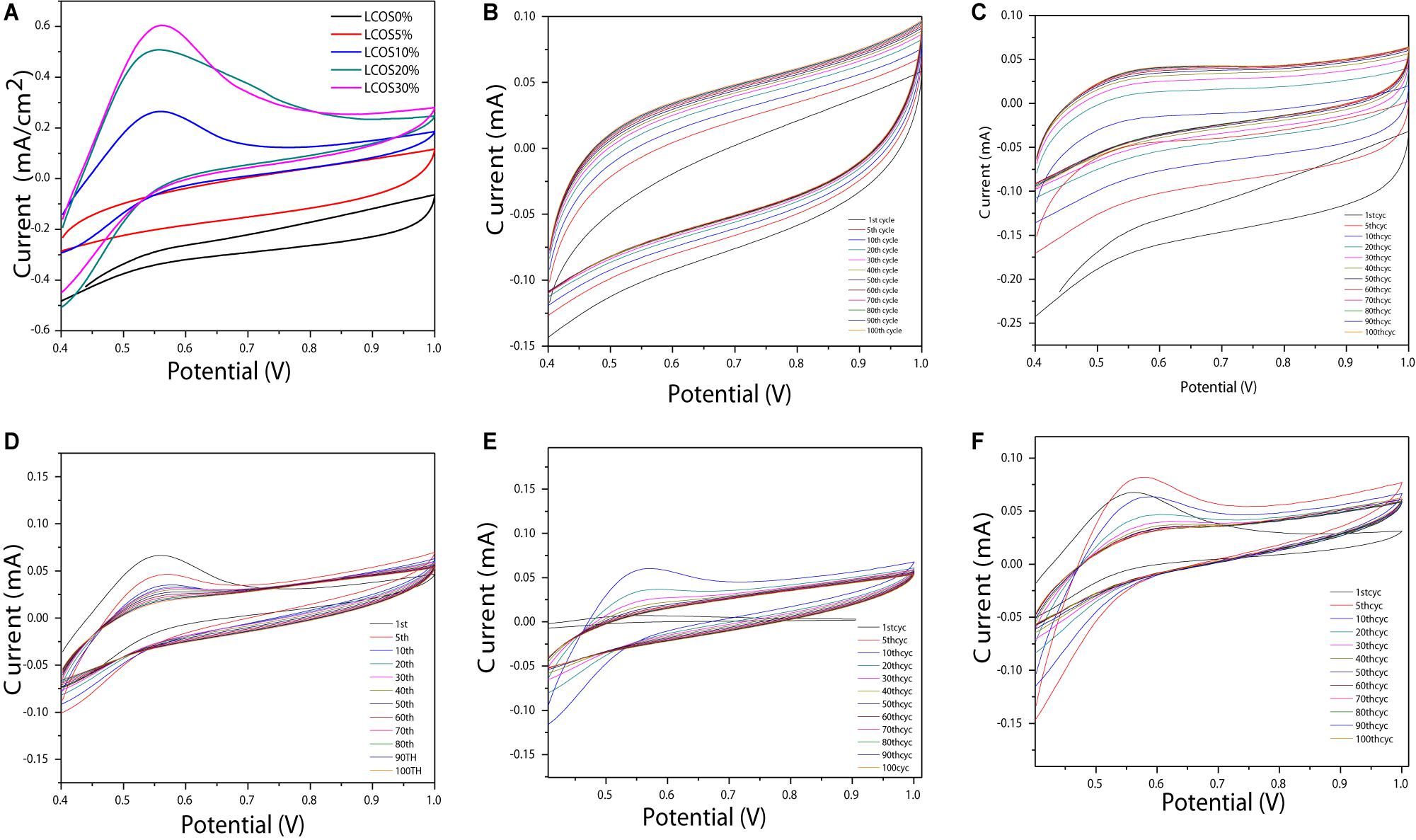
Figure 3. (A) CV plots versus RHE of the LSCO-coated electrodes in 0.5 M H2SO4 electrolyte recorded at 50 mV/s and 25°C and normalized to the geometric area of the electrode. Overlay CV plots of the 1st, 5th, 10th, 20th, 30th, 40th, 50th, 60th, 70th, 80th, 90th, and 100th cycles of the (B) LSCO0%, (C) LSCO5%, (D) LSCO10%, (E) LSCO20%, and (F) LSCO30% at 0.5 M H2SO4 electrolyte during repetitive potential cycles at 20 mV/s and 25°C.
To find out charge accumulation at the electrode/catalyst interface and to evaluate electrode capacity, capacitance measurements were conducted. The two electrochemical contribution faradic and proton adsorption processes are not integrated at the chosen potential range from 0.4 to 1 V. To avoid faradic contribution, we calculated specific capacitance (C, F/g) here for all LSCO catalysts by integrating the CV curves between 0.4 and 1 V vs RHE at scan rates of 5, 10, 20, 50, 70, and 100 mV/s using Eq. 1:
where γ is the scan rate (V/s), m is the mass in grams of the catalyst deposited on the working electrode, E1 and E2 are limits of potentials of the integration curves, and is the integration of the CV curve. From the calculated specific capacitance values tabulated in Table 1, we can see that the capacitance values decrease for all five LSCO catalysts with an increasing scan rate. This observation indicates that the double layer formation is more consistent in a quasi-stationary mode (Sugimoto et al., 2006; Devadas et al., 2011). The diffusion of active species can occur even in miniature pores, thereby all of the active sites of the LSCO samples contribute to the double layer formation. However, at a high scan rate, the charge accumulation occurs only on the active surface sites. The diffusion effect limits the migration of electrolytic ions and causes some active surface areas to be inaccessible for charge storage.
