 Weiyong Cui
Weiyong Cui Xueli Song2
Xueli Song2 Ye Chen
Ye Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mater. , 12 March 2020
Sec. Colloidal Materials and Interfaces
Volume 7 - 2020 | https://doi.org/10.3389/fmats.2020.00047
This article is part of the Research Topic Surface Chemistry of Flotation View all 12 articles
To investigate the effect of hydration behavior on the fluorapatite structure, single H2O molecule and three-layer water cluster adsorptions on the fluorapatite (001) surface were performed by means of density functional theory. The results show that a single H2O molecule can form stable chemisorption structures with the fluorapatite (001) surface in the form of single-site, two-site, and three-site adsorption and that the corresponding adsorption energies are 64.817, 98.712, and 139.620 kJ/mol, respectively. The interacting length of the Ca atom and the O atom of the H2O molecule is close to the length of the Ca–O bond in the bulk, and their overlap is mainly contributed by the O 2p and Ca 4s states. The fluorapatite (001) surface shows serious hydration reconstruction after adsorbing three layers of water molecules; these atoms in the surface layer are highly distorted, and the Ca and the PO4 are shifted in opposite directions along the z-axis direction. Further analysis shows that these surface Ca atoms are critical to the hydration behaviors of the transition area, as they can bind strongly to the H2O molecules, with the newly formed Ca–O bonds being between 2.164 and 2.486 Å.
Apatites have attracted more and more attention due to their complex structures and unique properties. On the one hand, because they are the most important bone material, the study of apatites occupies a huge market in the field of life science (Palazzo et al., 2007; Rey et al., 2014; Okada and Matsumoto, 2015); on the other hand, there are many valuable elements in them, such as rare earths, uranium, and thorium, which play significant roles in the development of future energy, high-end equipment, and other advancements of various key technologies (Li et al., 2014; Zhang, 2014; Emsbo et al., 2015). However, we should not ignore the fact that apatites, and mostly fluorapatite, are the main phosphorus-bearing minerals and play an irreplaceable role in industrial and agricultural production (Scholz et al., 2014). Data reveals that more than 90% of the fluorapatite resource is used to produce fertilizer, with other applications being in animal feed, detergents, food and beverages, and water treatment (Rawashdeh and Maxwell, 2011). Not all phosphate ores can be used directly, and this is becoming more and more of an issue as rich fluorapatite resources become even scarcer. With the further depletion of the apatite resources, billions of tons of apatite ores need to be pretreated by flotation. It is reported that more than half of the world’s marketable phosphate is concentrated via the flotation process, which takes place at the complex solid, water, and gas phase interface (Santos et al., 2010).
The existence of a water environment is a necessity for flotation, and the hydration behavior of the surface is very important. However, corresponding research is still needed, and progress has been slow. Due to the development of modern microscopic detection equipment, this trend has improved dramatically. Park et al. (2005) studied the structure of the Durango fluorapatite (100)–water interface with high-resolution X-ray reflectivity and found that the presence of a layered interfacial water structure exhibits two distinct water layers and that the heights of the first and second layers are 2.64 and 4.17 Å, respectively. Dan et al. (2006) conducted research on calcium surface sites at aqueous fluorapatite by means of 1H and 31P MAS NMR. Meanwhile, Pareek et al. (2008, 2009) made a systematic study of the role of water in the surface relaxation of the fluorapatite (100) surface with grazing incidence x-ray diffraction. In recent years, with the continuous progress of computer technology, the density functional theory method (DFT), which can be used to describe collector-mineral adsorption at the molecular level in 3D space, has triggered intense research and is providing valuable primary information on various interface systems (Mkhonto and de Leeuw, 2002; Rulis et al., 2004, 2007; Haverty et al., 2005; Chappell et al., 2008; Menéndez-Proupin et al., 2011). Mkhonto and de Leeuw (2002) studied the effect of water on the surface structure and morphology of fluorapatite and found that the hydration of the surfaces occurs by physisorption and shows Langmuir behavior. Mkhonto and de Leeuw (2002) studied the interaction of (001) carbonated hydroxylapatite surfaces with water. They found that a single molecule of water can be strongly adsorbed by the apatite surface and that the carbonate ion may lower both the adsorbate/surface interaction energy and the energy needed to deform the COHAp surface and the water molecule. However, many questions relating to the details of water adsorption on the fluorapatite surface at the molecular level remain without clear answers.
In this paper, the fluorapatite (001) surface was chosen to investigate the interactions between the fluorapatite (001) surface and single H2O molecule as well as water cluster, respectively. Different configurations were optimized, and the mechanism of water adsorptions on the fluorapatite (001) surface and their influence are discussed. The results provide an important insight into the structure of and enable investigation of flotation reagent interactions on the fluorapatite surface, which may aid current experimental efforts to understand the flotation process further.
Calculations for the fluorapatite (001) surface and single H2O molecule adsorptions were performed within Cambridge Serial Total Energy Package (CASTEP) (Clark et al., 2005). For the former calculations, the ultrasoft pseudo-potential was employed to describe the electron–ion interactions. The exchange correlation function used in the present study was the generalized gradient approximation (GGA), which was extended by the Perdew-Wang generalized-gradient approximation (PW91). The plane-wave cutoff of 400 eV was chosen, and the Brillouin zone was sampled with k-points of a 2 × 2 × 2 grid, which were found to be enough for the system. The convergence tolerances were set as follows: the maximum displacement was 0.002 Å, the maximum force was 0.08 eV⋅Å–1, the maximum energy change was 2.0 × 10–5 eV⋅atom–1, the maximum stress was 0.1 GPa, and the SCF convergence tolerance was 2.0 × 10–6 eV⋅atom–1. The valence electron configurations considered in all the studies were H 1s1, O 2s2 2p4, F 2s2 2p5, P 3s2 3p3, Ca 3s2 3p6 4s2.
The bulk crystal structure of the fluorapatite was obtained from the American Mineralogist Crystal Structure Database (AMCSD). The lattice parameters were calculated as a = b = 9.457 Å and c = 6.883 Å, which were in good agreement with the experimental results (a = b = 9.375 Å and c = 6.887 Å) (Comodi et al., 2001). As shown in Figure 1, each fluorapatite bulk possesses two Ca5(PO4)3F structures along the (001) plane, with the stacking sequence of Ca-Ca3(PO4)3F-Ca-Ca-Ca3(PO4)3F-Ca. These adjacent Ca-1 and Ca-2 sites are rather different in that the columnar Ca-1 site is coordinated to nine oxygen atoms in the arrangement of a tricapped trigonal prism, while the mirror Ca-2 site is in the Ca3(PO4)3F layer and bonds to six oxygen atoms and the column anion F (Hughes and Rakovan, 2002). However, the oxygen atoms are not very different, and they are marked for the sake of data discussion. The most stable surface was chosen, though there are different kinds of terminations along the (001) plane (Qiu et al., 2017). After testing, the (22) fluorapatite (001) surface structure with fifteen atomic layers was constructed, and the five bottom-most atomic layers were fixed to the bulk (see Figure 2). The vacuum gap of 30 Å in the z-direction was sufficient for our investigation.