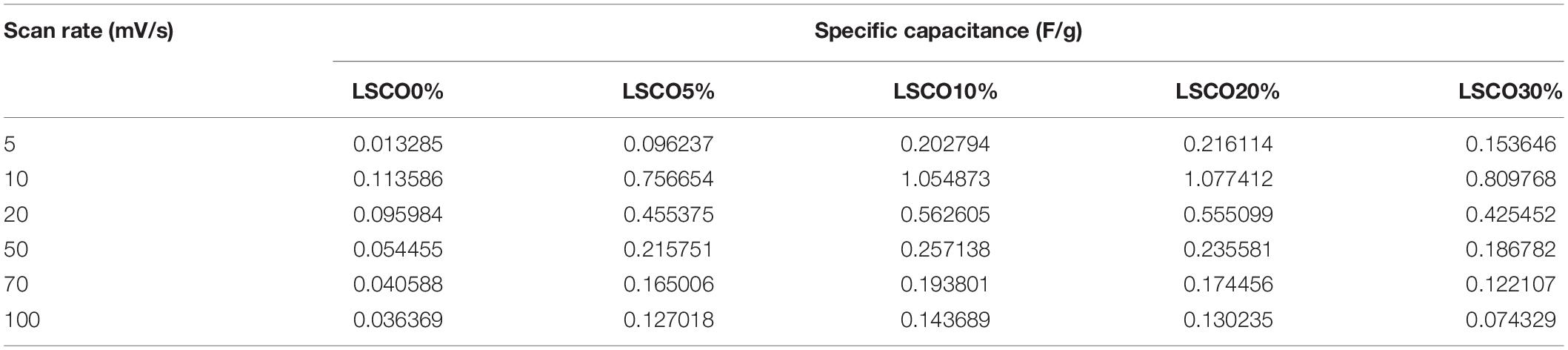
Table 1. Calculated specific capacitance of the LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% samples at scan rates of 5, 10, 20, 50, 70, and 100 mV/s.
The calculated specific capacitance values of the LSCO0% and LSCO5% samples are lower than those obtained for the LSCO10% and LSCO30% samples, while those from the LSCO20% sample showed the highest values. Overall, the specific capacitance values of our LSCO samples are in the order of LSCO20% > LSCO30% > LSCO10% > LSCO5% > LSCO0%. Specific capacitance indicates the stability of the catalysts on the electrode surface or due to morphology change making the catalysts less accessible to charges (Ardizzone et al., 1990). The increase of capacitance can also be correlated to the higher oxidation state of metallic ions in metal oxides (Ardizzone and Trasatti, 1996). Therefore, to improve the charge accumulation, it is important to increase the number of active sites and their accessibilities. For our LSCO samples, the presence of a well-defined morphology and he miniature size of the formed submicron particles refers to the high concentration of active sites at the electrolyte/catalyst interface with a large oxidation state change, and therefore improved capacitive properties (Mohan and Mao, 2018).
Generally, the number of catalyst active sites or active surface areas are considered proportional to voltammetry charges q∗ (Ardizzone et al., 1990; Wu et al., 2011). Calculation of q∗ is determined following Eq. 2 as reported by Audichon et al. (2014), Mohan and Mao (2018), Mao et al. (2019).
Briefly, q∗ value is the average of anodic and cathodic charges measured between 0.4 and 1 V vs RHE. The calculated q∗ values for the LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% samples are summarized in Table 2.
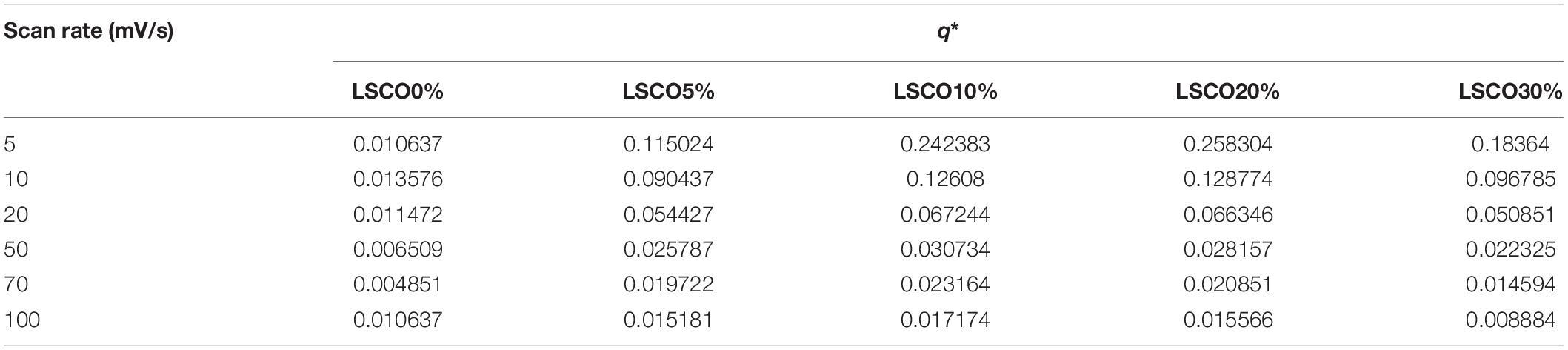
Table 2. Voltammetry charges (q*) of the LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% samples at scan rates of 5, 10, 20, 50, 70, and 100 mV/s.
Ardizzone and co-workers established two relations as following Eqs 3 and 4 which depend on the basis of diffusion species phenomena at the interface of the electrode/electrolyte depending on the scan rate (Ardizzone et al., 1990).
Following Eqs 3 and 4, the total charges (q∗total) and the most accessible charges (q∗outer) were determined when the scan rate values tend to 0 and ∞, respectively (Cheng et al., 2009).
where C1 and C2 are constants, v is the scan rate, and q∗ are the average charges calculated for different scan rates included between 5 to 100 mV/s. The q∗total and q∗outer values for crystalline oxides samples are obtained by linear part extrapolations of the curves presented in Figure 4 and are summarized in Table 3.