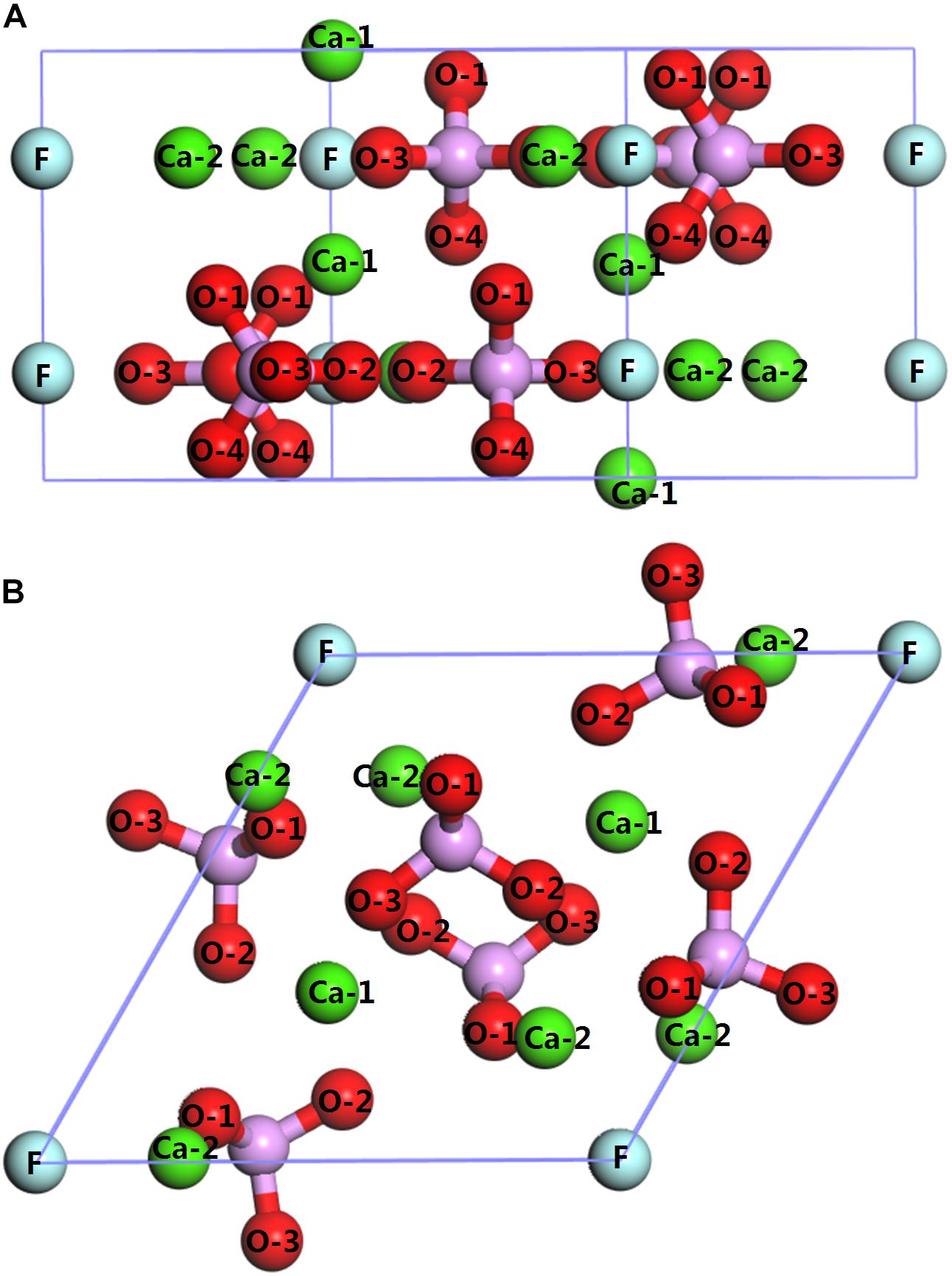
Figure 1. Crystal structure of fluorapatite: (A) side view (perpendicular to the fluorapatite (001) surface); (B) top view (parallel to the fluorapatite (001) surface).
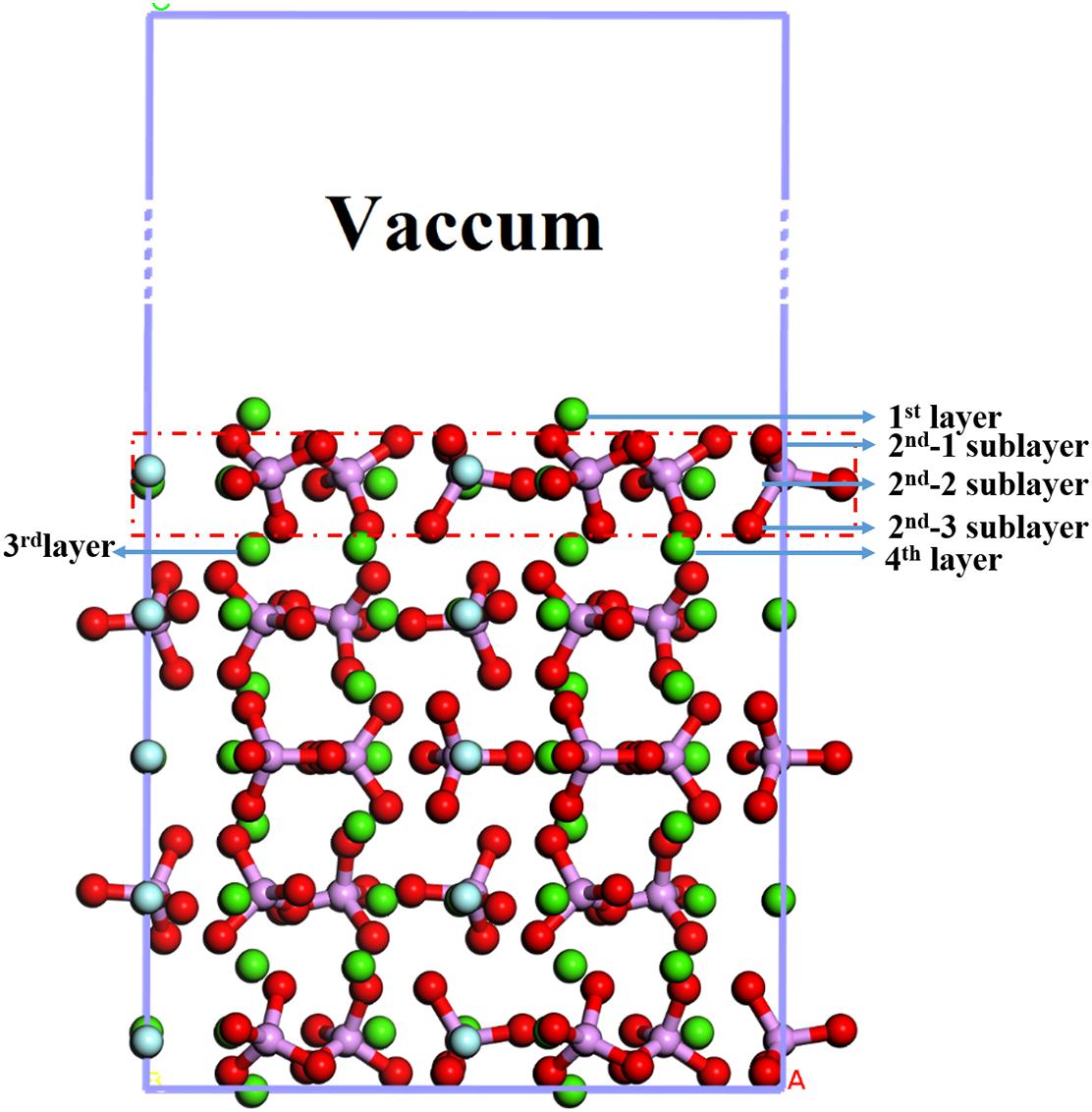
Figure 2. Structure of the fluorapatite (001) surface.
The optimization of a single H2O molecule was performed in a 10 × 10 × 10Å cubic cell, and the calculated results (see Figure 3A) are close to the experimental observations (Zhao et al., 2014; Chen et al., 2017). Adsorption clusters of three layers of water molecules (see Figure 3B) were optimized with the same condition, which was used to simulate the natural state of water.