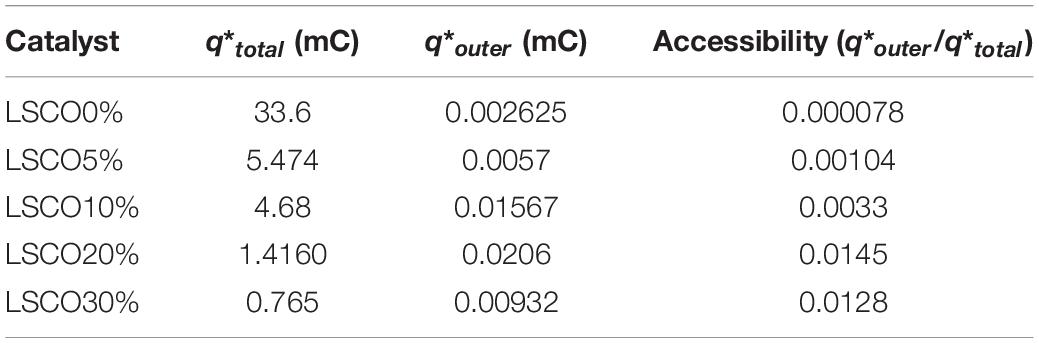
Table 3. Comparison between the total charge, outer charge, and charge accessibility of the LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% samples.
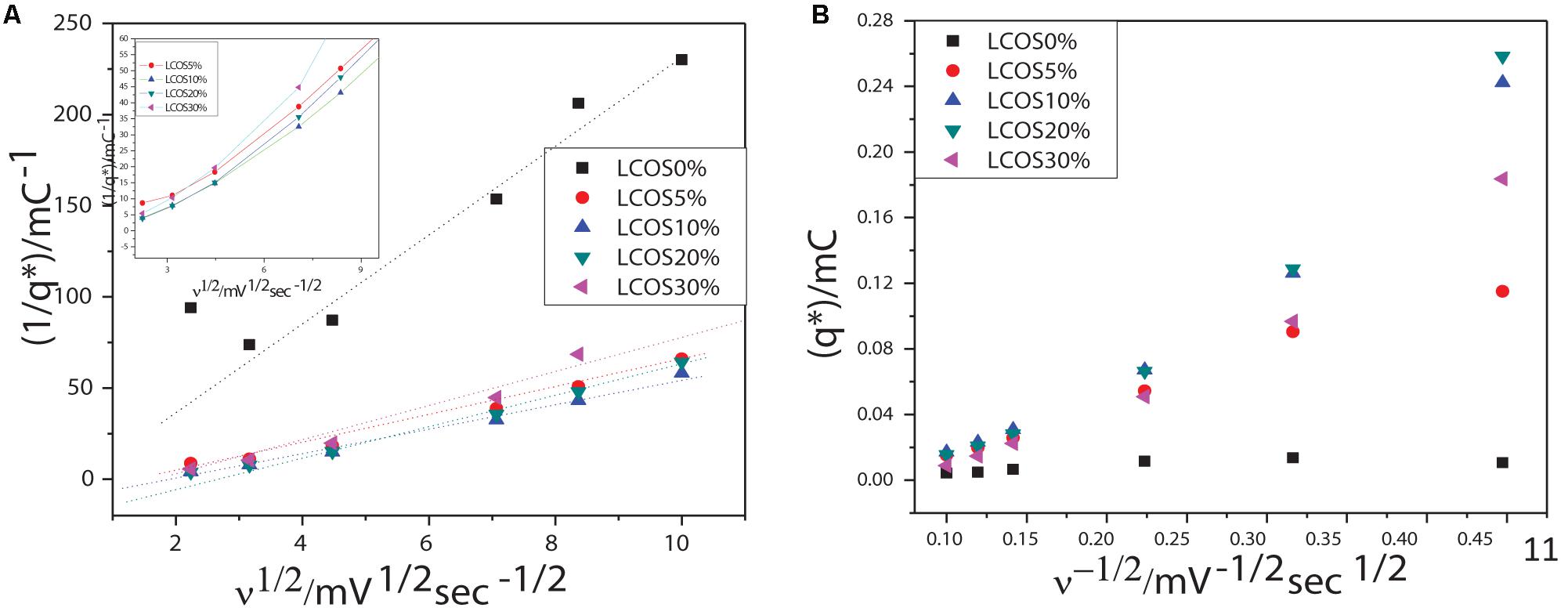
Figure 4. Scan rate dependency of voltammetric charges: extrapolations of (A) the total charges (q*total) and (B) the most accessible charges (q*outer) of the LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% samples.
The calculated total charges q∗total of the LSCO30% (0.765 mC) and LSCO20% (1.416 mC) samples synthesized by the combined sol–gel and molten-salt process are the lowest compared with the LSCO0% (33.6 mC), LSCO5% (5.47 mC), and LSCO10% (4.68 mC) samples. The supposition established from the capacitance measurements therefore confirms that the number of active sites increases and promotes the charge accumulation properties of the catalyst when the mean crystallite size decreases. The highest value for the most accessible charge q∗outer was obtained for the LSCO20% sample (0.0206 mC), followed by the LSCO10% (0.0156 mC), LSCO30% (0.009 mC), LSCO5% (0.005 mC) samples, and then the LSCO0% sample (0.0026 mC). The synthesized LSCO20% sample has allowed one to obtain a higher number of accessible active sites in the catalytic layer, which is probably due to the higher concentration of active sites on the surface and the stability of the catalyst at the electrode surface. Moreover, from the obtained charge values, the active site’s accessibility was evaluated by the ratio q∗outer/q∗total (Table 2). The highest active site’s accessibility was obtained from the LSCO20% sample (0.0145) and followed by the LSCO30% (0.0128), LSCO10% (0.0033), and LSCO5% (0.00104) samples, whereas the one with the lowest q∗outer/q∗total ratio was measured from the LSCO0% sample (0.000078).