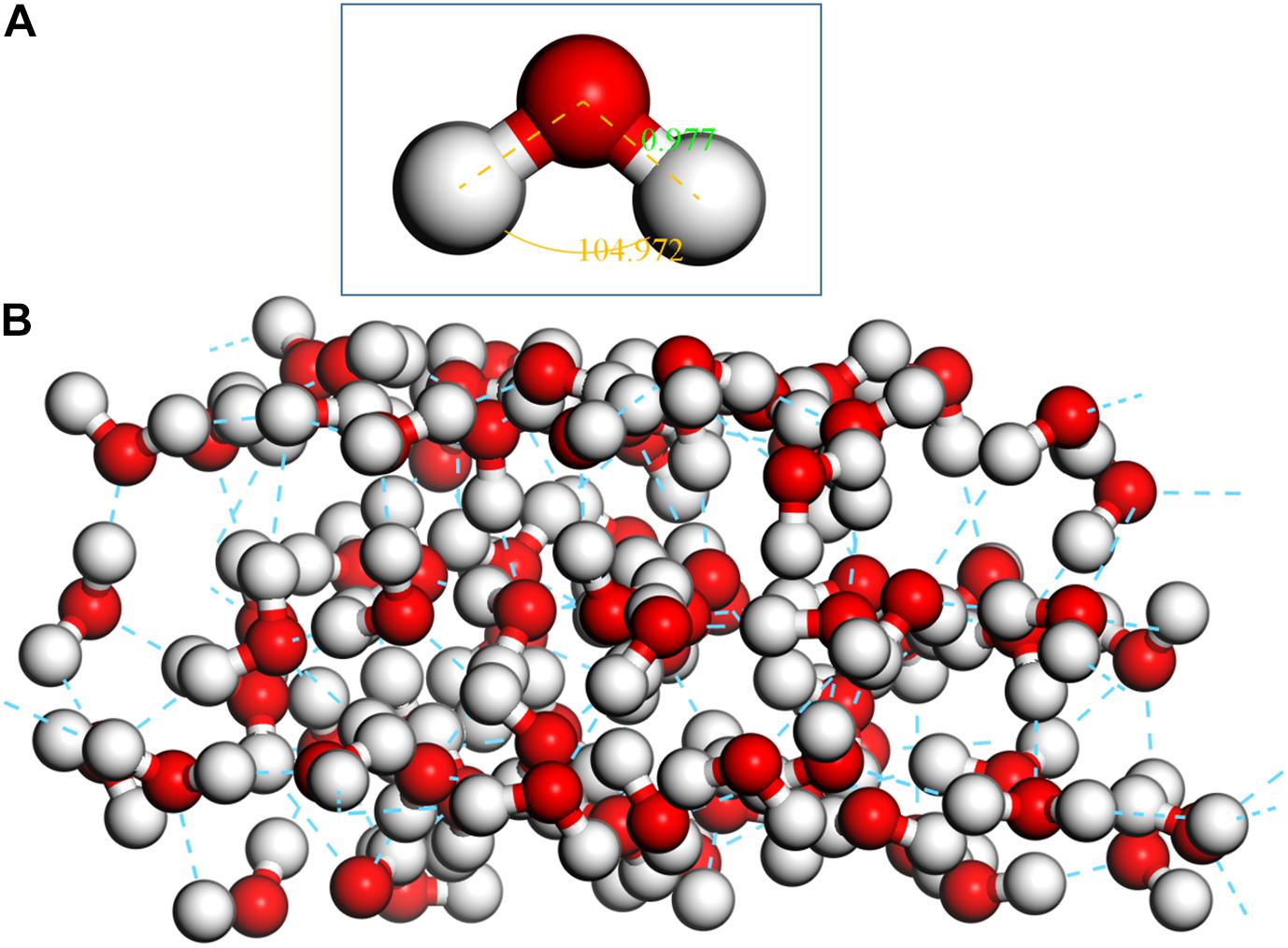
Figure 3. Optimized single H2O molecule and three-layer water cluster, (A) single H2O molecule; (B) three-layer water cluster.
The adsorption energy of H2O on the fluorapatite surface is calculated by the following equation (Zhao et al., 2015):
where Eads is the adsorption energy, EH2O is the energy of the H2O calculated in a cubic cell, Esurface is the energy of the fluorapatite slab, and EH2O/surface is the energy of the fluorapatite slab with adsorbed H2O.
As shown in Figure 2, the exposed atoms of the fluorapatite (001) surface can be divided into three parts. The Ca-1 atoms occupy the first layer, and the other atoms in the second layer can be divided into two parts, the O-1 atom in the first sublayer and Ca-2, P, O-2, and O-3 atoms in the second sublayer. So from top to bottom are Ca-1 atoms, O-1 atoms, and then Ca-2, P, O-2, and O-3 atoms. Usually, the tetrahedral PO4-group is considered indivisible in reactions, so the potential centers are divided into four parts, Ca-1, Ca-2, PO4-group, and F sites. It is worth noting that the F and Ca-2 atoms are not as active as other atoms, such as Ca-1 and O atoms, during the single H2O molecule adsorption; the optimized stable configurations are shown in Figure 4.
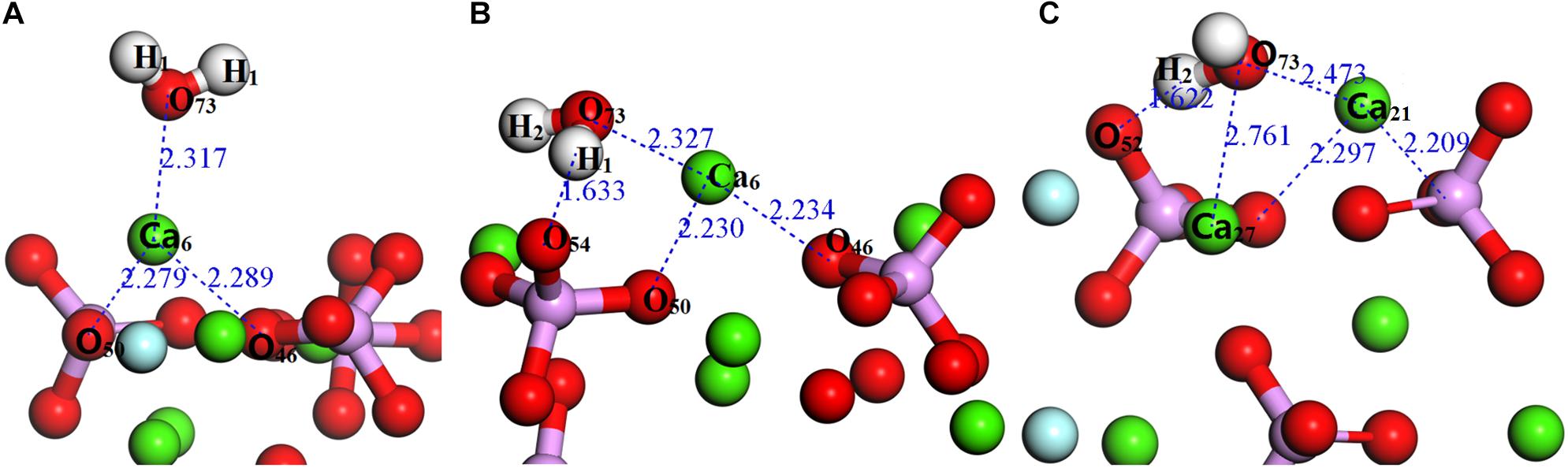
Figure 4. Configurations of a single H2O molecule on the fluorapatite (001) surface: (A) single-adsorption; (B) double-site adsorption; (C): three-site adsorption.
As seen in Figure 4A, the length between O73 and Ca6 is 2.317 Å, which is close to that of the Ca–O bond in the bulk. Meanwhile, the calculated Eads is −64.817 kJ/mol, confirming that the interaction between the Ca and O atoms is rather strong. As shown in Table 1, the O73 atom exhibits the ability to gain electrons, and its 2p state increases from 5.13 to 5.16e, while the Ca6 atom shows some electron-donating behavior and the main electron loss orbital is its 4s state (from 2.20 to 2.12e). The configuration of the H2O molecule changes very little, which suggests that this kind of adsorption is insufficient to initiate the decomposition of the H2O molecule.
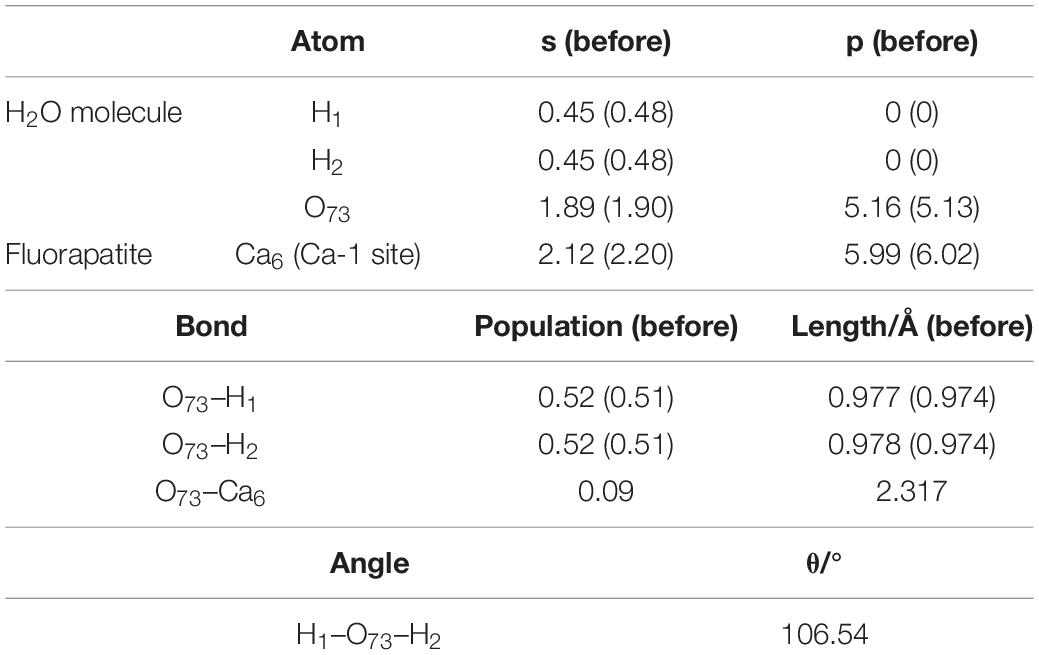
Table 1. Charge transfer and bond properties of single-site adsorption.