Repetitive CV measurement was achieved to assess these oxide samples’ electrochemical activity and stability (Cheng et al., 2009). As shown in Figure 3, 100 voltammetric cycles were carried out between 0.4 and 1 V vs RHE in 0.5 mol l–1 H2SO4 at 20 mV s–1 with the aim of evaluating these catalysts’ aging during a long-term test through their current density evolution.
For the five LSCO catalyst samples (Figure 3) no modification of the CV shapes appears during the long term 100 cycle test. This observation indicates that no alteration of the particles’ structure, which composed the catalytic layer, occurs during the test. However, the current densities decrease during the first cycles and finally tend to stabilize. The fact that the cathodic and anodic current densities do not evolve drastically and no extra peaks appear reveals a high stability of the obtained LSCO oxide samples after heat treatment under variable transient conditions. The corresponding CVs of the synthesized LSCO samples overlap between the 5th and 10th cycles (Figure 5). The evolution of the current during the test could arise from a slight loss of active sites in the catalytic layer, which could contribute to a small catalytic performance decrease.
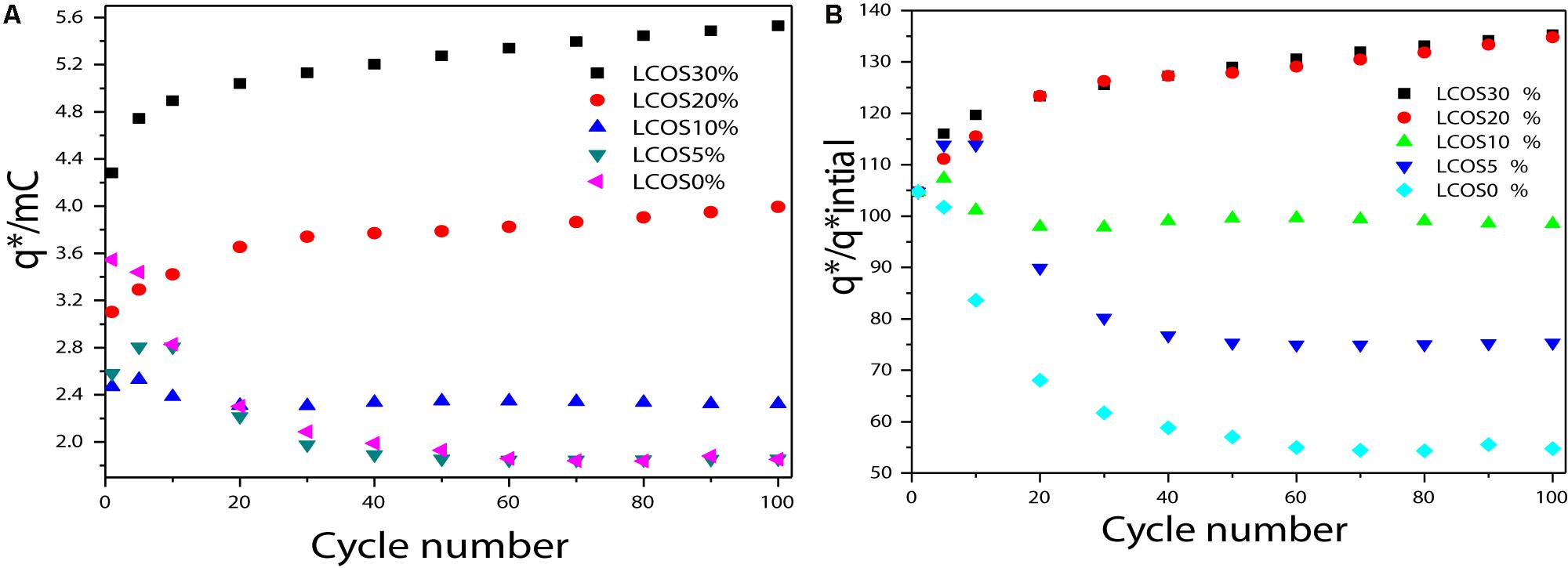
Figure 5. (A) Charge (q*) evolution and (B) q*/q*initial ratio of the LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% electrocatalysts during a stability test measurement.
For different cycles of this stability test, the charge values (q∗) were measured to quantify the active-sites losses and the degradation of the electrocatalytic performances (Figure 5). According to the agreement with the CV observations, the calculated charges decrease in the first activation time (first 5–10 cycles). For the LSCO30%, LSCO20% and LSCO10% samples, the decrease takes place until the 10th cycle whereas for the LSCO5% and LSCO0% catalysts, it is less significant and occurs only for the first five cycles.
As the charges are considered to be proportional to the number of active sites, the q∗/q∗initial ratio as a function of the number of cycles may be used to evaluate the active site losses (Figure 5). The remaining active sites after 50 cycles for the LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% samples are 84, 93, 92, 86, and 92% of the initial charges, respectively. As the CV shapes remain the same along the durability test, the charge ratio evolution could be attributed to the degradation of the catalytic layers, due to a slight erosion of the catalyst at the interface with the electrolyte (Da Silva et al., 1997).