For further study, DOS analysis of the single-site H2O adsorption on the fluorapatite (001) surface is presented. As revealed in Figure 5, the DOS of the H2O molecule has a distinct shift to the lower energy area after absorbing on the (001) surface of fluorapatite. For the Ca6, the peak in the conduction band contributed by the 3s state has been greatly weakened. Significant overlap is seen in the range from 2 to 3.5 eV, which is contributed by the O 2p and Ca 3s states. In addition, some bonding interactions also can be observed in the range from −8 to −4eV and 2 to 3eV.
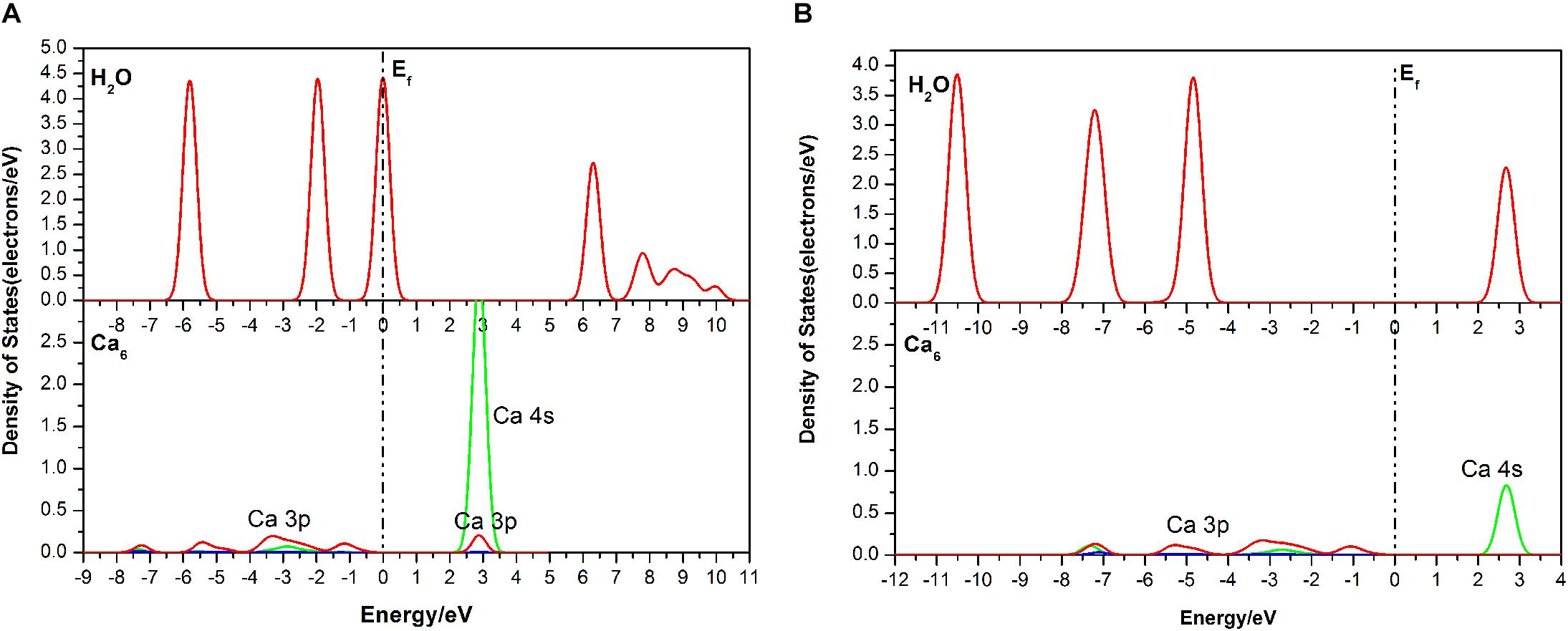
Figure 5. Densities of states (DOS) of the corresponding atoms in the single-site adsorption: (A) before adsorption; (B) after adsorption.
Figure 4B shows the double-site adsorption configuration of the H2O molecule on the fluorapatite (001) surface. It is found that strong bonding interaction exists between O73 and Ca6, with a distance of 2.327 Å (see Table 2). Meanwhile, a hydrogen bond is also observed with a H1 … O54 distance of 1.633 Å, and the corresponding angle of O73–H2–O54 is 160.89°. In addition, the bonding length of O73 and H1, O73, and H2 in the H2O molecule is increased from 0.974 to 1.038 Å and 0.9780 Å, respectively, and the calculated Eads is −98.712 kJ/mol, which indicates that the H2O molecule can be tightly adsorbed on the fluorapatite (001) surface. Further analysis shows that there are electron transfers between O73 and Ca6 atoms and H1 and O54 and that the O73 and H1 gain electrons from 5.13 to 5.19e and 0.48 to 0.55e by their 2p state and 1s state, respectively.
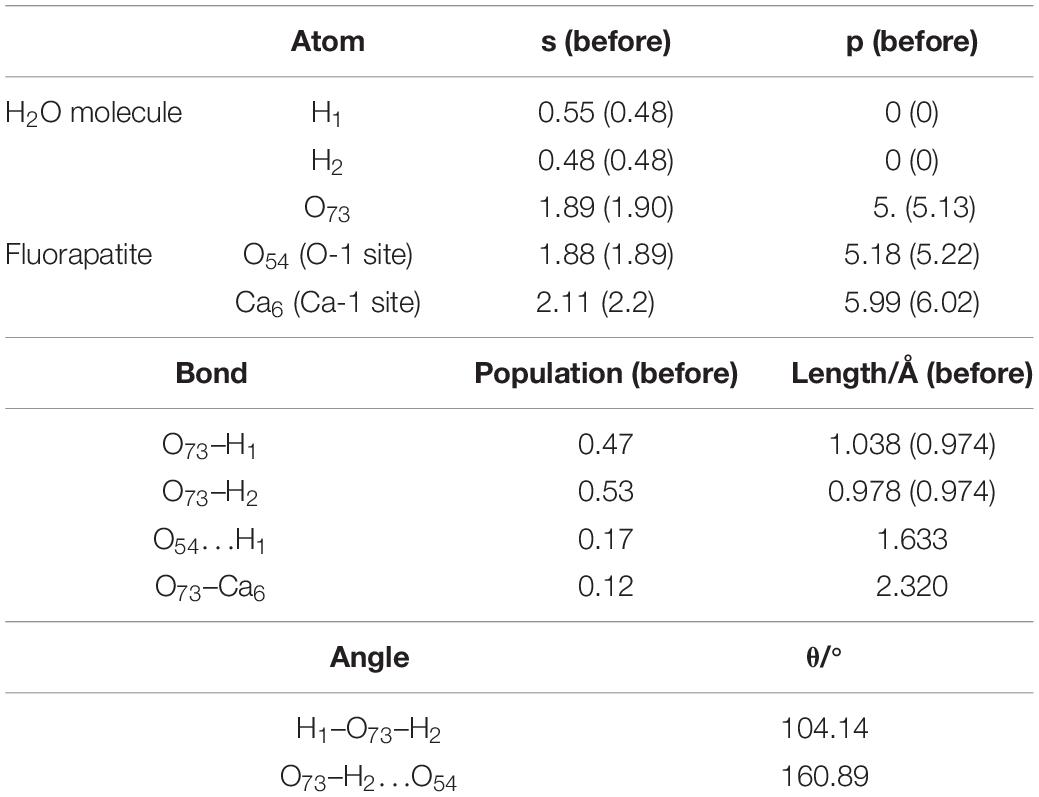
Table 2. Charge transfer and bond properties of the double-site adsorption.
As seen in Figure 6, the DOS of the H2O molecule has the same distinct shift to the left and the structure changes greatly, with the valence bands mixing together from −6.51 to 0.05 eV, indicating strong charge transfer during the adsorption process. The apparent change for the Ca6 is these peaks in the conduction band, which are contributed by 3p and 3d states. For the O54, all of the bands move to the lower energy area. Meanwhile, the parts near the Fermi level contributed by the 2p state broaden, but the peak next to the Fermi level shows a decrease from 2.5 to 2 eV. The potential bonding interactions both for the H2O–Ca6 and H2O–O54 systems are distributed near the top of the valence band.
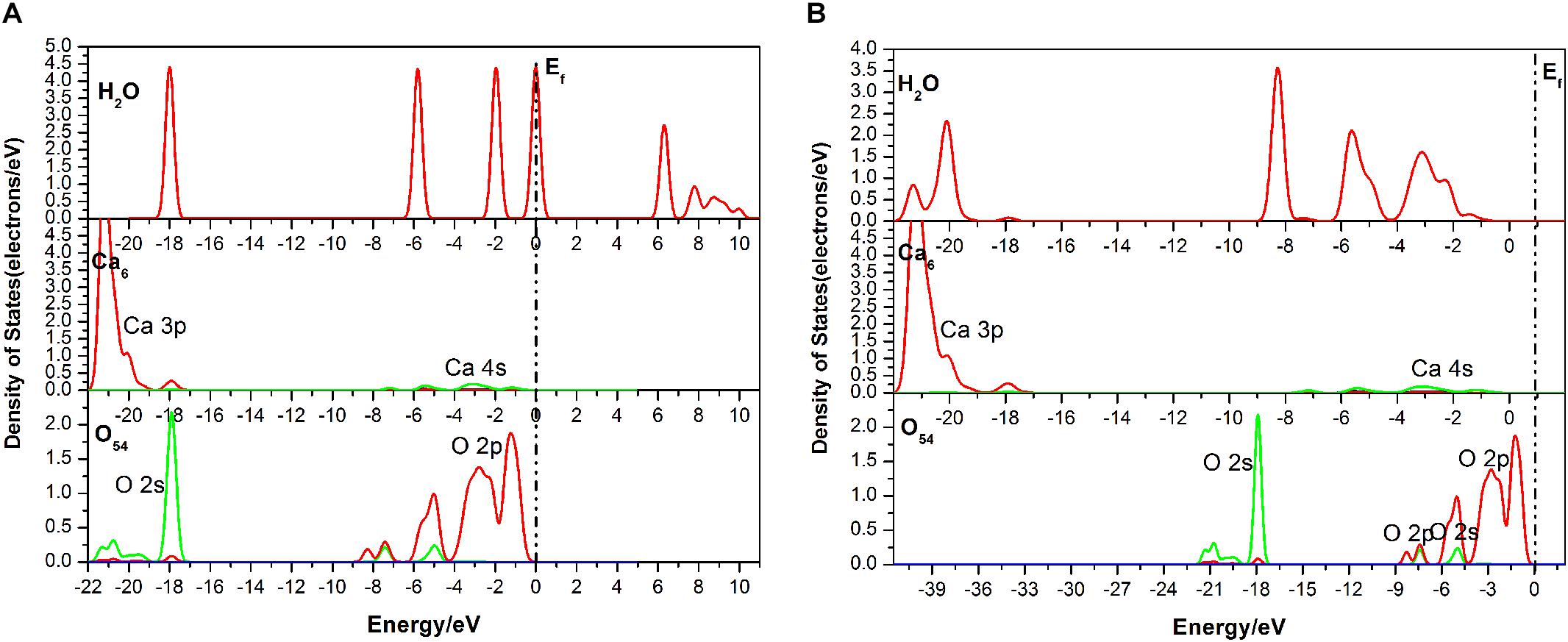
Figure 6. Densities of states (DOS) of the corresponding atoms of the double-site adsorption: (A) before adsorption; (B) after adsorption.
As shown in Figure 4C, the length of O73 and Ca12, Ca27 is 2.473 Å and 2.761 Å, and the obvious hydrogen bond of H2 and O52 is 1.622 Å. Our calculated result shows that the Eads is as large as −139.620 kJ/mol, which indicates that this configuration is very stable. Table 3 shows that the 1s state of the H1 and H2 atoms in the H2O increases from 0.52 to 0.44e and 0.38e, respectively, and that the O73 atom gains electrons mainly by the 2p state from 5.13 to 5.19e. At the fluorapatite (001) surface, the Ca21 in the Ca-1 site gains more charge and its value increase from 1.66 to 1.73e, while the 3s and 3p states lose 0.04 and 0.07e electrons, respectively. The change in the O–H bond length in the H2O seems more apparent than in the other two configurations, but the results are still very minor; too small to cause the H2O molecule to decompose. The angles of O73–H2…O52 are 135.16°.
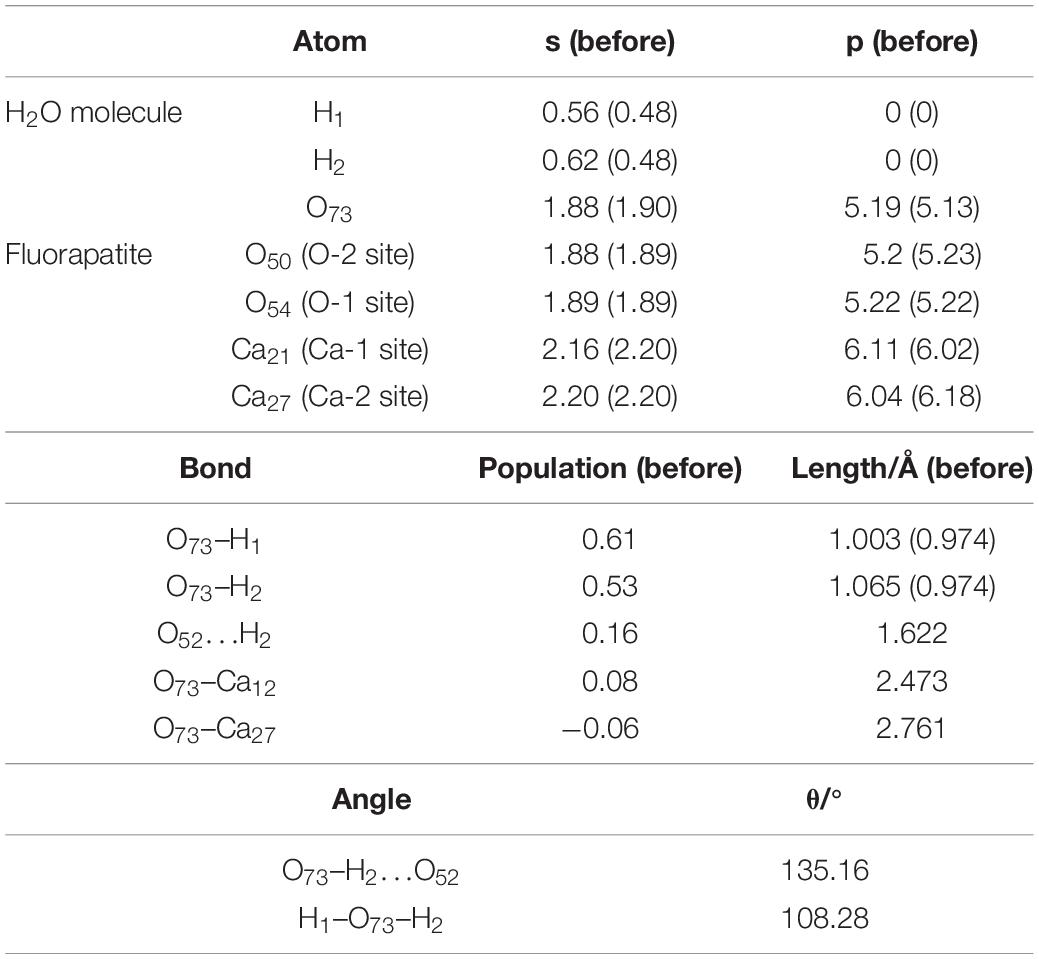
Table 3. Charge transfer and bond properties of the three-site adsorption.
It is seen in Figure 7 that the DOS of the H2O molecule has a distinct shift to the left, and the structure change greatly too, which indicates a strong charge transfer during the adsorption process. The obvious change of the Ca21 is that its peak at 2.7 eV contributed by the 4s state is weakened. For the Ca27, its valence band, which is contributed by the 3p state, is broadened, and the peak at 20.5 eV is weakened from 7.32 to 5386 eV. The top valence band of the O52, which is contributed by its 2p state, becomes wider and moves away from the Fermi level, which may affect its chemical properties. Meanwhile, the peak next to the Fermi level drops to 0.75 eV.