The OER activity of the LSCO samples was evaluated primarily by rotating ring disk electrode voltammetry (RRDE) at a 0.05 mV/s scan rate and 1600 rpm rotation rate. This scan rate is slow enough for steady-state behavior at the electrode surface, and the rotation rate is sufficiently fast to aid in product removal and limit bubble formation from evolved O2 at the electrode surface. As shown in Figure 6A, the horizontal dashed line at 10 mA cm–2 per geometric area is a significant figure of merit for the electroactive catalyst (Weber and Dignam, 1984; Matsumoto and Sato, 1986; Gorlin and Jaramillo, 2010; Walter et al., 2010). To evaluate the electrocatalytic properties of the oxide materials toward the OER as well as their behavior during this reaction, linear sweep voltammetry measurements were performed. For our LSCO samples (Figure 6A), current was first normalized to the geometric surface area of the working electrode to compare the catalytic performance of the LSCO samples as the same masses were deposited on the electrode. It demonstrated that the LSCO20% and LSCO30% samples have better electrocatalytic efficiency for OER than the LSCO0%, LSCO5%, and LSCO10% samples. Specifically, the obtained current densities of the LSCO0%, LSCO5%, LSCO 10%, LSCO20%, and LSCO30% samples are 55.28, 60.29, 60.6, 60.8, and 61.7 mA, respectively. The zoom parts of overpotential (Figure 6B) and disk current (Figure 6C) give a clearer demonstration of the trend.
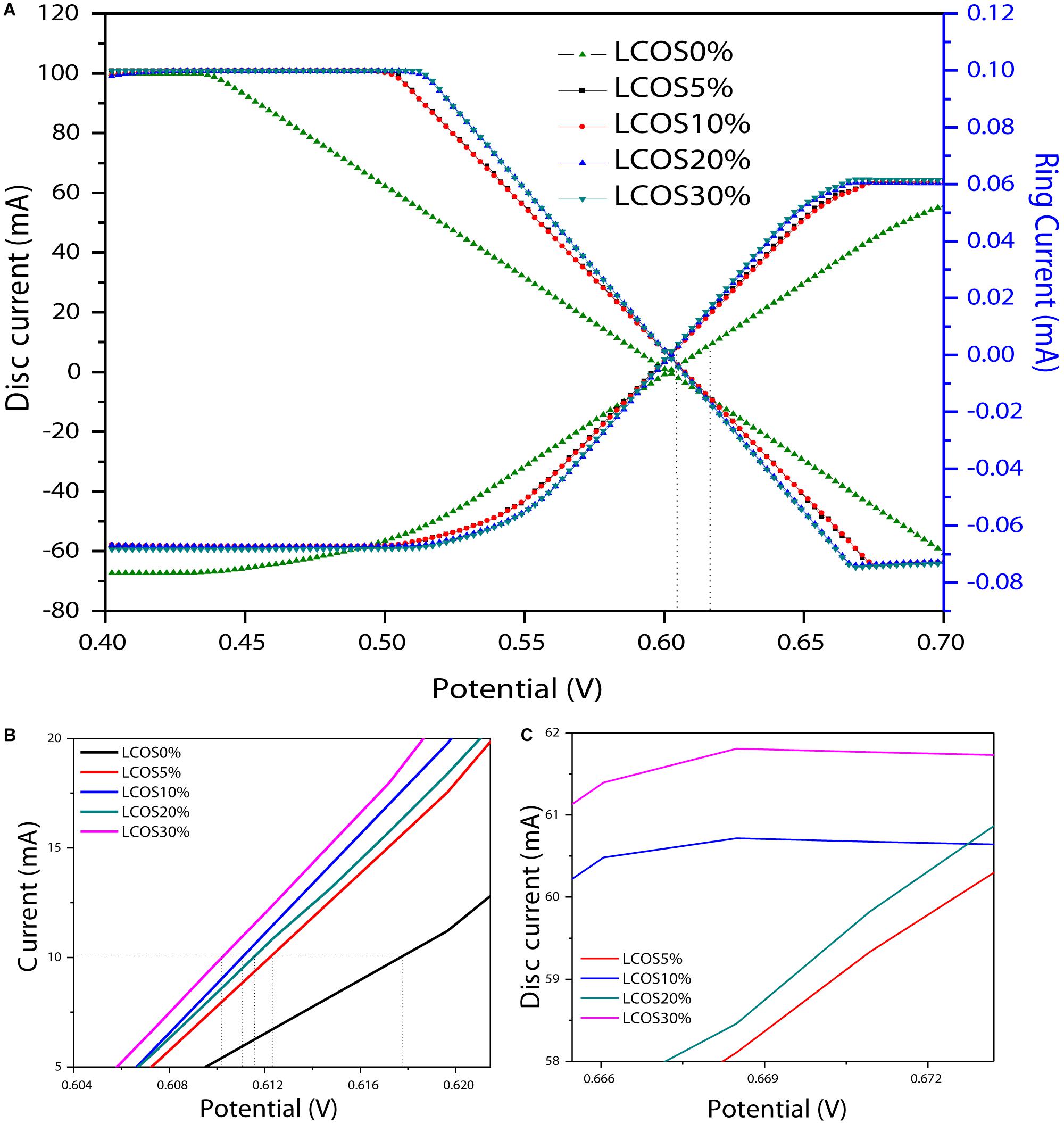
Figure 6. (A) Linear sweep voltammetry on RRDE (rotating ring disc electrode) shows OER activity of the LSCO0%, LSCO5%, LSCO10%, LSCO20% and LSCO30% samples at 5 mV/sec in 0.5M H2SO4. Zoomed parts of (A): (B) potential range and (C) disc current.
The electrochemically active surface area (ECSA) of the LSCO samples was estimated from the electrochemical double-layer capacitance of the catalytic surface (Trasatti and Petrii, 1991). The electrochemical capacitance was determined by measuring the non-Faradaic capacitive current associated with double-layer charging from the scan-rate dependence of CVs shown in Figure 3 (Trasatti and Petrii, 1991; Benck et al., 2012; Ouyang et al., 2019; Wang et al., 2020). Ideal catalysts for OER should have low overpotentials, be stable over time, and have high specific activity (or low surface area) (Mahmood et al., 2013). Electrochemically active surface area was calculated based on ECSA = Cdl/Cs where Cdl and Cs are double layer capacitance and specific capacitance, respectively.