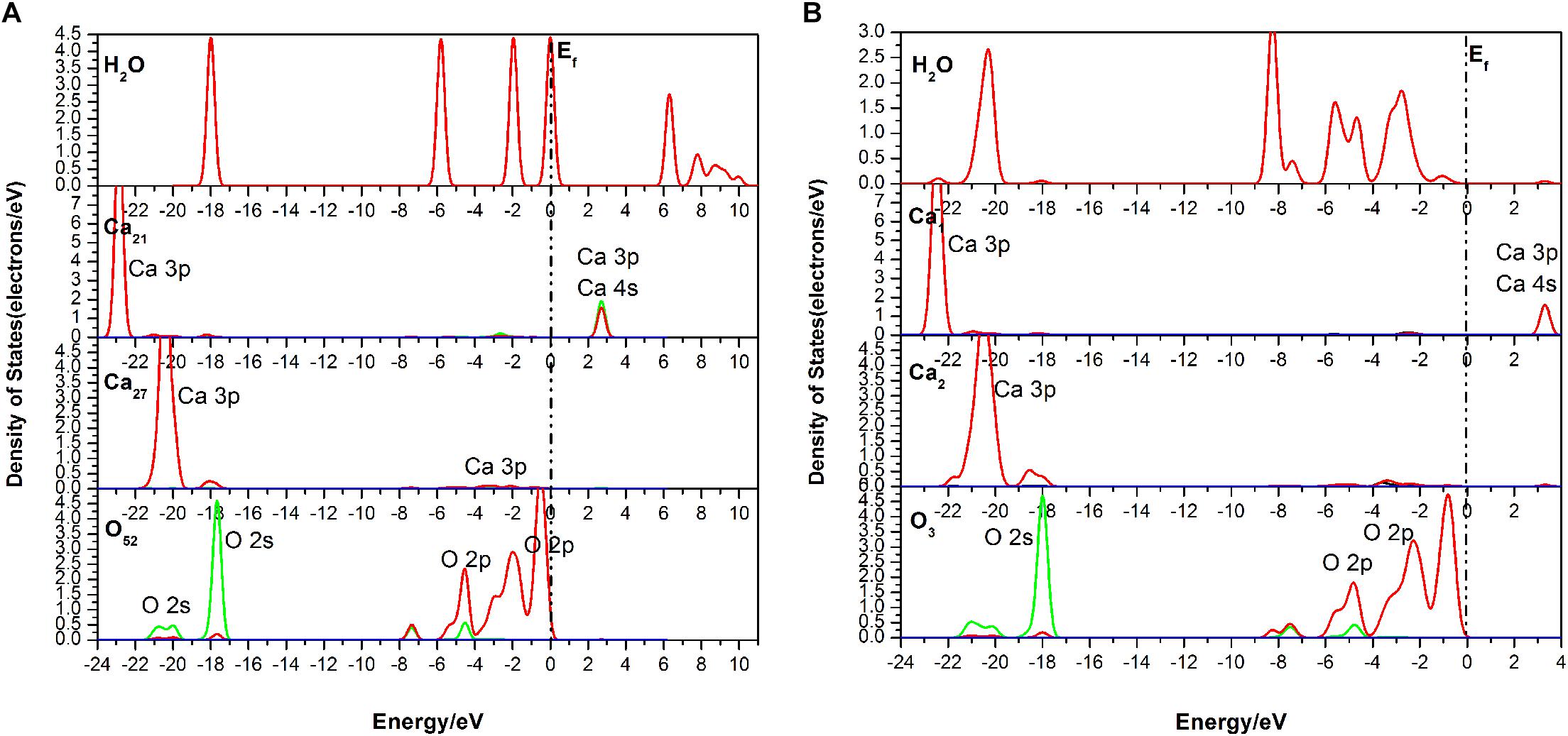
Figure 7. Densities of states (DOS) of the corresponding atoms of the three-site adsorption: (A) before adsorption; (B) after adsorption.
The adsorption configuration of three layers of water molecules on the fluorapatite (001) surface was optimized and is shown in Figure 8. It is an interesting phenomenon that a transitional interfacial layer is formed containing water molecules and Ca atoms, which suggests that strong hydration occurs in this region and that the fluorapatite (001) surface undergoes a drastic reconstitution. The whole area can be divided into four parts: the upmost layer is near-free water, the second is a tight adsorption layer, the third is a chaotic adsorption layer, and finally, the fourth layer is the mineral structure.
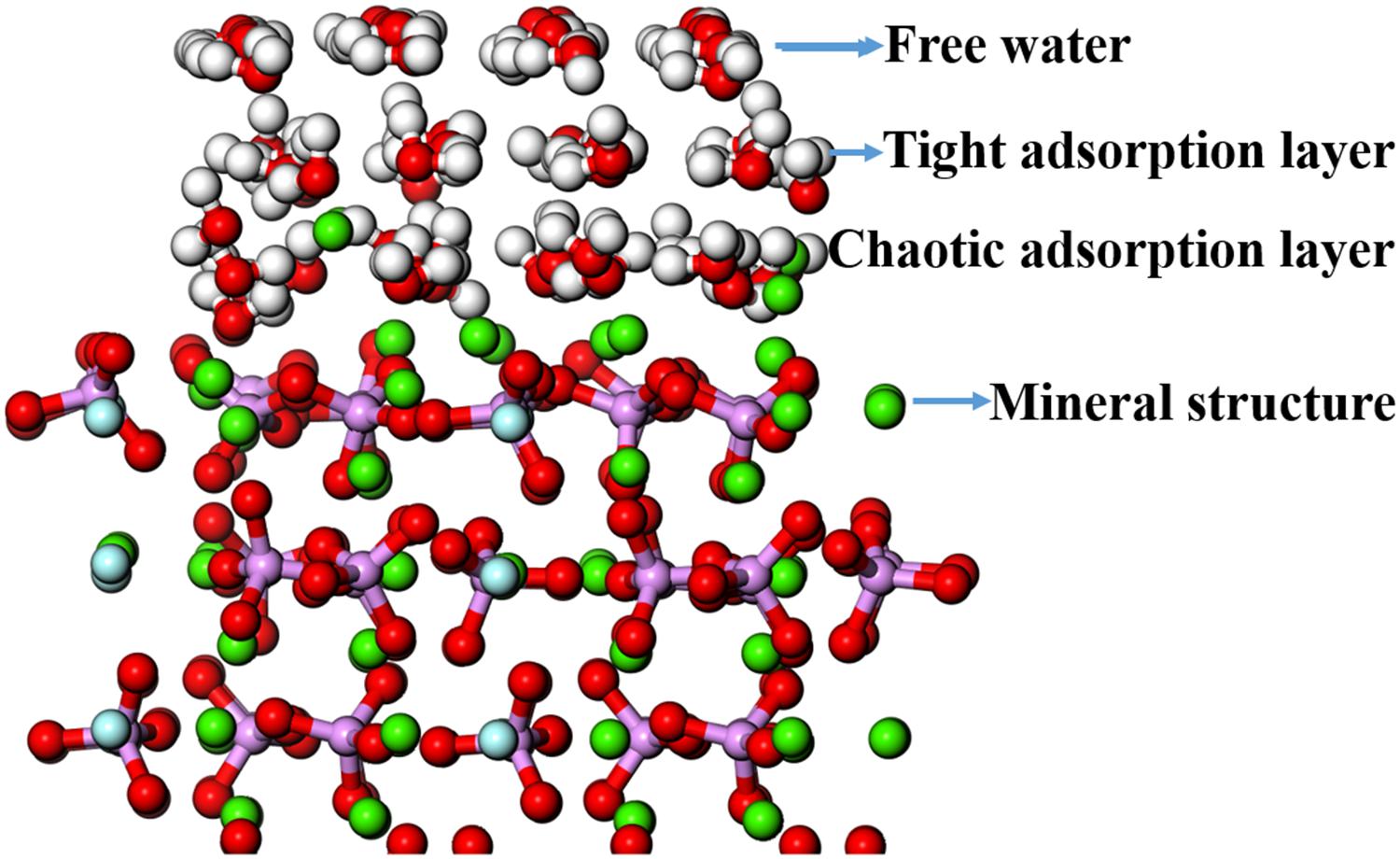
Figure 8. Adsorption configuration of three layers of water molecules on the fluorapatite (001) surface.
Table 4 shows the atomic displacement of the fluorapatite (001) surface. The calculation formula (Cui and Liu, 2010) is as follows:
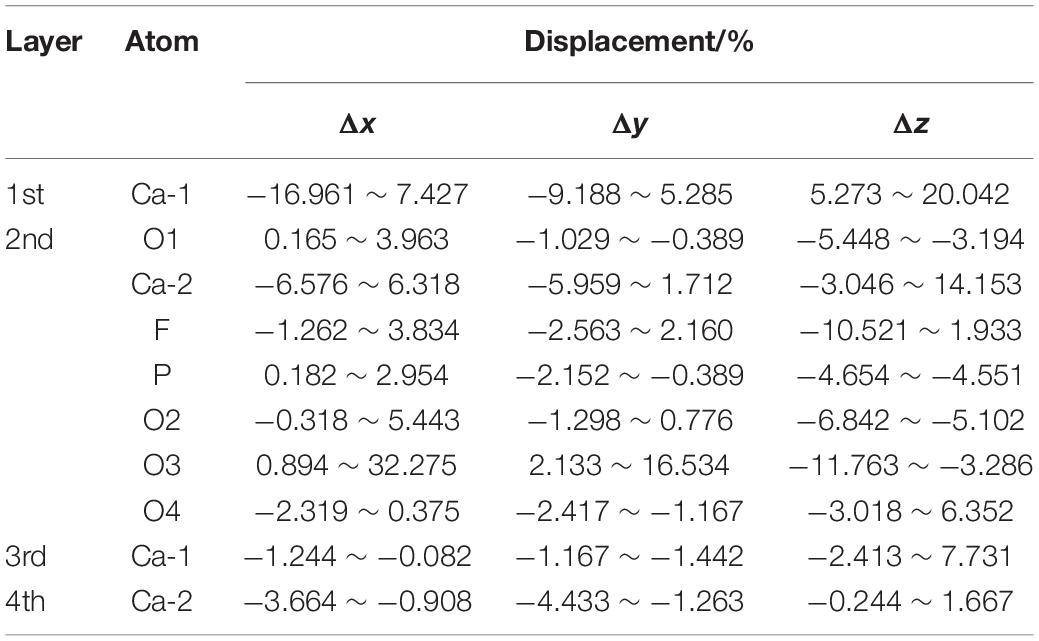
Table 4. Atomic displacements of the fluorapatite (001) surface before and after adsorption.
Here, △di represents the relative atomic coordinate shift of O, P, Ca, and F atoms; dbefore is the corresponding pre-adsorption atomic coordinate; dafter represents the coordinate after adsorption; a refers to the bulk lattice parameter along the x-axis before geometry optimization.
The relative atomic displacements of the fluorapatite (001) surface are presented in Table 4. It is found that the Ca-1 sites in the first layer have the largest distorted displacements and that the corresponding movements along the x-, y-, and z-axis directions are −16.961% ∼ 7.427%, −9.188% ∼ 5.285%, and 5.273% ∼ 20.042%. The Ca-2 sites in the second layer possess the second-largest distorted displacements, and the main movement is −3.046% ∼ 14.153% in the z-axis direction. The PO4 group moves toward the vacuum region strongly in the negative z-axis direction, which is in the opposite direction from the previous two Ca sites. The Ca-1 sites in both the third and fourth layers are quite stable, and their displacements along the z-axis direction are about 3 and 1%, respectively.
As shown in Figure 8, the displacement of the PO4 group seems not to be large; however, the Ca-1 atoms have entered the water environment, and the Ca-2 atoms also show significant upward displacement, which means that the fluorapatite (001) surface is severely dissociated. This result can well explain the source of Ca2+ in slurry. However, we have noted that some experimental reports on the adsorption behavior on the fluorapatite (100) surface (Park et al., 2005; Pareek et al., 2007, 2008) seem a little different from our calculations, though the adsorption behaviors of the two surfaces are not much different. This discrepancy may be attributable to the dissolution of sensitive Ca-1 atoms during sample preparation. As shown in Table 5, if we take away the chaotic adsorption layer, then the result will be very close to the experimental value.
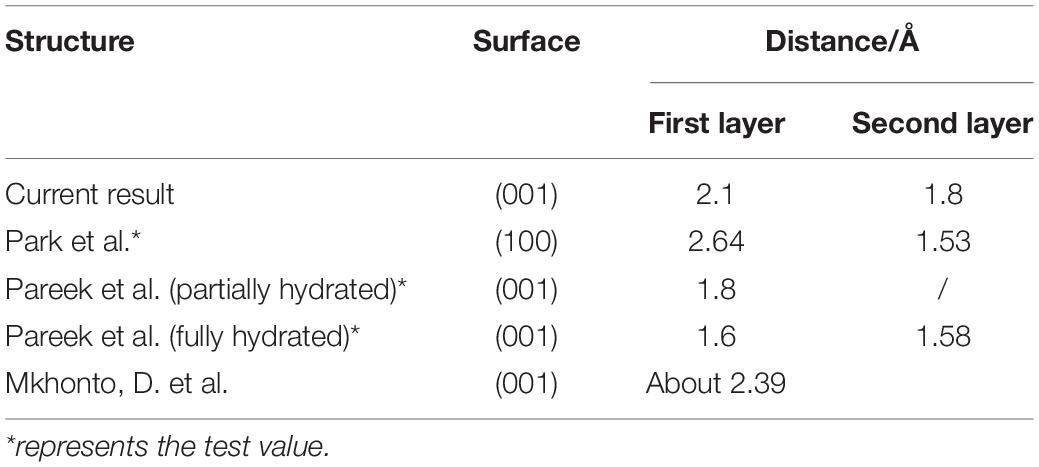
Table 5. Summary of the current calculation and previous experimental results of the layer spacing of adsorbing water.
The bindings of the mineral structure and the chaotic adsorption layer are mainly contributed by the Ca-2 atoms with the Ca–O ionic bond. Each Ca atom is bonded to six O atoms, half of which come from the H2O molecule, and the length of these newly formed bonds is 2.215∼ 2.439 Å. In addition, a few weak hydrogen bond interactions can be seen, which are contributed by the O atoms from PO4 groups and the H atoms from H2O molecules; their length is between 2.164 and 2.472 Å. The interactions in the chaotic adsorption layer are mainly due to the Ca–O ionic bond and the hydrogen bond; the latter comes from the H2O molecule. Each Ca is surrounded by six H2O molecules, which usually contain three molecules from the tight sorption layer. The lengths of the Ca–O bond and hydrogen bond are between 2.237 and 2.486 Å and from 1.903 to 1.956 Å, respectively. The lengths of the hydrogen bonds of the tight adsorption layer and free water layer are not much different and are basically between 1.913 and 1.967 Å; however, the lengths of the interlaminar hydrogen bonds of the latter are slightly greater than those of the former.
To investigate the effect of hydration behavior on the structure of fluorapatite, single H2O molecule and three-layer cluster adsorptions on the fluorapatite (001) surface were performed by means of density functional theory. The conclusions can be summarized as follows:
(1) The H2O molecule can form stable chemisorption structures with the fluorapatite (001) surface in the form of single-site, two-site, and three-site adsorption, each of which is associated with surface Ca atoms, especially those at the Ca-1 sites; the corresponding adsorption energies are 64.817, 98.712, and 139.620 kJ/mol. Ca atom can bind with the O of the H2O molecule, with ionic interaction mainly contributed by the O 2p and Ca 4s states, and the length is about 2.3 Å, which is close to that of the Ca–O bond in the fluorapatite bulk.
(2) The adsorption configuration of a three-layer water cluster on the fluorapatite (001) surface shows serious hydration reconstruction at the interface and a transitional interfacial area containing water molecules where Ca atoms are formed. The lower layer of the transition area is dominated by Ca–O ionic bonding and the upper parts by hydrogen bonding. Further analysis shows that these six-coordinated Ca atoms are critical to the hydration behaviors of the transition area, as they can bind strongly to the H2O molecules, with the newly formed Ca–O bonds being between 2.164 and 2.486 Å.
The datasets generated for this study are available on request to the corresponding authors.
JC: conceptualization and scheme. YC, WC, and XS: data curation. WC, XS, YL, YC, and CZ: formal analysis. JC, YC, and WC: investigation. JC: project administration. WC: writing–original draft.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was funded by the National Natural Science Foundation of People’s Republic of China (NSFC 51864003 and NSFC 51874106), Guangxi Natural Science Foundation (2018GXNSFAA050127 and 2018GXNSFAA281355), and Guangxi Key Laboratory of Processing for Non-ferrous Metals and Featured Materials (GXYSYF1811). The authors are thankful for this support.