The roughness factor (RF) is calculated by dividing the estimated ECSA by the geometric area of the electrode, 0.5024 cm2. Table 4 shows the obtained ECSA and roughness factor values for all five LSCO catalyst samples. Electrochemically active surface area value of the LSCO20% sample is 0.0903 cm2, which is higher than the LSCO10% (0.0717 cm2), LSCO30% (0.791 cm2), LSCO5% (0.0426 cm2), and LSCO0% (0.4538 cm2) samples. Roughness factor values of the LSCO0% (0.0903) and LSCO5% (0.0849) are lower than the LSCO10% (0.1428), LSCO20% (0.1799), and LSCO30% (0.1576) samples.
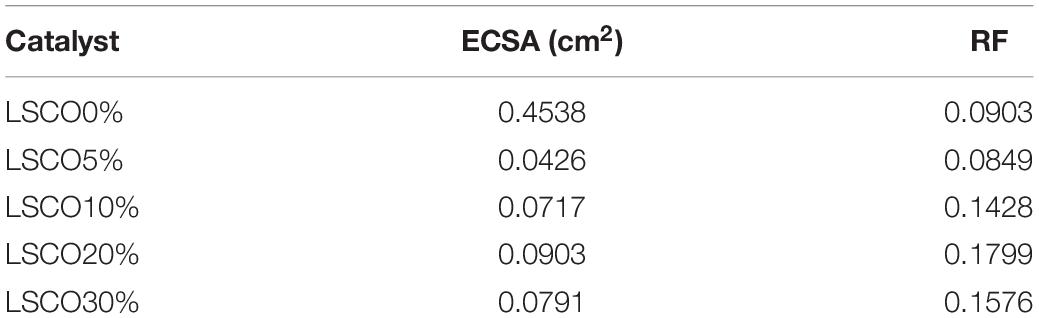
Table 4. Comparison between ECSA and RF values of the LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% samples.
In this paper, we report a combined sol–gel and molten salt synthesis (MSS) procedure to synthesize LSCO0%, LSCO5%, LSCO10%, LSCO20%, and LSCO30% samples. Moreover, we have demonstrated the electrocatalytic comparisons between the LSCO samples as electrode materials in terms of specific capacitance, total charge, and charge accessibility. Cyclic voltammetry and a LSV on rotation ring disk electrode measurements show the enhanced electrocatalytic activity of LSCO. The resulting current density could be a function of both the surface area and the morphology of the LSCO samples used and a combination of both faradaic and non-faradaic procedure.
All datasets generated for this study are included in the article/supplementary material.
SM conducted the experiments. YM initiated the ideas and led the effort. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
YM would like to thank the financial support by the National Science Foundation under CHE (award #1710160 and #1952803) and the IIT start-up funds.
Agilandeswari, K., and Ruban Kumar, A. (2014). Synthesis, characterization, microstructure, optical and magnetic properties of strontium cobalt carbonate precursor and Sr2Co2O5 oxide material. Superl. Microstruct. 68, 27–37. doi: 10.1016/j.spmi.2014.01.002
Ardizzone, S., Fregonara, G., and Trasatti, S. (1990). “Inner” and “Outer” Active Surface of RuO2 Electrodes. Electrochim. Acta 35, 263–267. doi: 10.1016/0013-4686(90)85068-x
Ardizzone, S., and Trasatti, S. (1996). Interfacial Properties of Oxides with Technological Impact in Electrochemistry. Adv. Colloid Interf. Sci. 64, 173–251. doi: 10.1016/0001-8686(95)00286-3
Armelao, L., Barreca, D., Bottaro, G., and Gasparotto, A. (2005). Hybrid Chemical Vapor Deposition/Sol-Gel Route in the Preparation of Nanophasic LaCoO3 Films Chem. Mater 17, 427–433. doi: 10.1021/cm0489643
Audichon, T., Mayousse, E., Morisset, S., Morais, C., Comminges, C., Napporn, T. W., et al. (2014). Electroactivity of RuO2-IrO2 mixed nanocatalysts toward the oxygen evolution reaction in a water electrolyzer supplied by a solar profile. Int. J. Hydrogen Energy 39 16785–16796. doi: 10.1016/j.ijhydene.2014.07.170
Benck, J. D., Chen, Z., Kuritzky, L. Y., Forman, A. J., and Jaramillo, T. F. (2012). Amorphous molybdenum sulfide catalysts for electrochemical hydrogen production: insights into the origin of their catalytic activity. Acs Catalysis 2, 1916–1923. doi: 10.1021/cs300451q
Bockris, J. O. M., and Otagawa, T. (1984). The electrocatalysis of oxygen evolution on perovskites. J. Electrochem. Soc. 131, 290–302. doi: 10.1149/1.2115565
Cheng, C., Zhang, L., Zhang, Y., and Jiang, S. (2008). Synthesis of LaCoO3 nano-powders by aqueous gel-casting for intermediate temperature solid oxide fuel cells. Solid State Ionics 179, 282–289. doi: 10.1016/j.ssi.2008.01.080
Cheng, J., Zhang, H., Chen, G., and Zhang, Y. (2009). Study of IrxRu1–xO2 oxides as anodic electrocatalysts for solid polymer electrolyte water electrolysis. Electrochim. Acta 54, 6250–6256. doi: 10.1016/j.electacta.2009.05.090
Cui, C., Gan, L., Heggen, M., Rudi, S., and Strasser, P. (2013). Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis. Nat. Mater. 12, 765–711.
Da Silva, L. A., Alves, V. A., Trasatti, S., and Boodts, J. F. C. (1997). Surface and Electrocatalytic Properties of Ternary Oxides Ir0.3Ti(0.7–x)PtxO2. Oxygen Evolution from Acidic Solution. J. Electroanal. Chem. 427, 97–104. doi: 10.1016/s0022-0728(97)83088-4
de la Cruz, R. G., Falcon, H., Pena, M., and Fierro, J. (2001). Role of bulk and surface structures of La1-xSrxNiO3 perovskite-type oxides in methane combustion. Appl. Cataly. B Environ. 33, 45–55. doi: 10.1016/s0926-3373(01)00157-6
Devadas, A., Baranton, S., Napporn, T. W., and Coutanceau, C. (2011). Tailoring of RuO2 nanoparticles by microwave assisted “Instant method” for energy storage applications. Power Sourc. 196, 4044–4053. doi: 10.1016/j.jpowsour.2010.11.149
Gasteiger, H. A., Kocha, S. S., Sompalli, B., and Wagner, F. T. (2005). Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Cataly. B Environ. 56, 9–35. doi: 10.1016/j.apcatb.2004.06.021
Gorlin, Y., and Jaramillo, T. F. (2010). A bifunctional nonprecious metal catalyst for oxygen reduction and water oxidation. J. Am. Chem. Soc. 132, 13612–13614. doi: 10.1021/ja104587v
Gupta, G., Slanac, D. A., Kumar, P., Wiggins-Camacho, J. D., Wang, X., Swinnea, S., et al. (2009). Highly stable and active Pt-Cu oxygen reduction electrocatalysts based on mesoporous graphitic carbon supports. Chem. Mater. 21, 4515–4526. doi: 10.1021/cm901203n
Hong, W. T., Risch, M., Stoerzinger, K. A., Grimaud, A., Suntivich, J., and Shao-Horn, Y. (2015). Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 8, 1404–1427. doi: 10.1039/c4ee03869j
Huynh, M. H. V., and Meyer, T. J. (2007). Proton-coupled electron transfer. Chem. Rev. 107, 5004–5064.
Jasem, S. M., and Tseung, A. C. C. (1979). A potentiostatic pulse study of oxygen evolution on Teflon-bonded nickel-cobalt oxide electrodes. J. Electrochem. Soc 126, 1353–1360. doi: 10.1149/1.2129276
JO’M, B., Otagawa, T., and Young, V. (1983). Solid state surface studies of the electrocatalysis of oxygen evolution on perovskites. J. Electroanaly. Chem. Interf. Electrochem. 150, 633–643. doi: 10.1016/s0022-0728(83)80243-5
Jörissen, L. (2006). Bifunctional oxygen/air electrodes. J. Power Sourc. 155, 23–32. doi: 10.1016/j.jpowsour.2005.07.038
Kaituo, W., Wu, X., Wu, W., and Li, Y. (2014). Synthesis of perovskite LaCoO3 by thermal decomposition of oxalates: Phase evolution and kinetics of the thermal transformation of the precursor. Ceramics Int. 40, 5997–6004. doi: 10.1016/j.ceramint.2013.11.048
Khalil, M. S. (2003). Synthesis, X-ray, infrared spectra and electrical conductivity of La/Ba-CoO3 systems. Mater. Sci. Eng. A 352, 64–70. doi: 10.1016/s0921-5093(02)00557-9
Lee, S. H., Lee, J. Y., and Park, Y. M. (2006). Complete oxidation of methane and CO at low temperature over LaCoO3 prepared by spray-freezing/freeze-drying method. Catal. Today 117, 376–381. doi: 10.1016/j.cattod.2006.05.035
Li, F., Xianghua, Y., Liying, C., Hongjun, P., and Xinquan, X. (2002). Solid-State Synthesis of LaCoO3 Perovskite Nanocrystals. J. Am. Ceram. Soc. 85, 2177–2180.
Loi, M. A., and Hummelen, J. C. (2013). Hybrid solar cells: perovskites under the sun. Nat. Mater. 12, 1087–1089.
Luo, W.-L., and Liu, W. (2007). Combustion synthesis and characterization of porous perovskite catalysts. J. Chem. Sci. 119, 237–241. doi: 10.1007/s12039-007-0031-7
Mahmood, M. F., Warsi, M. N., and Ashiq, M. (2013). Ishaq, Substitution of La and Fe with Dy and Mn in multiferroic La1-xDyxFe1-yMnyO3 nanocrystallites. J. Magn. Magn. Mater. 327, 64–70. doi: 10.1016/j.jmmm.2012.09.033
Mao, L., Mohan, S., and Mao, Y. (2019). Delafossite CuMnO2 as an Efficient Bifunctional Oxygen and Hydrogen Evolution Reaction Electrocatalyst for Water Splitting. J. Electrochem. Soc. 166, H233–H242.
Matsumoto, Y., and Sato, E. (1986). Electrocatalytic properties of transition metal oxides for oxygen evolution reaction. Mater. Chem. Phys. 14, 397–426. doi: 10.1016/0254-0584(86)90045-3
McCrory, C. C. L., Jung, S., Peters, J. C., and Jaramillo, T. F. (2013). Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977–16987. doi: 10.1021/ja407115p
Meadowcroft, D. (1970). Low-cost oxygen electrode material. Nature 226, 847–848. doi: 10.1038/226847a0
Mefford, J. T., Hardin, W. G., Dai, S., Johnston, K. P., and Stevenson, K. J. (2014). Anion charge storage through oxygen intercalation in LaMnO3 perovskite pseudocapacitor electrodes. Nat. Mater. 13, 726–732. doi: 10.1038/nmat4000
Mefford, J. T., Rong, X., Abakumov, A. M., Hardin, W. G., Dai, S., Kolpak, A. M., et al. (2016). Water electrolysis on La1-xSrxCoO3-δ perovskite electrocatalysts. Nat. Commun. 7:11053.
Mohan, S., and Mao, Y. (2018). Dependence of (photo)electrochemical properties on geometry factors of hydrothermally synthesized delafossite copper gallium oxide CuGaO2 toward oxygen evolution reaction. J. Electrochem. Soc. 165, H607–H613.
Mueller, D. N., Machala, M. L., Bluhm, H., and Chueh, W. C. (2015). Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 6:6097.