Chappell, H., Duer, M., Groom, N., Pickard, C., and Bristowe, P. (2008). Probing the surface structure of hydroxyapatite using NMR spectroscopy and first principles calculations. Phys. Chem. Chem. Phys. 10, 600–606. doi: 10.1039/B714512H
Chen, J., Chen, Y., Long, X., and Li, Y. (2017). DFT study of coadsorption of water and oxygen on galena (PbS) surface: an insight into the oxidation mechanism of galena. Appl. Surf. Sci. 420, 714–719. doi: 10.1016/j.apsusc.2017.05.199
Clark, S. J., Segall, M. D., Pickard, C. J., Hasnip, P. J., Probert, M. I. J., Refson, K., et al. (2005). First principles methods using CASTEP. Z. Krist. 220, 567–570. doi: 10.1524/zkri.220.5.567.65075
Comodi, P., Liu, Y., Zanazzi, P. F., and Montagnoli, M. (2001). Structural and vibrational behaviour of– uorapatite with pressure. Part I: in situ single-crystal X-ray diffraction investigation. Phys. Chem. Miner. 28, 219–224.
Cui, J., and Liu, W. (2010). First-principles study of the (001) surface of cubic BiAlO3. Phys. B Condens. Matter. 405, 4687–4690. doi: 10.1016/j.physb.2010.08.063
Dan, E. S., Jarlbring, M., Antzutkin, O. N., and Forsling, W. (2006). A spectroscopic study of calcium surface sites and adsorbed iron species at aqueous fluorapatite by means of 1H and 31P MAS NMR. Langmuir 22:11060. doi: 10.1021/la0602158
Emsbo, P., McLaughlin, P. I., Breit, G. N., du Bray, E. A., and Koenig, A. E. (2015). Rare earth elements in sedimentary phosphate deposits: solution to the global REE crisis? Gondwana Res. 27, 776–785. doi: 10.1016/j.gr.2014.10.008
Haverty, D., Tofail, S. A. M., Stanton, K. T., and McMonagle, J. B. (2005). Structure and stability of hydroxyapatite: density functional calculation and rietveld analysis. Phys. Rev. B 71:094103. doi: 10.1103/PhysRevB.71.094103
Hughes, J. M., and Rakovan, J. (2002). The crystal structure of apatite, Ca5(PO4)3(F,OH,Cl). Rev. Miner. Geochem. 48, 1–12. doi: 10.2138/rmg.2002.48.1
Li, X., Zeng, H., Teng, L., and Chen, H. (2014). Comparative investigation on the crystal structure and cell behavior of rare-earth doped fluorescent apatite nanocrystals. Mater. Lett. 125, 78–81. doi: 10.1016/j.matlet.2014.03.151
Menéndez-Proupin, E., Cervantes-Rodríguez, S., Osorio-Pulgar, R., Franco-Cisterna, M., Camacho-Montes, H., and Fuentes, M. E. (2011). Computer simulation of elastic constants of hydroxyapatite and fluorapatite. J. Mech. Behav. Biomed. Mater. 4, 1011–1020. doi: 10.1016/j.jmbbm.2011.03.001
Mkhonto, D., and de Leeuw, N. H. (2002). A computer modelling study of the effect of water on the surface structure and morphology of fluorapatite: introducing a Ca10(PO4)6F2 potential model. J. Mater. Chem. 12, 2633–2642. doi: 10.1039/b204111
Okada, M., and Matsumoto, T. (2015). Synthesis and modification of apatite nanoparticles for use in dental and medical applications. Jpn. Dent. Sci. Rev. 51, 85–95. doi: 10.1016/j.jdsr.2015.03.004
Palazzo, B., Iafisco, M., Laforgia, M., Margiotta, N., Natile, G., Bianchi, C. L., et al. (2007). Biomimetic hydroxyapatite-drug nanocrystals as potential bone substitutes with antitumor drug delivery properties. Adv. Funct. Mater. 17, 2180–2188. doi: 10.1002/adfm.200600361
Pareek, A., Torrelles, X., Angermund, K., Rius, J., Magdans, U., and Gies, H. (2008). Structure of interfacial water on fluorapatite (100) surface. Langmuir 24, 2459–2464. doi: 10.1021/la701929p
Pareek, A., Torrelles, X., Angermund, K., Rius, J., Magdans, U., and Gies, H. (2009). Competitive adsorption of glycine and water on the fluorapatite (100) surface. Langmuir 25, 1453–1458. doi: 10.1021/la802706y
Pareek, A., Torrelles, X., Rius, J., Magdans, U., and Gies, H. (2007). Role of water in the surface relaxation of the fluorapatite (100) surface by grazing incidence x-ray diffraction. Phys. Rev. B Condens. Matter Mater. Phys. 75, 1–6. doi: 10.1103/PhysRevB.75.035418
Park, C., Fenter, P., Zhang, Z., Cheng, L., and Sturchio, N. C. (2005). Structure of the fluorapatite (100)-water interface by high-resolution X-ray reflectivity. Am. Miner. 89, 1647–1654. doi: 10.2138/am-2004-11-1209
Qiu, Y.-Q., Cui, W.-Y., Li, L.-J., Ye, J.-J., Wang, J., and Zhang, Q. (2017). Structural, electronic properties with different terminations for fluorapatite (001) surface: a first-principles investigation. Comput. Mater. Sci. 126, 132–138. doi: 10.1016/j.commatsci.2016.09.027
Rawashdeh, R., and Maxwell, P. (2011). The evolution and prospects of the phosphate industry. Miner. Econ. 24, 15–27. doi: 10.1007/s13563-011-0003-8
Rey, C., Combes, C., Drouet, C., Cazalbou, S., and Sarda, S. (2014). Surface properties of biomimetic nanocrystalline apatites; applications in biomaterials. Prog. Cryst. Growth Charact. Mater. 60, 63–73. doi: 10.1016/j.pcrysgrow.2014.09.005
Rulis, P., Ouyang, L., and Ching, W. Y. (2004). Electronic structure and bonding in calcium apatite crystals: hydroxyapatite, fluorapatite, chlorapatite, and bromapatite. Phys. Rev. B Condens. Matter Mater. Phys. 70, 1–8. doi: 10.1103/PhysRevB.70.155104
Rulis, P., Ouyang, L., and Ching, W. Y. (2007). Electronic structure, bonding, charge distribution, and x-ray absorption spectra of the (001) surfaces of fluorapatite and hydroxyapatite from first principles. Phys. Rev. B. Condens. Matter 24, 1–8. doi: 10.1103/PhysRevB.76.245410
Santos, M. A., Santana, R. C., Capponi, F., Ataíde, C. H., and Barrozo, M. A. S. (2010). Effect of ionic species on the performance of apatite flotation. Sep. Purif. Technol. 76, 15–20. doi: 10.1016/j.seppur.2010.09.014
Scholz, R. W., Roy, A. H., Brand, F. S., and Hellums, D. T. (eds) (2014). Sustainable Phosphorus Management. Berlin: Springer, doi: 10.1007/978-94-007-7250-2
Zhang, P. (2014). Comprehensive recovery and sustainable development of phosphate resources. Proc. Eng. 83, 37–51. doi: 10.1016/j.proeng.2014.09.010
Zhao, C., Chen, J., Li, Y., Huang, D. W., and Li, W. (2015). DFT study of interactions between calcium hydroxyl ions and pyrite, marcasite, pyrrhotite surfaces. Appl. Surf. Sci. 355, 577–581. doi: 10.1016/j.apsusc.2015.07.081
Keywords: fluorapatite, hydration behavior, density-functional theory, water cluster, transitional interfacial layer
Citation: Cui W, Song X, Chen J, Chen Y, Li Y and Zhao C (2020) Adsorption Behaviors of Different Water Structures on the Fluorapatite (001) Surface: A DFT Study. Front. Mater. 7:47. doi: 10.3389/fmats.2020.00047
Received: 18 December 2019; Accepted: 12 February 2020;
Published: 12 March 2020.
Edited by:
Zhiyong Gao, Central South University, ChinaReviewed by:
Xuming Wang, The University of Utah, United StatesCopyright © 2020 Cui, Song, Chen, Chen, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianhua Chen, amhjaGVuQGd4dS5lZHUuY24=; Ye Chen, ZmJ5MThAMTI2LmNvbQ==; eWVjaGVuQGd4dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.