Neburchilov, V., Wang, H., Martin, J. J., and Qu, W. (2010). A review on air cathodes for zinc-air fuel cells. J. Power Sourc. 195, 1271–1291. doi: 10.1016/j.jpowsour.2009.08.100
Nitadori, T., Kurihara, S., and Misono, M. (1986). Catalytic properties of La1-xA’xMnO3 (A’ = Sr, Ce, Hf). J. Cataly. 98, 221–228. doi: 10.1016/0021-9517(86)90310-6
Nitadori, T., and Misono, M. (1985). Catalytic Properties of La1-xA’xFeO3 (A’ = Sr, Ce) and La1-xCexCoO3. J. Catalys. 93, 459–466. doi: 10.1016/0021-9517(85)90193-9
Ouyang, T., Ye, Y., Wu, C., Xiao, K., and Liu, Z. (2019). Heterostructures Composed of N-Doped Carbon Nanotubes Encapsulating Cobalt and β-Mo2C Nanoparticles as Bifunctional Electrodes for Water Splitting. Angew. Chem. Int. Ed. Engl. 58, 4923–4928. doi: 10.1002/anie.201814262
Patel, F., and Patel, S. (2012). Carbon monoxide oxidation on LaCoO3 perovskite type catalysts prepared by reactive grinding. Res. J. Recent Sci. 1, 152–159.
Pena, M., and Fierro, J. (2001). Chemical structures and performance of perovskite oxides. Chem. Rev. 101, 1981–2018. doi: 10.1021/cr980129f
Rousseau, F., Nikravech, M., Benabdelmoumène, L., and Guyon, C. (2007). Electrochemical studies on Sr doped LaMnO3 and LaCoO3 layers synthesized in a low-pressure plasma reactor equipped with a convergent nozzle. J. Appl. Electrochem. 37, 95–101. doi: 10.1007/s10800-006-9214-z
Royer, S., Duprez, D., Can, F., Courtois, X., Batiot-Dupeyrat, C., Laassiri, S., et al. (2014). Perovskites as substitutes of noble metals for heterogeneous catalysis: dream or reality. Chem. Rev. 114, 10292–10368. doi: 10.1021/cr500032a
Schaak, R. E., and Mallouk, T. E. (2002). Perovskites by design: a toolbox of solid-state reactions. Chem. Mater. 14, 1455–1471. doi: 10.1021/cm010689m
Singh, C., and Rakesh, M. (2009). Preparation and characterization of nickel doped, A and B site LaCoO3 perovskite. Indian J. Eng. Mater. Sci. 16, 288–290.
Slanac, D. A., Hardin, W. G., Johnston, K. P., and Stevenson, K. J. (2012). Atomic ensemble and electronic effects in Ag-rich AgPd nanoalloy catalysts for oxygen reduction in alkaline media. J. Am. Chem. Soc. 134, 9812–9819. doi: 10.1021/ja303580b
Sugimoto, W., Yokoshima, K., Murakami, Y., and Takasu, Y. (2006). Charge Storage Mechanism of Nanostructured Anhydrous and Hydrous Ruthenium-Based Oxides. Electrochim. Acta 52, 1742–1748. doi: 10.1016/j.electacta.2006.02.054
Trasatti, S., and Petrii, O. (1991). Real surface area measurements in electrochemistry. Pure Appl. Chem. 63, 711–734. doi: 10.1351/pac199163050711
Vojvodic, A., and Nørskov, J. K. (2011). Optimizing perovskites for the water-splitting reaction. Science 334, 1355–1356. doi: 10.1126/science.1215081
Walter, M. G., Warren, E. L., McKone, J. R., Boettcher, S. W., Mi, Q., Santori, E. A., et al. (2010). Solar water splitting cells. Chem. Rev. 110, 6446–6473.
Wang, X., Ouyang, T., Wang, L., Zhong, J., and Liu, Z. (2020). Surface Reorganization on Electrochemically-Induced Zn-Ni-Co Spinel Oxides for Enhanced Oxygen Electrocatalysis. Angew. Chem. Int. Ed. Engl. 59, 6492–6499. doi: 10.1002/anie.202000690
Weber, M. F., and Dignam, M. J. (1984). Efficiency of splitting water with semiconducting photoelectrodes. J. Electrochem. Soc. 131, 1258–1265. doi: 10.1149/1.2115797
Wu, X., Tayal, J., Basu, S., and Scott, K. (2011). Nano-Crystalline RuxSn1-xO2 Powder Catalysts for Oxygen Evolution Reaction in Proton Exchange Membrane Water Electrolysers. Int. J. Hydrogen Energy 36, 14796–14804. doi: 10.1016/j.ijhydene.2011.01.067
Yeo, B. S., and Bell, A. T. (2011). Enhanced activity of gold-supported cobalt oxide for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 133, 5587–5593. doi: 10.1021/ja200559j
Zhu, J., Li, H., Zhong, L., Xiao, P., Xu, X., Yang, X., et al. (2014). Perovskite oxides: preparation, characterizations, and applications in heterogeneous catalysis. ACS Catal 4, 2917–2940.
Keywords: LaCoO3, sol-gel, molten-salt synthesis, electrocatalysis, OER, Sr2+-doped
Citation: Mohan S and Mao Y (2020) Molten Salt Synthesized Submicron Perovskite La1–xSrxCoO3 Particles as Efficient Electrocatalyst for Water Electrolysis. Front. Mater. 7:259. doi: 10.3389/fmats.2020.00259
Received: 08 January 2020; Accepted: 15 July 2020;
Published: 22 September 2020.
Edited by:
Zhenhai Xia, University of North Texas, United StatesReviewed by:
Tianhua Zhou, Fujian Institute of Research on the Structure of Matter (CAS), ChinaCopyright © 2020 Mohan and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanbing Mao, eW1hbzE3QGlpdC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.